Sequential Interferon β-Cisplatin Treatment Enhances the Surface Exposure of Calreticulin in Cancer Cells via an Interferon Regulatory Factor 1-Dependent Manner

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Treatments
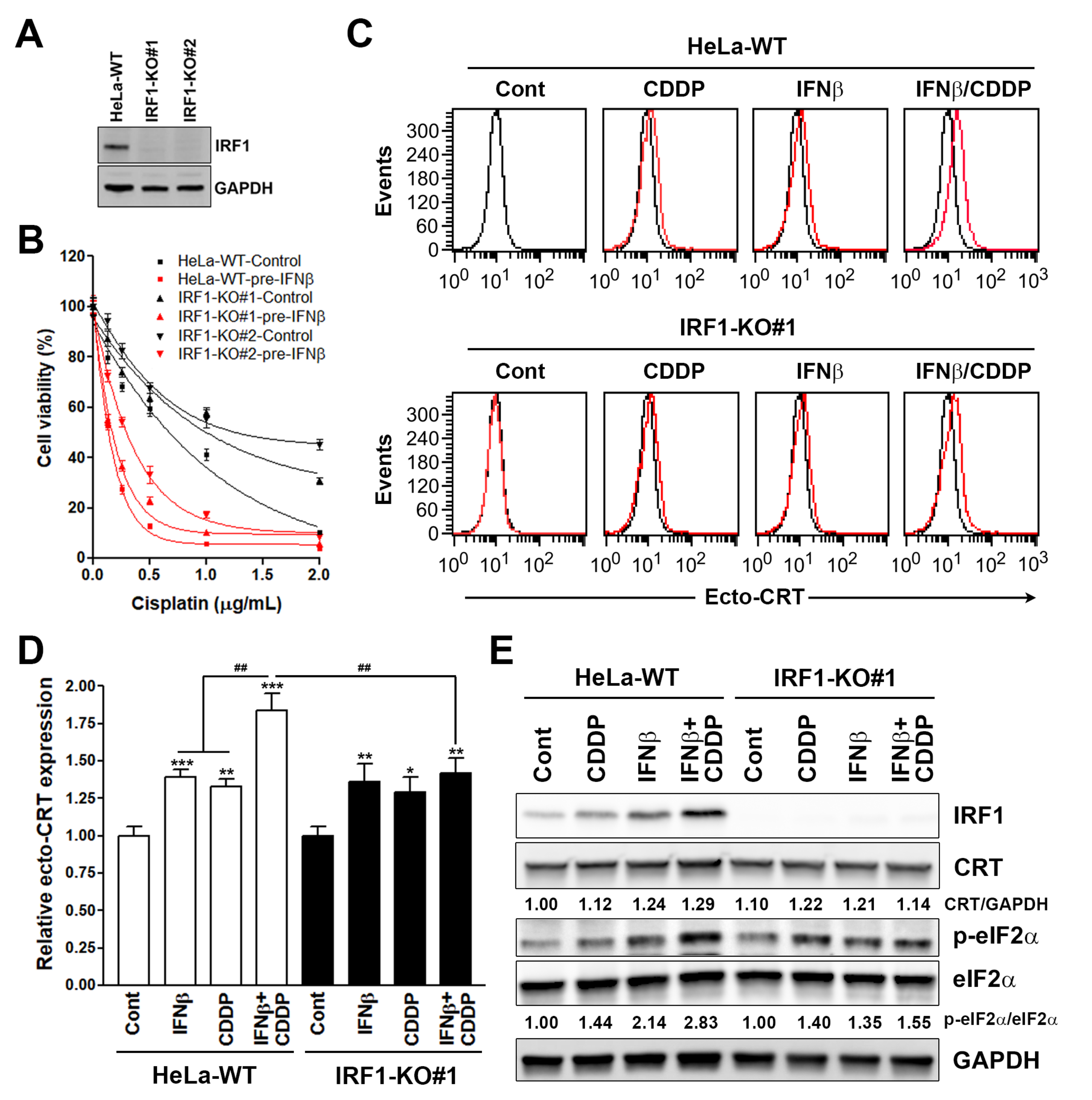
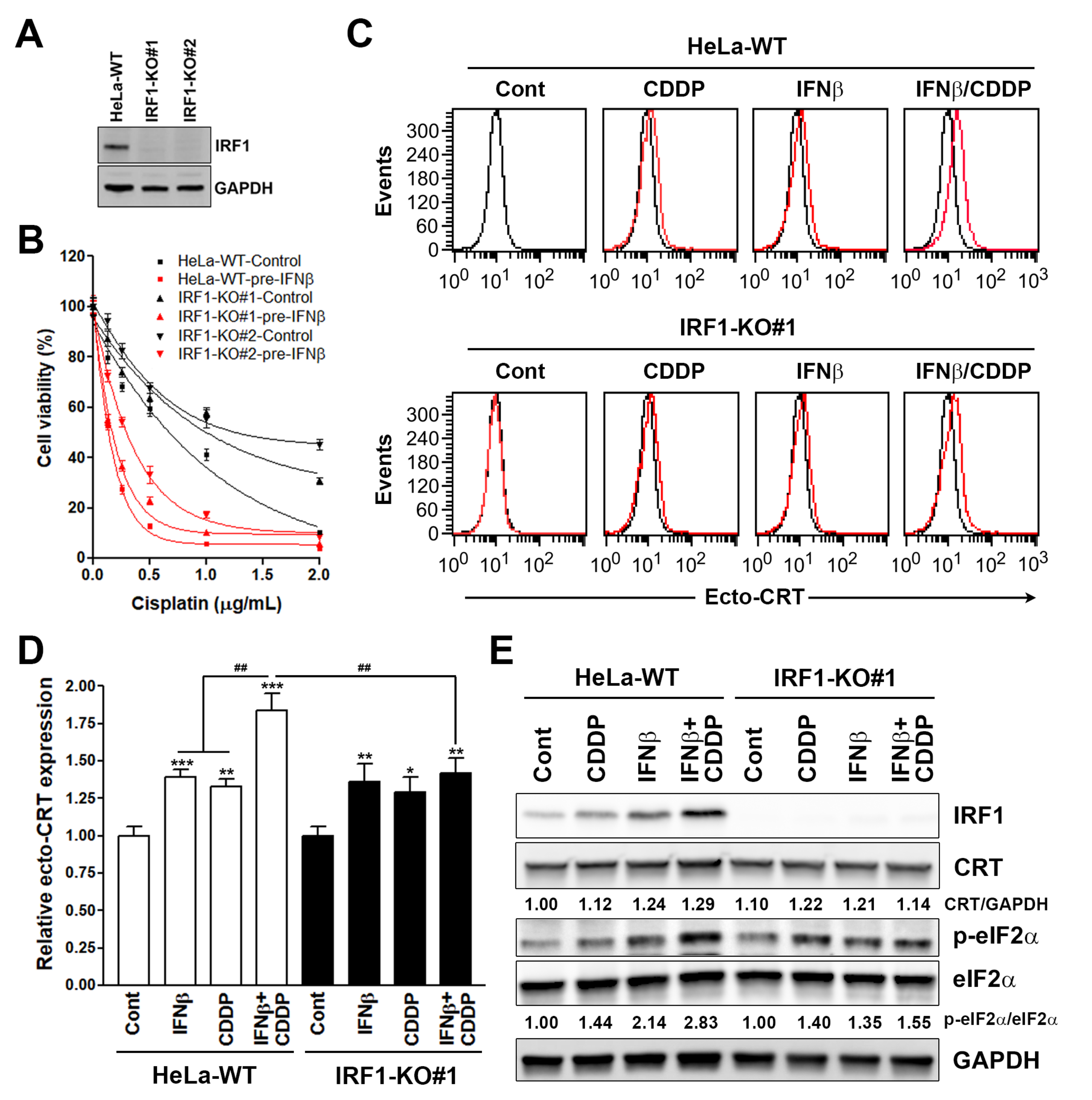
2.4. Establishment of Interferon Regulatory Factor 1 (IRF1)-Knockout Cells
2.5. siRNA Knockdown Analysis
2.6. Flow Cytometry
2.7. Cell Viability Assay
2.8. Western Blot Analysis
2.9. Bioinformatics Analysis of Public Data
2.10. Statistical Analysis
3. Results
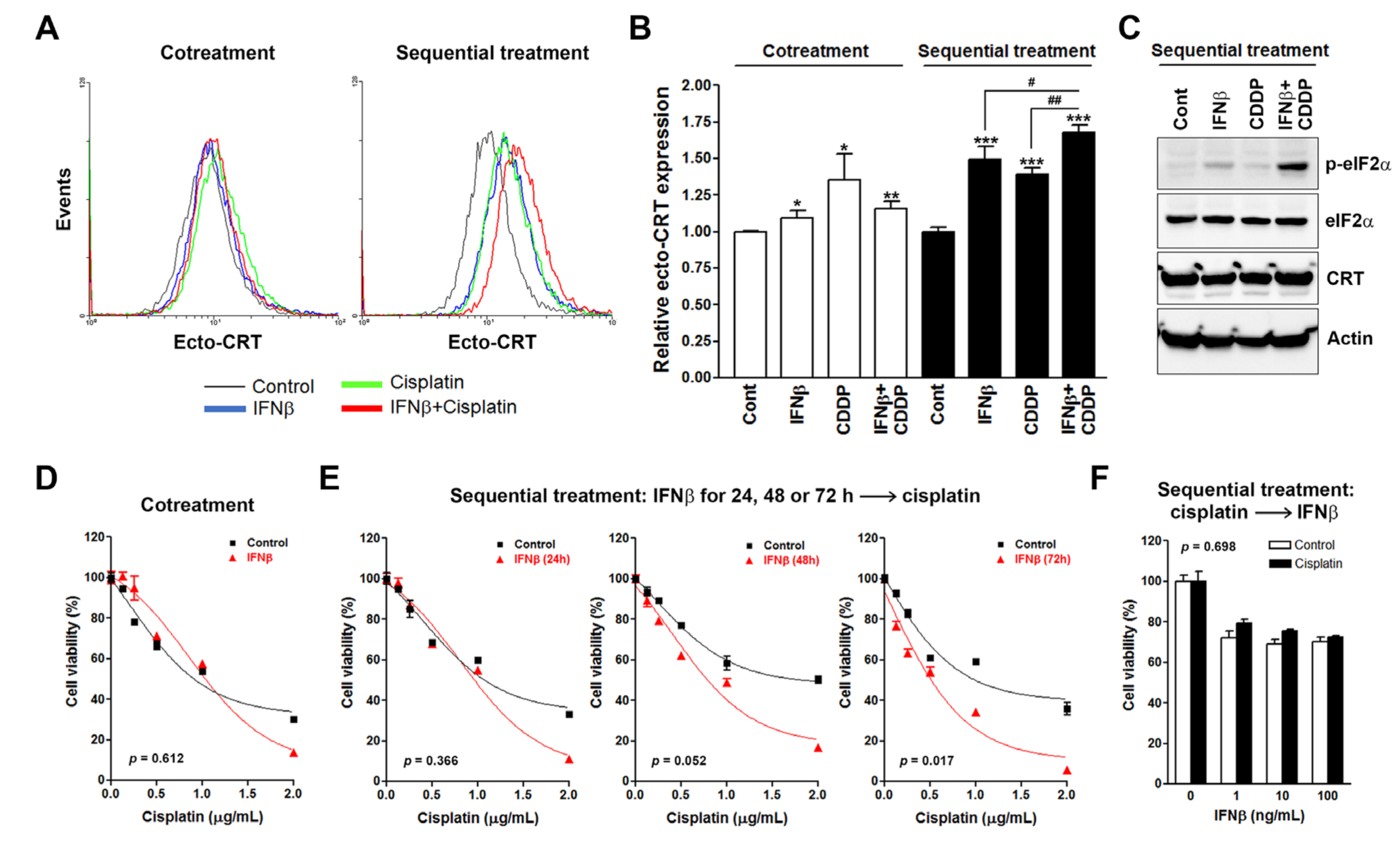
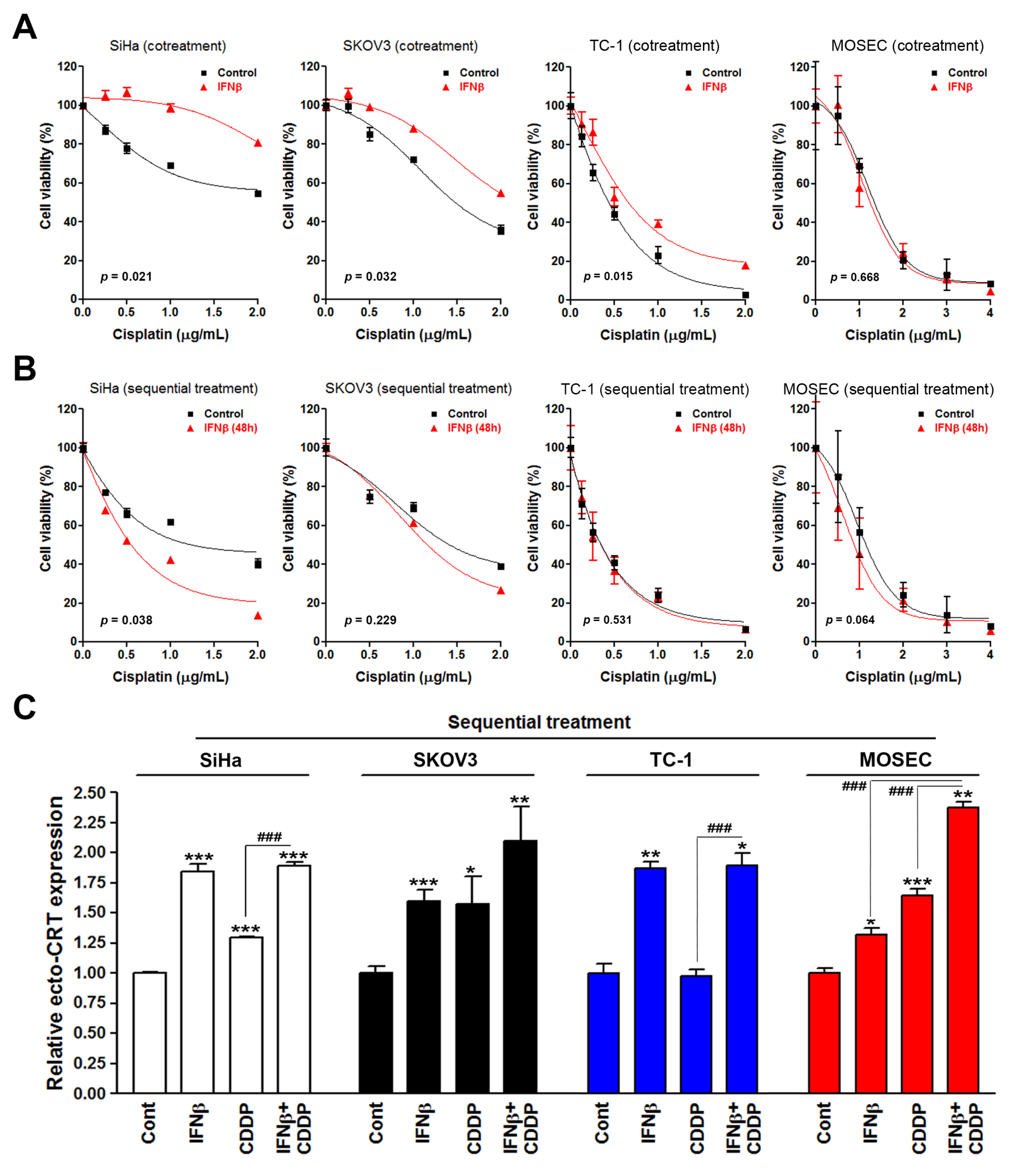
3.1. Sequential Interferon β (IFNβ) and Cisplatin Treatment Enhances the Surface Calreticulin (CRT) Exposure in Cancer Cells
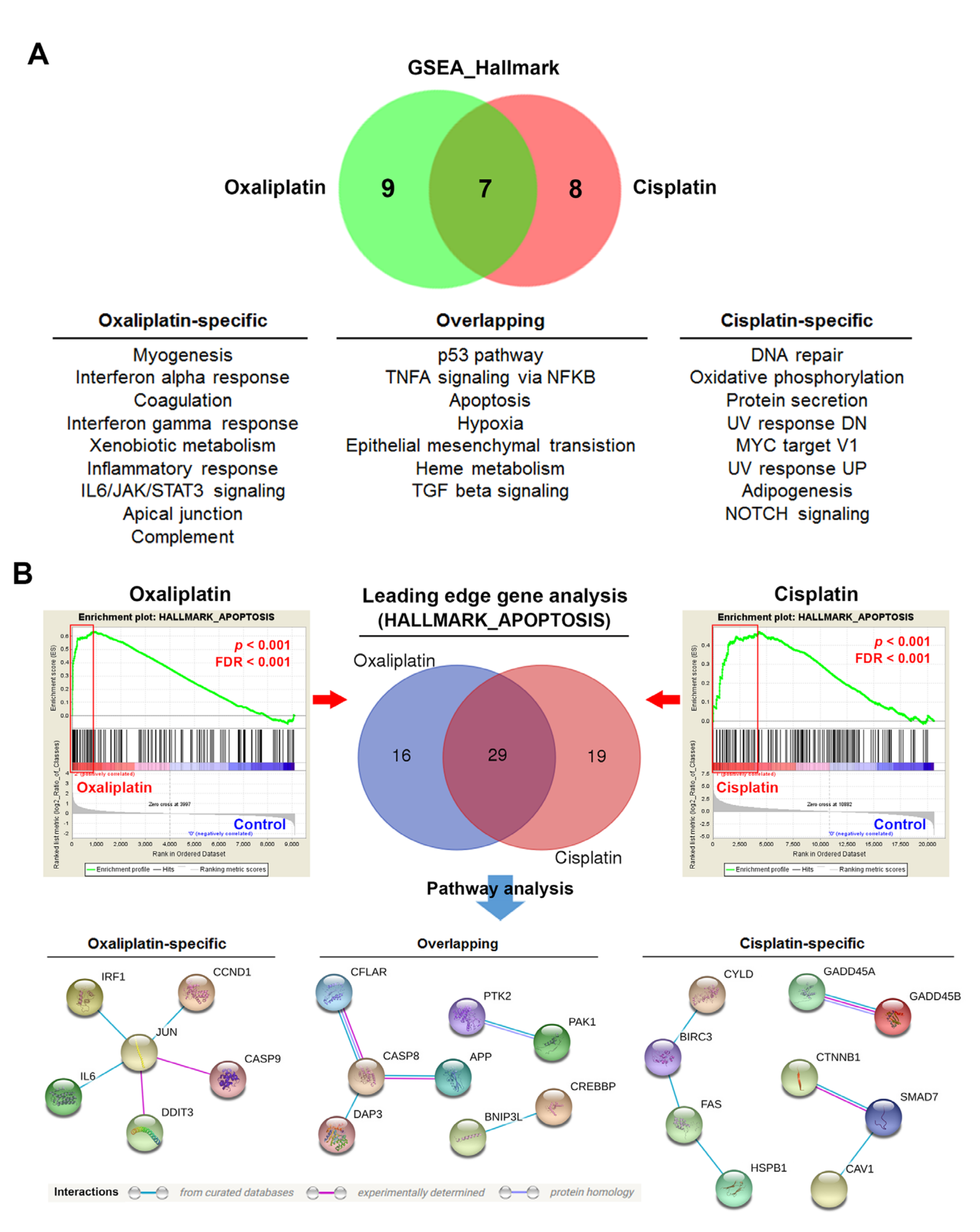
3.2. Gene Set Enrichment Analysis (GSEA) Identifies Potential Molecular Determinants for Oxaliplatin-Induced Immunogenic Cell Death (ICD)
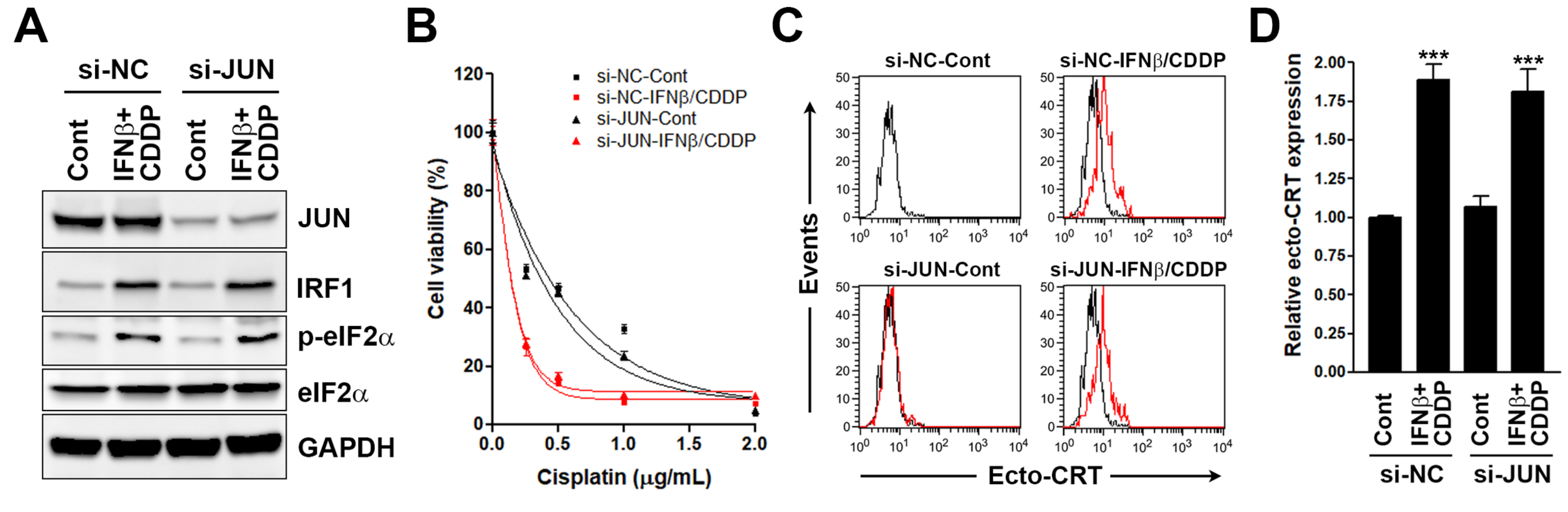
3.3. Interferon Regulatory Factor 1 (IRF1) Is Required for the Surface Calreticulin (CRT) Exposure Induced by Sequential Interferon β (IFNβ) and Cisplatin Treatment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Yatim, N.; Cullen, S.; Albert, M.L. Dying cells actively regulate adaptive immune responses. Nat. Rev. Immunol. 2017, 17, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol Immunother 2012, 61, 215–221. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.; et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef] [Green Version]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Obeid, M.; Panaretakis, T.; Joza, N.; Tufi, R.; Tesniere, A.; van Endert, P.; Zitvogel, L.; Kroemer, G. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ. 2007, 14, 1848–1850. [Google Scholar] [CrossRef]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Aaes, T.L.; Kaczmarek, A.; Delvaeye, T.; De Craene, B.; De Koker, S.; Heyndrickx, L.; Delrue, I.; Taminau, J.; Wiernicki, B.; De Groote, P.; et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell Rep. 2016, 15, 274–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, H.; Tani, K. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ. 2014, 21, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Panaretakis, T.; Joza, N.; Modjtahedi, N.; Tesniere, A.; Vitale, I.; Durchschlag, M.; Fimia, G.M.; Kepp, O.; Piacentini, M.; Froehlich, K.U.; et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ. 2008, 15, 1499–1509. [Google Scholar] [CrossRef] [Green Version]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, p.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Ma, Y.; Baracco, E.E.; Sistigu, A.; Enot, D.P.; Pietrocola, F.; Yang, H.; Adjemian, S.; Chaba, K.; Semeraro, K.; et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 2015, 350, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.; Kepp, O.; Schlemmer, F.; Adjemian, S.; Tailler, M.; Shen, S.; Michaud, M.; Menger, L.; Gdoura, A.; Tajeddine, N.; et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene 2011, 30, 1147–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010, 29, 482–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebe, C.; Demontoux, L.; Pilot, T.; Ghiringhelli, F. Platinum Derivatives Effects on Anticancer Immune Response. Biomolecules 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remedios, C.; et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef]
- Cirone, M.; Garufi, A.; Di Renzo, L.; Granato, M.; Faggioni, A.; D’Orazi, G. Zinc supplementation is required for the cytotoxic and immunogenic effects of chemotherapy in chemoresistant p53-functionally deficient cells. Oncoimmunology 2013, 2, e26198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aranda, F.; Bloy, N.; Pesquet, J.; Petit, B.; Chaba, K.; Sauvat, A.; Kepp, O.; Khadra, N.; Enot, D.; Pfirschke, C.; et al. Immune-dependent antineoplastic effects of cisplatin plus pyridoxine in non-small-cell lung cancer. Oncogene 2015, 34, 3053–3062. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.Y.; Guarnieri, F.G.; Staveley-O’Carroll, K.F.; Levitsky, H.I.; August, J.T.; Pardoll, D.M.; Wu, T.C. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996, 56, 21–26. [Google Scholar] [PubMed]
- Roby, K.F.; Taylor, C.C.; Sweetwood, J.P.; Cheng, Y.; Pace, J.L.; Tawfik, O.; Persons, D.L.; Smith, P.G.; Terranova, P.F. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis 2000, 21, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Wang, Y.; Zhu, J.; Jin, L.; Liu, B.; Xia, K.; Wang, J.; Gao, J.; Liang, C.; Tao, H. Autophagy inhibitor enhance ZnPc/BSA nanoparticle induced photodynamic therapy by suppressing PD-L1 expression in osteosarcoma immunotherapy. Biomaterials 2019, 192, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Brun, Y.F.; Varma, R.; Hector, S.M.; Pendyala, L.; Tummala, R.; Greco, W.R. Simultaneous modeling of concentration-effect and time-course patterns in gene expression data from microarrays. Cancer Genomics Proteomics 2008, 5, 43–53. [Google Scholar]
- Jovanovic, K.K.; Tanic, M.; Ivanovic, I.; Gligorijevic, N.; Dojcinovic, B.P.; Radulovic, S. Cell cycle, apoptosis, cellular uptake and whole-transcriptome microarray gene expression analysis of HeLa cells treated with a ruthenium(II)-arene complex with an isoquinoline-3-carboxylic acid ligand. J. Inorg Biochem 2016, 163, 362–373. [Google Scholar] [CrossRef]
- Hussner, J.; Ameling, S.; Hammer, E.; Herzog, S.; Steil, L.; Schwebe, M.; Niessen, J.; Schroeder, H.W.; Kroemer, H.K.; Ritter, C.A.; et al. Regulation of interferon-inducible proteins by doxorubicin via interferon gamma-Janus tyrosine kinase-signal transducer and activator of transcription signaling in tumor cells. Mol. Pharmacol 2012, 81, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; van Endert, P.; et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009, 28, 578–590. [Google Scholar] [CrossRef]
- Kepp, O.; Galluzzi, L.; Martins, I.; Schlemmer, F.; Adjemian, S.; Michaud, M.; Sukkurwala, A.Q.; Menger, L.; Zitvogel, L.; Kroemer, G. Molecular determinants of immunogenic cell death elicited by anticancer chemotherapy. Cancer Metastasis Rev. 2011, 30, 61–69. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Honda, K.; Yanai, H.; Takaoka, A.; Taniguchi, T. Regulation of the type I IFN induction: A current view. Int. Immunol. 2005, 17, 1367–1378. [Google Scholar] [CrossRef] [Green Version]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [Green Version]
- Borden, E.C. Interferons alpha and beta in cancer: Therapeutic opportunities from new insights. Nat. Rev. Drug Discov. 2019, 18, 219–234. [Google Scholar] [CrossRef]
- Jakimovski, D.; Kolb, C.; Ramanathan, M.; Zivadinov, R.; Weinstock-Guttman, B. Interferon beta for Multiple Sclerosis. Cold Spring Harb Perspect Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, S.; Hata, H.; Homma, E.; Shimizu, H. Sequential local injection of low-dose interferon-beta for maintenance therapy in stage II and III melanoma: A single-institution matched case-control study. Oncology 2012, 82, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, T.; Kayama, T.; Nishikawa, R.; Takahashi, H.; Hashimoto, N.; Takahashi, J.; Aoki, T.; Sugiyama, K.; Ogura, M.; Natsume, A.; et al. A multicenter phase I trial of combination therapy with interferon-beta and temozolomide for high-grade gliomas (INTEGRA study): The final report. J. Neurooncol. 2011, 104, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Colman, H.; Berkey, B.A.; Maor, M.H.; Groves, M.D.; Schultz, C.J.; Vermeulen, S.; Nelson, D.F.; Mehta, M.P.; Yung, W.K.; Radiation Therapy Oncology Group. Phase II Radiation Therapy Oncology Group trial of conventional radiation therapy followed by treatment with recombinant interferon-beta for supratentorial glioblastoma: Results of RTOG 9710. Int. J. Radiat Oncol. Biol. Phys. 2006, 66, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Borden, E.C.; Jacobs, B.; Hollovary, E.; Rybicki, L.; Elson, P.; Olencki, T.; Triozzi, P. Gene regulatory and clinical effects of interferon beta in patients with metastatic melanoma: A phase II trial. J. Interferon Cytokine Res. 2011, 31, 433–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voelter-Mahlknecht, S.; Mahlknecht, U.; Letzel, S.; Fierlbeck, G. Phase 2 trial of the continuous IV administration of interferon-beta in patients with disseminated malignant melanoma. Skinmed 2006, 5, 271–276. [Google Scholar] [CrossRef]
- Boultwood, J.; Fidler, C.; Lewis, S.; MacCarthy, A.; Sheridan, H.; Kelly, S.; Oscier, D.; Buckle, V.J.; Wainscoat, J.S. Allelic loss of IRF1 in myelodysplasia and acute myeloid leukemia: Retention of IRF1 on the 5q- chromosome in some patients with the 5q- syndrome. Blood 1993, 82, 2611–2616. [Google Scholar] [CrossRef] [Green Version]
- Nozawa, H.; Oda, E.; Nakao, K.; Ishihara, M.; Ueda, S.; Yokochi, T.; Ogasawara, K.; Nakatsuru, Y.; Shimizu, S.; Ohira, Y.; et al. Loss of transcription factor IRF-1 affects tumor susceptibility in mice carrying the Ha-ras transgene or nullizygosity for p53. Genes Dev. 1999, 13, 1240–1245. [Google Scholar] [CrossRef] [Green Version]
- Tan, R.S.; Taniguchi, T.; Harada, H. Identification of the lysyl oxidase gene as target of the antioncogenic transcription factor, IRF-1, and its possible role in tumor suppression. Cancer Res. 1996, 56, 2417–2421. [Google Scholar]
- Tanaka, N.; Ishihara, M.; Kitagawa, M.; Harada, H.; Kimura, T.; Matsuyama, T.; Lamphier, M.S.; Aizawa, S.; Mak, T.W.; Taniguchi, T. Cellular commitment to oncogene-induced transformation or apoptosis is dependent on the transcription factor IRF-1. Cell 1994, 77, 829–839. [Google Scholar] [CrossRef]
- Tanaka, N.; Ishihara, M.; Lamphier, M.S.; Nozawa, H.; Matsuyama, T.; Mak, T.W.; Aizawa, S.; Tokino, T.; Oren, M.; Taniguchi, T. Cooperation of the tumour suppressors IRF-1 and p53 in response to DNA damage. Nature 1996, 382, 816–818. [Google Scholar] [CrossRef] [PubMed]
- Walch-Ruckheim, B.; Pahne-Zeppenfeld, J.; Fischbach, J.; Wickenhauser, C.; Horn, L.C.; Tharun, L.; Buttner, R.; Mallmann, P.; Stern, P.; Kim, Y.J.; et al. STAT3/IRF1 Pathway Activation Sensitizes Cervical Cancer Cells to Chemotherapeutic Drugs. Cancer Res. 2016, 76, 3872–3883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavan, S.; Olivero, M.; Cora, D.; Di Renzo, M.F. IRF-1 expression is induced by cisplatin in ovarian cancer cells and limits drug effectiveness. Eur. J. Cancer 2013, 49, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Mosig, R.; Moshier, E.; Pereira, E.; Rahaman, J.; Prasad-Hayes, M.; Halpert, R.; Billaud, J.N.; Dottino, P.; Martignetti, J.A. Interferon regulatory factor 1 is an independent predictor of platinum resistance and survival in high-grade serous ovarian carcinoma. Gynecol. Oncol. 2014, 134, 591–598. [Google Scholar] [CrossRef]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2alpha kinases: Their structures and functions. Cell Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef]
- Giglio, P.; Gagliardi, M.; Tumino, N.; Antunes, F.; Smaili, S.; Cotella, D.; Santoro, C.; Bernardini, R.; Mattei, M.; Piacentini, M.; et al. PKR and GCN2 stress kinases promote an ER stress-independent eIF2alpha phosphorylation responsible for calreticulin exposure in melanoma cells. Oncoimmunology 2018, 7, e1466765. [Google Scholar] [CrossRef] [Green Version]
- Kirchhoff, S.; Koromilas, A.E.; Schaper, F.; Grashoff, M.; Sonenberg, N.; Hauser, H. IRF-1 induced cell growth inhibition and interferon induction requires the activity of the protein kinase PKR. Oncogene 1995, 11, 439–445. [Google Scholar]
- Obeid, M. ERP57 membrane translocation dictates the immunogenicity of tumor cell death by controlling the membrane translocation of calreticulin. J. Immunol. 2008, 181, 2533–2543. [Google Scholar] [CrossRef] [Green Version]
- Coe, H.; Jung, J.; Groenendyk, J.; Prins, D.; Michalak, M. ERp57 modulates STAT3 signaling from the lumen of the endoplasmic reticulum. J. Biol. Chem. 2010, 285, 6725–6738. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yamazaki, T.; Pietrocola, F.; Zhou, H.; Zitvogel, L.; Ma, Y.; Kroemer, G. Improvement of immunogenic chemotherapy by STAT3 inhibition. Oncoimmunology 2016, 5, e1078061. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Ren, H.; Wang, X.; Liu, P.; Yan, F.; Jiang, W.; Li, Y.; Li, J.; Gribben, J.G.; Jia, L.; et al. Rituximab-induced HMGB1 release is associated with inhibition of STAT3 activity in human diffuse large B-cell lymphoma. Oncotarget 2015, 6, 27816–27831. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yamazaki, T.; Pietrocola, F.; Zhou, H.; Zitvogel, L.; Ma, Y.; Kroemer, G. STAT3 Inhibition Enhances the Therapeutic Efficacy of Immunogenic Chemotherapy by Stimulating Type 1 Interferon Production by Cancer Cells. Cancer Res. 2015, 75, 3812–3822. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Top 5 Hallmarks | Number of Genes in Pathway | Number of Pathway Genes Differentially Expressed (% of Total) | NES 1 | p Value | FDR 2 |
|---|---|---|---|---|---|
| Cisplatin | |||||
| Angiogenesis | 36 | 9 (25%) | 1.13 | 0.109 | 1.000 |
| Allograft rejection | 199 | 71 (36%) | 1.04 | 0.210 | 1.000 |
| KRAS signal DN | 187 | 88 (47%) | 1.03 | 0.247 | 1.000 |
| Inflammatory response | 197 | 83 (42%) | 1.03 | 0.257 | 1.000 |
| Spermatogenesis | 131 | 56 (43%) | 1.01 | 0.402 | 1.000 |
| Doxorubicin | |||||
| Interferon alpha response | 91 | 48 (53%) | 2.72 | <0.001 | <0.001 |
| Interferon beta response | 193 | 101 (52%) | 2.69 | <0.001 | <0.001 |
| TFNA signaling via NFKB | 198 | 92 (46%) | 2.27 | <0.001 | <0.001 |
| Inflammatory response | 198 | 78 (39%) | 2.06 | <0.001 | <0.001 |
| Complement | 197 | 68 (35%) | 1.93 | <0.001 | <0.001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, P.-M.; Hsieh, Y.-Y.; Du, J.-L.; Yen, S.-C.; Hung, C.-F. Sequential Interferon β-Cisplatin Treatment Enhances the Surface Exposure of Calreticulin in Cancer Cells via an Interferon Regulatory Factor 1-Dependent Manner. Biomolecules 2020, 10, 643. https://doi.org/10.3390/biom10040643
Yang P-M, Hsieh Y-Y, Du J-L, Yen S-C, Hung C-F. Sequential Interferon β-Cisplatin Treatment Enhances the Surface Exposure of Calreticulin in Cancer Cells via an Interferon Regulatory Factor 1-Dependent Manner. Biomolecules. 2020; 10(4):643. https://doi.org/10.3390/biom10040643
Chicago/Turabian StyleYang, Pei-Ming, Yao-Yu Hsieh, Jia-Ling Du, Shih-Chieh Yen, and Chien-Fu Hung. 2020. "Sequential Interferon β-Cisplatin Treatment Enhances the Surface Exposure of Calreticulin in Cancer Cells via an Interferon Regulatory Factor 1-Dependent Manner" Biomolecules 10, no. 4: 643. https://doi.org/10.3390/biom10040643