
An Overview of Mitochondrial Protein Defects in Neuromuscular Diseases

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Overview of NMDs’ Clinical Features
3. Role of Mitochondria in Neuromuscular Diseases
4. Alteration of the Respiratory Chain Enzymes in NMDs
5. Alterations of Mitochondrial Enzymes Impair Neuromuscular Functions

{kind=link}
{kind=link}
{kind=link}
| Gene | Protein Name | Associated Neuromuscular Phenotypes | Pathogenic Mutations | Reference(s) |
|---|---|---|---|---|
| OPA1 | Optic atrophy 1 | Excercise intollerance, ataxia, and ophtalmoplegia | R455M (c.G1334A) S545R (c.C1635G) Q297X (c.C889T) A357T (c.G1069A) | [199,200] |
| MFN2 | Mitofusin 2 | Type 2 Charcot–Marie–Tooth neuropathy, motor neuropathy, muscle weakness, and atrophy | R95W (c.C280T) R280H (c.G839A) | [202,203] |
| ACO2 | Aconitase | Truncal hypotonia, muscle atrophy, and seizures | R607C (c.1819T9 P712l (c.C2135T) | [205] |
| MDH2 | Malate dehydrogenase 2 | Muscle weakness, muscle atrophy, and severe hypotonia | P133L (c.C398T) P207L (c.C620T) G199Afs*10 (c596delG) | [206,207] |
| CPTII | Carnitine palmitoyl transferase II | Severe infantile hepatocardiomuscular disease and myopathy | S113L (c.S113L) P227L (c.C1196T) K414TfsX7 (c.1238_1239delAG) K642Tfsx6 (c.1926_1935DEL) | [208,209,210] |
| SCAD | Short-chain acylCoA dehydrogenase | Hypotonia, seizures, progressive myopathy, cardiomyopathy, and progressive external ophtalmoplegia | G209S (c.G625A) R171W (c.C511T) | [211] |
| VLCAD | Very-long-chain acylCoA dehydrogenase | Early onset cardiac and skeletal myopathy | F418L (c.T1372C) G401A (c.G1322A) E454K (c.G1600A) R575Q (c.G1844A) | [211] |
| ETFDH | Electron transfer flavoprotein dehydrogenase | Myopathy, dysphagia and respiratory failure, and multiple acyl-CoA dehydrogenase deficiency | A187V (c.C560T) D511N (c.G1531A) | [212,213] |
| ECHS1 | Enoyl-CoA hydratase | Early onset Leigh-like syndrome, dystonia, and ataxia syndrome | A158D (c.C473A) Q159R (c.A476G) V82L (c.G244T) | [214] |
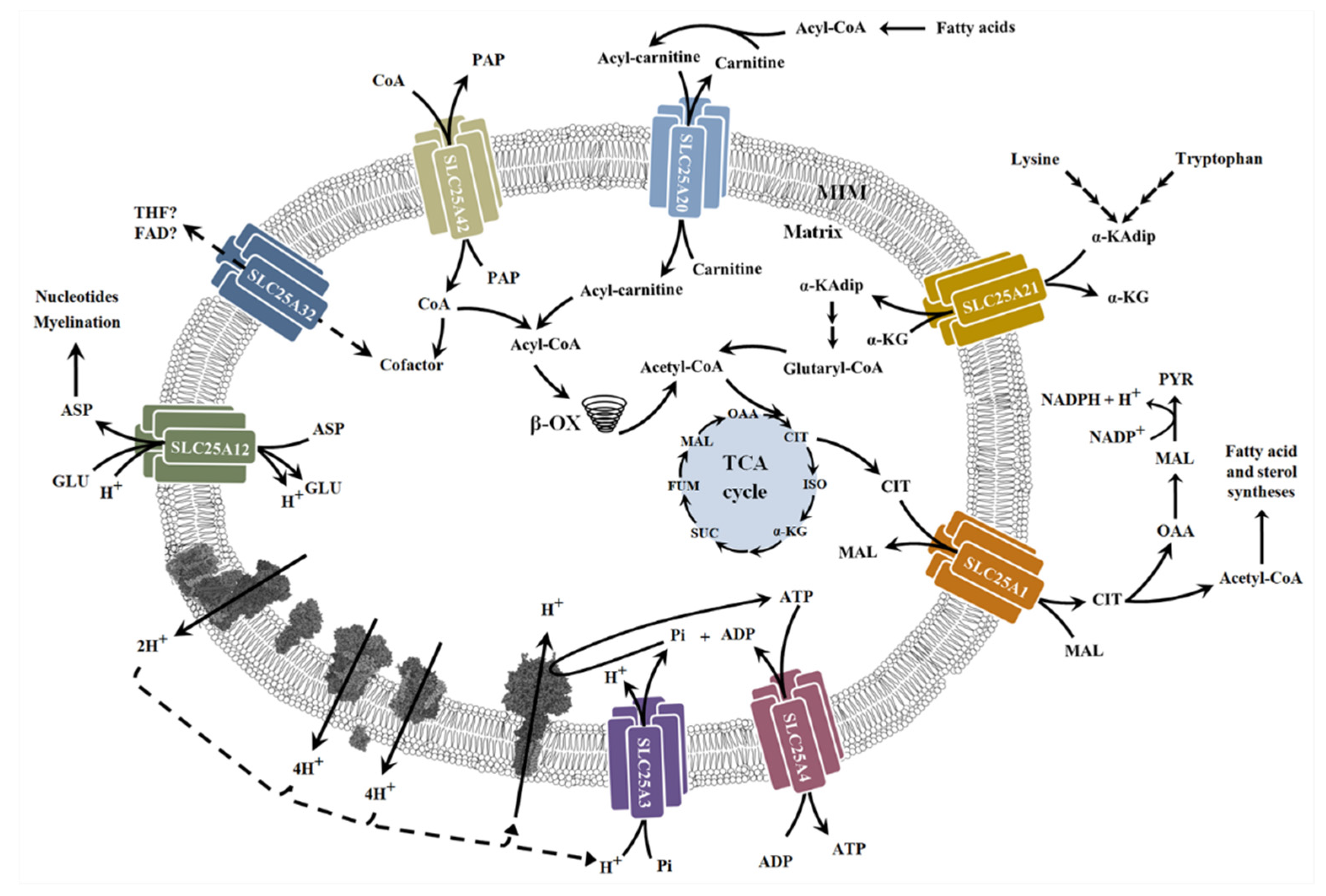
6. Alterations of Mitochondrial Carriers and NMDs
7. Diagnosis and Treatments
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Ach | acetylcholine |
| ALS | amyotrophic lateral sclerosis |
| ASOs | antisense oligonucleotides |
| BCS1L | ubiquinol-cytochrome c reductase complex chaperone |
| CMS | congenital myasthenic syndrome |
| CMT | Charcot–Marie–Tooth disease |
| DMPK | myotonic dystrophy protein kinase |
| DOA | dominant optic atrophy FSHD, facioscapulohumeral muscular dystrophy |
| HCMP | hypertrophic cardiomyopathy |
| IBM | inclusion body myositis |
| LS | Leigh syndrome |
| MADD | multiple acyl-CoA dehydrogenase deficiency |
| MD | muscular dystrophy |
| MELAS | mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes |
| MERRF | myoclonic epilepsy and ragged-red fibers |
| MILS | maternally inherited Leigh syndrome |
| NCLA | neonatal cardiomyopathy with lactic acidosis |
| NMD | neuromuscular disease NMJ, neuromuscular junction |
| PCH | pontocerebellar hypoplasia |
| SCAD | short-chain acyl-CoA dehydrogenase |
| SDH | succinate dehydrogenase |
| SMN | survivor motor neuron |
References
- Bonne, G.; Rivier, F.; Hamroun, D. The 2018 Version of the Gene Table of Monogenic Neuromuscular Disorders (Nuclear Genome). Neuromuscul. Disord. NMD 2017, 27, 1152–1183. [Google Scholar] [CrossRef] [PubMed]
- Vasli, N.; Laporte, J. Impacts of Massively Parallel Sequencing for Genetic Diagnosis of Neuromuscular Disorders. Acta Neuropathol. 2013, 125, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Scotton, C.; Passarelli, C.; Neri, M.; Ferlini, A. Biomarkers in Rare Neuromuscular Diseases. Exp. Cell Res. 2014, 325, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Benditt, J.O.; Boitano, L.J. Pulmonary Issues in Patients with Chronic Neuromuscular Disease. Am. J. Respir. Crit. Care Med. 2013, 187, 1046–1055. [Google Scholar] [CrossRef]
- Pleasure, D. Advances in Translational Research in Neuromuscular Diseases. Arch. Neurol. 2011, 68, 429–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantó-Santos, J.; Grau-Junyent, J.M.; Garrabou, G. The Impact of Mitochondrial Deficiencies in Neuromuscular Diseases. Antioxidants 2020, 9, 964. [Google Scholar] [CrossRef] [PubMed]
- Gatchel, R.J.; Robert, C.; Landers, N.; Hulla, R. Neuromuscular Diseases. In Encyclopedia of Behavioral Medicine; Gellman, M., Turner, J.R., Eds.; Springer: New York, NY, USA, 2016; pp. 1–4. ISBN 978-1-4614-6439-6. [Google Scholar]
- Bos, I.; Kuks, J.B.M.; Wynia, K. Development and Testing Psychometric Properties of an ICF-Based Health Measure: The Neuromuscular Disease Impact Profile. J. Rehabil. Med. 2015, 47, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grumati, P.; Coletto, L.; Sabatelli, P.; Cescon, M.; Angelin, A.; Bertaggia, E.; Blaauw, B.; Urciuolo, A.; Tiepolo, T.; Merlini, L.; et al. Autophagy Is Defective in Collagen VI Muscular Dystrophies, and Its Reactivation Rescues Myofiber Degeneration. Nat. Med. 2010, 16, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Irwin, W.A.; Bergamin, N.; Sabatelli, P.; Reggiani, C.; Megighian, A.; Merlini, L.; Braghetta, P.; Columbaro, M.; Volpin, D.; Bressan, G.M.; et al. Mitochondrial Dysfunction and Apoptosis in Myopathic Mice with Collagen VI Deficiency. Nat. Genet. 2003, 35, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Sussman, M. Duchenne Muscular Dystrophy. JAAOS—J. Am. Acad. Orthop. Surg. 2002, 10, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Lunt, P.W.; Jardine, P.E.; Koch, M.; Maynard, J.; Osborn, M.; Williams, M.; Harper, P.S.; Upadhyaya, M. Phenotypic-Genotypic Correlation Will Assist Genetic Counseling in 4q35-Facioscapulohumeral Muscular Dystrophy. Muscle Nerve. Suppl. 1995, 18, S103–S109. [Google Scholar] [CrossRef]
- Wijmenga, C.; Deaven, L.; Frants, R.R. Dinucleotide Repeat Polymorphism Adjacent to the ANT1 Gene on 4q35. Nucleic Acids Res. 1992, 20, 1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turki, A.; Hayot, M.; Carnac, G.; Pillard, F.; Passerieux, E.; Bommart, S.; De Mauverger, E.R.; Hugon, G.; Pincemail, J.; Pietri, S.; et al. Functional Muscle Impairment in Facioscapulohumeral Muscular Dystrophy Is Correlated with Oxidative Stress and Mitochondrial Dysfunction. Free Radic. Biol. Med. 2012, 53, 1068–1079. [Google Scholar] [CrossRef]
- Udd, B.; Krahe, R. The Myotonic Dystrophies: Molecular, Clinical, and Therapeutic Challenges. Lancet Neurol. 2012, 11, 891–905. [Google Scholar] [CrossRef]
- Wojciechowska, M.; Sobczak, K.; Kozlowski, P.; Sedehizadeh, S.; Wojtkowiak-Szlachcic, A.; Czubak, K.; Markus, R.; Lusakowska, A.; Kaminska, A.; Brook, J.D. Quantitative Methods to Monitor RNA Biomarkers in Myotonic Dystrophy. Sci. Rep. 2018, 8, 5885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbreath, H.R.; Castro, D.; Iannaccone, S.T. Congenital Myopathies and Muscular Dystrophies. Neurol. Clin. 2014, 32, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Ibarra, M.C.A.; Malicdan, M.C.V.; Murayama, K.; Ichihara, Y.; Kikuchi, H.; Nonaka, I.; Noguchi, S.; Hayashi, Y.K.; Nishino, I. Central Core Disease Is Due to RYR1 Mutations in More than 90% of Patients. Brain 2006, 129, 1470–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, C.W.; Schmidt, J.; Lünemann, J.D. Immune and Myodegenerative Pathomechanisms in Inclusion Body Myositis. Ann. Clin. Transl. Neurol. 2017, 4, 422–445. [Google Scholar] [CrossRef]
- Schmidt, J. Current Classification and Management of Inflammatory Myopathies. J. Neuromuscul. Dis. 2018, 5, 109–129. [Google Scholar] [CrossRef]
- Nishimune, H.; Shigemoto, K. Practical Anatomy of the Neuromuscular Junction in Health and Disease. Neurol. Clin. 2018, 36, 231–240. [Google Scholar] [CrossRef]
- Juel, V.C. Clinical neurophysiology of neuromuscular junction disease. Handb. Clin. Neurol. 2019, 161, 291–303. [Google Scholar] [PubMed]
- Gilhus, N.E.; Verschuuren, J.J. Myasthenia Gravis: Subgroup Classification and Therapeutic Strategies. Lancet Neurol. 2015, 14, 231–240. [Google Scholar] [CrossRef]
- Meriggioli, M.N.; Sanders, D.B. Autoimmune Myasthenia Gravis: Emerging Clinical and Biological Heterogeneity. Lancet Neurol. 2009, 8, 475–490. [Google Scholar] [CrossRef] [Green Version]
- Titulaer, M.J.; Lang, B.; Verschuuren, J.J.G.M. Lambert-Eaton Myasthenic Syndrome: From Clinical Characteristics to Therapeutic Strategies. Lancet Neurol. 2011, 10, 1098–1107. [Google Scholar] [CrossRef]
- Wirtz, P.W.; Wintzen, A.R.; Verschuuren, J.J. Lambert-Eaton Myasthenic Syndrome Has a More Progressive Course in Patients with Lung Cancer. Muscle Nerve 2005, 32, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Mahadeva, B.; Phillips, L.H.; Juel, V.C. Autoimmune Disorders of Neuromuscular Transmission. Semin. Neurol. 2008, 28, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.G. Congenital Myasthenic Syndromes in 2018. Curr. Neurol. Neurosci. Rep. 2017, 18, 46. [Google Scholar] [CrossRef] [Green Version]
- Engel, A.G.; Shen, X.M.; Selcen, D.; Sine, S.M. Congenital Myasthenic Syndromes: Pathogenesis, Diagnosis, and Treatment. Lancet Neurol. 2015, 14, 420–434. [Google Scholar] [CrossRef] [Green Version]
- Foster, L.A.; Salajegheh, M.K. Motor Neuron Disease: Pathophysiology, Diagnosis, and Management. Am. J. Med. 2019, 132, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Primer 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Salameh, J.S.; Brown, R.H.; Berry, J.D. Amyotrophic Lateral Sclerosis: Review. Semin. Neurol. 2015, 35, 469–476. [Google Scholar] [CrossRef]
- Vande Velde, C.; Miller, T.M.; Cashman, N.R.; Cleveland, D.W. Selective Association of Misfolded ALS-Linked Mutant SOD1 with the Cytoplasmic Face of Mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 4022–4027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onodera, K.; Shimojo, D.; Ishihara, Y.; Yano, M.; Miya, F.; Banno, H.; Kuzumaki, N.; Ito, T.; Okada, R.; De Araújo Herculano, B.; et al. Unveiling Synapse Pathology in Spinal Bulbar Muscular Atrophy by Genome-Wide Transcriptome Analysis of Purified Motor Neurons Derived from Disease Specific IPSCs. Mol. Brain 2020, 13, 18. [Google Scholar] [CrossRef] [Green Version]
- Spinal Muscular Atrophy. Neurol Clin. 2015, 33, 831–846. [CrossRef] [PubMed] [Green Version]
- Katsetos, C.D.; Koutzaki, S.; Melvin, J.J. Mitochondrial Dysfunction in Neuromuscular Disorders. Semin. Pediatr. Neurol. 2013, 20, 202–215. [Google Scholar] [CrossRef]
- Mary, P.; Servais, L.; Vialle, R. Neuromuscular Diseases: Diagnosis and Management. Orthop. Traumatol. Surg. Res. 2018, 104, S89–S95. [Google Scholar] [CrossRef]
- Anderson, A.J.; Jackson, T.D.; Stroud, D.A.; Stojanovski, D. Mitochondria—Hubs for Regulating Cellular Biochemistry: Emerging Concepts and Networks. Open Biol. 2019, 9, 190126. [Google Scholar] [CrossRef] [Green Version]
- Romanello, V.; Sandri, M. The Connection between the Dynamic Remodeling of the Mitochondrial Network and the Regulation of Muscle Mass. Cell. Mol. Life Sci. 2021, 78, 1305–1328. [Google Scholar] [CrossRef]
- Zemirli, N.; Morel, E.; Molino, D. Mitochondrial Dynamics in Basal and Stressful Conditions. Int. J. Mol. Sci. 2018, 19, 564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Nottia, M.; Verrigni, D.; Torraco, A.; Rizza, T.; Bertini, E.; Carrozzo, R. Mitochondrial Dynamics: Molecular Mechanisms, Related Primary Mitochondrial Disorders and Therapeutic Approaches. Genes 2021, 12, 247. [Google Scholar] [CrossRef]
- Bartsakoulia, M.; Pyle, A.; Troncoso-Chandía, D.; Vial-Brizzi, J.; Paz-Fiblas, M.V.; Duff, J.; Griffin, H.; Boczonadi, V.; Lochmüller, H.; Kleinle, S.; et al. A Novel Mechanism Causing Imbalance of Mitochondrial Fusion and Fission in Human Myopathies. Hum. Mol. Genet. 2018, 27, 1186–1195. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Balancing Mitochondrial Biogenesis and Mitophagy to Maintain Energy Metabolism Homeostasis. Cell Death Differ. 2015, 22, 1399–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, Z.; Fu, T.; Kelly, D.P.; Vega, R.B. Skeletal Muscle Mitochondrial Remodeling in Exercise and Diseases. Cell Res. 2018, 28, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Calvo, C.; Suhm, T.; Büttner, S.; Ott, M. The Basic Machineries for Mitochondrial Protein Quality Control. Mitochondrion 2020, 50, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Kovács, R. Mitochondria and Neuronal Activity. Am. J. Physiol.—Cell Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef]
- Sorrentino, V.; Menzies, K.J.; Auwerx, J. Repairing Mitochondrial Dysfunction in Disease. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 353–389. [Google Scholar] [CrossRef]
- Nascimento, A.; Ortez, C.; Jou, C.; O’Callaghan, M.; Ramos, F.; Garcia-Cazorla, À. Neuromuscular Manifestations in Mitochondrial Diseases in Children. Semin. Pediatr. Neurol. 2016, 23, 290–305. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.P.; Foletta, V.C.; Snow, R.J.; Wadley, G.D. Skeletal Muscle Mitochondria: A Major Player in Exercise, Health and Disease. Biochim. Biophys. Acta—Gen. Subj. 2014, 1840, 1276–1284. [Google Scholar] [CrossRef]
- Siciliano, G.; Chico, L.; Lo Gerfo, A.; Simoncini, C.; Schirinzi, E.; Ricci, G. Exercise-Related Oxidative Stress as Mechanism to Fight Physical Dysfunction in Neuromuscular Disorders. Front. Physiol. 2020, 11, 451. [Google Scholar] [CrossRef]
- Mensch, A.; Zierz, S. Cellular Stress in the Pathogenesis of Muscular Disorders-From Cause to Consequence. Int. J. Mol. Sci. 2020, 21, 5830. [Google Scholar] [CrossRef]
- Afroze, D.; Kumar, A. ER Stress in Skeletal Muscle Remodeling and Myopathies. FEBS J. 2019, 286, 379–398. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, L.R.; Calap-Quintana, P.; Lapeña-Luzón, T.; Pallardó, F.V.; Schneuwly, S.; Navarro, J.A.; Gonzalez-Cabo, P. Oxidative Stress Modulates Rearrangement of Endoplasmic Reticulum-Mitochondria Contacts and Calcium Dysregulation in a Friedreich’s Ataxia Model. Redox Biol. 2020, 37, 101762. [Google Scholar] [CrossRef]
- Reid, R.A.; Moyle, J.; Mitchell, P. Synthesis of Adenosine Triphosphate by a Protonmotive Force in Rat Liver Mitochondria. Nature 1966, 212, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Capaldi, R.A.; Aggeler, R. Mechanism of the F1F0-Type ATP Synthase, a Biological Rotary Motor. Trends Biochem. Sci. 2002, 27, 154–160. [Google Scholar] [CrossRef]
- Sousa, J.S.; D’Imprima, E.; Vonck, J. Mitochondrial Respiratory Chain Complexes. In Membrane Protein Complexes: Structure and Function. Subcellular Biochemistry; Harris, J., Boekema, E., Eds.; Springer: Singapore, 2018; Volume 87. [Google Scholar]
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef]
- Brandt, U. A Two-State Stabilization-Change Mechanism for Proton-Pumping Complex I. Biochim. Biophys. Acta 2011, 1807, 1364–1369. [Google Scholar] [CrossRef] [Green Version]
- Hirst, J.; Roessler, M.M. Energy Conversion, Redox Catalysis and Generation of Reactive Oxygen Species by Respiratory Complex I. Biochim. Biophys. Acta 2016, 1857, 872–883. [Google Scholar] [CrossRef] [Green Version]
- Rutter, J.; Winge, D.R.; Schiffman, J.D. Succinate Dehydrogenase—Assembly, Regulation and Role in Human Disease. Mitochondrion 2010, 10, 393–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, S.; Lee, J.W.; Okada, K.; Lee, J.K.; Iwata, M.; Rasmussen, B.; Link, T.A.; Ramaswamy, S.; Jap, B.K. Complete Structure of the 11-Subunit Bovine Mitochondrial Cytochrome Bc1 Complex. Science 1998, 281, 64–71. [Google Scholar] [CrossRef]
- Brand, M.D. The Sites and Topology of Mitochondrial Superoxide Production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [Green Version]
- Zong, S.; Wu, M.; Gu, J.; Liu, T.; Guo, R.; Yang, M. Structure of the Intact 14-Subunit Human Cytochrome c Oxidase. Cell Res. 2018, 28, 1026–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, M.R. Mitochondrial Myopathies: Genetic Mechanisms. Arch. Neurol. 1998, 55, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.B.; Chinnery, P.F. The Dynamics of Mitochondrial DNA Heteroplasmy: Implications for Human Health and Disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef]
- Rygiel, K.A.; Picard, M.; Turnbull, D.M. The Ageing Neuromuscular System and Sarcopenia: A Mitochondrial Perspective. J. Physiol. 2016, 594, 4499–4512. [Google Scholar] [CrossRef]
- Iommarini, L.; Calvaruso, M.A.; Kurelac, I.; Gasparre, G.; Porcelli, A.M. Complex i Impairment in Mitochondrial Diseases and Cancer: Parallel Roads Leading to Different Outcomes. Int. J. Biochem. Cell Biol. 2013, 45, 47–63. [Google Scholar] [CrossRef]
- Nardin, R.A.; Johns, D.R. Mitochondrial Dysfunction and Neuromuscular Disease. Muscle Nerve 2001, 24, 170–191. [Google Scholar] [CrossRef]
- Metodiev, M.D.; Gerber, S.; Hubert, L.; Delahodde, A.; Chretien, D.; Gérard, X.; Amati-Bonneau, P.; Giacomotto, M.C.; Boddaert, N.; Kaminska, A.; et al. Mutations in the Tricarboxylic Acid Cycle Enzyme, Aconitase 2, Cause Either Isolated or Syndromic Optic Neuropathy with Encephalopathy and Cerebellar Atrophy. J. Med. Genet. 2014, 51, 834–838. [Google Scholar] [CrossRef]
- Fernández-Silva, P.; Enriquez, J.A.; Montoya, J. Replication and Transcription of Mammalian Mitochondrial DNA. Exp. Physiol. 2003, 88, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, M.E.; Hepple, R.T. Mitochondrial Mechanisms of Neuromuscular Junction Degeneration with Aging. Cells 2020, 9, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanagat, J.; Cao, Z.; Pathare, P.; Aiken, J.M. Mitochondrial DNA Deletion Mutations Colocalize with Segmental Electron Transport System Abnormalities, Muscle Fiber Atrophy, Fiber Splitting, and Oxidative Damage in Sarcopenia. FASEB J. 2001, 15, 322–332. [Google Scholar] [CrossRef]
- Hoefs, S.J.G.; Rodenburg, R.J.; Smeitink, J.A.M.; van den Heuvel, L.P. Molecular Base of Biochemical Complex I Deficiency. Mitochondrion 2012, 12, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.H.; Willems, P.H.G.M.; Smeitink, J.A.M. Monogenic Mitochondrial Disorders. N. Engl. J. Med. 2012, 366, 1132–1141. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Hong, D.; Zhang, W.; Yao, S.; Qi, X.; Lv, H.; Zheng, R.; Feng, L.; Huang, Y.; Yuan, Y.; et al. Mutations in Mitochondrially Encoded Complex i Enzyme as the Second Common Cause in a Cohort of Chinese Patients with Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis and Stroke-like Episodes. J. Hum. Genet. 2011, 56, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Villanueva-Paz, M.; Povea-Cabello, S.; Villalón-García, I.; Suárez-Rivero, J.M.; Álvarez-Córdoba, M.; de la Mata, M.; Talaverón-Rey, M.; Jackson, S.; Sánchez-Alcázar, J.A. Pathophysiological Characterization of MERRF Patient-Specific Induced Neurons Generated by Direct Reprogramming. Biochim. Biophys. Acta—Mol. Cell Res. 2019, 1866, 861–881. [Google Scholar] [CrossRef]
- DiMauro, S.; Schon, E.A. Mitochondrial Disorders in the Nervous System. Annu. Rev. Neurosci. 2008, 31, 91–123. [Google Scholar] [CrossRef] [PubMed]
- Van Rahden, V.A.; Fernandez-Vizarra, E.; Alawi, M.; Brand, K.; Fellmann, F.; Horn, D.; Zeviani, M.; Kutsche, K. Mutations in NDUFB11, Encoding a Complex I Component of the Mitochondrial Respiratory Chain, Cause Microphthalmia with Linear Skin Defects Syndrome. Am. J. Hum. Genet. 2015, 96, 640–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haack, T.B.; Haberberger, B.; Frisch, E.-M.; Wieland, T.; Iuso, A.; Gorza, M.; Strecker, V.; Graf, E.; Mayr, J.A.; Herberg, U.; et al. Molecular Diagnosis in Mitochondrial Complex I Deficiency Using Exome Sequencing. J. Med. Genet. 2012, 49, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Bénit, P.; Slama, A.; Cartault, F.; Giurgea, I.; Chretien, D.; Lebon, S.; Marsac, C.; Munnich, A.; Rötig, A.; Rustin, P. Mutant NDUFS3 Subunit of Mitochondrial Complex I Causes Leigh Syndrome. J. Med. Genet. 2004, 41, 14–17. [Google Scholar] [CrossRef] [PubMed]
- González-Quintana, A.; García-Consuegra, I.; Belanger-Quintana, A.; Serrano-Lorenzo, P.; Lucia, A.; Blázquez, A.; Docampo, J.; Ugalde, C.; Morán, M.; Arenas, J.; et al. Novel Ndufa13 Mutations Associated with Oxphos Deficiency and Leigh Syndrome: A Second Family Report. Genes 2020, 11, 855. [Google Scholar] [CrossRef]
- Spiegel, R.; Shaag, A.; Mandel, H.; Reich, D.; Penyakov, M.; Hujeirat, Y.; Saada, A.; Elpeleg, O.; Shalev, S.A. Mutated NDUFS6 Is the Cause of Fatal Neonatal Lactic Acidemia in Caucasus Jews. Eur. J. Hum. Genet. EJHG 2009, 17, 1200–1203. [Google Scholar] [CrossRef] [Green Version]
- Schuelke, M.; Smeitink, J.; Mariman, E.; Loeffen, J.; Plecko, B.; Trijbels, F.; Stöckler-Ipsiroglu, S.; van den Heuvel, L. Mutant NDUFV1 Subunit of Mitochondrial Complex I Causes Leukodystrophy and Myoclonic Epilepsy. Nat. Genet. 1999, 21, 260–261. [Google Scholar] [CrossRef] [PubMed]
- Bugiani, M.; Invernizzi, F.; Alberio, S.; Briem, E.; Lamantea, E.; Carrara, F.; Moroni, I.; Farina, L.; Spada, M.; Donati, M.A.; et al. Clinical and Molecular Findings in Children with Complex I Deficiency. Biochim. Biophys. Acta BBA—Bioenerg. 2004, 1659, 136–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeffen, J.L.; Smeitink, J.A.; Trijbels, J.M.; Janssen, A.J.; Triepels, R.H.; Sengers, R.C.; van den Heuvel, L.P. Isolated Complex I Deficiency in Children: Clinical, Biochemical and Genetic Aspects. Hum. Mutat. 2000, 15, 123–134. [Google Scholar] [CrossRef]
- Simon, D.K.; Friedman, J.; Breakefield, X.O.; Jankovic, J.; Brin, M.F.; Provias, J.; Bressman, S.B.; Charness, M.E.; Tarsy, D.; Johns, D.R.; et al. A Heteroplasmic Mitochondrial Complex I Gene Mutation in Adult-Onset Dystonia. Neurogenetics 2003, 4, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Kirby, D.M.; McFarland, R.; Ohtake, A.; Dunning, C.; Ryan, M.T.; Wilson, C.; Ketteridge, D.; Turnbull, D.M.; Thorburn, D.R.; Taylor, R.W. Mutations of the Mitochondrial ND1 Gene as a Cause of MELAS. J. Med. Genet. 2004, 41, 784–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFarland, R.; Kirby, D.M.; Fowler, K.J.; Ohtake, A.; Ryan, M.T.; Amor, D.J.; Fletcher, J.M.; Dixon, J.W.; Collins, F.A.; Turnbull, D.M.; et al. De Novo Mutations in the Mitochondrial ND3 Gene as a Cause of Infantile Mitochondrial Encephalopathy and Complex I Deficiency. Ann. Neurol. 2004, 55, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Lertrit, P.; Noer, A.S.; Jean-Francois, M.J.; Kapsa, R.; Dennett, X.; Thyagarajan, D.; Lethlean, K.; Byrne, E.; Marzuki, S. A New Disease-Related Mutation for Mitochondrial Encephalopathy Lactic Acidosis and Strokelike Episodes (MELAS) Syndrome Affects the ND4 Subunit of the Respiratory Complex I. Am. J. Hum. Genet. 1992, 51, 457–468. [Google Scholar]
- Liolitsa, D.; Rahman, S.; Benton, S.; Carr, L.J.; Hanna, M.G. Is the Mitochondrial Complex I ND5 Gene a Hot-Spot for MELAS Causing Mutations? Ann. Neurol. 2003, 53, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Ravn, K.; Wibrand, F.; Hansen, F.J.; Horn, N.; Rosenberg, T.; Schwartz, M. An MtDNA Mutation, 14453G-->A, in the NADH Dehydrogenase Subunit 6 Associated with Severe MELAS Syndrome. Eur. J. Hum. Genet. EJHG 2001, 9, 805–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Moreira, D.; Ugalde, C.; Smeets, R.; Rodenburg, R.J.T.; Lopez-Laso, E.; Ruiz-Falco, M.L.; Briones, P.; Martin, M.A.; Smeitink, J.A.M.; Arenas, J. X-Linked NDUFA1 Gene Mutations Associated with Mitochondrial Encephalomyopathy. Ann. Neurol. 2007, 61, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Hoefs, S.J.G.; Dieteren, C.E.J.; Distelmaier, F.; Janssen, R.J.R.J.; Epplen, A.; Swarts, H.G.P.; Forkink, M.; Rodenburg, R.J.; Nijtmans, L.G.; Willems, P.H.; et al. NDUFA2 Complex I Mutation Leads to Leigh Disease. Am. J. Hum. Genet. 2008, 82, 1306–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoefs, S.J.G.; van Spronsen, F.J.; Lenssen, E.W.H.; Nijtmans, L.G.; Rodenburg, R.J.; Smeitink, J.A.M.; van den Heuvel, L.P. NDUFA10 Mutations Cause Complex I Deficiency in a Patient with Leigh Disease. Eur. J. Hum. Genet. 2011, 19, 270–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, I.; Hershkovitz, E.; Shaag, A.; Edvardson, S.; Saada, A.; Elpeleg, O. Mitochondrial Complex I Deficiency Caused by a Deleterious NDUFA11 Mutation. Ann. Neurol. 2008, 63, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, E.; Rodenburg, R.J.; van den Brand, M.; Thomsen, L.L.; Duno, M.; Batbayli, M.; Wibrand, F.; Nijtmans, L. Respiratory Chain Complex I Deficiency Due to NDUFA12 Mutations as a New Cause of Leigh Syndrome. J. Med. Genet. 2011, 48, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Angebault, C.; Charif, M.; Guegen, N.; Piro-Megy, C.; Mousson de Camaret, B.; Procaccio, V.; Guichet, P.-O.; Hebrard, M.; Manes, G.; Leboucq, N.; et al. Mutation in NDUFA13/GRIM19 Leads to Early Onset Hypotonia, Dyskinesia and Sensorial Deficiencies, and Mitochondrial Complex I Instability. Hum. Mol. Genet. 2015, 24, 3948–3955. [Google Scholar] [CrossRef] [Green Version]
- Alston, C.L.; Howard, C.; Oláhová, M.; Hardy, S.A.; He, L.; Murray, P.G.; O’Sullivan, S.; Doherty, G.; Shield, J.P.H.; Hargreaves, I.P.; et al. A Recurrent Mitochondrial p.Trp22Arg NDUFB3 Variant Causes a Distinctive Facial Appearance, Short Stature and a Mild Biochemical and Clinical Phenotype. J. Med. Genet. 2016, 53, 634–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piekutowska-Abramczuk, D.; Assouline, Z.; Mataković, L.; Feichtinger, R.G.; Koňařiková, E.; Jurkiewicz, E.; Stawiński, P.; Gusic, M.; Koller, A.; Pollak, A.; et al. NDUFB8 Mutations Cause Mitochondrial Complex I Deficiency in Individuals with Leigh-like Encephalomyopathy. Am. J. Hum. Genet. 2018, 102, 460–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haack, T.B.; Madignier, F.; Herzer, M.; Lamantea, E.; Danhauser, K.; Invernizzi, F.; Koch, J.; Freitag, M.; Drost, R.; Hillier, I.; et al. Mutation Screening of 75 Candidate Genes in 152 Complex I Deficiency Cases Identifies Pathogenic Variants in 16 Genes Including NDUFB9. J. Med. Genet. 2012, 49, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friederich, M.W.; Erdogan, A.J.; Coughlin, C.R., II; Elos, M.T.; Jiang, H.; O’Rourke, C.P.; Lovell, M.A.; Wartchow, E.; Gowan, K.; Chatfield, K.C.; et al. Mutations in the Accessory Subunit NDUFB10 Result in Isolated Complex I Deficiency and Illustrate the Critical Role of Intermembrane Space Import for Complex I Holoenzyme Assembly. Hum. Mol. Genet. 2017, 26, 702–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinson, K.; Kovacs-Nagy, R.; Õiglane-Shlik, E.; Pajusalu, S.; Nõukas, M.; Wintjes, L.T.; van den Brandt, F.C.A.; Brink, M.; Acker, T.; Ahting, U.; et al. Diverse Phenotype in Patients with Complex I Deficiency Due to Mutations in NDUFB11. Eur. J. Med. Genet. 2019, 62, 103572. [Google Scholar] [CrossRef] [PubMed]
- Shehata, B.M.; Cundiff, C.A.; Lee, K.; Sabharwal, A.; Lalwani, M.K.; Davis, A.K.; Agrawal, V.; Sivasubbu, S.; Iannucci, G.J.; Gibson, G. Exome Sequencing of Patients with Histiocytoid Cardiomyopathy Reveals a de Novo NDUFB11 Mutation That Plays a Role in the Pathogenesis of Histiocytoid Cardiomyopathy. Am. J. Med. Genet. A 2015, 167, 2114–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alahmad, A.; Nasca, A.; Heidler, J.; Thompson, K.; Oláhová, M.; Legati, A.; Lamantea, E.; Meisterknecht, J.; Spagnolo, M.; He, L.; et al. Bi-Allelic Pathogenic Variants in NDUFC2 Cause Early-Onset Leigh Syndrome and Stalled Biogenesis of Complex I. EMBO Mol. Med. 2020, 12, e12619. [Google Scholar] [CrossRef]
- Bénit, P.; Chretien, D.; Kadhom, N.; de Lonlay-Debeney, P.; Cormier-Daire, V.; Cabral, A.; Peudenier, S.; Rustin, P.; Munnich, A.; Rötig, A. Large-Scale Deletion and Point Mutations of the Nuclear NDUFV1 and NDUFS1 Genes in Mitochondrial Complex I Deficiency. Am. J. Hum. Genet. 2001, 68, 1344–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeffen, J.; Elpeleg, O.; Smeitink, J.; Smeets, R.; Stöckler-Ipsiroglu, S.; Mandel, H.; Sengers, R.; Trijbels, F.; van den Heuvel, L. Mutations in the Complex I NDUFS2 Gene of Patients with Cardiomyopathy and Encephalomyopathy. Ann. Neurol. 2001, 49, 195–201. [Google Scholar] [CrossRef]
- Budde, S.M.S.; van den Heuvel, L.P.W.J.; Janssen, A.J.; Smeets, R.J.P.; Buskens, C.A.F.; DeMeirleir, L.; Van Coster, R.; Baethmann, M.; Voit, T.; Trijbels, J.M.F.; et al. Combined Enzymatic Complex I and III Deficiency Associated with Mutations in the Nuclear Encoded NDUFS4 Gene. Biochem. Biophys. Res. Commun. 2000, 275, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Smeitink, J.; van den Heuvel, L. Human Mitochondrial Complex I in Health and Disease. Am. J. Hum. Genet. 1999, 64, 1505–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeffen, J.; Smeitink, J.; Triepels, R.; Smeets, R.; Schuelke, M.; Sengers, R.; Trijbels, F.; Hamel, B.; Mullaart, R.; van den Heuvel, L. The First Nuclear-Encoded Complex I Mutation in a Patient with Leigh Syndrome. Am. J. Hum. Genet. 1998, 63, 1598–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bénit, P.; Beugnot, R.; Chretien, D.; Giurgea, I.; De Lonlay-Debeney, P.; Issartel, J.-P.; Corral-Debrinski, M.; Kerscher, S.; Rustin, P.; Rötig, A.; et al. Mutant NDUFV2 Subunit of Mitochondrial Complex I Causes Early Onset Hypertrophic Cardiomyopathy and Encephalopathy. Hum. Mutat. 2003, 21, 582–586. [Google Scholar] [CrossRef]
- Nouws, J.; Wibrand, F.; van den Brand, M.; Venselaar, H.; Duno, M.; Lund, A.M.; Trautner, S.; Nijtmans, L.; Ostergard, E. A Patient with Complex I Deficiency Caused by a Novel ACAD9 Mutation Not Responding to Riboflavin Treatment. JIMD Rep. 2014, 12, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repp, B.M.; Mastantuono, E.; Alston, C.L.; Schiff, M.; Haack, T.B.; Rötig, A.; Ardissone, A.; Lombès, A.; Catarino, C.B.; Diodato, D.; et al. Clinical, Biochemical and Genetic Spectrum of 70 Patients with ACAD9 Deficiency: Is Riboflavin Supplementation Effective? Orphanet J. Rare Dis. 2018, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Fragaki, K.; Chaussenot, A.; Boutron, A.; Bannwarth, S.; Cochaud, C.; Richelme, C.; Sacconi, S.; Paquis-Flucklinger, V. Severe Defect in Mitochondrial Complex I Assembly with Mitochondrial DNA Deletions in ACAD9-Deficient Mild Myopathy. Muscle Nerve 2017, 55, 919–922. [Google Scholar] [CrossRef]
- Haack, T.B.; Danhauser, K.; Haberberger, B.; Hoser, J.; Strecker, V.; Boehm, D.; Uziel, G.; Lamantea, E.; Invernizzi, F.; Poulton, J.; et al. Exome Sequencing Identifies ACAD9 Mutations as a Cause of Complex I Deficiency. Nat. Genet. 2010, 42, 1131–1134. [Google Scholar] [CrossRef] [PubMed]
- Garone, C.; Donati, M.A.; Sacchini, M.; Garcia-Diaz, B.; Bruno, C.; Calvo, S.; Mootha, V.K.; Dimauro, S. Mitochondrial Encephalomyopathy Due to a Novel Mutation in ACAD9. JAMA Neurol. 2013, 70, 1177–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo, S.E.; Tucker, E.J.; Compton, A.G.; Kirby, D.M.; Crawford, G.; Burtt, N.P.; Rivas, M.; Guiducci, C.; Bruno, D.L.; Goldberger, O.A.; et al. High-Throughput, Pooled Sequencing Identifies Mutations in NUBPL and FOXRED1 in Human Complex I Deficiency. Nat. Genet. 2010, 42, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Protasoni, M.; Bruno, C.; Donati, M.A.; Mohamoud, K.; Severino, M.; Allegri, A.; Robinson, A.J.; Reyes, A.; Zeviani, M.; Garone, C. Novel Compound Heterozygous Pathogenic Variants in Nucleotide-Binding Protein like Protein (NUBPL) Cause Leukoencephalopathy with Multi-Systemic Involvement. Mol. Genet. Metab. 2020, 129, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Dunning, C.J.R.; McKenzie, M.; Sugiana, C.; Lazarou, M.; Silke, J.; Connelly, A.; Fletcher, J.M.; Kirby, D.M.; Thorburn, D.R.; Ryan, M.T. Human CIA30 Is Involved in the Early Assembly of Mitochondrial Complex I and Mutations in Its Gene Cause Disease. EMBO J. 2007, 26, 3227–3237. [Google Scholar] [CrossRef] [Green Version]
- Ogilvie, I.; Kennaway, N.G.; Shoubridge, E.A. A Molecular Chaperone for Mitochondrial Complex I Assembly Is Mutated in a Progressive Encephalopathy. J. Clin. Investig. 2005, 115, 2784–2792. [Google Scholar] [CrossRef] [PubMed]
- Saada, A.; Vogel, R.O.; Hoefs, S.J.; van den Brand, M.A.; Wessels, H.J.; Willems, P.H.; Venselaar, H.; Shaag, A.; Barghuti, F.; Reish, O.; et al. Mutations in NDUFAF3 (C3ORF60), Encoding an NDUFAF4 (C6ORF66)-Interacting Complex I Assembly Protein, Cause Fatal Neonatal Mitochondrial Disease. Am. J. Hum. Genet. 2009, 84, 718–727. [Google Scholar] [CrossRef] [Green Version]
- Saada, A.; Edvardson, S.; Rapoport, M.; Shaag, A.; Amry, K.; Miller, C.; Lorberboum-Galski, H.; Elpeleg, O. C6ORF66 Is an Assembly Factor of Mitochondrial Complex I. Am. J. Hum. Genet. 2008, 82, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.-E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A Mitochondrial Protein Compendium Elucidates Complex I Disease Biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Alston, C.L.; Veling, M.T.; Heidler, J.; Taylor, L.S.; Alaimo, J.T.; Sung, A.Y.; He, L.; Hopton, S.; Broomfield, A.; Pavaine, J.; et al. Pathogenic Bi-Allelic Mutations in NDUFAF8 Cause Leigh Syndrome with an Isolated Complex I Deficiency. Am. J. Hum. Genet. 2020, 106, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Kremer, L.S.; Bader, D.M.; Mertes, C.; Kopajtich, R.; Pichler, G.; Iuso, A.; Haack, T.B.; Graf, E.; Schwarzmayr, T.; Terrile, C.; et al. Genetic Diagnosis of Mendelian Disorders via RNA Sequencing. Nat. Commun. 2017, 8, 15824. [Google Scholar] [CrossRef]
- Sánchez-Caballero, L.; Ruzzenente, B.; Bianchi, L.; Assouline, Z.; Barcia, G.; Metodiev, M.D.; Rio, M.; Funalot, B.; van den Brand, M.A.M.; Guerrero-Castillo, S.; et al. Mutations in Complex I Assembly Factor TMEM126B Result in Muscle Weakness and Isolated Complex I Deficiency. Am. J. Hum. Genet. 2016, 99, 208–216. [Google Scholar] [CrossRef] [Green Version]
- Alston, C.L.; Compton, A.G.; Formosa, L.E.; Strecker, V.; Oláhová, M.; Haack, T.B.; Smet, J.; Stouffs, K.; Diakumis, P.; Ciara, E.; et al. Biallelic Mutations in TMEM126B Cause Severe Complex I Deficiency with a Variable Clinical Phenotype. Am. J. Hum. Genet. 2016, 99, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Hoekstra, A.S.; Bayley, J.P. The Role of Complex II in Disease. Biochim. Biophys. Acta—Bioenerg. 2013, 1827, 543–551. [Google Scholar] [CrossRef] [Green Version]
- Al Khazal, F.; Holte, M.N.; Bolon, B.; White, T.A.; LeBrasseur, N.; Maher, L.J. A Conditional Mouse Model of Complex II Deficiency Manifesting as Leigh-like Syndrome. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 13189–13201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parfait, B.; Chretien, D.; Rötig, A.; Marsac, C.; Munnich, A.; Rustin, P. Compound Heterozygous Mutations in the Flavoprotein Gene of the Respiratory Chain Complex II in a Patient with Leigh Syndrome. Hum. Genet. 2000, 106, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Levitas, A.; Muhammad, E.; Harel, G.; Saada, A.; Caspi, V.C.; Manor, E.; Beck, J.C.; Sheffield, V.; Parvari, R. Familial Neonatal Isolated Cardiomyopathy Caused by a Mutation in the Flavoprotein Subunit of Succinate Dehydrogenase. Eur. J. Hum. Genet. 2010, 18, 1160–1165. [Google Scholar] [CrossRef]
- Ghezzi, D.; Goffrini, P.; Uziel, G.; Horvath, R.; Klopstock, T.; Lochmüller, H.; D’Adamo, P.; Gasparini, P.; Strom, T.M.; Prokisch, H.; et al. SDHAF1, Encoding a LYR Complex-II Specific Assembly Factor, Is Mutated in SDH-Defective Infantile Leukoencephalopathy. Nat. Genet. 2009, 41, 654–656. [Google Scholar] [CrossRef] [PubMed]
- Bouzidi, M.F.; Schägger, H.; Collombet, J.M.; Carrier, H.; Flocard, F.; Quard, S.; Mousson, B.; Godinot, C. Decreased Expression of Ubiquinol-Cytochrome c Reductase Subunits in Patients Exhibiting Mitochondrial Myopathy with Progressive Exercise Intolerance. Neuromuscul. Disord. NMD 1993, 3, 599–604. [Google Scholar] [CrossRef]
- Keightley, J.A.; Anitori, R.; Burton, M.D.; Quan, F.; Buist, N.R.; Kennaway, N.G. Mitochondrial Encephalomyopathy and Complex III Deficiency Associated with a Stop-Codon Mutation in the Cytochrome b Gene. Am. J. Hum. Genet. 2000, 67, 1400–1410. [Google Scholar] [CrossRef] [Green Version]
- Andreu, A.L.; Checcarelli, N.; Iwata, S.; Shanske, S.; DiMauro, S. A Missense Mutation in the Mitochondrial Cytochrome b Gene in a Revisited Case with Histiocytoid Cardiomyopathy. Pediatr. Res. 2000, 48, 311–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peruzzo, R.; Corrà, S.; Costa, R.; Brischigliaro, M.; Varanita, T.; Biasutto, L.; Rampazzo, C.; Ghezzi, D.; Leanza, L.; Zoratti, M.; et al. Exploiting Pyocyanin to Treat Mitochondrial Disease Due to Respiratory Complex III Dysfunction. Nat. Commun. 2021, 12, 2103. [Google Scholar] [CrossRef] [PubMed]
- Meunier, B.; Fisher, N.; Ransac, S.; Mazat, J.P.; Brasseur, G. Respiratory Complex III Dysfunction in Humans and the Use of Yeast as a Model Organism to Study Mitochondrial Myopathy and Associated Diseases. Biochim. Biophys. Acta—Bioenerg. 2013, 1827, 1346–1361. [Google Scholar] [CrossRef] [Green Version]
- De Coo, I.F.M.; Renier, W.O.; Ruitenbeek, W.; Ter Laak, H.J.; Bakker, M.; Schägger, H.; Van Oost, B.A.; Smeets, H.J.M. A 4-Base Pair Deletion in the Mitochondrial Cytochrome b Gene Associated with Parkinsonism/MELAS Overlap Syndrome. Ann. Neurol. 1999, 45, 130–133. [Google Scholar] [CrossRef]
- Blázquez, A.; Gil-Borlado, M.C.; Morán, M.; Verdú, A.; Cazorla-Calleja, M.R.; Martín, M.A.; Arenas, J.; Ugalde, C. Infantile Mitochondrial Encephalomyopathy with Unusual Phenotype Caused by a Novel BCS1L Mutation in an Isolated Complex III-Deficient Patient. Neuromuscul. Disord. NMD 2009, 19, 143–146. [Google Scholar] [CrossRef]
- Tegelberg, S.; Tomašić, N.; Kallijärvi, J.; Purhonen, J.; Elmér, E.; Lindberg, E.; Nord, D.G.; Soller, M.; Lesko, N.; Wedell, A.; et al. Respiratory Chain Complex III Deficiency Due to Mutated BCS1L: A Novel Phenotype with Encephalomyopathy, Partially Phenocopied in a Bcs1l Mutant Mouse Model. Orphanet J. Rare Dis. 2017, 12, 73. [Google Scholar] [CrossRef] [Green Version]
- Invernizzi, F.; Tigano, M.; Dallabona, C.; Donnini, C.; Ferrero, I.; Cremonte, M.; Ghezzi, D.; Lamperti, C.; Zeviani, M. A Homozygous Mutation in LYRM7/MZM1L Associated with Early Onset Encephalopathy, Lactic Acidosis, and Severe Reduction of Mitochondrial Complex III Activity. Hum. Mutat. 2013, 34, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- Morino, H.; Miyamoto, R.; Ohnishi, S.; Maruyama, H.; Kawakami, H. Exome Sequencing Reveals a Novel TTC19 Mutation in an Autosomal Recessive Spinocerebellar Ataxia Patient. BMC Neurol. 2014, 14, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanschers, B.F.J.; Szklarczyk, R.; van den Brand, M.A.M.; Jonckheere, A.; Suijskens, J.; Smeets, R.; Rodenburg, R.J.; Stephan, K.; Helland, I.B.; Elkamil, A.; et al. A Mutation in the Human CBP4 Ortholog UQCC3 Impairs Complex III Assembly, Activity and Cytochrome b Stability. Hum. Mol. Genet. 2014, 23, 6356–6365. [Google Scholar] [CrossRef] [Green Version]
- Gusic, M.; Schottmann, G.; Feichtinger, R.G.; Du, C.; Scholz, C.; Wagner, M.; Mayr, J.A.; Lee, C.Y.; Yépez, V.A.; Lorenz, N.; et al. Bi-Allelic UQCRFS1 Variants Are Associated with Mitochondrial Complex III Deficiency, Cardiomyopathy, and Alopecia Totalis. Am. J. Hum. Genet. 2020, 106, 102–111. [Google Scholar] [CrossRef]
- Barel, O.; Shorer, Z.; Flusser, H.; Ofir, R.; Narkis, G.; Finer, G.; Shalev, H.; Nasasra, A.; Saada, A.; Birk, O.S. Mitochondrial Complex III Deficiency Associated with a Homozygous Mutation in UQCRQ. Am. J. Hum. Genet. 2008, 82, 1211–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoubridge, E.A. Cytochrome c Oxidase Deficiency. Am. J. Med. Genet. 2001, 106, 46–52. [Google Scholar] [CrossRef]
- Herrero-Martín, M.D.; Pineda, M.; Briones, P.; López-Gallardo, E.; Carreras, M.; Benac, M.; Angel Idoate, M.; Vilaseca, M.A.; Artuch, R.; López-Pérez, M.J.; et al. A New Pathologic Mitochondrial DNA Mutation in the Cytochrome Oxidase Subunit I (MT-CO1). Hum. Mutat. 2008, 29, E112–E122. [Google Scholar] [CrossRef] [PubMed]
- Poole, O.V.; Everett, C.M.; Gandhi, S.; Marino, S.; Bugiardini, E.; Woodward, C.; Lam, A.; Quinlivan, R.; Hanna, M.G.; Pitceathly, R.D.S. Adult-Onset Leigh Syndrome Linked to the Novel Stop Codon Mutation m.6579G>A in MT-CO1. Mitochondrion 2019, 47, 294–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, L.; Piga, D.; Lamantea, E.; Carrara, F.; Uziel, G.; Cudia, P.; Zani, A.; Farina, L.; Morandi, L.; Mora, M.; et al. Identification of Novel Mutations in Five Patients with Mitochondrial Encephalomyopathy. Biochim. Biophys. Acta 2009, 1787, 491–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamperti, C.; Diodato, D.; Lamantea, E.; Carrara, F.; Ghezzi, D.; Mereghetti, P.; Rizzi, R.; Zeviani, M. MELAS-like Encephalomyopathy Caused by a New Pathogenic Mutation in the Mitochondrial DNA Encoded Cytochrome c Oxidase Subunit I. Neuromuscul. Disord. NMD 2012, 22, 990–994. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.M.; Taylor, R.W.; Johnson, M.A.; Chinnery, P.F.; Chrzanowska-Lightowlers, Z.M.A.; Andrews, R.M.; Nelson, I.P.; Wood, N.W.; Lamont, P.J.; Hanna, M.G.; et al. An MtDNA Mutation in the Initiation Codon of the Cytochrome C Oxidase Subunit II Gene Results in Lower Levels of the Protein and a Mitochondrial Encephalomyopathy. Am. J. Hum. Genet. 1999, 64, 1330–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, L.J.C.; Dai, P.; Tan, D.; Lipson, M.; Grix, A.; Sifry-Platt, M.; Gropman, A.; Chen, T.J. Severe Lactic Acidosis Caused by a Novel Frame-Shift Mutation in Mitochondrial-Encoded Cytochrome c Oxidase Subunit II. Am. J. Med. Genet. 2001, 102, 95–99. [Google Scholar] [CrossRef]
- Roos, S.; Sofou, K.; Hedberg-Oldfors, C.; Kollberg, G.; Lindgren, U.; Thomsen, C.; Tulinius, M.; Oldfors, A. Mitochondrial Complex IV Deficiency Caused by a Novel Frameshift Variant in MT-CO2 Associated with Myopathy and Perturbed Acylcarnitine Profile. Eur. J. Hum. Genet. 2019, 27, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Taanman, J.W.; Cooper, J.M.; Nelson, I.; Hargreaves, I.; Meunier, B.; Hanna, M.G.; García, J.J.; Capaldi, R.A.; Lake, B.D.; et al. A Missense Mutation of Cytochrome Oxidase Subunit II Causes Defective Assembly and Myopathy. Am. J. Hum. Genet. 1999, 65, 1030–1039. [Google Scholar] [CrossRef] [Green Version]
- Mkaouar-Rebai, E.; Ellouze, E.; Chamkha, I.; Kammoun, F.; Triki, C.; Fakhfakh, F. Molecular-Clinical Correlation in a Family with a Novel Heteroplasmic Leigh Syndrome Missense Mutation in the Mitochondrial Cytochrome c Oxidase III Gene. J. Child Neurol. 2011, 26, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Horváth, R.; Scharfe, C.; Hoeltzenbein, M.; Do, B.H.; Schröder, C.; Warzok, R.; Vogelgesang, S.; Lochmüller, H.; Müller-Höcker, J.; Gerbitz, K.D.; et al. Childhood Onset Mitochondrial Myopathy and Lactic Acidosis Caused by a Stop Mutation in the Mitochondrial Cytochrome c Oxidase III Gene. J. Med. Genet. 2002, 39, 812–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, M.G.; Nelson, I.P.; Rahman, S.; Lane, R.J.M.; Land, J.; Heales, S.; Cooper, M.J.; Schapira, A.H.V.; Morgan-Hughes, J.A.; Wood, N.W. Cytochrome c Oxidase Deficiency Associated with the First Stop-Codon Point Mutation in Human MtDNA. Am. J. Hum. Genet. 1998, 63, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marotta, R.; Chin, J.; Kirby, D.M.; Chiotis, M.; Cook, M.; Collins, S.J. Novel Single Base Pair COX III Subunit Deletion of Mitochondrial DNA Associated with Rhabdomyolysis. J. Clin. Neurosci. 2011, 18, 290–292. [Google Scholar] [CrossRef]
- Tiranti, V.; Hoertnagel, K.; Carrozzo, R.; Galimberti, C.; Munaro, M.; Granatiero, M.; Zelante, L.; Gasparini, P.; Marzella, R.; Rocchi, M.; et al. Mutations of SURF-1 in Leigh Disease Associated with Cytochrome c Oxidase Deficiency. Am. J. Hum. Genet. 1998, 63, 1609–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, C.; Biancheri, R.; Garavaglia, B.; Biedi, C.; Rossi, A.; Lamba, L.D.; Bado, M.; Greco, M.; Zeviani, M.; Minetti, C. A Novel Mutation in the Surf1 Gene in a Child with Leigh Disease, Peripheral Neuropathy, and Cytochrome-c Oxidase Deficiency. J. Child Neurol. 2002, 17, 233–236. [Google Scholar] [CrossRef]
- Kose, M.; Canda, E.; Kagnici, M.; Aykut, A.; Adebali, O.; Durmaz, A.; Bircan, A.; Diniz, G.; Eraslan, C.; Kose, E.; et al. SURF1 Related Leigh Syndrome: Clinical and Molecular Findings of 16 Patients from Turkey. Mol. Genet. Metab. Rep. 2020, 25, 100657. [Google Scholar] [CrossRef] [PubMed]
- Echaniz-Laguna, A.; Ghezzi, D.; Chassagne, M.; Mayençon, M.; Padet, S.; Melchionda, L.; Rouvet, I.; Lannes, B.; Bozon, D.; Latour, P.; et al. SURF1 Deficiency Causes Demyelinating Charcot-Marie-Tooth Disease. Neurology 2013, 81, 1523–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brischigliaro, M.; Zeviani, M. Cytochrome c Oxidase Deficiency. Biochim. Biophys. Acta—Bioenerg. 2021, 1862, 148335. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, L.C.; Sue, M.C.; Davidson, M.M.; Tanji, K.; Nishino, I.; Sadlock, J.E.; Krishna, S.; Walker, W.; Selby, J.; Glerum, D.M.; et al. Fatal Infantile Cardioencephalomyopathy with COX Deficiency and Mutations in SCO2, a COX Assembly Gene. Nat. Genet. 1999, 23, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Rebelo, A.P.; Saade, D.; Pereira, C.V.; Farooq, A.; Huff, T.C.; Abreu, L.; Moraes, C.T.; Mnatsakanova, D.; Mathews, K.; Yang, H.; et al. SCO2 Mutations Cause Early-Onset Axonal Charcot-Marie-Tooth Disease Associated with Cellular Copper Deficiency. Brain 2018, 141, 662–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dautant, A.; Meier, T.; Hahn, A.; Tribouillard-Tanvier, D.; di Rago, J.P.; Kucharczyk, R. ATP Synthase Diseases Of Mitochondrial Genetic Origin. Front. Physiol. 2018, 9, 329. [Google Scholar] [CrossRef] [PubMed]
- Childs, A.M.; Hutchin, T.; Pysden, K.; Highet, L.; Bamford, J.; Livingston, J.; Crow, Y.J. Variable Phenotype Including Leigh Syndrome with a 9185T>C Mutation in the MTATP6 Gene. Neuropediatrics 2007, 38, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Felhi, R.; Mkaouar-Rebai, E.; Sfaihi-Ben Mansour, L.; Alila-Fersi, O.; Tabebi, M.; Ben Rhouma, B.; Ammar, M.; Keskes, L.; Hachicha, M.; Fakhfakh, F. Mutational Analysis in Patients with Neuromuscular Disorders: Detection of Mitochondrial Deletion and Double Mutations in the MT-ATP6 Gene. Biochem. Biophys. Res. Commun. 2016, 473, 61–66. [Google Scholar] [CrossRef]
- Ware, S.M.; El-Hassan, N.; Kahler, S.G.; Zhang, Q.; Ma, Y.-W.; Miller, E.; Wong, B.; Spicer, R.L.; Craigen, W.J.; Kozel, B.A.; et al. Infantile Cardiomyopathy Caused by a Mutation in the Overlapping Region of Mitochondrial ATPase 6 and 8 Genes. J. Med. Genet. 2009, 46, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Imai, A.; Fujita, S.; Kishita, Y.; Kohda, M.; Tokuzawa, Y.; Hirata, T.; Mizuno, Y.; Harashima, H.; Nakaya, A.; Sakata, Y.; et al. Rapidly Progressive Infantile Cardiomyopathy with Mitochondrial Respiratory Chain Complex V Deficiency Due to Loss of ATPase 6 and 8 Protein. Int. J. Cardiol. 2016, 207, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Kytövuori, L.; Lipponen, J.; Rusanen, H.; Komulainen, T.; Martikainen, M.H.; Majamaa, K. A Novel Mutation m.8561C>G in MT-ATP6/8 Causing a Mitochondrial Syndrome with Ataxia, Peripheral Neuropathy, Diabetes Mellitus, and Hypergonadotropic Hypogonadism. J. Neurol. 2016, 263, 2188–2195. [Google Scholar] [CrossRef]
- Galimberti, C.A.; Diegoli, M.; Sartori, I.; Uggetti, C.; Brega, A.; Tartara, A.; Arbustini, E. Brain Pseudoatrophy and Mental Regression on Valproate and a Mitochondrial DNA Mutation. Neurology 2006, 67, 1715–1717. [Google Scholar] [CrossRef]
- Auré, K.; Dubourg, O.; Jardel, C.; Clarysse, L.; Sternberg, D.; Fournier, E.; Laforêt, P.; Streichenberger, N.; Petiot, P.; Gervais-Bernard, H.; et al. Episodic Weakness Due to Mitochondrial DNA MT-ATP6/8 Mutations. Neurology 2013, 81, 1810–1818. [Google Scholar] [CrossRef] [PubMed]
- Tansel, T.; Paçal, F.; Ustek, D. A Novel ATP8 Gene Mutation in an Infant with Tetralogy of Fallot. Cardiol. Young 2014, 24, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, A.I.; Hogeveen, M.; Nijtmans, L.G.J.; van den Brand, M.A.M.; Janssen, A.J.M.; Diepstra, J.H.S.; van den Brandt, F.C.A.; van den Heuvel, L.P.; Hol, F.A.; Hofste, T.G.J.; et al. A Novel Mitochondrial ATP8 Gene Mutation in a Patient with Apical Hypertrophic Cardiomyopathy and Neuropathy. J. Med. Genet. 2008, 45, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Thelen, M.P.; Wirth, B.; Kye, M.J. Mitochondrial Defects in the Respiratory Complex I Contribute to Impaired Translational Initiation via ROS and Energy Homeostasis in SMA Motor Neurons. Acta Neuropathol. Commun. 2020, 8, 223. [Google Scholar] [CrossRef] [PubMed]
- Nukui, T.; Matsui, A.; Niimi, H.; Yamamoto, M.; Matsuda, N.; Piao, J.L.; Noguchi, K.; Kitajima, I.; Nakatsuji, Y. Cerebrospinal Fluid ATP as a Potential Biomarker in Patients with Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Stroke like Episodes (MELAS). Mitochondrion 2020, 50, 145–148. [Google Scholar] [CrossRef]
- Van Vranken, J.G.; Bricker, D.K.; Dephoure, N.; Gygi, S.P.; Cox, J.E.; Thummel, C.S.; Rutter, J. SDHAF4 Promotes Mitochondrial Succinate Dehydrogenase Activity and Prevents Neurodegeneration. Cell Metab. 2014, 20, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Rizwan, M.; Rasheed, H.A.; Tarjan, G. Succinate Dehydrogenase Complex: An Updated Review. Arch. Pathol. Lab. Med. 2018, 142, 1564–1570. [Google Scholar] [CrossRef] [Green Version]
- Birch-Machin, M.A.; Taylor, R.W.; Cochran, B.; Ackrell, B.A.C.; Turnbull, D.M. Late-Onset Optic Atrophy, Ataxia, and Myopathy Associated with a Mutation of a Complex II Gene. Ann. Neurol. 2000, 48, 330–335. [Google Scholar] [CrossRef]
- Fernández-Vizarra, E.; Zeviani, M. Nuclear Gene Mutations as the Cause of Mitochondrial Complex III Deficiency. Front. Genet. 2015, 6, 134. [Google Scholar] [CrossRef]
- Morgan-hughes, J.A.; Hayes, D.J.; Clark, J.B.; Landon, D.N.; Swash, M.; Stark, R.J.; Rudge, P. Mitochondrial Encephalomyopathies: Biochemical Studies in Two Cases Revealing Defects in the Respiratory Chain. Brain 1982, 105, 553–582. [Google Scholar] [CrossRef]
- Darley Usmar, V.M.; Kennaway, N.G.; Buist, N.R.M.; Capaldi, R.A. Deficiency in Ubiquinone Cytochrome c Reductase in a Patient with Mitochondrial Myopathy and Lactic Acidosis. Proc. Natl. Acad. Sci. USA 1983, 80, 5103–5106. [Google Scholar] [CrossRef] [Green Version]
- Rana, M.; De Coo, I.; Diaz, F.; Smeets, H.; Moraes, C.T. An Out-of-Frame Cytochrome b Gene Deletion from a Patient with Parkinsonism Is Associated with Impaired Complex III Assembly and an Increase in Free Radical Production. Ann. Neurol. 2000, 48, 774–781. [Google Scholar] [CrossRef]
- Ghezzi, D.; Arzuffi, P.; Zordan, M.; Da Re, C.; Lamperti, C.; Benna, C.; D’Adamo, P.; Diodato, D.; Costa, R.; Mariotti, C.; et al. Mutations in TTC19 Cause Mitochondrial Complex III Deficiency and Neurological Impairment in Humans and Flies. Nat. Genet. 2011, 43, 259–263. [Google Scholar] [CrossRef] [PubMed]
- De Lonlay, P.; Valnot, I.; Barrientos, A.; Gorbatyuk, M.; Tzagoloff, A.; Taanman, J.W.; Benayoun, E.; Chrétien, D.; Kadhom, N.; Lombès, A.; et al. A Mutant Mitochondrial Respiratory Chain Assembly Protein Causes Complex III Deficiency in Patients with Tubulopathy, Encephalopathy and Liver Failure. Nat. Genet. 2001, 29, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Morán, M.; Marín-Buera, L.; Carmen Gil-Borlado, M.; Rivera, H.; Blázquez, A.; Seneca, S.; Vázquez-López, M.; Arenas, J.; Martín, M.A.; Ugalde, C. Cellular Pathophysiological Consequences of BCS1L Mutations in Mitochondrial Complex III Enzyme Deficiency. Hum. Mutat. 2010, 31, 930–941. [Google Scholar] [CrossRef] [Green Version]
- Zierz, C.M.; Baty, K.; Blakely, E.L.; Hopton, S.; Falkous, G.; Schaefer, A.M.; Hadjivassiliou, M.; Sarrigiannis, P.G.; Ng, Y.S.; Taylor, R.W. A Novel Pathogenic Variant in MT-CO2 Causes an Isolated Mitochondrial Complex IV Deficiency and Late-Onset Cerebellar Ataxia. J. Clin. Med. 2019, 8, 789. [Google Scholar] [CrossRef] [Green Version]
- Brix, N.; Jensen, J.M.; Pedersen, I.S.; Ernst, A.; Frost, S.; Bogaard, P.; Petersen, M.B.; Bender, L. Mitochondrial Disease Caused by a Novel Homozygous Mutation (Gly106del) in the SCO1 Gene. Neonatology 2019, 116, 290–294. [Google Scholar] [CrossRef]
- Jaksch, M.; Paret, C.; Stucka, R.; Horn, N.; Muller-Höcker, J.; Horvath, R.; Trepesch, N.; Stecker, G.; Freisinger, P.; Thirion, C.; et al. Cytochrome c Oxidase Deficiency Due to Mutations in SCO2, Encoding a Mitochondrial Copper-Binding Protein, Is Rescued by Copper in Human Myoblasts. Hum. Mol. Genet. 2001, 10, 3025–3035. [Google Scholar] [CrossRef] [Green Version]
- Verdijk, R.M.; De Krijger, R.; Schoonderwoerd, K.; Tiranti, V.; Smeets, H.; Govaerts, L.C.P.; De Coo, R. Phenotypic Consequences of a Novel SCO2 Gene Mutation. Am. J. Med. Genet. A 2008, 146, 2822–2827. [Google Scholar] [CrossRef]
- Matsuyama, S.; Reed, J.C. Mitochondria-Dependent Apoptosis and Cellular PH Regulation. Cell Death Differ. 2000, 7, 1155–1165. [Google Scholar] [CrossRef] [Green Version]
- Sunitha, B.; Gayathri, N.; Kumar, M.; Keshava Prasad, T.S.; Nalini, A.; Padmanabhan, B.; Srinivas Bharath, M.M. Muscle Biopsies from Human Muscle Diseases with Myopathic Pathology Reveal Common Alterations in Mitochondrial Function. J. Neurochem. 2016, 138, 174–191. [Google Scholar] [CrossRef] [Green Version]
- Alexiou, A.; Nizami, B.; Khan, F.I.; Soursou, G.; Vairaktarakis, C.; Chatzichronis, S.; Tsiamis, V.; Manztavinos, V.; Yarla, N.S.; Ashraf, G.M. Mitochondrial Dynamics and Proteins Related to Neurodegenerative Diseases. Curr. Protein Pept. Sci. 2017, 19, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Frattaruolo, L.; Brindisi, M.; Curcio, R.; Marra, F.; Dolce, V.; Cappello, A.R. Targeting the Mitochondrial Metabolic Network: A Promising Strategy in Cancer Treatment. Int. J. Mol. Sci. 2020, 21, 6014. [Google Scholar] [CrossRef]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 Play Distinct Roles in Mitochondrial Fusion Reactions via GTPase Activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear Gene OPA1, Encoding a Mitochondrial Dynamin-Related Protein, Is Mutated in Dominant Optic Atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, Encoding a Dynamin-Related GTPase, Is Mutated in Autosomal Dominant Optic Atrophy Linked to Chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Shankar, S.P.; Biousse, V.; Miller, N.R.; Bean, L.J.H.; Coffee, B.; Hegde, M.; Newman, N.J. Genetic Screening for OPA1 and OPA3 Mutations in Patients with Suspected Inherited Optic Neuropathies. Ophthalmology 2011, 118, 558–563. [Google Scholar] [CrossRef] [Green Version]
- Maresca, A.; la Morgia, C.; Caporali, L.; Valentino, M.L.; Carelli, V. The Optic Nerve: A “Mito-Window” on Mitochondrial Neurodegeneration. Mol. Cell. Neurosci. 2013, 55, 62–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu-Wai-Man, P.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer-Grumbach, M.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-System Neurological Disease Is Common in Patients with OPA1 Mutations. Brain 2010, 133, 771–786. [Google Scholar] [CrossRef]
- Amati-Bonneau, P.; Valentino, M.L.; Reynier, P.; Gallardo, M.E.; Bornstein, B.; Boissière, A.; Campos, Y.; Rivera, H.; De La Aleja, J.G.; Carroccia, R.; et al. OPA1 Mutations Induce Mitochondrial DNA Instability and Optic Atrophy “plus” Phenotypes. Brain 2008, 131, 338–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurá, M.; Pipis, M.; Rossor, A.M.; Reilly, M.M. Charcot-Marie-Tooth Disease and Related Disorders: An Evolving Landscape. Curr. Opin. Neurol. 2019, 32, 641–650. [Google Scholar] [CrossRef]
- Rocha, A.G.; Franco, A.; Krezel, A.M.; Rumsey, J.M.; Alberti, J.M.; Knight, W.C.; Biris, N.; Zacharioudakis, E.; Janetka, J.W.; Baloh, R.H.; et al. MFN2 Agonists Reverse Mitochondrial Defects in Preclinical Models of Charcot-Marie-Tooth Disease Type 2A. Science 2018, 360, 336–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beutner, G.; Alavian, K.N.; Jonas, E.A.; Porter, G.A. The Mitochondrial Permeability Transition Pore and ATP Synthase. Handb. Exp. Pharmacol. 2017, 240, 21–46. [Google Scholar] [CrossRef] [PubMed]
- Sadat, R.; Barca, E.; Masand, R.; Donti, T.R.; Naini, A.; De Vivo, D.C.; DiMauro, S.; Hanchard, N.A.; Graham, B.H. Functional Cellular Analyses Reveal Energy Metabolism Defect and Mitochondrial DNA Depletion in a Case of Mitochondrial Aconitase Deficiency. Mol. Genet. Metab. 2016, 118, 28–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ait-El-Mkadem, S.; Dayem-Quere, M.; Gusic, M.; Chaussenot, A.; Bannwarth, S.; François, B.; Genin, E.C.; Fragaki, K.; Volker-Touw, C.L.M.; Vasnier, C.; et al. Mutations in MDH2, Encoding a Krebs Cycle Enzyme, Cause Early-Onset Severe Encephalopathy. Am. J. Hum. Genet. 2017, 100, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Rustin, P.; Bourgeron, T.; Parfait, B.; Chretien, D.; Munnich, A.; Rötig, A. Inborn Errors of the Krebs Cycle: A Group of Unusual Mitochondrial Diseases in Human. Biochim. Biophys. Acta—Mol. Basis Dis. 1997, 1361, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Wieser, T. Carnitine Palmitoyltransferase II Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Bouchireb, K.; Teychene, A.M.; Rigal, O.; De Lonlay, P.; Valayannopoulos, V.; Gaudelus, J.; Sellier, N.; Bonnefont, J.P.; Brivet, M.; De Pontual, L. Post-Mortem MRI Reveals CPT2 Deficiency after Sudden Infant Death. Eur. J. Pediatr. 2010, 169, 1561–1563. [Google Scholar] [CrossRef]
- Boemer, F.; Deberg, M.; Schoos, R.; Caberg, J.H.; Gaillez, S.; Dugauquier, C.; Delbecque, K.; François, A.; Maton, P.; Demonceau, N.; et al. Diagnostic Pitfall in Antenatal Manifestations of CPT II Deficiency. Clin. Genet. 2016, 89, 193–197. [Google Scholar] [CrossRef]
- Mathur, A.; Sims, H.F.; Gopalakrishnan, D.; Gibson, B.; Rinaldo, P.; Vockley, J.; Hug, G.; Strauss, A.W. Molecular Heterogeneity in Very-Long-Chain Acyl-CoA Dehydrogenase Deficiency Causing Pediatric Cardiomyopathy and Sudden Death. Circulation 1999, 99, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Macchione, F.; Salviati, L.; Bordugo, A.; Vincenzi, M.; Camilot, M.; Teofoli, F.; Pancheri, E.; Zordan, R.; Bertolin, C.; Rossi, S.; et al. Multiple Acyl-COA Dehydrogenase Deficiency in Elderly Carriers. J. Neurol. 2020, 267, 1414–1419. [Google Scholar] [CrossRef] [PubMed]
- Grünert, S.C. Clinical and Genetical Heterogeneity of Late-Onset Multiple Acyl-Coenzyme A Dehydrogenase Deficiency. Orphanet J. Rare Dis. 2014, 9, 117. [Google Scholar] [CrossRef] [Green Version]
- Ronchi, D.; Monfrini, E.; Bonato, S.; Mancinelli, V.; Cinnante, C.; Salani, S.; Bordoni, A.; Ciscato, P.; Fortunato, F.; Villa, M.; et al. Dystonia-Ataxia Syndrome with Permanent Torsional Nystagmus Caused by ECHS1 Deficiency. Ann. Clin. Transl. Neurol. 2020, 7, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, R.; Pines, O.; Ta-Shma, A.; Burak, E.; Shaag, A.; Halvardson, J.; Edvardson, S.; Mahajna, M.; Zenvirt, S.; Saada, A.; et al. Infantile Cerebellar-Retinal Degeneration Associated with a Mutation in Mitochondrial Aconitase, ACO2. Am. J. Hum. Genet. 2012, 90, 518–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizo, J.; Xu, J. The Synaptic Vesicle Release Machinery. Annu. Rev. Biophys. 2015, 44, 339–367. [Google Scholar] [CrossRef] [PubMed]
- Ugur, B.; Bao, H.; Stawarski, M.; Duraine, L.R.; Zuo, Z.; Lin, Y.Q.; Neely, G.G.; Macleod, G.T.; Chapman, E.R.; Bellen, H.J. The Krebs Cycle Enzyme Isocitrate Dehydrogenase 3A Couples Mitochondrial Metabolism to Synaptic Transmission. Cell Rep. 2017, 21, 3794–3806. [Google Scholar] [CrossRef] [Green Version]
- Debashree, B.; Kumar, M.; Keshava Prasad, T.S.; Natarajan, A.; Christopher, R.; Nalini, A.; Bindu, P.S.; Gayathri, N.; Srinivas Bharath, M.M. Mitochondrial Dysfunction in Human Skeletal Muscle Biopsies of Lipid Storage Disorder. J. Neurochem. 2018, 145, 323–341. [Google Scholar] [CrossRef]
- Vockley, J.; Whiteman, D.A.H. Defects of Mitochondrial β-Oxidation: A Growing Group of Disorders. Neuromuscul. Disord. 2002, 12, 235–246. [Google Scholar] [CrossRef]
- Laforêt, P.; Vianey-Saban, C. Disorders of Muscle Lipid Metabolism: Diagnostic and Therapeutic Challenges. Neuromuscul. Disord. NMD 2010, 20, 693–700. [Google Scholar] [CrossRef]
- Joshi, P.R.; Young, P.; Deschauer, M.; Zierz, S. Expanding Mutation Spectrum in CPT II Gene: Identification of Four Novel Mutations. J. Neurol. 2013, 260, 1412–1414. [Google Scholar] [CrossRef]
- Dimauro, S.; Dimauro, P.M.M. Muscle Carnitine Palmityltransferase Deficiency and Myoglobinuria. Science 1973, 182, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Watson, K.S.; Boukhloufi, I.; Bowerman, M.; Parson, S.H. The Relationship between Body Composition, Fatty Acid Metabolism and Diet in Spinal Muscular Atrophy. Brain Sci. 2021, 11, 131. [Google Scholar] [CrossRef]
- van Maldegem, B.T.; Duran, M.; Wanders, R.J.A.; Niezen-Koning, K.E.; Hogeveen, M.; Ijlst, L.; Waterham, H.R.; Wijburg, F.A. Clinical, Biochemical, and Genetic Heterogeneity in Short-Chain Acyl-Coenzyme A Dehydrogenase Deficiency. JAMA 2006, 296, 943. [Google Scholar] [CrossRef] [Green Version]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J.A. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leslie, N.D.; Valencia, C.A.; Strauss, A.W.; Zhang, K. Very Long-Chain Acyl-Coenzyme a Dehydrogenase Deficiency; University of Washington: Seattle, WA, USA, 2021. [Google Scholar]
- Peters, H.; Buck, N.; Wanders, R.; Ruiter, J.; Waterham, H.; Koster, J.; Yaplito-Lee, J.; Ferdinandusse, S.; Pitt, J. ECHS1 Mutations in Leigh Disease: A New Inborn Error of Metabolism Affecting Valine Metabolism. Brain J. Neurol. 2014, 137, 2903–2908. [Google Scholar] [CrossRef] [Green Version]
- Peters, H.; Ferdinandusse, S.; Ruiter, J.P.; Wanders, R.J.A.; Boneh, A.; Pitt, J. Metabolite Studies in HIBCH and ECHS1 Defects: Implications for Screening. Mol. Genet. Metab. 2015, 115, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F. The Mitochondrial Transporter Family SLC25: Identification, Properties and Physiopathology. Mol. Aspects Med. 2013, 34, 465–484. [Google Scholar] [CrossRef] [PubMed]
- Carrisi, C.; Antonucci, D.; Lunetti, P.; Migoni, D.; Girelli, C.R.; Dolce, V.; Fanizzi, F.P.; Benedetti, M.; Capobianco, L. Transport of Platinum Bonded Nucleotides into Proteoliposomes, Mediated by Drosophila Melanogaster Thiamine Pyrophosphate Carrier Protein (DmTpc1). J. Inorg. Biochem. 2014, 130, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Curcio, R.; Lunetti, P.; Zara, V.; Ferramosca, A.; Marra, F.; Fiermonte, G.; Cappello, A.R.; De Leonardis, F.; Capobianco, L.; Dolce, V. Drosophila Melanogaster Mitochondrial Carriers: Similarities and Differences with the Human Carriers. Int. J. Mol. Sci. 2020, 21, 6052. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F.; Scarcia, P.; Monné, M. Diseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: A Review. Biomolecules 2020, 10, 655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaouch, A.; Porcelli, V.; Cox, D.; Edvardson, S.; Scarcia, P.; De Grassi, A.; Pierri, C.L.; Cossins, J.; Laval, S.H.; Griffin, H.; et al. Mutations in the Mitochondrial Citrate Carrier SLC25A1 Are Associated with Impaired Neuromuscular Transmission. J. Neuromuscul. Dis. 2014, 1, 75–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pop, A.; Williams, M.; Struys, E.A.; Monné, M.; Jansen, E.E.W.; De Grassi, A.; Kanhai, W.A.; Scarcia, P.; Ojeda, M.R.F.; Porcelli, V.; et al. An Overview of Combined D-2- and L-2-Hydroxyglutaric Aciduria: Functional Analysis of CIC Variants. J. Inherit. Metab. Dis. 2018, 41, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Finsterer, J. Congenital Myasthenic Syndromes. Orphanet J. Rare Dis. 2019, 14, 57. [Google Scholar] [CrossRef]
- Balaraju, S.; Töpf, A.; McMacken, G.; Kumar, V.P.; Pechmann, A.; Roper, H.; Vengalil, S.; Polavarapu, K.; Nashi, S.; Mahajan, N.P.; et al. Congenital Myasthenic Syndrome with Mild Intellectual Disability Caused by a Recurrent SLC25A1 Variant. Eur. J. Hum. Genet. 2020, 28, 373–377. [Google Scholar] [CrossRef] [Green Version]
- Mayr, J.A.; Merkel, O.; Kohlwein, S.D.; Gebhardt, B.R.; Böhles, H.; Fötschl, U.; Koch, J.; Jaksch, M.; Lochmüller, H.; Horváth, R.; et al. Mitochondrial Phosphate-Carrier Deficiency: A Novel Disorder of Oxidative Phosphorylation. Am. J. Hum. Genet. 2007, 80, 478–484. [Google Scholar] [CrossRef] [Green Version]
- Mayr, J.A.; Zimmermann, F.A.; Horváth, R.; Schneider, H.C.; Schoser, B.; Holinski-Feder, E.; Czermin, B.; Freisinger, P.; Sperl, W. Deficiency of the Mitochondrial Phosphate Carrier Presenting as Myopathy and Cardiomyopathy in a Family with Three Affected Children. Neuromuscul. Disord. 2011, 21, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Bhoj, E.J.; Li, M.; Ahrens-Nicklas, R.; Pyle, L.C.; Wang, J.; Zhang, V.W.; Clarke, C.; Wong, L.J.; Sondheimer, N.; Ficicioglu, C.; et al. Pathologic variants of the mitochondrial phosphate carrier SLC25A3: Two new patients and expansion of the cardiomyopathy/skeletal myopathy phenotype with and without lactic acidosis. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2015; Volume 19. [Google Scholar]
- Kwong, J.Q.; Davis, J.; Baines, C.P.; Sargent, M.A.; Karch, J.; Wang, X.; Huang, T.; Molkentin, J.D. Genetic Deletion of the Mitochondrial Phosphate Carrier Desensitizes the Mitochondrial Permeability Transition Pore and Causes Cardiomyopathy. Cell Death Differ. 2014, 21, 1209–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorgoglione, R.; Porcelli, V.; Santoro, A.; Daddabbo, L.; Vozza, A.; Monné, M.; Di Noia, M.A.; Palmieri, L.; Fiermonte, G.; Palmieri, F. The Human Uncoupling Proteins 5 and 6 (UCP5/SLC25A14 and UCP6/SLC25A30) Transport Sulfur Oxyanions, Phosphate and Dicarboxylates. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 724–733. [Google Scholar] [CrossRef]
- Fontanesi, F.; Palmieri, L.; Scarcia, P.; Lodi, T.; Donnini, C.; Limongelli, A.; Tiranti, V.; Zeviani, M.; Ferrero, I.; Viola, A.M. Mutations in AAC2, Equivalent to Human AdPEO-Associated ANT1 Mutations, Lead to Defective Oxidative Phosphorylation in Saccharomyces Cerevisiae and Affect Mitochondrial DNA Stability. Hum. Mol. Genet. 2004, 13, 923–934. [Google Scholar] [CrossRef]
- Palmieri, L.; Alberio, S.; Pisano, I.; Lodi, T.; Meznaric-Petrusa, M.; Zidar, J.; Santoro, A.; Scarcia, P.; Fontanesi, F.; Lamantea, E.; et al. Complete Loss-of-Function of the Heart/Muscle-Specific Adenine Nucleotide Translocator Is Associated with Mitochondrial Myopathy and Cardiomyopathy. Hum. Mol. Genet. 2005, 14, 3079–3088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, K.; Majd, H.; Dallabona, C.; Reinson, K.; King, M.S.; Alston, C.L.; He, L.; Lodi, T.; Jones, S.A.; Fattal-Valevski, A.; et al. Recurrent De Novo Dominant Mutations in SLC25A4 Cause Severe Early-Onset Mitochondrial Disease and Loss of Mitochondrial DNA Copy Number. Am. J. Hum. Genet. 2016, 99, 860–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, M.S.; Thompson, K.; Hopton, S.; He, L.; Kunji, E.R.S.; Taylor, R.W.; Ortiz-Gonzalez, X.R. Expanding the Phenotype of de Novo SLC25A4 -Linked Mitochondrial Disease to Include Mild Myopathy. Neurol. Genet. 2018, 4, e256. [Google Scholar] [CrossRef] [Green Version]
- Profilo, E.; Peña-Altamira, L.E.; Corricelli, M.; Castegna, A.; Danese, A.; Agrimi, G.; Petralla, S.; Giannuzzi, G.; Porcelli, V.; Sbano, L.; et al. Down-Regulation of the Mitochondrial Aspartate-Glutamate Carrier Isoform 1 AGC1 Inhibits Proliferation and N-Acetylaspartate Synthesis in Neuro2A Cells. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1422–1435. [Google Scholar] [CrossRef]
- Diane Shelton, G.; Minor, K.M.; Li, K.; Naviaux, J.C.; Monk, J.; Wang, L.; Guzik, E.; Guo, L.T.; Porcelli, V.; Gorgoglione, R.; et al. A Mutation in the Mitochondrial Aspartate/Glutamate Carrier Leads to a More Oxidizing Intramitochondrial Environment and an Inflammatory Myopathy in Dutch Shepherd Dogs. J. Neuromuscul. Dis. 2019, 6, 485–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanley, C.A.; Hale, D.E.; Berry, G.T.; Deleeuw, S.; Boxer, J.; Bonnefont, J.P. Brief Report: A Deficiency of Carnitine-Acylcarnitine Translocase in the Inner Mitochondrial Membrane. N. Engl. J. Med. 1992, 327, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Indiveri, C.; Iacobazzi, V.; Tonazzi, A.; Giangregorio, N.; Infantino, V.; Convertini, P.; Console, L.; Palmieri, F. The Mitochondrial Carnitine/Acylcarnitine Carrier: Function, Structure and Physiopathology. Mol. Aspects Med. 2011, 32, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Boczonadi, V.; King, M.S.; Smith, A.C.; Olahova, M.; Bansagi, B.; Roos, A.; Eyassu, F.; Borchers, C.; Ramesh, V.; Lochmüller, H.; et al. Mitochondrial Oxodicarboxylate Carrier Deficiency Is Associated with Mitochondrial DNA Depletion and Spinal Muscular Atrophy–like Disease. Genet. Med. 2018, 20, 1224–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiff, M.; Veauville-Merllié, A.; Su, C.H.; Tzagoloff, A.; Rak, M.; Ogier de Baulny, H.; Boutron, A.; Smedts-Walters, H.; Romero, N.B.; Rigal, O.; et al. SLC25A32 Mutations and Riboflavin-Responsive Exercise Intolerance. N. Engl. J. Med. 2016, 374, 795–797. [Google Scholar] [CrossRef] [Green Version]
- Hellebrekers, D.M.E.I.; Sallevelt, S.C.E.H.; Theunissen, T.E.J.; Hendrickx, A.T.M.; Gottschalk, R.W.; Hoeijmakers, J.G.J.; Habets, D.D.; Bierau, J.; Schoonderwoerd, K.G.; Smeets, H.J.M. Novel SLC25A32 Mutation in a Patient with a Severe Neuromuscular Phenotype. Eur. J. Hum. Genet. 2017, 25, 886–888. [Google Scholar] [CrossRef]
- Shamseldin, H.E.; Smith, L.L.; Kentab, A.; Alkhalidi, H.; Summers, B.; Alsedairy, H.; Xiong, Y.; Gupta, V.A.; Alkuraya, F.S. Mutation of the Mitochondrial Carrier SLC25A42 Causes a Novel Form of Mitochondrial Myopathy in Humans. Hum. Genet. 2015, 135, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Almannai, M.; Alasmari, A.; Alqasmi, A.; Faqeih, E.; Al Mutairi, F.; Alotaibi, M.; Samman, M.M.; Eyaid, W.; Aljadhai, Y.I.; Shamseldin, H.E.; et al. Expanding the Phenotype of SLC25A42-Associated Mitochondrial Encephalomyopathy. Clin. Genet. 2018, 93, 1097–1102. [Google Scholar] [CrossRef]
- Aldosary, M.; Baselm, S.; Abdulrahim, M.; Almass, R.; Alsagob, M.; AlMasseri, Z.; Huma, R.; AlQuait, L.; Al-Shidi, T.; Al-Obeid, E.; et al. SLC25A42-Associated Mitochondrial Encephalomyopathy: Report of Additional Founder Cases and Functional Characterization of a Novel Deletion. JIMD Rep. 2021, 60, 75–87. [Google Scholar] [CrossRef]
- Van Dijk, T.; Rudnik-Schöneborn, S.; Senderek, J.; Hajmousa, G.; Mei, H.; Dusl, M.; Aronica, E.; Barth, P.; Baas, F. Pontocerebellar Hypoplasia with Spinal Muscular Atrophy (PCH1): Identification of SLC25A46 Mutations in the Original Dutch PCH1 Family. Brain 2017, 140, e46. [Google Scholar] [CrossRef]
- Ali, M.S.; Suda, K.; Kowada, R.; Ueoka, I.; Yoshida, H.; Yamaguchi, M. Neuron-Specific Knockdown of Solute Carrier Protein SLC25A46a Induces Locomotive Defects, an Abnormal Neuron Terminal Morphology, Learning Disability, and Shortened Lifespan. IBRO Rep. 2020, 8, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F. Mitochondrial Transporters of the SLC25 Family and Associated Diseases: A Review. J. Inherit. Metab. Dis. 2014, 37, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Dolce, V.; Cappello, A.R.; Capobianco, L. Mitochondrial Tricarboxylate and Dicarboxylate-Tricarboxylate Carriers: From Animals to Plants. IUBMB Life 2014, 66, 462–471. [Google Scholar] [CrossRef]
- Edvardson, S.; Porcelli, V.; Jalas, C.; Soiferman, D.; Kellner, Y.; Shaag, A.; Korman, S.H.; Pierri, C.L.; Scarcia, P.; Fraenkel, N.D.; et al. Agenesis of Corpus Callosum and Optic Nerve Hypoplasia Due to Mutations in SLC25A1 Encoding the Mitochondrial Citrate Transporter. J. Med. Genet. 2013, 50, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, M.; Zhang, L.; Shi, Y.; Zhao, L.; Wu, B.; Li, X.; Zhou, S. A Case Report of an Intermediate Phenotype between Congenital Myasthenic Syndrome and D-2- and L-2-Hydroxyglutaric Aciduria Due to Novel SLC25A1 Variants. BMC Neurol. 2020, 20, 278. [Google Scholar] [CrossRef]
- Dolce, V.; Fiermonte, G.; Palmieri, F. Tissue-Specific Expression of the Two Isoforms of the Mitochondrial Phosphate Carrier in Bovine Tissues. FEBS Lett. 1996, 399, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Fiermonte, G.; Dolce, V.; Palmieri, F. Expression in Escherichia Coli, Functional Characterization, and Tissue Distribution of Isoforms A and B of the Phosphate Carrier from Bovine Mitochondria. J. Biol. Chem. 1998, 273, 22782–22787. [Google Scholar] [CrossRef] [Green Version]
- Fiermonte, G.; De Leonardis, F.; Todisco, S.; Palmieri, L.; Lasorsa, F.M.; Palmieri, F. Identification of the Mitochondrial ATP-Mg/Pi Transporter. Bacterial Expression, Reconstitution, Functional Characterization, and Tissue Distribution. J. Biol. Chem. 2004, 279, 30722–30730. [Google Scholar] [CrossRef] [Green Version]
- Fiermonte, G.; Palmieri, L.; Dolce, V.; Lasorsa, F.M.; Palmieri, F.; Runswick, M.J.; Walker, J.E. The Sequence, Bacterial Expression, and Functional Reconstitution of the Rat Mitochondrial Dicarboxylate Transporter Cloned via Distant Homologs in Yeast and Caenorhabditis Elegans. J. Biol. Chem. 1998, 273, 24754–24759. [Google Scholar] [CrossRef] [Green Version]
- Raho, S.; Capobianco, L.; Malivindi, R.; Vozza, A.; Piazzolla, C.; De Leonardis, F.; Gorgoglione, R.; Scarcia, P.; Pezzuto, F.; Agrimi, G.; et al. KRAS-Regulated Glutamine Metabolism Requires UCP2-Mediated Aspartate Transport to Support Pancreatic Cancer Growth. Nat. Metab. 2020, 2, 1373–1381. [Google Scholar] [CrossRef]
- Stepien, G.; Torroni, A.; Chung, A.B.; Hodge, J.A.; Wallaces, D.C. Differential Expression of Adenine Nucleotide Translocator Isoforms in Mammalian Tissues and during Muscle Cell Differentiation. J. Biol. Chem. 1992, 267, 14592–14597. [Google Scholar] [CrossRef]
- Li, Y.; Cappello, A.R.; Muto, L.; Martello, E.; Madeo, M.; Curcio, R.; Lunetti, P.; Raho, S.; Zaffino, F.; Frattaruolo, L.; et al. Functional Characterization of the Partially Purified Sac1p Independent Adenine Nucleotide Transport System (ANTS) from Yeast Endoplasmic Reticulum. J. Biochem. 2018, 164, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Farina, F.; Alberti, A.; Breuil, N.; Bolotin-Fukuhara, M.; Pinto, M.; Culetto, E. Differential Expression Pattern of the Four Mitochondrial Adenine Nucleotide Transporter Ant Genes and Their Roles during the Development of Caenorhabditis Elegans. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2008, 237, 1668–1681. [Google Scholar] [CrossRef]
- di Punzio, G.; Di Noia, M.A.; Delahodde, A.; Sellem, C.; Donnini, C.; Palmieri, L.; Lodi, T.; Dallabona, C. A Yeast-Based Screening Unravels Potential Therapeutic Molecules for Mitochondrial Diseases Associated with Dominant ANT1 Mutations. Int. J. Mol. Sci. 2021, 22, 4461. [Google Scholar] [CrossRef]
- Palmieri, L.; Pardo, B.; Lasorsa, F.M.; Del Arco, A.; Kobayashi, K.; Iijima, M.; Runswick, M.J.; Walker, J.E.; Saheki, T.; Satrústegui, J.; et al. Citrin and Aralar1 Are Ca(2+)-Stimulated Aspartate/Glutamate Transporters in Mitochondria. EMBO J. 2001, 20, 5060–5069. [Google Scholar] [CrossRef]
- Lunetti, P.; Marsano, R.M.; Curcio, R.; Dolce, V.; Fiermonte, G.; Cappello, A.R.; Marra, F.; Moschetti, R.; Li, Y.; Aiello, D.; et al. The Mitochondrial Aspartate/Glutamate Carrier (AGC or Aralar1) Isoforms in D. Melanogaster: Biochemical Characterization, Gene Structure, and Evolutionary Analysis. Biochim. Biophys. Acta—Gen. Subj. 2021, 1865, 129854. [Google Scholar] [CrossRef]
- Pfeiffer, B.; Sen, K.; Kaur, S.; Pappas, K. Expanding Phenotypic Spectrum of Cerebral Aspartate-Glutamate Carrier Isoform 1 (AGC1) Deficiency. Neuropediatrics 2020, 51, 160–163. [Google Scholar] [CrossRef]
- Wibom, R.; Lasorsa, F.M.; Töhönen, V.; Barbaro, M.; Sterky, F.H.; Kucinski, T.; Naess, K.; Jonsson, M.; Pierri, C.L.; Palmieri, F.; et al. AGC1 Deficiency Associated with Global Cerebral Hypomyelination. N. Engl. J. Med. 2009, 361, 489–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, M.J.; Li, D.; Gai, X.; McCormick, E.; Place, E.; Lasorsa, F.M.; Otieno, F.G.; Hou, C.; Kim, C.E.; Abdel-Magid, N.; et al. AGC1 deficiency causes infantile epilepsy, abnormal myelination, and reduced N-acetylaspartate. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2014; Volume 14. [Google Scholar]
- Fiermonte, G.; Dolce, V.; Palmieri, L.; Ventura, M.; Runswick, M.J.; Palmieri, F.; Walker, J.E. Identification of the Human Mitochondrial Oxodicarboxylate Carrier. Bacterial Expression, Reconstitution, Functional Characterization, Tissue Distribution, and Chromosomal Location. J. Biol. Chem. 2001, 276, 8225–8230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Titus, S.A.; Moran, R.G. Retrovirally Mediated Complementation of the GlyB Phenotype. Cloning of a Human Gene Encoding the Carrier for Entry of Folates into Mitochondria. J. Biol. Chem. 2000, 275, 36811–36817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spaan, A.N.; Ijlst, L.; Van Roermund, C.W.T.; Wijburg, F.A.; Wanders, R.J.A.; Waterham, H.R. Identification of the Human Mitochondrial FAD Transporter and Its Potential Role in Multiple Acyl-CoA Dehydrogenase Deficiency. Mol. Genet. Metab. 2005, 86, 441–447. [Google Scholar] [CrossRef]
- Fiermonte, G.; Paradies, E.; Todisco, S.; Marobbio, C.M.T.; Palmieri, F. A Novel Member of Solute Carrier Family 25 (SLC25A42) Is a Transporter of Coenzyme A and Adenosine 3′,5′-Diphosphate in Human Mitochondria. J. Biol. Chem. 2009, 284, 18152–18159. [Google Scholar] [CrossRef] [Green Version]
- Iuso, A.; Alhaddad, B.; Weigel, C.; Kotzaeridou, U.; Mastantuono, E.; Schwarzmayr, T.; Graf, E.; Terrile, C.; Prokisch, H.; Strom, T.M.; et al. A homozygous splice site mutation in SLC25A42, encoding the mitochondrial transporter of coenzyme a, causes metabolic crises and epileptic encephalopathy. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2019; Volume 44. [Google Scholar]
- Vozza, A.; De Leonardis, F.; Paradies, E.; De Grassi, A.; Pierri, C.L.; Parisi, G.; Marobbio, C.M.T.; Lasorsa, F.M.; Muto, L.; Capobianco, L.; et al. Biochemical Characterization of a New Mitochondrial Transporter of Dephosphocoenzyme A in Drosophila Melanogaster. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Hammer, M.B.; Ding, J.; Mochel, F.; Eleuch-Fayache, G.; Charles, P.; Coutelier, M.; Gibbs, J.R.; Arepalli, S.K.; Chong, S.B.; Hernandez, D.G.; et al. SLC25A46 Mutations Associated with Autosomal Recessive Cerebellar Ataxia in North African Families. Neurodegener. Dis. 2017, 17, 208–212. [Google Scholar] [CrossRef]
- Sulaiman, R.A.; Patel, N.; Alsharif, H.; Arold, S.T.; Alkuraya, F.S. A Novel Mutation in SLC25A46 Causes Optic Atrophy and Progressive Limb Spasticity, with No Cerebellar Atrophy or Axonal Neuropathy. Clin. Genet. 2017, 92, 230–231. [Google Scholar] [CrossRef] [PubMed]
- Abrams, A.J.; Hufnagel, R.B.; Rebelo, A.; Zanna, C.; Patel, N.; Gonzalez, M.A.; Campeanu, I.J.; Griffin, L.B.; Groenewald, S.; Strickland, A.V.; et al. Mutations in SLC25A46, Encoding a UGO1-like Protein, Cause an Optic Atrophy Spectrum Disorder. Nat. Genet. 2015, 47, 926–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janer, A.; Prudent, J.; Paupe, V.; Fahiminiya, S.; Majewski, J.; Sgarioto, N.; Des Rosiers, C.; Forest, A.; Lin, Z.Z.-Y.; Gingras, A.A.-C.; et al. SLC 25A46 Is Required for Mitochondrial Lipid Homeostasis and Cristae Maintenance and Is Responsible for Leigh Syndrome. EMBO Mol. Med. 2016, 8, 1019–1038. [Google Scholar] [CrossRef]
- Suda, K.; Ueoka, I.; Azuma, Y.; Muraoka, Y.; Yoshida, H.; Yamaguchi, M. Novel Drosophila Model for Mitochondrial Diseases by Targeting of a Solute Carrier Protein SLC25A46. Brain Res. 2018, 1689, 30–44. [Google Scholar] [CrossRef]
- Scoto, M.; Finkel, R.; Mercuri, E.; Muntoni, F. Genetic Therapies for Inherited Neuromuscular Disorders. Lancet Child Adolesc. Health 2018, 2, 600–609. [Google Scholar] [CrossRef]
- Heaton, R.; Millichap, L.; Saleem, F.; Gannon, J.; Begum, G.; Hargreaves, I.P. Current Biochemical Treatments of Mitochondrial Respiratory Chain Disorders. Expert Opin. Orphan Drugs 2019, 7, 277–285. [Google Scholar] [CrossRef]
- Rai, P.K.; Russell, O.M.; Lightowlers, R.N.; Turnbull, D.M. Potential Compounds for the Treatment of Mitochondrial Disease. Br. Med. Bull. 2015, 116, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Glover, E.I.; Martin, J.; Maher, A.; Thornhill, R.E.; Moran, G.R.; Tarnopolsky, M.A. A Randomized Trial of Coenzyme Q10 in Mitochondrial Disorders. Muscle Nerve 2010, 42, 739–748. [Google Scholar] [CrossRef]
- Montenegro, L.; Turnaturi, R.; Parenti, C.; Pasquinucci, L. Idebenone: Novel Strategies to Improve Its Systemic and Local Efficacy. Nanomaterials 2018, 8, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, D.S. Review of Clinical Trials for Mitochondrial Disorders: 1997-2012. Neurotherapeutics 2013, 10, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, D.; Catteruccia, M.; Piemonte, F.; Pastore, A.; Tozzi, G.; Dionisi-Vici, C.; Pontrelli, G.; Corsetti, T.; Livadiotti, S.; Kheifets, V.; et al. EPI-743 Reverses the Progression of the Pediatric Mitochondrial Disease—Genetically Defined Leigh Syndrome. Mol. Genet. Metab. 2012, 107, 383–388. [Google Scholar] [CrossRef]
- Enns, G.M.; Kinsman, S.L.; Perlman, S.L.; Spicer, K.M.; Abdenur, J.E.; Cohen, B.H.; Amagata, A.; Barnes, A.; Kheifets, V.; Shrader, W.D.; et al. Initial Experience in the Treatment of Inherited Mitochondrial Disease with EPI-743. Mol. Genet. Metab. 2012, 105, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamaguchi, H.; Kikusato, M.; Hashizume, O.; Nagatoishi, S.; Matsuo, A.; Sato, T.; Kudo, T.; Matsuhashi, T.; Murayama, K.; et al. Mitochonic Acid 5 Binds Mitochondria and Ameliorates Renal Tubular and Cardiac Myocyte Damage. J. Am. Soc. Nephrol. JASN 2016, 27, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Matsuhashi, T.; Sato, T.; Kanno, S.-I.; Suzuki, T.; Matsuo, A.; Oba, Y.; Kikusato, M.; Ogasawara, E.; Kudo, T.; Suzuki, K.; et al. Mitochonic Acid 5 (MA-5) Facilitates ATP Synthase Oligomerization and Cell Survival in Various Mitochondrial Diseases. EBioMedicine 2017, 20, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikawa, Y.; Izumi, R.; Koide, M.; Hagiwara, Y.; Kanzaki, M.; Suzuki, N.; Kikuchi, K.; Matsuhashi, T.; Akiyama, Y.; Ichijo, M.; et al. Mitochondrial Dysfunction Underlying Sporadic Inclusion Body Myositis Is Ameliorated by the Mitochondrial Homing Drug MA-5. PLoS ONE 2020, 15, e0231064. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, M.; Martin, D.A.; Hedlund, Z.; Jonsson, M.; Von, U.D.; Wedell, A. The Ketogenic Diet Compensates for AGC1 Deficiency and Improves Myelination. Epilepsia 2015, 56, 176–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernsen, P.L.; Gabreëls, F.J.; Ruitenbeek, W.; Sengers, R.C.; Stadhouders, A.M.; Renier, W.O. Successful Treatment of Pure Myopathy, Associated with Complex I Deficiency, with Riboflavin and Carnitine. Arch. Neurol. 1991, 48, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Artuch, R.; Vilaseca, M.A.; Pineda, M. Biochemical Monitoring of the Treatment in Paediatric Patients with Mitochondrial Disease. J. Inherit. Metab. Dis. 1998, 21, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Gimenes, A.C.; Bravo, D.M.; Nápolis, L.M.; Mello, M.T.; Oliveira, A.S.B.; Neder, J.A.; Nery, L.E. Effect of L-Carnitine on Exercise Performance in Patients with Mitochondrial Myopathy. Braz. J. Med. Biol. Res. Rev. Bras. Pesqui. Medicas E Biol. 2015, 48, 354–362. [Google Scholar] [CrossRef] [Green Version]
- Schoch, K.M.; Miller, T.M. Antisense Oligonucleotides: Translation from Mouse Models to Human Neurodegenerative Diseases. Neuron 2017, 94, 1056–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laing, N. Neuromuscular Disorders Genetics: What Is the Best That We Can Do? Neuromuscul. Disord. 2017, 27, S245. [Google Scholar] [CrossRef]
- Saraiva, J.; Nobre, R.J.; Pereira de Almeida, L. Gene Therapy for the CNS Using AAVs: The Impact of Systemic Delivery by AAV9. J. Controlled Release 2016, 241, 94–109. [Google Scholar] [CrossRef]
- Duan, D. Considerations on Preclinical Neuromuscular Disease Gene Therapy Studies. In Muscle Gene Therapy; Springer International Publishing: Cham, Switzerland, 2019; pp. 291–326. ISBN 978-3-030-03095-7. [Google Scholar]
- Di Meo, I.; Marchet, S.; Lamperti, C.; Zeviani, M.; Viscomi, C. AAV9-Based Gene Therapy Partially Ameliorates the Clinical Phenotype of a Mouse Model of Leigh Syndrome. Gene Ther. 2017, 24, 661–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, C.V.; Peralta, S.; Arguello, T.; Bacman, S.R.; Diaz, F.; Moraes, C.T. Myopathy Reversion in Mice after Restauration of Mitochondrial Complex I. EMBO Mol. Med. 2020, 12, e10674. [Google Scholar] [CrossRef]
- Yang, L.; Slone, J.; Li, Z.; Lou, X.; Hu, Y.-C.; Queme, L.F.; Jankowski, M.P.; Huang, T. Systemic Administration of AAV-Slc25a46 Mitigates Mitochondrial Neuropathy in Slc25a46-/- Mice. Hum. Mol. Genet. 2020, 29, 649–661. [Google Scholar] [CrossRef]
- Liufu, T.; Wang, Z. Treatment for Mitochondrial Diseases. Rev. Neurosci. 2021, 32, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Tinker, R.J.; Lim, A.Z.; Stefanetti, R.J.; McFarland, R. Current and Emerging Clinical Treatment in Mitochondrial Disease. Mol. Diagn. Ther. 2021, 25, 181–206. [Google Scholar] [CrossRef] [PubMed]
- Pitceathly, R.D.S.; Keshavan, N.; Rahman, J.; Rahman, S. Moving towards Clinical Trials for Mitochondrial Diseases. J. Inherit. Metab. Dis. 2021, 44, 22–41. [Google Scholar] [CrossRef] [PubMed]



| Gene/Protein | Function | Associated Neuromuscular Phenotype(s) | Pathogenic Mutant(s) | Reference(s) |
|---|---|---|---|---|
| Complex I Subunits | ||||
| MTDN1 | H+ translocation | MELAS, dystonia, spasticity, and myopathy | T164A (m.A3796G) G131S (m.3697A) E214K (m.G3946A) Y215H (m.T3949C) | [86,87] |
| MTDN3 | H+ translocation | Leigh syndrome | S34P (m.T10158C) | [88] |
| MTDN4 | Putative proton channel | MELAS | T109A (m.A11084G) | [89] |
| MTDN5 | Putative proton channel | MELAS | E145G (m.A12770G) M237L (m:A13045C) | [90] |
| MTDN6 | H+ translocation | MELAS | A74V (m.G14453A) | [91] |
| NDUFA1 | Assembly/stability | Leigh syndrome, hypotonia, nystagmus, and decreasedreflexes | G8R (c.G22C) R37S (c.G215C) | [92] |
| NDUFA2 | Assembly/stability | Leigh syndrome, hypertrophic cardiomyopathy, and developmental delay | Exon 2 Skipping (c.G208A+5) | [93] |
| NDUFA6 | Assembly/stability | Intrauterine growth retardation, respiratory insufficiency, and lactic acidosis | C115Y (c.G344A) | [82] |
| NDUFA8 | Assembly/stability | Severe neonatal hypotonia, dysmorphic features, and epilepsy | E190K (c.G325A) A224V (c.C671T) | [84] |
| NDUFA10 | Assembly/stability | Leigh syndrome and delayed psychomotor development | M1? (c.A1G) Q42R (c.A425G) | [94] |
| NDUFA11 | Assembly/stability | Hypertrophic cardiomyopathy and no motor development | Exon 2 skipping (IVS1_5 GtoA) | [95] |
| NDUFA12 | Assembly/stability | Leigh syndrome, progressive loss of motor abilities, scoliosis, and dystonia | R60X (c.C178T) | [96] |
| NDUFA13 | Assembly/stability | Delayed development, hypotonia, abnormal eye movements, and poor feeding | R57H (c.G170A) | [97] |
| NDUFB3 | Assembly/stability | Myopathy, hypotonia, developmental delay, and lactic acidosis | G70X (c.G208T) W20R (c.T64C) | [79,98] |
| NDUFB8 | Assembly/stability | Leigh syndrome, respiratory failure, seizures, hypotonia, cardiac hypertrophy, failure to thrive, and severely delayed psychomotor development | P76Q (c.C277A) C144W (c.C432G) | [99] |
| NDUFB9 | Assembly/stability | Progressive hypotonia | R47L (c.G140T) L64P (c.T191C) | [100] |
| NDUFB10 | Assembly/stability | Cardiomyopathy and lactic acidosis | E70X (c.206_207insT) C107S (c.T319C) | [101] |
| NDUFB11 | Assembly/stability | Hypertrophic cardiomyopathy and lactic acidosis | W85X (c.G254A) S96P (c.C286T) Y108X (c.T320G) P110S (c.C328T) | [102,103] |
| NDUFC2 | Assembly/stability | Leigh syndrome | H58L (c.A173T) c.346_*7del | [104] |
| NDUFS1 | NADH oxidation | Growth retardation, axial hypotonia, and dystonia | R241W (c.C829T) D252G (c.A863G) M707V (c.T2227G) | [105] |
| NDUFS2 | NADH oxidation | Neonatal lactic acidosis and hypertrophic cardiomyopathy | R228Q (c.G283A) P229Q (c.C686A) S413P (c.T1237C) | [106] |
| NDUFS3 | NADH oxidation | Leigh syndrome, severe axial dystonia with oral and pharyngeal motor dysfunction, dysphagia, and tetraparetic syndrome | T145I (c.C434T) R199W (c.C595T) | [80] |
| NDUFS4 | Assembly/stability | Muscular hypotonia, absence of visual and auditive attention, and cardiac defects | T96X (c.delG289) R106X (c.C316T) | [107] |
| NDUFS6 | Assembly/stability | Fatal infantile lactic acidosis | C115Y (c.G344A) | [82] |
| NDUFS7 | NADH oxidation | Leigh syndrome, feeding problems, dysarthria, and ataxia | V122M (c.G384A) | [108] |
| NDUFS8 | NADH oxidation | Leigh syndrome, poor feeding, and episodes of apnea and cyanosis | P79L (c.C236T) R102H (c.G305A) | [109] |
| NDUFV2 | NADH oxidation | Hypertrophic cardiomyopathy and truncal hypotonia | Exon 2 skipping (c.IVS2+5_+8del) | [110] |
| Complex I Assembly Proteins | ||||
| ACAD9 | Assembly | Myalgia, hypotonia, hypertrophic cardiomyopathy, and severe lactic acidosis | L98S (c.T223A) A220V (c.C659T) R414C (c.C1240T) R532T (c.C1594T) | [111,112,113,114,115] |
| FOXRED1 | Assembly | Leigh syndrome, congenital lactic acidosis, athetoid movements of the limbs in early childhood, and hypotonia | Q323X (c.C694T) N430S (c.A1289G) | [116] |
| NUBPL | Assembly | Developmental delay, short stature, myopathy, nystagmus, and ataxia | G56R (c.G166A) L104P (c.T311C) F242L (c.C726G) | [117] |
| NDUFAF1 | Assembly | Hypertrophic cardiomyopathy, developmental delay, lactic acidosis, and hypotonia | T207P (c.A1001C) K253R (c.A1140G) | [118] |
| NDUFAF2 | Assembly | Ataxia, lethargy, nystagmus, and hypotonia | R45X (c.C182T) | [119] |
| NDUFAF3 | Assembly | Axial hypotonia, no eye contact, and wide anterior fontanel | G77R (c.G229C) R122P (c.G365C) | [120] |
| NDUFAF4 | Assembly | Cardiomyopathy | L65P (c.T194C) | [120,121] |
| NDUFAF6 | Assembly | Focal seizures, decreased movement and strength, ataxia, lactic acidosis, and Leigh syndrome | Q99R (c.A296G) | [122] |
| NDUFAF8 | Assembly | Leigh syndrome | F18SfsX32 (C.45_52Ddup) F55L (c.C165G) | [123] |
| TIMMDC1 | Assembly | Infantile-onset hypotonia, delayed or minimal psychomotor development, dysmetria, dyskinetic movements, nystagmus, and Leigh syndrome | R255X (c.C673T) | [124] |
| TMEM126B | Assembly | Exercise intolerance, muscle weakness, myalgia, and hypertrophic cardiomyopathy | Q70X (c.C208T) D133N (c.G397A) G212V (c.G635T) | [125,126] |
| Complex II Subunits | ||||
| SDHA | Succinate oxidation | Leigh syndrome and neonatal dilated cardiomyopathy | G355E (c.G1700A) A524V (c.C1607T) | [127,128,129,130] |
| SDHAF1 | Assembly factor | Spastic quadriplegia and psychomotor regression | R55P (c.G164C) G355E (c.G1700A) | [131] |
| Complex III Subunits | ||||
| MTCYB | Catalytic subunit | Exercise intolerance, encephalomyopathy, and cardiomyopathy | G166X (m.G15242A) G166E (m.G15243A) G251C (m.G15498A) G290D (m.G15615A) L13fsX50 (m.1478del4) | [132,133,134,135,136,137] |
| BCS1L | Assembly factor | Muscle weakness, Leigh syndrome, and myopathy | T50A (c.A148G) D103IfsX8 (c.A306T) E133DfsX23 (c.399delA) | [136,138,139] |
| LYRM7 | Chaperon protein | Progressive weakness, severe psychomotor regression, generalized hypotonia, inability to walk, and severe spastic tetraparesis | D25N (c.G73A) | [140] |
| TTC19 | Assembly factor | Ataxia and spastic paraparesis | Q173X (c.C517T) L219X (c.T656G) Q277X (c.C829T) | [131,141] |
| UQCC3 | Assembly factor | Hypotonia, delayed development, and lactic acidosis | V20E (c.T59A) | [142] |
| UQCRFS1 | Catalytic subunit | Cardiomyopathy | V14D(c.T41A) R204X (c.C610T) V72_81del10 (c.G215-1C) | [143] |
| UQCRQ | Binds and stabilization of cytochrome c | Leigh-like syndrome, severe psychomotor retardation, dystomia, and ataxia | S45F (c.C208T) | [144] |
| Complex IV Subunits | ||||
| MT-CO1 | Reduction of O2 to H2O | MELAS, myopathy, myoglobinuria, motor neuron disease, exercise intolerance, epilepsy, and Leigh syndrome | G226X (m.G6578A) Q232K (m.C6597A) K265fs271X (m.A6698del) G351D (m.G6955A) | [145,146,147,148,149] |
| MT-CO2 | Acceptor of electrons from cyt c | Encephalomyopathy, myopathy, and hypertrophic cardiomyopathy | M1T (m.T7587C) K29M (m.T7671A) M153X (m.8042delAT) L168X (m.8088delT) | [150,151,152,153] |
| MT-CO3 | Putative oxygen uptake regulator | Myopathy, muscle weakness, exercise intollerance, and seizures | W58X (m.G9379A) V91A (m.T9478C) P118QfsX124 (m.9559del) W248X (m.G9952A) | [154,155,156,157] |
| SURF-1 | Assembly factor | Leigh syndrome, Charcot–Marie–Tooth disease | P119L (c.C356T) N178fsX8 (c.531_534del) R192W (c.C574T) Q251X (c.C751T) | [158,159,160,161] |
| SCO-1 | Assembly factor | Hypotonia and cardiomyopathy | G124E (c.C385G) Q251X (c.C751T) | [162] |
| SCO-2 | Metallochaperone | Fatal infantile cardioencephalomyopathy, hypotonia, HCMP, and Charcot–Marie–Tooth disease | Q53X (c.158T) E140K (c.G418A) P169T (c.C505A) S225F (c.C674T) | [163,164] |
| Complex V Subunits | ||||
| MT-ATP6 | Participation in the unidirectional H+ transfer | MILS, ataxia, and Charcot–Marie–Tooth disease | I24T (m.T8597C) P136S (m.C8932T) | [165,166,167,168,169,170] |
| MT-ATP8 | Assembly/stability | Epilepsy, tetralogy of Fallot, weakness, infantile cardiomyopathy, hypertrophic cardiomyopathy, and ataxia | P39L (m.C8481T) W55R (m.T8528C) W55X (m.G8529A) | [168,169,170,171,172,173,174] |
| Gene | Protein Name | Associated Neuromuscular Phenotype(s) | Pathogenic Mutation(s) | Reference(s) |
|---|---|---|---|---|
| SLC25A1 | Citrate carrier (CIC) | Congenital myastenic syndrome | R247Q (c.G740A) G130D (c.G389G) R282H (c.G845G) R210X (c.C628C) V49M (c.G145GA) | [233,234,235,236] |
| SCL25A3 | Phosphate carrier (PIC) | Cardiac and skeletal myopathy | c.158-9A>G G72E (c.G215A) L200W (c.T599G) G296_S300delinsQIP (c.886-898delins7) | [237,238,239,240] |
| SLC25A4 | ADP/ATP carrier isoform 1 (AAC1) | Adult onset progressive external ophtalmoplegia, adult onset cardiomyopathy, skeletal myopathy, and childhood onset mild myopathy | A80H (c.G239A) R235G (c.C703G) K33Q (c.A97T) A123D (c.C368A) | [231,241,242,243,244,245] |
| SLC25A12 | Aspartate/glutamate carrier isoform 1 (AGC1) | Hypotonia, epilepsy, hypomyelination, muscle weakness, and inflammatory myopathy | R252Q (c.G1058A) Q590R (c.A1769G) T444I (c.C1331T) | [246,247] |
| SLC25A20 | Carnitine/acylcarnitine carrier (CAC) | Muscle weakness, hypotonia, cardiomyopathy, and respiratory distress | Q238R (c.A713G) R275Q (c.G824A) | [248,249] |
| SLC25A21 | 2-Oxodicarboxylate carrier (ODC1) | Spinal motor atrophy-like disease | K232R (c.A695G) | [250] |
| SLC25A32 | Folate transporter (MFT) | Excercise intolerance, early onset ataxia, myoclonic dysarthria, and muscle weakness | c.-264_31delins14 W142X (c.G425A) R147L (c.G440A) | [251,252] |
| SLC25A42 | Coenzyme A transporter | Developmental delay, encephalomyopathy, mild and severe motor impairment, and myopathy | N291D (c.A871G) c.380 +2T>A F173_175RFS (c.522_524del) | [253,254,255] |
| SLC25A46 | SLC25A46 | Type 2 Charcot–Marie–Tooth neuropathy, Leigh syndrome, and progressive myoclonic ataxia | T142I (c.C425T) R246X (c.A736T) L348P (c.T1022C) R340C (c.C1018T) E335D (c.A1005T) L138R (c.T413G) | [256,257] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marra, F.; Lunetti, P.; Curcio, R.; Lasorsa, F.M.; Capobianco, L.; Porcelli, V.; Dolce, V.; Fiermonte, G.; Scarcia, P. An Overview of Mitochondrial Protein Defects in Neuromuscular Diseases. Biomolecules 2021, 11, 1633. https://doi.org/10.3390/biom11111633
Marra F, Lunetti P, Curcio R, Lasorsa FM, Capobianco L, Porcelli V, Dolce V, Fiermonte G, Scarcia P. An Overview of Mitochondrial Protein Defects in Neuromuscular Diseases. Biomolecules. 2021; 11(11):1633. https://doi.org/10.3390/biom11111633
Chicago/Turabian StyleMarra, Federica, Paola Lunetti, Rosita Curcio, Francesco Massimo Lasorsa, Loredana Capobianco, Vito Porcelli, Vincenza Dolce, Giuseppe Fiermonte, and Pasquale Scarcia. 2021. "An Overview of Mitochondrial Protein Defects in Neuromuscular Diseases" Biomolecules 11, no. 11: 1633. https://doi.org/10.3390/biom11111633