
Understanding the Role of the Gut Microbiome and Microbial Metabolites in Non-Alcoholic Fatty Liver Disease: Current Evidence and Perspectives

 , , , ,
, , , ,
Abstract
:1. Introduction
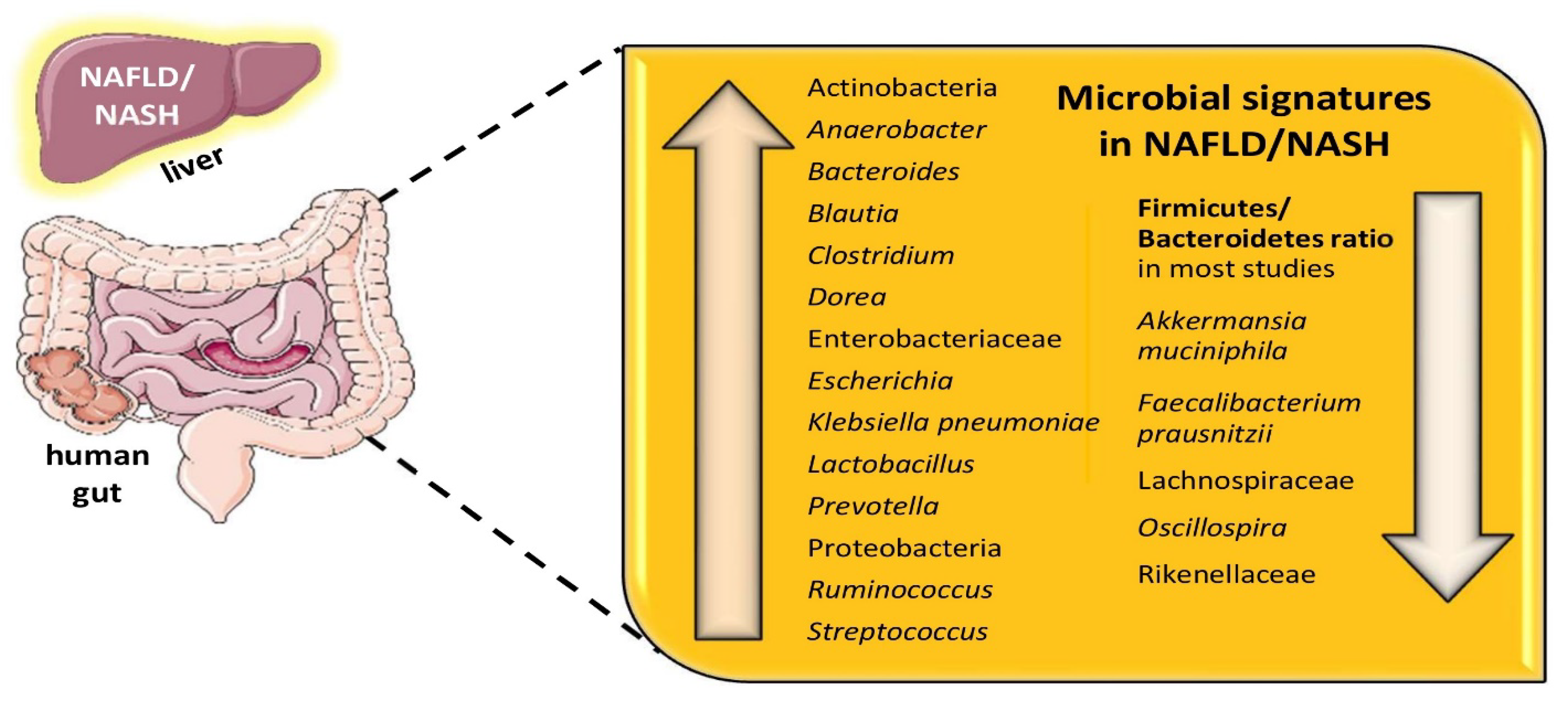
2. NAFLD, Gut Dysbiosis, and Microbial Signatures

{kind=link}
{kind=link}
{kind=link}
| Animal Studies | ||
|---|---|---|
| Study, Year | Animal Model | Remarks |
| Rahman et al., 2016 [44] | Knockout mice of the F11 receptor gene, a gene conferring a junctional adhesion molecule A, implicated in derangement in intestinal permeability | ↑ Firmicutes ↑ Proteobacteria |
| Pierantonelli et al., 2017 [45] | NLRP3 Knockout mice | ↓ Gram negative species ↓ Bacterial translocation after treatment with antibiotics |
| Llorente et al., 2017 [46] | Sublytic Atp4aSl/Sl mice treated with PPIs | ↑Enterococcus faecalis with PPIs |
| Gart et al., 2018 [47] | Leiden mice | Variations in gut microbiota, non-specific |
| Schneider et al., 2019 [48] | Rats with methionine-choline deficient diet-induced NASH | ↓ Gut microbiota diversity |
| Petrov et al., 2019 [49] | GF-HFD not responders | ↑ Desulfovibrio ↑Oscillospira ↓ Bacteroides ↓ Oribacterium |
| Chen et al., 2019 [50] | Knockout SIRT3 HFD mice | ↑ Desulfovibrio ↓ Oscillibacter ↓ Alloprevotella |
| De Sant’Ana et al., 2019 [51] | Knockout mice (caspases 1/11 and NLRP3 HFD) | ↑ Proteobacteria ↑ F/B ratio |
| Ahmad et al., 2020 [52] | Mice C57BL/6J HFD | Alterations in Prevotellaceae UCG-003, Ruminococcaceae UCG-005, Desulfovibrio, the Lachnospiraceae NK4A136 group, Lactobacillus and Akkermansia |
| Cavallari et al., 2020 [53] | NOD2 Knockout mice | ↑ Clostridiales ↓ Erysipelotrichaceae |
| Zhang et al., 2021 [54] | Mice, C57BL/6 male, high-fat, high-cholesterol diet | ↑ Mucispirillum ↑ Desulfovibrio ↑ Anaerotruncus ↑ Desulfovibrionaceae ↓ Bifidobacterium ↓ Bacteroides |
| Human Studies | ||||
|---|---|---|---|---|
| Study, Year | Population | Lab Techniques | Microbiome | Remarks |
| Belgaumkar et al., 2016 [55] | NAFLD as described by serum cytokeratin 18, 18 patients who underwent laparoscopic sleeve gastrectomy (UK) | Serum: Liquid chromatography tandem-mass spectometry for BA | No bacteria were further detected | Total BA did not change; ↓ primary glycine- and taurine-conjugated BA, ↓ cholic acid, and ↑ secondary BA, ↑ glycine-conjugated urodeoxycholic acid over the study period. These changes are associated with reduction in insulin resistance, pro-inflammatory cytokines, and CK-18 levels |
| Boursier et al., 2016 [56] | Biopsy-proven NAFLD among 57 patients (France, USA) | Fecal Microbiome: 16S rRNA gene Sequencing | Patients with NASH and F2≥2: ↑ Bacteroides ↓ Prevotella. Patients with F2 ≥ 2: ↑↑ Ruminococcus | NASH was related to ↑ Bacteroides, while significant fibrosis to ↑↑ Ruminococcus |
| Loomba et al., 2017 [57] | Biopsy-proven NAFLD among 86 patients (USA) | Fecal Microbiome: Whole-genome shotgun sequencing of DNA from feces | Patients with NAFLD: ↑ Proteobacteria ↑ Firmicutes Patients with NAFLD and ≤F2: ↑ Eubacterium rectale ↑ Bacteroides vulgatus Patients with NAFLD and >F2: ↑ Bacteroides vulgaris ↑ Escherischia coli | Patients with NAFLD and ≤F2: ↑ Lactate ↑ Acetate ↑ Formate Patients with NAFLD and >F2: ↑ Butyrate ↑ D-Lactate ↑ Propionate ↑ Succinate |
| Del Chierico et al., 2017 [36] | NAFLD in 61 patients and 51 non-NAFLD controls (Italy) | Fecal Microbiome: rRNA Sequencing Serum metabolites: GC/MS | Patients with NAFLD: ↑ Actinobacteria ↓ Bacteroidetes ↑ Ruminococcus ↑ Blautia ↑ Dorea ↓ Oscillospira ↓ Rikenellaceae | Patients with NAFLD: ↑ 2-butanone ↑ 1-pentanol ↑ 4-methyl-2-pentanone |
| Puri et al., 2018 [58] | Biopsy-proven NAFLD among 86 patients and 24 non-NAFLD controls (USA) | Serum metabolites: LC/MS | No bacteria were further detected | Patients with NAFLD and ≥F2: ↑ conjugated cholate ↓ ratio of total secondary to primary BAs Patients with NASH had ↑↑ total conjugated primary BAs when compared to controls |
| Hoyles et al., 2018 [59] | Biopsy-proven NAFLD among 56 patients (UK, Italy, France) | Fecal Microbiome: Shotgun Metagenomic Sequencing Serum and urine metabolites: LC/MS | Among patients with steatosis: ↑ Proteobacteria ↑ Actinobacteria | Among patients with steatosis: -Serum BCAAs: ↑ leucine ↑ valine ↑ isoleucine ↑ phenylacetic acid -Urine: ↑ choline |
| Caussy et al., 2018 [60] | Discovery cohort of 156 twins Validation cohort of Biopsy-proven NAFLD among 156 patients (USA, France) | Fecal Microbiome: Whole Shotgun Metagenomics Sequencing Liver: MRI-PDFF; MRE Serum metabolites: LC/MS; GC/MS | Patients with NAFLD and >F2: ↑ Furmicutes ↑ Bacteroidetes ↑ Proteobacteria | 56 metabolites had a relationship with hepatic fibrosis, among which 3-(4-hydroxyphenyl) lactate, N-formylmethionine, phenyllactate, mannitol, allantoine and N-(2-furoyl) glycine were the most abundant 3-(4-hydroxyphenyl) lactate was ↑↑ in liver fibrosis and steatosis |
| Caussy et al., 2019 [61] | Cross-sectional; 203 participants including NAFLD and healthy controls (USA) | Fecal Microbiome: 16S rRNA Sequencing Liver: MRI/MRE | Patients with NAFLD and cirrhosis: ↑ Enterobacteriaceae ↑ Streptococcus ↑ Gallibacterium ↑ Megasphaera A trend towards Gram negative species in advanced fibrosis was reported | No metabolites were further detected |
| Lee et al., 2020 [62] | Biopsy-proven NAFLD among 171 patients and 31 non-NAFLD controls (USA, Korea) | Fecal Microbiome: 16S rRNA Sequencing | Patients with NAFLD and >F2, non-obese: ↑ Ruminococcaceae ↑ Veillonellaceae | Patients with NAFLD and >F2, non-obese: ↑ BA ↑ Propionate in feces |
| Adams et al., 2020 [63] | Biopsy-proven NAFLD among 67 patients and 55 non-NAFLD controls (USA) | Fecal Microbiome: 16S rRNA Sequencing Serum and fecal metabolites: HPLC/MS | Patients with NAFLD and >F2: ↑ Firmicutes ↑ Proteobacteria ↑ Actinobacteria ↓ Bacteriodetes ↑ Actinomycetaceae ↓ Lachnospiraceae | Patients with NAFLD and >F2: ↑ BA in serum and feces |
| Masarone et al., 2021 [64] | Biopsy-proven NAFLD among 144 patients (Italy) | Serum metabolites: GC/MS | No bacteria were further detected | Patients with NAFLD and >F2: ↑ Glycocholic acid ↑ Taurocholic acid ↑ Phenylalanine ↑ BCAAs ↓ Glutathione |
| Nimer et al., 2021 [65] | Biopsy-proven NAFLD among 102 patients and 50 non-NAFLD controls (USA) | Plasma BA metabolites: LC/MS | No bacteria were further detected | Patients with NAFLD and >F2: ↑↑ Plasma 7-keto-DCA levels Some glycine conjugated forms of BAs ↑↑ in more advanced stages of NAFLD |
3. Microbiome-Derived Compounds in the Pathogenesis of NAFLD
3.1. SCFAs
3.2. Endotoxins
3.3. Bile Acids
3.4. Choline and TMAO
3.5. Ammonia
4. Therapeutic Interventions Related to Gut Microbiota for NAFLD
4.1. Diet
4.2. Exercise
4.3. Bariatric Surgery
4.4. Probiotics
4.5. Prebiotics
4.6. Synbiotics
4.7. Antibiotics
4.8. FMT
4.9. Bacteriophages
5. Limitations of the Studies
6. Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Perumpail, B.J.; Khan, M.A.; Yoo, E.R.; Cholankeril, G.; Kim, D.; Ahmed, A. Clinical epidemiology and disease burden of nonalcoholic fatty liver disease. World J. Gastroenterol. 2017, 23, 8263–8276. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagaria, S.P.; Ray, P.S.; Sim, M.S.; Ye, X.; Shamonki, J.M.; Cui, X.; Giuliano, A.E. Personalizing breast cancer staging by the inclusion of ER, PR, and HER2. JAMA Surg. 2014, 149, 125–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welzel, T.M.; Graubard, B.I.; Quraishi, S.; Zeuzem, S.; Davila, J.A.; El-Serag, H.B.; McGlynn, K.A. Population-attributable fractions of risk factors for hepatocellular carcinoma in the United States. Am. J. Gastroenterol. 2013, 108, 1314–1321. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.R.; Nguyen, M.H.; Lim, J.K. Hepatocellular carcinoma in patients with non-alcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 8294–8303. [Google Scholar] [CrossRef] [PubMed]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef]
- Jadhav, K.; Cohen, T.S. Can You Trust Your Gut? Implicating a Disrupted Intestinal Microbiome in the Progression of NAFLD/NASH. Front. Endocrinol. 2020, 11, 592157. [Google Scholar] [CrossRef]
- Eslam, M.; George, J. MAFLD: A holistic view to redefining fatty liver disease. J. Hepatol. 2021, 74, 983–985. [Google Scholar] [CrossRef]
- Ye, Q.; Zou, B.; Yeo, Y.H.; Li, J.; Huang, D.Q.; Wu, Y.; Yang, H.; Liu, C.; Kam, L.Y.; Tan, X.X.E.; et al. Global prevalence, incidence, and outcomes of non-obese or lean non-alcoholic fatty liver disease: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2020, 5, 739–752. [Google Scholar] [CrossRef]
- Trépo, E.; Nahon, P.; Bontempi, G.; Valenti, L.; Falleti, E.; Nischalke, H.D.; Hamza, S.; Corradini, S.G.; Burza, M.A.; Guyot, E.; et al. Association between the PNPLA3 (rs738409 C>G) variant and hepatocellular carcinoma: Evidence from a meta-analysis of individual participant data. Hepatology 2014, 59, 2170–2177. [Google Scholar] [CrossRef]
- Falleti, E.; Fabris, C.; Cmet, S.; Cussigh, A.; Bitetto, D.; Fontanini, E.; Fornasiere, E.; Bignulin, S.; Fumolo, E.; Bignulin, E.; et al. PNPLA3 rs738409C/G polymorphism in cirrhosis: Relationship with the aetiology of liver disease and hepatocellular carcinoma occurrence. Liver Int. 2011, 31, 1137–1143. [Google Scholar] [CrossRef]
- Chalasani, N.; Guo, X.; Loomba, R.; Goodarzi, M.O.; Haritunians, T.; Kwon, S.; Cui, J.; Taylor, K.D.; Wilson, L.; Cummings, O.W.; et al. Genome-wide association study identifies variants associated with histologic features of nonalcoholic Fatty liver disease. Gastroenterology 2010, 139, 1567–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [Green Version]
- Petta, S.; Miele, L.; Bugianesi, E.; Cammà, C.; Rosso, C.; Boccia, S.; Cabibi, D.; Di Marco, V.; Grimaudo, S.; Grieco, A.; et al. Glucokinase regulatory protein gene polymorphism affects liver fibrosis in non-alcoholic fatty liver disease. PLoS ONE 2014, 9, e87523. [Google Scholar] [CrossRef] [Green Version]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230.e1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, L.H.S.; Wong, S.H. Microbiota, Obesity and NAFLD. Adv. Exp. Med. Biol. 2018, 1061, 111–125. [Google Scholar] [CrossRef]
- Abdelmalek, M.F.; Liu, C.; Shuster, J.; Nelson, D.R.; Asal, N.R. Familial aggregation of insulin resistance in first-degree relatives of patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2006, 4, 1162–1169. [Google Scholar] [CrossRef]
- Loomba, R.; Hwang, S.J.; O’Donnell, C.J.; Ellison, R.C.; Vasan, R.S.; D’Agostino, R.B., Sr.; Liang, T.J.; Fox, C.S. Parental obesity and offspring serum alanine and aspartate aminotransferase levels: The Framingham heart study. Gastroenterology 2008, 134, 953–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilar, L.; Oliveira, C.P.; Faintuch, J.; Mello, E.S.; Nogueira, M.A.; Santos, T.E.; Alves, V.A.; Carrilho, F.J. High-fat diet: A trigger of non-alcoholic steatohepatitis? Preliminary findings in obese subjects. Nutrition 2008, 24, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef] [Green Version]
- Zelber-Sagi, S.; Nitzan-Kaluski, D.; Goldsmith, R.; Webb, M.; Blendis, L.; Halpern, Z.; Oren, R. Long term nutritional intake and the risk for non-alcoholic fatty liver disease (NAFLD): A population based study. J. Hepatol. 2007, 47, 711–717. [Google Scholar] [CrossRef]
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Araya, J.; Rodrigo, R.; Videla, L.A.; Thielemann, L.; Orellana, M.; Pettinelli, P.; Poniachik, J. Increase in long-chain polyunsaturated fatty acid n-6/n-3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin. Sci. 2004, 106, 635–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortez-Pinto, H.; Jesus, L.; Barros, H.; Lopes, C.; Moura, M.C.; Camilo, M.E. How different is the dietary pattern in non-alcoholic steatohepatitis patients? Clin. Nutr. 2006, 25, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Vallianou, N.; Liu, J.; Dalamaga, M. What are the key points in the association between the gut microbiome and nonalcoholic fatty liver disease? Metabol. Open 2019, 1, 9–10. [Google Scholar] [CrossRef]
- Vallianou, N.; Stratigou, T.; Christodoulatos, G.S.; Dalamaga, M. Understanding the Role of the Gut Microbiome and Microbial Metabolites in Obesity and Obesity-Associated Metabolic Disorders: Current Evidence and Perspectives. Curr. Obes. Rep. 2019, 8, 317–332. [Google Scholar] [CrossRef]
- Hoozemans, J.; de Brauw, M.; Nieuwdorp, M.; Gerdes, V. Gut Microbiome and Metabolites in Patients with NAFLD and after Bariatric Surgery: A Comprehensive Review. Metabolites 2021, 11, 353. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [Green Version]
- Arab, J.P.; Karpen, S.J.; Dawson, P.A.; Arrese, M.; Trauner, M. Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology 2017, 65, 350–362. [Google Scholar] [CrossRef]
- Jiang, W.; Wu, N.; Wang, X.; Chi, Y.; Zhang, Y.; Qiu, X.; Hu, Y.; Li, J.; Liu, Y. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci. Rep. 2015, 5, 8096. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Gerbes, A.; Zoulim, F.; Tilg, H.; Dufour, J.F.; Bruix, J.; Paradis, V.; Salem, R.; Peck-Radosavljevic, M.; Galle, P.R.; Greten, T.F.; et al. Gut roundtable meeting paper: Selected recent advances in hepatocellular carcinoma. Gut 2018, 67, 380–388. [Google Scholar] [CrossRef]
- Satapathy, S.K.; Banerjee, P.; Pierre, J.F.; Higgins, D.; Dutta, S.; Heda, R.; Khan, S.D.; Mupparaju, V.K.; Mas, V.; Nair, S.; et al. Characterization of Gut Microbiome in Liver Transplant Recipients with Nonalcoholic Steatohepatitis. Transplant. Direct 2020, 6, e625. [Google Scholar] [CrossRef] [PubMed]
- Del Chierico, F.; Nobili, V.; Vernocchi, P.; Russo, A.; De Stefanis, C.; Gnani, D.; Furlanello, C.; Zandonà, A.; Paci, P.; Capuani, G.; et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology 2017, 65, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Cope, K.; Risby, T.H.; Diehl, A.M. Obesity and female gender increase breath ethanol concentration: Potential implications for the pathogenesis of nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2001, 96, 1200–1204. [Google Scholar] [CrossRef]
- Rao, R.K.; Seth, A.; Sheth, P. Recent Advances in Alcoholic Liver Disease I. Role of intestinal permeability and endotoxemia in alcoholic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G881–G884. [Google Scholar] [CrossRef]
- Xu, J.; Lai, K.K.Y.; Verlinsky, A.; Lugea, A.; French, S.W.; Cooper, M.P.; Ji, C.; Tsukamoto, H. Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. J. Hepatol. 2011, 55, 673–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Medeiros, I.C.; de Lima, J.G. Is nonalcoholic fatty liver disease an endogenous alcoholic fatty liver disease?—A mechanistic hypothesis. Med. Hypotheses 2015, 85, 148–152. [Google Scholar] [CrossRef]
- Schwenger, K.J.; Clermont-Dejean, N.; Allard, J.P. The role of the gut microbiome in chronic liver disease: The clinical evidence revised. JHEP Rep. 2019, 1, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Roy, T.; Llopis, M.; Lepage, P.; Bruneau, A.; Rabot, S.; Bevilacqua, C.; Martin, P.; Philippe, C.; Walker, F.; Bado, A.; et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2013, 62, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G.; et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 675–688.e677. [Google Scholar] [CrossRef]
- Rahman, K.; Desai, C.; Iyer, S.S.; Thorn, N.E.; Kumar, P.; Liu, Y.; Smith, T.; Neish, A.S.; Li, H.; Tan, S.; et al. Loss of Junctional Adhesion Molecule a Promotes Severe Steatohepatitis in Mice on a Diet High in Saturated Fat, Fructose, and Cholesterol. Gastroenterology 2016, 151, 733–746.e712. [Google Scholar] [CrossRef] [Green Version]
- Pierantonelli, I.; Rychlicki, C.; Agostinelli, L.; Giordano, D.M.; Gaggini, M.; Fraumene, C.; Saponaro, C.; Manghina, V.; Sartini, L.; Mingarelli, E.; et al. Lack of NLRP3-inflammasome leads to gut-liver axis derangement, gut dysbiosis and a worsened phenotype in a mouse model of NAFLD. Sci. Rep. 2017, 7, 12200. [Google Scholar] [CrossRef]
- Llorente, C.; Jepsen, P.; Inamine, T.; Wang, L.; Bluemel, S.; Wang, H.J.; Loomba, R.; Bajaj, J.S.; Schubert, M.L.; Sikaroodi, M.; et al. Gastric acid suppression promotes alcoholic liver disease by inducing overgrowth of intestinal Enterococcus. Nat. Commun. 2017, 8, 837. [Google Scholar] [CrossRef]
- Gart, E.; Souto Lima, E.; Schuren, F.; de Ruiter, C.G.F.; Attema, J.; Verschuren, L.; Keijer, J.; Salic, K.; Morrison, M.C.; Kleemann, R. Diet-Independent Correlations between Bacteria and Dysfunction of Gut, Adipose Tissue, and Liver: A Comprehensive Microbiota Analysis in Feces and Mucosa of the Ileum and Colon in Obese Mice with NAFLD. Int. J. Mol. Sci. 2018, 20, 1. [Google Scholar] [CrossRef] [Green Version]
- Schneider, K.M.; Mohs, A.; Kilic, K.; Candels, L.S.; Elfers, C.; Bennek, E.; Schneider, L.B.; Heymann, F.; Gassler, N.; Penders, J.; et al. Intestinal Microbiota Protects against MCD Diet-Induced Steatohepatitis. Int. J. Mol. Sci. 2019, 20, 308. [Google Scholar] [CrossRef] [Green Version]
- Petrov, P.D.; García-Mediavilla, M.V.; Guzmán, C.; Porras, D.; Nistal, E.; Martínez-Flórez, S.; Castell, J.V.; González-Gallego, J.; Sánchez-Campos, S.; Jover, R. A Network Involving Gut Microbiota, Circulating Bile Acids, and Hepatic Metabolism Genes That Protects Against Non-Alcoholic Fatty Liver Disease. Mol. Nutr. Food Res. 2019, 63, e1900487. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Hui, S.; Lang, H.; Zhou, M.; Zhang, Y.; Kang, C.; Zeng, X.; Zhang, Q.; Yi, L.; Mi, M. SIRT3 Deficiency Promotes High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease in Correlation with Impaired Intestinal Permeability through Gut Microbial Dysbiosis. Mol. Nutr. Food Res. 2019, 63, e1800612. [Google Scholar] [CrossRef]
- de Sant’Ana, L.P.; Ribeiro, D.J.S.; Martins, A.M.A.; Dos Santos, F.N.; Corrêa, R.; Almeida, R.D.N.; Eberlin, M.N.; Maurice, C.F.; Magalhães, K.G. Absence of the Caspases 1/11 Modulates Liver Global Lipid Profile and Gut Microbiota in High-Fat-Diet-Induced Obese Mice. Front. Immunol. 2019, 10, 2926. [Google Scholar] [CrossRef]
- Ahmad, M.I.; Ijaz, M.U.; Hussain, M.; Haq, I.U.; Zhao, D.; Li, C. High-Fat Proteins Drive Dynamic Changes in Gut Microbiota, Hepatic Metabolome, and Endotoxemia-TLR-4-NFκB-Mediated Inflammation in Mice. J. Agric. Food Chem. 2020, 68, 11710–11725. [Google Scholar] [CrossRef]
- Cavallari, J.F.; Pokrajac, N.T.; Zlitni, S.; Foley, K.P.; Henriksbo, B.D.; Schertzer, J.D. NOD2 in hepatocytes engages a liver-gut axis to protect against steatosis, fibrosis, and gut dysbiosis during fatty liver disease in mice. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E305–E314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Coker, O.O.; Chu, E.S.; Fu, K.; Lau, H.C.H.; Wang, Y.X.; Chan, A.W.H.; Wei, H.; Yang, X.; Sung, J.J.Y.; et al. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut 2021, 70, 761–774. [Google Scholar] [CrossRef]
- Belgaumkar, A.P.; Vincent, R.P.; Carswell, K.A.; Hughes, R.D.; Alaghband-Zadeh, J.; Mitry, R.R.; le Roux, C.W.; Patel, A.G. Changes in Bile Acid Profile After Laparoscopic Sleeve Gastrectomy are Associated with Improvements in Metabolic Profile and Fatty Liver Disease. Obes. Surg. 2016, 26, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [Green Version]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e1055. [Google Scholar] [CrossRef]
- Puri, P.; Daita, K.; Joyce, A.; Mirshahi, F.; Santhekadur, P.K.; Cazanave, S.; Luketic, V.A.; Siddiqui, M.S.; Boyett, S.; Min, H.K.; et al. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology 2018, 67, 534–548. [Google Scholar] [CrossRef]
- Hoyles, L.; Fernández-Real, J.M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H.; et al. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 2018, 24, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Caussy, C.; Hsu, C.; Lo, M.T.; Liu, A.; Bettencourt, R.; Ajmera, V.H.; Bassirian, S.; Hooker, J.; Sy, E.; Richards, L.; et al. Link between gut-microbiome derived metabolite and shared gene-effects with hepatic steatosis and fibrosis in NAFLD. Hepatology 2018, 68, 918–932. [Google Scholar] [CrossRef] [Green Version]
- Caussy, C.; Tripathi, A.; Humphrey, G.; Bassirian, S.; Singh, S.; Faulkner, C.; Bettencourt, R.; Rizo, E.; Richards, L.; Xu, Z.Z.; et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat. Commun. 2019, 10, 1406. [Google Scholar] [CrossRef]
- Lee, G.; You, H.J.; Bajaj, J.S.; Joo, S.K.; Yu, J.; Park, S.; Kang, H.; Park, J.H.; Kim, J.H.; Lee, D.H.; et al. Distinct signatures of gut microbiome and metabolites associated with significant fibrosis in non-obese NAFLD. Nat. Commun. 2020, 11, 4982. [Google Scholar] [CrossRef]
- Adams, L.A.; Wang, Z.; Liddle, C.; Melton, P.E.; Ariff, A.; Chandraratna, H.; Tan, J.; Ching, H.; Coulter, S.; de Boer, B.; et al. Bile acids associate with specific gut microbiota, low-level alcohol consumption and liver fibrosis in patients with non-alcoholic fatty liver disease. Liver Int. 2020, 40, 1356–1365. [Google Scholar] [CrossRef]
- Masarone, M.; Troisi, J.; Aglitti, A.; Torre, P.; Colucci, A.; Dallio, M.; Federico, A.; Balsano, C.; Persico, M. Untargeted metabolomics as a diagnostic tool in NAFLD: Discrimination of steatosis, steatohepatitis and cirrhosis. Metabolomics 2021, 17, 12. [Google Scholar] [CrossRef]
- Nimer, N.; Choucair, I.; Wang, Z.; Nemet, I.; Li, L.; Gukasyan, J.; Weeks, T.L.; Alkhouri, N.; Zein, N.; Tang, W.H.W.; et al. Bile acids profile, histopathological indices and genetic variants for non-alcoholic fatty liver disease progression. Metabolism 2021, 116, 154457. [Google Scholar] [CrossRef]
- Wolever, T.M.; Brighenti, F.; Royall, D.; Jenkins, A.L.; Jenkins, D.J. Effect of rectal infusion of short chain fatty acids in human subjects. Am. J. Gastroenterol. 1989, 84, 1027–1033. [Google Scholar] [PubMed]
- McNeil, N.I. The contribution of the large intestine to energy supplies in man. Am. J. Clin. Nutr. 1984, 39, 338–342. [Google Scholar] [CrossRef]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Besten, G.; Bleeker, A.; Gerding, A.; van Eunen, K.; Havinga, R.; van Dijk, T.H.; Oosterveer, M.H.; Jonker, J.W.; Groen, A.K.; Reijngoud, D.J.; et al. Short-Chain Fatty Acids Protect Against High-Fat Diet-Induced Obesity via a PPARγ-Dependent Switch From Lipogenesis to Fat Oxidation. Diabetes 2015, 64, 2398–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, R.A.; You, S.H.; Zhou, J.; Siddique, M.M.; Bay, B.H.; Zhu, X.; Privalsky, M.L.; Cheng, S.Y.; Stevens, R.D.; Summers, S.A.; et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J. Clin. Investig. 2012, 122, 2428–2438. [Google Scholar] [CrossRef] [Green Version]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772. [Google Scholar] [CrossRef] [Green Version]
- Musso, G.; Gambino, R.; Cassader, M. Obesity, diabetes, and gut microbiota: The hygiene hypothesis expanded? Diabetes Care 2010, 33, 2277–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svegliati-Baroni, G.; Saccomanno, S.; Rychlicki, C.; Agostinelli, L.; De Minicis, S.; Candelaresi, C.; Faraci, G.; Pacetti, D.; Vivarelli, M.; Nicolini, D.; et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011, 31, 1285–1297. [Google Scholar] [CrossRef] [Green Version]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, H.; Li, X.; Weiszmann, J.; Wang, P.; Baribault, H.; Chen, J.L.; Tian, H.; Li, Y. Activation of G protein-coupled receptor 43 in adipocytes leads to inhibition of lipolysis and suppression of plasma free fatty acids. Endocrinology 2008, 149, 4519–4526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Jang, C.; Liu, J.; Uehara, K.; Gilbert, M.; Izzo, L.; Zeng, X.; Trefely, S.; Fernandez, S.; Carrer, A.; et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 2020, 579, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Iannucci, L.F.; Sun, J.; Singh, B.K.; Zhou, J.; Kaddai, V.A.; Lanni, A.; Yen, P.M.; Sinha, R.A. Short chain fatty acids induce UCP2-mediated autophagy in hepatic cells. Biochem. Biophys. Res. Commun. 2016, 480, 461–467. [Google Scholar] [CrossRef]
- den Besten, G.; Lange, K.; Havinga, R.; van Dijk, T.H.; Gerding, A.; van Eunen, K.; Müller, M.; Groen, A.K.; Hooiveld, G.J.; Bakker, B.M.; et al. Gut-derived short-chain fatty acids are vividly assimilated into host carbohydrates and lipids. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G900–G910. [Google Scholar] [CrossRef] [PubMed]
- Waldecker, M.; Kautenburger, T.; Daumann, H.; Busch, C.; Schrenk, D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J. Nutr. Biochem. 2008, 19, 587–593. [Google Scholar] [CrossRef]
- Juanola, O.; Ferrusquía-Acosta, J.; García-Villalba, R.; Zapater, P.; Magaz, M.; Marín, A.; Olivas, P.; Baiges, A.; Bellot, P.; Turon, F.; et al. Circulating levels of butyrate are inversely related to portal hypertension, endotoxemia, and systemic inflammation in patients with cirrhosis. FASEB J. 2019, 33, 11595–11605. [Google Scholar] [CrossRef] [PubMed]
- Zhai, S.; Qin, S.; Li, L.; Zhu, L.; Zou, Z.; Wang, L. Dietary butyrate suppresses inflammation through modulating gut microbiota in high-fat diet-fed mice. FEMS Microbiol. Lett. 2019, 366, fnz153. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R.; Szabo, G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2016, 64, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harte, A.L.; da Silva, N.F.; Creely, S.J.; McGee, K.C.; Billyard, T.; Youssef-Elabd, E.M.; Tripathi, G.; Ashour, E.; Abdalla, M.S.; Sharada, H.M.; et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J. Inflamm. 2010, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, N.N.; McGillicuddy, F.C.; Anderson, P.D.; Hinkle, C.C.; Shah, R.; Pruscino, L.; Tabita-Martinez, J.; Sellers, K.F.; Rickels, M.R.; Reilly, M.P. Experimental endotoxemia induces adipose inflammation and insulin resistance in humans. Diabetes 2010, 59, 172–181. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Nakatsu, G.; Zhang, X.; Yu, J. Pathophysiological mechanisms and therapeutic potentials of macrophages in non-alcoholic steatohepatitis. Expert Opin. Ther. Targets 2016, 20, 615–626. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Stienstra, R.; van Diepen, J.A.; Tack, C.J.; Zaki, M.H.; van de Veerdonk, F.L.; Perera, D.; Neale, G.A.; Hooiveld, G.J.; Hijmans, A.; Vroegrijk, I.; et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 15324–15329. [Google Scholar] [CrossRef] [Green Version]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Lee, H.E.; Lee, J.Y. A pharmacological inhibitor of NLRP3 inflammasome prevents non-alcoholic fatty liver disease in a mouse model induced by high fat diet. Sci. Rep. 2016, 6, 24399. [Google Scholar] [CrossRef]
- Csak, T.; Velayudham, A.; Hritz, I.; Petrasek, J.; Levin, I.; Lippai, D.; Catalano, D.; Mandrekar, P.; Dolganiuc, A.; Kurt-Jones, E.; et al. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G433–G441. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Martinez, I.; Santoro, N.; Chen, Y.; Hoque, R.; Ouyang, X.; Caprio, S.; Shlomchik, M.J.; Coffman, R.L.; Candia, A.; Mehal, W.Z. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Investig. 2016, 126, 859–864. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.; Chassaing, B.; Zhang, L.; San Yeoh, B.; Xiao, X.; Kumar, M.; Baker, M.T.; Cai, J.; Walker, R.; Borkowski, K.; et al. Microbiota-Dependent Hepatic Lipogenesis Mediated by Stearoyl CoA Desaturase 1 (SCD1) Promotes Metabolic Syndrome in TLR5-Deficient Mice. Cell Metab. 2015, 22, 983–996. [Google Scholar] [CrossRef] [Green Version]
- Moran-Ramos, S.; López-Contreras, B.E.; Canizales-Quinteros, S. Gut Microbiota in Obesity and Metabolic Abnormalities: A Matter of Composition or Functionality? Arch. Med. Res. 2017, 48, 735–753. [Google Scholar] [CrossRef]
- Yuan, L.; Bambha, K. Bile acid receptors and nonalcoholic fatty liver disease. World J. Hepatol. 2015, 7, 2811–2818. [Google Scholar] [CrossRef]
- Dawson, P.A.; Karpen, S.J. Intestinal transport and metabolism of bile acids. J. Lipid Res. 2015, 56, 1085–1099. [Google Scholar] [CrossRef] [Green Version]
- Ferslew, B.C.; Xie, G.; Johnston, C.K.; Su, M.; Stewart, P.W.; Jia, W.; Brouwer, K.L.; Barritt, A.S.t. Altered Bile Acid Metabolome in Patients with Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2015, 60, 3318–3328. [Google Scholar] [CrossRef] [Green Version]
- Appleby, R.N.; Moghul, I.; Khan, S.; Yee, M.; Manousou, P.; Neal, T.D.; Walters, J.R.F. Non-alcoholic fatty liver disease is associated with dysregulated bile acid synthesis and diarrhea: A prospective observational study. PLoS ONE 2019, 14, e0211348. [Google Scholar] [CrossRef] [Green Version]
- Yara, S.-I.; Ikegami, T.; Miyazaki, T.; Murakami, M.; Iwamoto, J.; Hirayama, T.; Kohjima, M.; Nakamuta, M.; Honda, A. Circulating bile acid profiles in Japanese patients with NASH. GastroHep 2019, 1, 302–310. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zheng, M.; Liu, J.; Luo, Y.; Yang, W.; Yang, J.; Liu, J.; Zhou, J.; Xu, C.; Zhao, F.; et al. Ratio of Conjugated Chenodeoxycholic to Muricholic Acids is Associated with Severity of Nonalcoholic Steatohepatitis. Obesity 2019, 27, 2055–2066. [Google Scholar] [CrossRef]
- Pols, T.W.; Noriega, L.G.; Nomura, M.; Auwerx, J.; Schoonjans, K. The bile acid membrane receptor TGR5: A valuable metabolic target. Dig. Dis. 2011, 29, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Fiorucci, S.; Biagioli, M.; Zampella, A.; Distrutti, E. Bile Acids Activated Receptors Regulate Innate Immunity. Front. Immunol. 2018, 9, 1853. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Keitel, V.; Donner, M.; Winandy, S.; Kubitz, R.; Häussinger, D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem. Biophys. Res. Commun. 2008, 372, 78–84. [Google Scholar] [CrossRef]
- Molinaro, A.; Wahlström, A.; Marschall, H.U. Role of Bile Acids in Metabolic Control. Trends Endocrinol. Metab. 2018, 29, 31–41. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Mehedint, M.G.; Zeisel, S.H. Choline’s role in maintaining liver function: New evidence for epigenetic mechanisms. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.M.; Vance, D.E. Reduction in VLDL, but not HDL, in plasma of rats deficient in choline. Biochem. Cell Biol. 1990, 68, 552–558. [Google Scholar] [CrossRef]
- Stephenson, K.; Kennedy, L.; Hargrove, L.; Demieville, J.; Thomson, J.; Alpini, G.; Francis, H. Updates on Dietary Models of Nonalcoholic Fatty Liver Disease: Current Studies and Insights. Gene Expr. 2018, 18, 5–17. [Google Scholar] [CrossRef]
- Dumas, M.E.; Barton, R.H.; Toye, A.; Cloarec, O.; Blancher, C.; Rothwell, A.; Fearnside, J.; Tatoud, R.; Blanc, V.; Lindon, J.C.; et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12511–12516. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.; Hazen, S.L. Microbiome, trimethylamine N-oxide, and cardiometabolic disease. Transl. Res. 2017, 179, 108–115. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.M.; Liu, Y.; Zhou, R.F.; Chen, X.L.; Wang, C.; Tan, X.Y.; Wang, L.J.; Zheng, R.D.; Zhang, H.W.; Ling, W.H.; et al. Associations of gut-flora-dependent metabolite trimethylamine-N-oxide, betaine and choline with non-alcoholic fatty liver disease in adults. Sci. Rep. 2016, 6, 19076. [Google Scholar] [CrossRef]
- León-Mimila, P.; Villamil-Ramírez, H.; Li, X.S.; Shih, D.M.; Hui, S.T.; Ocampo-Medina, E.; López-Contreras, B.; Morán-Ramos, S.; Olivares-Arevalo, M.; Grandini-Rosales, P.; et al. Trimethylamine N-oxide levels are associated with NASH in obese subjects with type 2 diabetes. Diabetes Metab. 2021, 47, 101183. [Google Scholar] [CrossRef]
- Barrea, L.; Annunziata, G.; Muscogiuri, G.; Di Somma, C.; Laudisio, D.; Maisto, M.; de Alteriis, G.; Tenore, G.C.; Colao, A.; Savastano, S. Trimethylamine-N-oxide (TMAO) as Novel Potential Biomarker of Early Predictors of Metabolic Syndrome. Nutrients 2018, 10, 1971. [Google Scholar] [CrossRef] [Green Version]
- De Chiara, F.; Thomsen, K.L.; Habtesion, A.; Jones, H.; Davies, N.; Gracia-Sancho, J.; Manicardi, N.; Hall, A.; Andreola, F.; Paish, H.L.; et al. Ammonia Scavenging Prevents Progression of Fibrosis in Experimental Nonalcoholic Fatty Liver Disease. Hepatology 2020, 71, 874–892. [Google Scholar] [CrossRef]
- De Chiara, F.; Heebøll, S.; Marrone, G.; Montoliu, C.; Hamilton-Dutoit, S.; Ferrandez, A.; Andreola, F.; Rombouts, K.; Grønbæk, H.; Felipo, V.; et al. Urea cycle dysregulation in non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 905–915. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, K.L.; De Chiara, F.; Rombouts, K.; Vilstrup, H.; Andreola, F.; Mookerjee, R.P.; Jalan, R. Ammonia: A novel target for the treatment of non-alcoholic steatohepatitis. Med. Hypotheses 2018, 113, 91–97. [Google Scholar] [CrossRef]
- Richardson, A.J.; McKain, N.; Wallace, R.J. Ammonia production by human faecal bacteria, and the enumeration, isolation and characterization of bacteria capable of growth on peptides and amino acids. BMC Microbiol. 2013, 13, 6. [Google Scholar] [CrossRef] [Green Version]
- Lykke Eriksen, P.; Sørensen, M.; Grønbæk, H.; Hamilton-Dutoit, S.; Vilstrup, H.; Thomsen, K.L. Non-alcoholic fatty liver disease causes dissociated changes in metabolic liver functions. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 551–560. [Google Scholar] [CrossRef]
- Begriche, K.; Massart, J.; Robin, M.A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Bak-Fredslund, K.P.; Lykke Eriksen, P.; Munk, O.L.; Villadsen, G.E.; Keiding, S.; Sørensen, M. Metabolic liver function in humans measured by 2-(18)F-fluoro-2-deoxy-D-galactose PET/CT-reproducibility and clinical potential. EJNMMI Res. 2017, 7, 71. [Google Scholar] [CrossRef] [Green Version]
- Vilstrup, H. Synthesis of urea after stimulation with amino acids: Relation to liver function. Gut 1980, 21, 990–995. [Google Scholar] [CrossRef] [Green Version]
- Anthony, P.P.; Ishak, K.G.; Nayak, N.C.; Poulsen, H.E.; Scheuer, P.J.; Sobin, L.H. The morphology of cirrhosis. Recommendations on definition, nomenclature, and classification by a working group sponsored by the World Health Organization. J. Clin. Pathol. 1978, 31, 395–414. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, K.L.; Grønbæk, H.; Glavind, E.; Hebbard, L.; Jessen, N.; Clouston, A.; George, J.; Vilstrup, H. Experimental nonalcoholic steatohepatitis compromises ureagenesis, an essential hepatic metabolic function. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G295–G301. [Google Scholar] [CrossRef] [Green Version]
- Eriksen, P.L.; Vilstrup, H.; Rigbolt, K.; Suppli, M.P.; Sørensen, M.; Heebøll, S.; Veidal, S.S.; Knop, F.K.; Thomsen, K.L. Non-alcoholic fatty liver disease alters expression of genes governing hepatic nitrogen conversion. Liver Int. 2019, 39, 2094–2101. [Google Scholar] [CrossRef]
- Pessayre, D.; Fromenty, B. NASH: A mitochondrial disease. J. Hepatol. 2005, 42, 928–940. [Google Scholar] [CrossRef]
- Marini, J.C.; Broussard, S.R. Hyperammonemia increases sensitivity to LPS. Mol. Genet. Metab. 2006, 88, 131–137. [Google Scholar] [CrossRef]
- Jover, R.; Rodrigo, R.; Felipo, V.; Insausti, R.; Sáez-Valero, J.; García-Ayllón, M.S.; Suárez, I.; Candela, A.; Compañ, A.; Esteban, A.; et al. Brain edema and inflammatory activation in bile duct ligated rats with diet-induced hyperammonemia: A model of hepatic encephalopathy in cirrhosis. Hepatology 2006, 43, 1257–1266. [Google Scholar] [CrossRef]
- Wright, G.; Davies, N.A.; Shawcross, D.L.; Hodges, S.J.; Zwingmann, C.; Brooks, H.F.; Mani, A.R.; Harry, D.; Stadlbauer, V.; Zou, Z.; et al. Endotoxemia produces coma and brain swelling in bile duct ligated rats. Hepatology 2007, 45, 1517–1526. [Google Scholar] [CrossRef]
- Wright, G.; Jalan, R. Ammonia and inflammation in the pathogenesis of hepatic encephalopathy: Pandora’s box? Hepatology 2007, 46, 291–294. [Google Scholar] [CrossRef]
- Häussinger, D.; Kircheis, G.; Fischer, R.; Schliess, F.; vom Dahl, S. Hepatic encephalopathy in chronic liver disease: A clinical manifestation of astrocyte swelling and low-grade cerebral edema? J. Hepatol. 2000, 32, 1035–1038. [Google Scholar] [CrossRef]
- Kimelberg, H.K. Astrocytic swelling in cerebral ischemia as a possible cause of injury and target for therapy. Glia 2005, 50, 389–397. [Google Scholar] [CrossRef]
- Rodrigo, R.; Cauli, O.; Gomez-Pinedo, U.; Agusti, A.; Hernandez-Rabaza, V.; Garcia-Verdugo, J.M.; Felipo, V. Hyperammonemia induces neuroinflammation that contributes to cognitive impairment in rats with hepatic encephalopathy. Gastroenterology 2010, 139, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Zemtsova, I.; Görg, B.; Keitel, V.; Bidmon, H.J.; Schrör, K.; Häussinger, D. Microglia activation in hepatic encephalopathy in rats and humans. Hepatology 2011, 54, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Cagnin, A.; Taylor-Robinson, S.D.; Forton, D.M.; Banati, R.B. In vivo imaging of cerebral “peripheral benzodiazepine binding sites” in patients with hepatic encephalopathy. Gut 2006, 55, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Balzano, T.; Dadsetan, S.; Forteza, J.; Cabrera-Pastor, A.; Taoro-Gonzalez, L.; Malaguarnera, M.; Gil-Perotin, S.; Cubas-Nuñez, L.; Casanova, B.; Castro-Quintas, A.; et al. Chronic hyperammonemia induces peripheral inflammation that leads to cognitive impairment in rats: Reversed by anti-TNF-α treatment. J. Hepatol. 2020, 73, 582–592. [Google Scholar] [CrossRef]
- Higarza, S.G.; Arboleya, S.; Gueimonde, M.; Gómez-Lázaro, E.; Arias, J.L.; Arias, N. Neurobehavioral dysfunction in non-alcoholic steatohepatitis is associated with hyperammonemia, gut dysbiosis, and metabolic and functional brain regional deficits. PLoS ONE 2019, 14, e0223019. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, V.; Betrapally, N.S.; Hylemon, P.B.; White, M.B.; Gillevet, P.M.; Unser, A.B.; Fagan, A.; Daita, K.; Heuman, D.M.; Zhou, H.; et al. Impaired Gut-Liver-Brain Axis in Patients with Cirrhosis. Sci. Rep. 2016, 6, 26800. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Kassam, Z.; Fagan, A.; Gavis, E.A.; Liu, E.; Cox, I.J.; Kheradman, R.; Heuman, D.; Wang, J.; Gurry, T.; et al. Fecal microbiota transplant from a rational stool donor improves hepatic encephalopathy: A randomized clinical trial. Hepatology 2017, 66, 1727–1738. [Google Scholar] [CrossRef]
- Vilstrup, H.; Amodio, P.; Bajaj, J.; Cordoba, J.; Ferenci, P.; Mullen, K.D.; Weissenborn, K.; Wong, P. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver. Hepatology 2014, 60, 715–735. [Google Scholar] [CrossRef]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.M.; Schwacha, H.; Steinbrückner, B.; Brinkmann, F.E.; Ditzen, A.K.; Aponte, J.J.; Pelz, K.; Berger, D.; Kist, M.; Blum, H.E. Small intestinal bacterial overgrowth in human cirrhosis is associated with systemic endotoxemia. Am. J. Gastroenterol. 2002, 97, 2364–2370. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Betrapally, N.S.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; White, M.B.; Unser, A.; Thacker, L.R.; Sanyal, A.J.; Kang, D.J.; et al. Salivary microbiota reflects changes in gut microbiota in cirrhosis with hepatic encephalopathy. Hepatology 2015, 62, 1260–1271. [Google Scholar] [CrossRef] [Green Version]
- Li, D.Y.; Yang, M.; Edwards, S.; Ye, S.Q. Nonalcoholic fatty liver disease: For better or worse, blame the gut microbiota? JPEN J. Parenter. Enteral. Nutr. 2013, 37, 787–793. [Google Scholar] [CrossRef] [Green Version]
- Hamurcu Varol, P.; Kaya, E.; Alphan, E.; Yilmaz, Y. Role of intensive dietary and lifestyle interventions in the treatment of lean nonalcoholic fatty liver disease patients. Eur. J. Gastroenterol. Hepatol. 2020, 32, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Savard, C.; Tartaglione, E.V.; Kuver, R.; Haigh, W.G.; Farrell, G.C.; Subramanian, S.; Chait, A.; Yeh, M.M.; Quinn, L.S.; Ioannou, G.N. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology 2013, 57, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.F.; Cotter, P.D.; Healy, S.; Marques, T.M.; O’Sullivan, O.; Fouhy, F.; Clarke, S.F.; O’Toole, P.W.; Quigley, E.M.; Stanton, C.; et al. Composition and energy harvesting capacity of the gut microbiota: Relationship to diet, obesity and time in mouse models. Gut 2010, 59, 1635–1642. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Bäckhed, F.; Fulton, L.; Gordon, J.I. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobsdottir, G.; Xu, J.; Molin, G.; Ahrné, S.; Nyman, M. High-fat diet reduces the formation of butyrate, but increases succinate, inflammation, liver fat and cholesterol in rats, while dietary fibre counteracts these effects. PLoS ONE 2013, 8, e80476. [Google Scholar] [CrossRef] [Green Version]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basaranoglu, M.; Basaranoglu, G.; Sabuncu, T.; Sentürk, H. Fructose as a key player in the development of fatty liver disease. World J. Gastroenterol. 2013, 19, 1166–1172. [Google Scholar] [CrossRef]
- Rojas, A.; González, I.; Rodríguez, B.; Romero, J.; Figueroa, H.; Llanos, J.; Morales, E.; Pérez-Castro, R. Evidence of involvement of the receptor for advanced glycation end-products (RAGE) in the adhesion of Helicobacter pylori to gastric epithelial cells. Microbes Infect. 2011, 13, 818–823. [Google Scholar] [CrossRef]
- Ciccocioppo, R.; Vanoli, A.; Klersy, C.; Imbesi, V.; Boccaccio, V.; Manca, R.; Betti, E.; Cangemi, G.C.; Strada, E.; Besio, R.; et al. Role of the advanced glycation end products receptor in Crohn’s disease inflammation. World J. Gastroenterol. 2013, 19, 8269–8281. [Google Scholar] [CrossRef]
- Leung, C.; Herath, C.B.; Jia, Z.; Andrikopoulos, S.; Brown, B.E.; Davies, M.J.; Rivera, L.R.; Furness, J.B.; Forbes, J.M.; Angus, P.W. Dietary advanced glycation end-products aggravate non-alcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 8026–8040. [Google Scholar] [CrossRef] [Green Version]
- Molloy, J.W.; Calcagno, C.J.; Williams, C.D.; Jones, F.J.; Torres, D.M.; Harrison, S.A. Association of coffee and caffeine consumption with fatty liver disease, nonalcoholic steatohepatitis, and degree of hepatic fibrosis. Hepatology 2012, 55, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Vitaglione, P.; Morisco, F.; Mazzone, G.; Amoruso, D.C.; Ribecco, M.T.; Romano, A.; Fogliano, V.; Caporaso, N.; D’Argenio, G. Coffee reduces liver damage in a rat model of steatohepatitis: The underlying mechanisms and the role of polyphenols and melanoidins. Hepatology 2010, 52, 1652–1661. [Google Scholar] [CrossRef] [PubMed]
- Cowan, T.E.; Palmnäs, M.S.; Yang, J.; Bomhof, M.R.; Ardell, K.L.; Reimer, R.A.; Vogel, H.J.; Shearer, J. Chronic coffee consumption in the diet-induced obese rat: Impact on gut microbiota and serum metabolomics. J. Nutr. Biochem. 2014, 25, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Oishi, K. Influence of coffee (Coffea arabica) and galacto-oligosaccharide consumption on intestinal microbiota and the host responses. FEMS Microbiol. Lett. 2013, 343, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, O.J.; Roderick, P.; Buchanan, R.; Fallowfield, J.A.; Hayes, P.C.; Parkes, J. Systematic review with meta-analysis: Coffee consumption and the risk of cirrhosis. Aliment. Pharmacol. Ther. 2016, 43, 562–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setiawan, V.W.; Wilkens, L.R.; Lu, S.C.; Hernandez, B.Y.; Le Marchand, L.; Henderson, B.E. Association of coffee intake with reduced incidence of liver cancer and death from chronic liver disease in the US multiethnic cohort. Gastroenterology 2015, 148, 118–125.e15. [Google Scholar] [CrossRef] [Green Version]
- Hodge, A.; Lim, S.; Goh, E.; Wong, O.; Marsh, P.; Knight, V.; Sievert, W.; de Courten, B. Coffee Intake Is Associated with a Lower Liver Stiffness in Patients with Non-Alcoholic Fatty Liver Disease, Hepatitis C, and Hepatitis B. Nutrients 2017, 9, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, D.B.; Jeong, H.W.; Cho, D.; Lee, B.J.; Lee, J.H.; Choi, J.Y.; Bae, I.H.; Lee, S.J. Fermented green tea extract alleviates obesity and related complications and alters gut microbiota composition in diet-induced obese mice. J. Med. Food 2015, 18, 549–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Farah, B.L.; Sinha, R.A.; Wu, Y.; Singh, B.K.; Bay, B.H.; Yang, C.S.; Yen, P.M. Epigallocatechin-3-gallate (EGCG), a green tea polyphenol, stimulates hepatic autophagy and lipid clearance. PLoS ONE 2014, 9, e87161. [Google Scholar] [CrossRef] [Green Version]
- Santamarina, A.B.; Carvalho-Silva, M.; Gomes, L.M.; Okuda, M.H.; Santana, A.A.; Streck, E.L.; Seelaender, M.; do Nascimento, C.M.; Ribeiro, E.B.; Lira, F.S.; et al. Decaffeinated green tea extract rich in epigallocatechin-3-gallate prevents fatty liver disease by increased activities of mitochondrial respiratory chain complexes in diet-induced obesity mice. J. Nutr. Biochem. 2015, 26, 1348–1356. [Google Scholar] [CrossRef] [Green Version]
- Pezeshki, A.; Safi, S.; Feizi, A.; Askari, G.; Karami, F. The Effect of Green Tea Extract Supplementation on Liver Enzymes in Patients with Nonalcoholic Fatty Liver Disease. Int. J. Prev. Med. 2016, 7, 28. [Google Scholar] [CrossRef]
- Sakata, R.; Nakamura, T.; Torimura, T.; Ueno, T.; Sata, M. Green tea with high-density catechins improves liver function and fat infiltration in non-alcoholic fatty liver disease (NAFLD) patients: A double-blind placebo-controlled study. Int. J. Mol. Med. 2013, 32, 989–994. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Li, S.; Li, J.; Wang, J.; Zhang, R.; Zhou, Y.; Yin, Q.; Zheng, Y.; Wang, F.; Xia, Y.; et al. Effects of Omega-3 Fatty Acid in Nonalcoholic Fatty Liver Disease: A Meta-Analysis. Gastroenterol. Res. Pract. 2016, 2016, 1459790. [Google Scholar] [CrossRef] [Green Version]
- Di Minno, M.N.; Russolillo, A.; Lupoli, R.; Ambrosino, P.; Di Minno, A.; Tarantino, G. Omega-3 fatty acids for the treatment of non-alcoholic fatty liver disease. World J. Gastroenterol. 2012, 18, 5839–5847. [Google Scholar] [CrossRef]
- Bashiardes, S.; Shapiro, H.; Rozin, S.; Shibolet, O.; Elinav, E. Non-alcoholic fatty liver and the gut microbiota. Mol. Metab. 2016, 5, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Carbajo-Pescador, S.; Porras, D.; García-Mediavilla, M.V.; Martínez-Flórez, S.; Juarez-Fernández, M.; Cuevas, M.J.; Mauriz, J.L.; González-Gallego, J.; Nistal, E.; Sánchez-Campos, S. Beneficial effects of exercise on gut microbiota functionality and barrier integrity, and gut-liver crosstalk in an in vivo model of early obesity and non-alcoholic fatty liver disease. Dis. Models Mech. 2019, 12, dmm039206. [Google Scholar] [CrossRef] [Green Version]
- Baker, C.J.; Martinez-Huenchullan, S.F.; D’Souza, M.; Xu, Y.; Li, M.; Bi, Y.; Johnson, N.A.; Twigg, S.M. Effect of exercise on hepatic steatosis: Are benefits seen without dietary intervention? A systematic review and meta-analysis. J. Diabetes 2021, 13, 63–77. [Google Scholar] [CrossRef]
- Huber, Y.; Pfirrmann, D.; Gebhardt, I.; Labenz, C.; Gehrke, N.; Straub, B.K.; Ruckes, C.; Bantel, H.; Belda, E.; Clément, K.; et al. Improvement of non-invasive markers of NAFLD from an individualised, web-based exercise program. Aliment. Pharmacol. Ther. 2019, 50, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Høier, B.; Rufener, N.; Bojsen-Møller, J.; Bangsbo, J.; Hellsten, Y. The effect of passive movement training on angiogenic factors and capillary growth in human skeletal muscle. J. Physiol. 2010, 588, 3833–3845. [Google Scholar] [CrossRef] [PubMed]
- Arulanandan, A.; Ang, B.; Bettencourt, R.; Hooker, J.; Behling, C.; Lin, G.Y.; Valasek, M.A.; Ix, J.H.; Schnabl, B.; Sirlin, C.B.; et al. Association Between Quantity of Liver Fat and Cardiovascular Risk in Patients With Nonalcoholic Fatty Liver Disease Independent of Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2015, 13, 1513–1520.e1511. [Google Scholar] [CrossRef] [PubMed]
- Hashida, R.; Kawaguchi, T.; Bekki, M.; Omoto, M.; Matsuse, H.; Nago, T.; Takano, Y.; Ueno, T.; Koga, H.; George, J.; et al. Aerobic vs. resistance exercise in non-alcoholic fatty liver disease: A systematic review. J. Hepatol. 2017, 66, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Alvarez, L.; Xu, H.; Martinez-Tellez, B. Influence of Exercise on the Human Gut Microbiota of Healthy Adults: A Systematic Review. Clin. Transl. Gastroenterol. 2020, 11, e00126. [Google Scholar] [CrossRef]
- Monda, V.; Villano, I.; Messina, A.; Valenzano, A.; Esposito, T.; Moscatelli, F.; Viggiano, A.; Cibelli, G.; Chieffi, S.; Monda, M.; et al. Exercise Modifies the Gut Microbiota with Positive Health Effects. Oxid. Med. Cell. Longev. 2017, 2017, 3831972. [Google Scholar] [CrossRef]
- Mailing, L.J.; Allen, J.M.; Buford, T.W.; Fields, C.J.; Woods, J.A. Exercise and the Gut Microbiome: A Review of the Evidence, Potential Mechanisms, and Implications for Human Health. Exerc. Sport Sci. Rev. 2019, 47, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Welly, R.J.; Liu, T.W.; Zidon, T.M.; Rowles, J.L., 3rd; Park, Y.M.; Smith, T.N.; Swanson, K.S.; Padilla, J.; Vieira-Potter, V.J. Comparison of Diet versus Exercise on Metabolic Function and Gut Microbiota in Obese Rats. Med. Sci. Sports Exerc. 2016, 48, 1688–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, W.; Penney, N.C.; Cronin, O.; Garcia-Perez, I.; Molloy, M.G.; Holmes, E.; Shanahan, F.; Cotter, P.D.; O’Sullivan, O. The microbiome of professional athletes differs from that of more sedentary subjects in composition and particularly at the functional metabolic level. Gut 2018, 67, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.M.; Mailing, L.J.; Niemiro, G.M.; Moore, R.; Cook, M.D.; White, B.A.; Holscher, H.D.; Woods, J.A. Exercise Alters Gut Microbiota Composition and Function in Lean and Obese Humans. Med. Sci. Sports Exerc. 2018, 50, 747–757. [Google Scholar] [CrossRef]
- Vallianou, N.; Dalamaga, M.; Stratigou, T.; Karampela, I.; Tsigalou, C. Do Antibiotics Cause Obesity Through Long-term Alterations in the Gut Microbiome? A Review of Current Evidence. Curr. Obes. Rep. 2021, 10, 244–262. [Google Scholar] [CrossRef]
- Munukka, E.; Ahtiainen, J.P.; Puigbó, P.; Jalkanen, S.; Pahkala, K.; Keskitalo, A.; Kujala, U.M.; Pietilä, S.; Hollmén, M.; Elo, L.; et al. Six-Week Endurance Exercise Alters Gut Metagenome That Is not Reflected in Systemic Metabolism in Over-weight Women. Front. Microbiol. 2018, 9, 2323. [Google Scholar] [CrossRef]
- Sjöström, L.; Narbro, K.; Sjöström, C.D.; Karason, K.; Larsson, B.; Wedel, H.; Lystig, T.; Sullivan, M.; Bouchard, C.; Carlsson, B.; et al. Effects of bariatric surgery on mortality in Swedish obese subjects. N. Engl. J. Med. 2007, 357, 741–752. [Google Scholar] [CrossRef] [Green Version]
- Adams, T.D.; Davidson, L.E.; Litwin, S.E.; Kim, J.; Kolotkin, R.L.; Nanjee, M.N.; Gutierrez, J.M.; Frogley, S.J.; Ibele, A.R.; Brinton, E.A.; et al. Weight and Metabolic Outcomes 12 Years after Gastric Bypass. N. Engl. J. Med. 2017, 377, 1143–1155. [Google Scholar] [CrossRef]
- Steenackers, N.; Vanuytsel, T.; Augustijns, P.; Tack, J.; Mertens, A.; Lannoo, M.; Van der Schueren, B.; Matthys, C. Adaptations in gastrointestinal physiology after sleeve gastrectomy and Roux-en-Y gastric bypass. Lancet Gastroenterol. Hepatol. 2021, 6, 225–237. [Google Scholar] [CrossRef]
- Karami, R.; Kermansaravi, M.; Pishgahroudsari, M.; Talebi, M.; Mohammadzadeh, N.; Pazouki, A. Changes in gut microbial flora after Roux-en-Y gastric bypass and sleeve gastrectomy and their effects on post-operative weight loss. Updates Surg. 2021, 73, 1493–1499. [Google Scholar] [CrossRef]
- Steinert, R.E.; Rehman, A.; Souto Lima, E.J.; Agamennone, V.; Schuren, F.H.J.; Gero, D.; Schreiner, P.; Vonlanthen, R.; Ismaeil, A.; Tzafos, S.; et al. Roux-en-Y gastric bypass surgery changes fungal and bacterial microbiota in morbidly obese patients—A pilot study. PLoS ONE 2020, 15, e0236936. [Google Scholar] [CrossRef]
- Farin, W.; Oñate, F.P.; Plassais, J.; Bonny, C.; Beglinger, C.; Woelnerhanssen, B.; Nocca, D.; Magoules, F.; Le Chatelier, E.; Pons, N.; et al. Impact of laparoscopic Roux-en-Y gastric bypass and sleeve gastrectomy on gut microbiota: A metagenomic comparative analysis. Surg. Obes. Relat. Dis. 2020, 16, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Palleja, A.; Kashani, A.; Allin, K.H.; Nielsen, T.; Zhang, C.; Li, Y.; Brach, T.; Liang, S.; Feng, Q.; Jørgensen, N.B.; et al. Roux-en-Y gastric bypass surgery of morbidly obese patients induces swift and persistent changes of the individual gut microbiota. Genome Med. 2016, 8, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Siles, M.; Duncan, S.H.; Garcia-Gil, L.J.; Martinez-Medina, M. Faecalibacterium prausnitzii: From microbiology to diagnostics and prognostics. ISME J. 2017, 11, 841–852. [Google Scholar] [CrossRef]
- Lenoir, M.; Martín, R.; Torres-Maravilla, E.; Chadi, S.; González-Dávila, P.; Sokol, H.; Langella, P.; Chain, F.; Bermúdez-Humarán, L.G. Butyrate mediates anti-inflammatory effects of Faecalibacterium prausnitzii in intestinal epithelial cells through Dact3. Gut Microbes 2020, 12, 1–16. [Google Scholar] [CrossRef]
- Zhang, M.; Zhou, L.; Wang, Y.; Dorfman, R.G.; Tang, D.; Xu, L.; Pan, Y.; Zhou, Q.; Li, Y.; Yin, Y.; et al. Faecalibacterium prausnitzii produces butyrate to decrease c-Myc-related metabolism and Th17 differentiation by inhibiting histone deacetylase 3. Int. Immunol. 2019, 31, 499–514. [Google Scholar] [CrossRef]
- Tabasi, M.; Eybpoosh, S.; Siadat, S.D.; Elyasinia, F.; Soroush, A.; Bouzari, S. Modulation of the Gut Microbiota and Serum Biomarkers After Laparoscopic Sleeve Gastrectomy: A 1-Year Follow-Up Study. Obes. Surg. 2021, 31, 1949–1956. [Google Scholar] [CrossRef]
- Tremaroli, V.; Karlsson, F.; Werling, M.; Ståhlman, M.; Kovatcheva-Datchary, P.; Olbers, T.; Fändriks, L.; le Roux, C.W.; Nielsen, J.; Bäckhed, F. Roux-en-Y Gastric Bypass and Vertical Banded Gastroplasty Induce Long-Term Changes on the Human Gut Microbiome Contributing to Fat Mass Regulation. Cell Metab. 2015, 22, 228–238. [Google Scholar] [CrossRef] [Green Version]
- Faria, S.L.; Santos, A.; Magro, D.O.; Cazzo, E.; Assalin, H.B.; Guadagnini, D.; Vieira, F.T.; Dutra, E.S.; Saad, M.J.A.; Ito, M.K. Gut Microbiota Modifications and Weight Regain in Morbidly Obese Women After Roux-en-Y Gastric Bypass. Obes. Surg. 2020, 30, 4958–4966. [Google Scholar] [CrossRef]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, D.E.; Fraser, C.M.; Palumbo, F.; Ravel, J.; Rowthorn, V.; Schwartz, J. Probiotics: Achieving a better regulatory fit. Food Drug Law J. 2014, 69, 237–272. [Google Scholar]
- Paolella, G.; Mandato, C.; Pierri, L.; Poeta, M.; Di Stasi, M.; Vajro, P. Gut-liver axis and probiotics: Their role in non-alcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 15518–15531. [Google Scholar] [CrossRef]
- Govender, M.; Choonara, Y.E.; Kumar, P.; du Toit, L.C.; van Vuuren, S.; Pillay, V. A review of the advancements in probiotic delivery: Conventional vs. non-conventional formulations for intestinal flora supplementation. AAPS PharmSciTech. 2014, 15, 29–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alisi, A.; Bedogni, G.; Baviera, G.; Giorgio, V.; Porro, E.; Paris, C.; Giammaria, P.; Reali, L.; Anania, F.; Nobili, V. Randomised clinical trial: The beneficial effects of VSL#3 in obese children with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2014, 39, 1276–1285. [Google Scholar] [CrossRef] [PubMed]
- Loguercio, C.; Federico, A.; Tuccillo, C.; Terracciano, F.; D’Auria, M.V.; De Simone, C.; Del Vecchio Blanco, C. Beneficial effects of a probiotic VSL#3 on parameters of liver dysfunction in chronic liver diseases. J. Clin. Gastroenterol. 2005, 39, 540–543. [Google Scholar] [CrossRef] [Green Version]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munukka, E.; Rintala, A.; Toivonen, R.; Nylund, M.; Yang, B.; Takanen, A.; Hänninen, A.; Vuopio, J.; Huovinen, P.; Jalkanen, S.; et al. Faecalibacterium prausnitzii treatment improves hepatic health and reduces adipose tissue inflammation in high-fat fed mice. ISME J. 2017, 11, 1667–1679. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Gallego, C.; Pohl, S.; Salminen, S.; De Vos, W.M.; Kneifel, W. Akkermansia muciniphila: A novel functional microbe with probiotic properties. Benef. Microbes 2016, 7, 571–584. [Google Scholar] [CrossRef]
- Stenman, L.K.; Burcelin, R.; Lahtinen, S. Establishing a causal link between gut microbes, body weight gain and glucose metabolism in humans—Towards treatment with probiotics. Benef. Microbes 2016, 7, 11–22. [Google Scholar] [CrossRef]
- Plovier, H.; Everard, A.; Druart, C.; Depommier, C.; Van Hul, M.; Geurts, L.; Chilloux, J.; Ottman, N.; Duparc, T.; Lichtenstein, L.; et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat. Med. 2017, 23, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Liu, W.; Wang, J.; Shi, J.; Sun, Y.; Wang, W.; Ning, G.; Liu, R.; Hong, J. Akkermansia muciniphila improves metabolic profiles by reducing inflammation in chow diet-fed mice. J. Mol. Endocrinol. 2017, 58, 1–14. [Google Scholar] [CrossRef]
- Gao, X.; Zhu, Y.; Wen, Y.; Liu, G.; Wan, C. Efficacy of probiotics in non-alcoholic fatty liver disease in adult and children: A meta-analysis of randomized controlled trials. Hepatol. Res. 2016, 46, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.Y.; Li, L.; Yu, C.H.; Shen, Z.; Chen, L.H.; Li, Y.M. Effects of probiotics on nonalcoholic fatty liver disease: A meta-analysis. World J. Gastroenterol. 2013, 19, 6911–6918. [Google Scholar] [CrossRef]
- Shavakhi, A.; Minakari, M.; Firouzian, H.; Assali, R.; Hekmatdoost, A.; Ferns, G. Effect of a Probiotic and Metformin on Liver Aminotransferases in Non-alcoholic Steatohepatitis: A Double Blind Randomized Clinical Trial. Int. J. Prev. Med. 2013, 4, 531–537. [Google Scholar]
- Vallianou, N.; Stratigou, T.; Christodoulatos, G.S.; Tsigalou, C.; Dalamaga, M. Probiotics, Prebiotics, Synbiotics, Postbiotics, and Obesity: Current Evidence, Controversies, and Perspectives. Curr. Obes. Rep. 2020, 9, 179–192. [Google Scholar] [CrossRef]
- Reijnders, D.; Goossens, G.H.; Hermes, G.D.; Neis, E.P.; van der Beek, C.M.; Most, J.; Holst, J.J.; Lenaerts, K.; Kootte, R.S.; Nieuwdorp, M.; et al. Effects of Gut Microbiota Manipulation by Antibiotics on Host Metabolism in Obese Humans: A Randomized Double-Blind Placebo-Controlled Trial. Cell Metab. 2016, 24, 63–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangarapu, V.; Ince, A.T.; Baysal, B.; Kayar, Y.; Kılıç, U.; Gök, Ö.; Uysal, Ö.; Şenturk, H. Efficacy of rifaximin on circulating endotoxins and cytokines in patients with nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2015, 27, 840–845. [Google Scholar] [CrossRef] [PubMed]
- van Nood, E.; Vrieze, A.; Nieuwdorp, M.; Fuentes, S.; Zoetendal, E.G.; de Vos, W.M.; Visser, C.E.; Kuijper, E.J.; Bartelsman, J.F.; Tijssen, J.G.; et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N. Engl. J. Med. 2013, 368, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Kelly, C.R.; Kim, A.M.; Laine, L.; Wu, G.D. The AGA’s Fecal Microbiota Transplantation National Registry: An Important Step Toward Understanding Risks and Benefits of Microbiota Therapeutics. Gastroenterology 2017, 152, 681–684. [Google Scholar] [CrossRef] [Green Version]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e917. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Pan, Q.; Shen, F.; Cao, H.X.; Ding, W.J.; Chen, Y.W.; Fan, J.G. Total fecal microbiota transplantation alleviates high-fat diet-induced steatohepatitis in mice via beneficial regulation of gut microbiota. Sci. Rep. 2017, 7, 1529. [Google Scholar] [CrossRef] [Green Version]
- Li, S.S.; Zhu, A.; Benes, V.; Costea, P.I.; Hercog, R.; Hildebrand, F.; Huerta-Cepas, J.; Nieuwdorp, M.; Salojärvi, J.; Voigt, A.Y.; et al. Durable coexistence of donor and recipient strains after fecal microbiota transplantation. Science 2016, 352, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Salzman, N.H.; Acharya, C.; Sterling, R.K.; White, M.B.; Gavis, E.A.; Fagan, A.; Hayward, M.; Holtz, M.L.; Matherly, S.; et al. Fecal Microbial Transplant Capsules Are Safe in Hepatic Encephalopathy: A Phase 1, Randomized, Placebo-Controlled Trial. Hepatology 2019, 70, 1690–1703. [Google Scholar] [CrossRef]
- Philips, C.A.; Pande, A.; Shasthry, S.M.; Jamwal, K.D.; Khillan, V.; Chandel, S.S.; Kumar, G.; Sharma, M.K.; Maiwall, R.; Jindal, A.; et al. Healthy Donor Fecal Microbiota Transplantation in Steroid-Ineligible Severe Alcoholic Hepatitis: A Pilot Study. Clin. Gastroenterol. Hepatol. 2017, 15, 600–602. [Google Scholar] [CrossRef]
- Duan, Y.; Llorente, C.; Lang, S.; Brandl, K.; Chu, H.; Jiang, L.; White, R.C.; Clarke, T.H.; Nguyen, K.; Torralba, M.; et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 2019, 575, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Górski, A.; Bollyky, P.L.; Przybylski, M.; Borysowski, J.; Międzybrodzki, R.; Jończyk-Matysiak, E.; Weber-Dąbrowska, B. Perspectives of Phage Therapy in Non-bacterial Infections. Front. Microbiol. 2018, 9, 3306. [Google Scholar] [CrossRef]
- Ramachandran, G.; Bikard, D. Editing the microbiome the CRISPR way. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spyrou, N.; Vallianou, N.; Kadillari, J.; Dalamaga, M. The interplay of obesity, gut microbiome and diet in the immune check point inhibitors therapy era. Semin. Cancer Biol. 2021, 73, 356–376. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallianou, N.; Christodoulatos, G.S.; Karampela, I.; Tsilingiris, D.; Magkos, F.; Stratigou, T.; Kounatidis, D.; Dalamaga, M. Understanding the Role of the Gut Microbiome and Microbial Metabolites in Non-Alcoholic Fatty Liver Disease: Current Evidence and Perspectives. Biomolecules 2022, 12, 56. https://doi.org/10.3390/biom12010056
Vallianou N, Christodoulatos GS, Karampela I, Tsilingiris D, Magkos F, Stratigou T, Kounatidis D, Dalamaga M. Understanding the Role of the Gut Microbiome and Microbial Metabolites in Non-Alcoholic Fatty Liver Disease: Current Evidence and Perspectives. Biomolecules. 2022; 12(1):56. https://doi.org/10.3390/biom12010056
Chicago/Turabian StyleVallianou, Natalia, Gerasimos Socrates Christodoulatos, Irene Karampela, Dimitrios Tsilingiris, Faidon Magkos, Theodora Stratigou, Dimitris Kounatidis, and Maria Dalamaga. 2022. "Understanding the Role of the Gut Microbiome and Microbial Metabolites in Non-Alcoholic Fatty Liver Disease: Current Evidence and Perspectives" Biomolecules 12, no. 1: 56. https://doi.org/10.3390/biom12010056