
The Structural–Functional Crosstalk of the Calsequestrin System: Insights and Pathological Implications

1
Department of Molecular Medicine, University of Pavia, 27100 Pavia, Italy
2
Laboratory of Molecular Cardiology, IRCCS ICS Maugeri, 27100 Pavia, Italy
3
Centro Nacional de Investigaciones Cardiovasculares Carlos III (CNIC), 28029 Madrid, Spain
*
Author to whom correspondence should be addressed.
Biomolecules 2023, 13(12), 1693; https://doi.org/10.3390/biom13121693
Submission received: 30 October 2023
/
Revised: 14 November 2023
/
Accepted: 21 November 2023
/
Published: 23 November 2023
(This article belongs to the Special Issue Ion Channels: Structure, Function, Regulatory Mechanisms and Roles in Physiology and Disease)
Abstract
:Calsequestrin (CASQ) is a key intra-sarcoplasmic reticulum Ca2+-handling protein that plays a pivotal role in the contraction of cardiac and skeletal muscles. Its Ca2+-dependent polymerization dynamics shape the translation of electric excitation signals to the Ca2+-induced contraction of the actin-myosin architecture. Mutations in CASQ are linked to life-threatening pathological conditions, including tubular aggregate myopathy, malignant hyperthermia, and Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT). The variability in the penetrance of these phenotypes and the lack of a clear understanding of the disease mechanisms associated with CASQ mutations pose a major challenge to the development of effective therapeutic strategies. In vitro studies have mainly focused on the polymerization and Ca2+-buffering properties of CASQ but have provided little insight into the complex interplay of structural and functional changes that underlie disease. In this review, the biochemical and structural natures of CASQ are explored in-depth, while emphasizing their direct and indirect consequences for muscle Ca2+ physiology. We propose a novel functional classification of CASQ pathological missense mutations based on the structural stability of the monomer, dimer, or linear polymer conformation. We also highlight emerging similarities between polymeric CASQ and polyelectrolyte systems, emphasizing the potential for the use of this paradigm to guide further research.
1. An Introduction to CASQ
1.1. The Right Buffer at the Right Place
For muscle sarcomere contraction to occur, large quantities of Ca2+ must flow out of the sarcoplasmic reticulum (SR) lumen into the cytosol, driven by a very steep electrochemical gradient (SR/cytosol Nernst potential for Ca2+ is approx. 125 mV in a non-contracting cell). The maintenance of this on-demand Ca2+ supply partially relies on the highly acidic calsequestrin (CASQ) protein, and its interaction with each member of the Calcium Release Unit (Figure 1): Junctin (JNT), Triadin (TRDN), and RyR [1,2,3]. CASQ is relatively small (45 kDa), and yet is the most prominently expressed protein within the lumen of the “junctional” sarcoplasmic reticulum (jSR) (up to 100 mg/mL) [4,5]. Abundant quantities of Ca2+ cooperatively bind to CASQ with low affinity, allowing for a rapid back-and-forth (with each contraction–relaxation cycle) exchange of massive quantities of Ca2+ between CASQ and the matrix of the jSR. In mammals, two CASQ genes are differentially expressed in skeletal fast-twitch fibers (CASQ1) and cardiac muscle (CASQ2), whereas both isoforms are equally represented in slow-twitch fibers [6,7]. Measurements in CASQ2-KO mice indicate that CASQ2 stores about 50% of the cardiac SR Ca2+ content [8] while maintaining a free calcium concentration of approximately 1 mM [9]. Similar studies in CASQ1-KO fast-twitch skeletal muscles indicate that the Ca2+ stored in CSQ1 contributes 75% of the released calcium [10], while still maintaining a free Ca2+ concentration of approximately 1 mM in the resting fiber.
1.2. CASQ Macro-Architecture(s)
The first electron micrographs of the jSR revealed an electron-dense filling able to assume the most diverse conformations from wire-shaped structures immediately beneath and parallel to the junctional membrane to intricately branched filaments and spherical bodies or puncta [11,12,13,14,15,16,17]. Imaging in vivo and cross-linking experiments support the notion that this material is Ca2+-complexed CASQ [18,19,20]. In addition, these structures disassemble and disperse under luminal Ca2+ depletion conditions [15,21], which correlates with the fact that the recombinant protein can organize in vitro into multiple architectures of varying compactness, hierarchically ordered in response to rising Ca2+ concentrations from micromolar levels up to 20 mM [3,16,17,22] (Figure 2). As higher order polymeric structures are formed, the capacity and cooperativity of Ca2+-binding events show a parallel, stepwise increase, which has consequences for the modulation of intra-SR Ca2+ storage and release. But how are CASQ multimers organized?
2. CASQ Multimeric Organization
2.1. CASQ Secondary and Tertiary Folding Is Sustained by Cations
A high abundance of carboxylates, carried by aspartic and glutamic acids, characterizes all CASQ isoforms, with the human skeletal and cardiac proteins featuring an isoelectric point of 4.0 and 4.2, respectively (Figure 2). Due to repulsion between negative charges, the polypeptide retains an extended, random coil conformation at low ionic strength (i.e., lower than that provided by 100 mM KCl) [23]. Multiple, monovalent, or divalent cations can likewise guide the folding of three negatively charged, nearly identical, thioredoxin domains (Figure 3A,B) [24,25,26,27,28], where numerous hydrophobic interactions hold the interior of the domains [29]. The minimal ionic concentration sustaining secondary and tertiary protein folding varies with the coordination number and ionic radius of the cation [23,24,25,27,28,29,30], with Ca2+ featuring the lowest effective concentrations, plus distinctive binding cooperativity even at modest ionic strength [23,25]. Low concentrations of monovalent cations (i.e., 85–150 mM KCl) have a cooperative effect on Ca2+-binding events at the early stages of protein folding [23,29,31]. It appears, however, that similar or higher abundances of monovalent ions inhibit compaction of the critical, dimerization-competent monomer, which is ultimately stabilized by a minimal amount of Ca2+ [32].
2.2. The Ca2+-Specific Dimerization Switch Is Salt-Sensitive
The first and obligatory step of CASQ supra-molecular assembly is the Ca2+-driven “front-to-front” dimerization through N-terminal domain swapping (Figure 3C,D) [29,32,33,34]. Upon Ca2+ binding, a few alternatively charged residues of the N-terminus flip and establish a hydrogen bonding network. In turn, this secures the extended conformation of the N-terminal tail over the surface of the opposite monomer (Figure 3C,D). Multiple intercalated Ca2+ ions bridge in between the abundant carboxylate groups exposed from each monomer’s surface, as revealed by equilibrium dialysis [4,32]. The comparison between different crystal structures of the CASQ dimers reveals that the inter-monomer space could also be filled by other cations than Ca2+. Nonetheless, only divalent ions engage the two monomers in a tight architecture, allowing more hydrophobic residues to line the inter-monomer interface (Figure 3E) [31,32,35,36].
2.3. Poorly Understood Ca2+-Dependent Mechanisms Drive CASQ Polymerization
The bottleneck of CASQ polymerization is proper tetramer assembly [4,32]. The ordered “back-to-back” tetramerization necessitates the intrinsically disordered C-terminal tail [37,38]. Truncation mutants lacking the C-terminus indeed behave as constitutive dimers in the solution and fail to multimerize upon Ca2+ addition [4]. It has been hypothesized that the strong negatively charged tail repels the formation of improperly positioned dimers, yet the exact mechanism driving physiological polymerization has not been elucidated. Interestingly, the main difference between skeletal and cardiac isoforms lies precisely in the C-terminal segment (Figure 4A). The longer and more negatively charged C-terminus of CASQ2 drives multimerization at higher Ca2+ concentrations (~2 mM) than CASQ1 (~0.7 mM) [37,38], and the swapping of this segment between isoforms causes the reciprocal exchange of their Ca2+-dependent polymerization kinetics [37]. As the coulombic properties of the C-terminal tail drive the specific conformational responsiveness to Ca2+, the other negative charges of the polypeptide shape the surface electric potential of the growing CASQ polymer, with CASQ1 polymers providing a more charged surface than CASQ2, onto which more Ca2+ ions can be adsorbed [4]. Since the dimer-to-tetramer transition, CASQ Ca2+ binding capacity and cooperativity increase; newly bound Ca2+ ions somehow stabilize further Ca2+-coordinating sites. The outcome is a multimeric species endowed with an enormous Ca2+-binding capacity; up to 60 Ca2+ ions are bound per molecule for human cardiac CASQ2, and up to 80 for the skeletal CASQ1 [4,29,32].
3. CASQ Polymer Dynamics Influence Ca2+ Release from jSR
3.1. CASQ Is a Dynamic System
While there is no question that CASQ forms polymers in vivo, the extent to which depolymerization–polymerization cycles occur during a single Excitation–Contraction Coupling (ECC) cycle is a matter of ongoing research. The shifts in the equilibrium among distinct CASQ quaternary states have huge functional implications both directly on the Ca2+ storage capacity and Ca2+ release kinetics, either indirectly through affecting the protein’s localization and interactome. In skeletal muscle, limited changes in intra-SR Ca2+ weaken CASQ1 architecture and increase its mobility within the longitudinal SR [16]. In cardiac muscle, beat-to-beat variations in CASQ2 multimerization status have not been experimentally verified, to our knowledge. Considering the effect of post-translational modifications (which will be discussed in Section 4), it seems likely that CASQ2 (with a measured in vitro Ca2+ affinity for the recombinant form of about 2 mM [4,29,37]) at least partially polymerizes in vivo at the physiological 1 mM intra-SR Ca2+. Further in silico studies exploring this possibility rendered that such quaternary state transitions would be important in shaping Ca2+ release termination and refractoriness [39]. Still, dissecting the direct effect of CASQ on RyR activity versus the indirect effects on intra-SR Ca2+ buffering (which in turn also affects RyR activity) remains a technically challenging question.
3.2. CASQ Ca2+-Driven States Physically Tune the Ca2+ Release Channel
Both in skeletal [16] and cardiac [40,41] muscles, CASQ interplays with the JNT-TRD-RyR Ca2+ Release Unit (CRU) (Figure 1) [42]. Two SR transmembrane proteins, TRDN and JNT, are essential, non-interchangeable mediators of the CASQ-dependent modulations of RyR activity [40,43,44,45,46]. JNT and TRDN both feature a single-transmembrane helical domain and a long, flexible, luminal domain exposing multiple KEKE (Lys-Glu-Lys-Glu) motifs, believed to be CASQ-anchoring points [3,47]. CASQ recruitment by TRDN and JNT is in bidirectional equilibrium with its Ca2+-dependent polymerization. Both JNT and TRD stabilize CASQ polymeric structures in vivo [1,48,49], whereas in vitro, varying stoichiometries of JNT shape CASQ2 polymerization dynamics [3,50]. On the other hand, the CASQ Ca2+-driven conformational state affects its affinity for JNT and TRD; C-terminally phosphorylated CASQ1, which polymerizes at lower Ca2+ than its unphosphorylated form, is more readily recruited by Junctin [3,51,52]. Similarly, the strength of CASQ–RyR interaction is also Ca2+-sensitive; it remains intact for free Ca2+ levels within the physiological SR range (0–1 mM) but diminishes at 5 mM Ca2+ [53]. Regarding the identification of the CASQ-binding region of RyR, it has been revealed that, when CASQ2 is co-expressed in HEK293 cells alongside RyR (HEK293 cells are heterologous expression systems which do not contain detectable levels of neither endogenous CASQ nor endogenous RyR), CASQ2 can directly bind the first luminal loop (residues 4521–4573) of the cardiac RyR2 [53]; yet, different in vitro tests could not identify a critical region for such binding [54]. Interestingly, the C-terminal domain of CASQ serves as a crucial mediator (although not the sole one) for its interaction with any component of the CRU [45,55]. The particular functional impact of CASQ on RyR activity is determined by the specific partner involved, where TRDN and JNT, despite their similarities, mediate distinct effects on the opening and closure of the RyR channel [49,56,57,58].
3.3. CASQ Polymerization Regulates ECC Indirectly by Modifying Free Intra-SR Ca2+
Bilayer [40,59] and in vitro [8,60] data suggest that, in the absence of CASQ, RyR activity is still sensitive to varying intra-SR Ca2+ levels. Cardiac RyRs are endowed with activating intra-SR Ca2+-binding sites [59], whereas skeletal RyRs feature a Ca2+-dependent inactivation site on their cytosolic side [61]. Whatever the mechanism of release modulation, what is clear is that CASQ-independent mechanisms exist intrinsic to the RyRs which are able to activate and terminate SR Ca2+ release. Moreover, intra-SR Ca2+ buffering via distinct CASQ quaternary states, together with proper CASQ retention at the jSR, are essential for indirectly shaping the physiological kinetics of jSR Ca2+ release and refractoriness (i.e., the ability for RyRs to activate a second time once they have been already active). Accordingly, CASQ2-related hereditary cardiac arrhythmias commonly feature an impaired release refractoriness [8], whereas mutations or deletions of skeletal CASQ1 have been linked to contraction defects, fatigue [16], and degenerative skeletal muscle diseases [15].
Finally, it must also be pointed out that CASQ is neither the only intra-SR Ca2+ buffer nor the only intra-SR Ca2+ release modulator. The histidine-rich calcium-binding protein (HRC) is a structurally distinct, high capacity, low Ca2+ affinity buffer (HRC KD = 1.9 mM [62]). Since no Ca2+-binding motifs are known within HRC, it is presumed that its acidic repeats constitute its Ca2+ binding sites [63] (HRC contains more acidic sites than CASQ). Functional studies in CASQ2/HRC double-KO mice have revealed that the absence of both proteins improves the cardiac arrhythmic phenotype vs. CASQ2-KO mice [64]. Since HRC, like CASQ, has the capability of binding TRDN in a Ca2+-dependent manner, a model has been proposed in which the CASQ2-dependent and HRC-dependent regulation mechanisms of SR Ca2+ release are complementary; TRDN would bind CASQ2 at low intra-SR Ca2+ levels (inhibiting the channel), and HRC would bind TRDN at high SR-free Ca2+ (priming the channel for further aperture) [64]. Besides these direct and indirect effects on SR Ca2+ release, HRC has been shown to modulate SR Ca2+ uptake in a SR Ca2+-dependent manner by directly binding to SERCA [65]. The phosphorylation of HRC S96 by Fam20C regulates the interactions of HRC with both TRDN and SERCA2a [66]. Consequently, the HRC mutation S96A (S81A in mice) has been linked to arrhythmias and sudden cardiac death [67].
4. Post-Translational Modifications Modulate the Responsivity of the CASQ System
4.1. CASQ Dynamics Are Post-Translationally Tuned
The Ca2+-binding and multimerization properties of CASQ are tightly and intimately related to its trafficking and retainment at the jSR. Both pathological missense mutations and N-terminal tags alter the protein’s physiological localization [68,69,70,71]. It stands to reason that post-translational modifications of CASQ, by shaping the kinetics of Ca2+-dependent quaternary assembly, also modulate CASQ trafficking and retention within the jSR. Multiple covalent modifications at various sites have been detected using mass-spectrometry including glycosylation, phosphorylation, and even the proteolysis of its N-terminal segment [34,72,73]. Above all, the favored modification hotspot is the C-terminus. This further highlights the critical and intertwined role of this disordered segment in modulating the protein’s Ca2+-related properties.
4.2. Phosphorylation
Phosphorylation at either Thr-353 by Casein Kinase 2 [51,52] or at Ser-248 and Ser-369 by Fam20C [21] increases the responsiveness to Ca2+ of recombinant skeletal CASQ1 [72]. Likewise, for cardiac CASQ2, the phosphorylation of Ser-385 by Fam20C increases its overall Ca2+-binding capacity [74], whereas the phosphorylation of both Ser-385 and Ser-393 by Casein Kinase 2 lowers the Ca2+ threshold for polymerization in vitro [72]. The augmented sensitivity of phosphorylated CASQ1 and CASQ2 to Ca2+ could be explained, in principle, by the higher chemical affinity of Ca2+ towards phosphate rather than carboxylate groups. This mechanism may not be unique to CASQ, as the phosphorylation of the highly flexible protein Casein increases its Ca2+-binding capacity, its solubility, and its ability to form Ca2+-containing colloidal suspensions [75,76]. It appears that the C-terminus of skeletal CASQ1 is mainly unphosphorylated in vivo, whereas variable amounts of C-terminally non-phosphorylated, singly phosphorylated, and doubly phosphorylated CASQ2 have been confirmed to co-exist in cardiomyocytes [72,77]. The relevance of skeletal CASQ phosphorylation in vivo, along with the effect of cardiac CASQ2 phosphorylation on the regulation of Ca2+ fluxes at the dyad, are still obscure aspects of CASQ biology.
4.3. Glycosylation
As for phosphorylation, cardiac CASQ2 in vivo exhibits a wide range of glycosylation levels, whereas skeletal CASQ1 primarily possesses a lone mannose group [72]. A paradigm for the role of glycosylation is given by research conducted on CASQ1. Significant co-translational glycosylation at a specific C-terminal Asn residue, with up to 8–9 mannose groups, hinders CASQ multimerization until the protein reaches the jSR lumen, where mannose trimming restores CASQ’s polymerization capacity so that polymerization-competent species are retained [78,79]. While bulky glycosyl chains hinder oligomerization, short mannose chains stabilize both dimerization and polymerization. This double effect is a consequence of the critical positioning of the glycosylation residue in between the C-terminus of the host monomer and the N-terminus of the dimerization partner. Mannosyl chains at the conserved Asn-316 residue hold the extended N-terminal swapped domains from one side, and physically constrain the sides of the growing polymer on the other side (Figure 4B,C) [72]. In the heart, the defective glycosylation of CASQ2 has been linked to acquired cardiac diseases; Man8,9-containing CASQ2 is highly increased in the heart failure of different etiologies, and it is worth mentioning that the protein is concomitantly more phosphorylated [80,81]. As far as genetic diseases are concerned, the mutated CASQ2-K206N (linked to Catecholaminergic Polymorphic Ventricular Tachycardia type 2) features a de novo N-glycosylation site, together with defective SR targeting and reduced Ca2+-binding capacity [82].
5. Molecular Patho-Physiological Implications of CASQ’s Defects
5.1. Uncertainties Surrounding the Biochemical Behavior of CASQ’s Pathological Variants
Skeletal CASQ1 pathological missense mutations in heterozygous conditions are linked to either malignant hyperthermia (CASQ1 M87T) or tubular aggregate myopathy (CASQ1 D44N, G103D, D244G, I387T) [15,83,84], whereas the numerous pathological mutations of the cardiac isoform lead to Catecholaminergic Polymorphic Ventricular Tachycardia type 2 (CPVT2) mainly in homozygosis [31,36,85]. CASQ1 and CASQ2 missense mutations are scattered across all three thioredoxin domains of CASQ and the mutated proteins do not share any common defect in their Ca2+-dependent polymerization properties [31,36,85,86]. Moreover, most of the published biochemical analyses on either the polymerization and/or Ca2+-buffering capabilities of recombinant CASQ missense pathological mutants [15,28,31,33,34,35,36,85,86,87,88,89,90] (Table 1) are sometimes difficult to interpret or in contrast with each other.
A first limitation that may have led to this situation is the previously described hyper sensitivity of CASQ to the ionic conditions [85,86]. Secondly, the very intimate relationship between Ca2+-binding events and polymerization complicates the interpretation of Ca2+-binding curves, particularly those obtained with methods such as MicroScale Thermophoresis, which are sensitive to changes in molecular size. A third limiting aspect involves the widely used turbidity assay, which cannot discriminate among multimerization kinetics and/or aggregation processes occurring in vivo. Different polymerization defects, for example, characterize CASQ1, D244G, and I385T mutants, either in their linear polymerization or in the formation of branching points [15]. In addition, the post-translational modifications of CASQs are not well accounted for in in vitro assays. On the road towards a pathophysiological understanding of CASQ mutations, it appears that the results obtained from the wealth of in vitro approaches should not be considered in isolation, but as part of a complex, still fragmented scenario.
5.2. Dimerization of CASQ Missense Variants May Underlie Distinct Penetrance Mechanisms
The variable penetrance of inherited CASQ2 mutations [31,36], when combined with the 100% penetrance of the dominant K180R CASQ2 [91], has recently led to the hypothesis that two distinct molecular processes, triggered by two distinct groups of CASQ2 mutations, may underlie the recessive and dominant forms of CPVT2 [31,89].
Starting from the above, we propose here the first functional classification of the pathological missense mutations of CASQs, which stems from the expected structural consequence on the stability of either the monomer, the dimer, or the recently crystallized linear polymer conformation [31] (Figure 4D, Table 1). For each characterized pathological mutation, we compiled its biochemical feature as described in published studies.
In Table 1, each column summarizes the biochemical data published for each of the most studied pathological mutants for either skeletal or cardiac CASQ. Mutants have been clustered and colored depending on the CASQ conformational state possibly affected (either the stability of the monomer, of the dimer, or of the polymer), and, within each structural group, ordered by the residue number. Lighter to darker colors reflect the quaternary state, from the monomer to the polymeric one, possibly affected by the mutation. CASQ2 pathological variants are colored from light blue to dark blue, and CASQ1 from light yellow to dark yellow, from the left to the right. Mutations altering the folding of the monomer encloses those mutation residues falling within the hydrophobic core of the thioredoxin domains, whereas mutations altering the inter-monomer dimerization or tetramerization interfaces have been included in the “Dimer”, “D/P”, or “Polymer” groups, respectively. An additional group named “P*” contains a mutants with supposed altered polymeric conformation. In the crystal structure by Titus et al. [31] (PDB ID: 6OWW) (Figure 4D), 28 was used as a model for the structural inspection and sorting of the mutations. Experiments are listed on the first column on the left and are also grouped based on the type of information given. Those experiments informing on the tertiary architecture folding and stability of the protein monomer in the absence of Ca2+ (tryptophan fluorescence, Circular Dichroism, degree of protection by trypsin digestion, melting temperature curve, mono-dispersion in Gel Filtration Chromatography) have been grouped under the name “tertiary structure”. Experiments analyzing the Ca2+-induced structural rearrangements and Ca2+-binding affinities are grouped under the name “Ca2+-dependent properties” (Ca2+-induced protection by trypsin digestion, Ca2+-induced changes in the Circular Dichroism spectrum and tryptophan fluorescence signal, Ca2+-induced increase in the apparent radius of gyration on Dynamic Light Scattering, appearance of a peak species in Gel Filtration Chromatography corresponding to the elution volume for a CASQ dimer, Ca2+-induced increase in the 350 nm turbidity, Ca2+-induced increase in the 350 nm turbidity of the protein pre-incubated with 2 mM Mg2+, Ca2+-induced increase in the high-molecular-weight species in Native-PAGE, Ca2+-binding curve measured either through MicroScale Thermophoresis or equilibrium dialysis). For each box, the experimental interpretation is summarized. More specifically, the behavior of each mutant is evaluated for each indicated experiment in terms of the overall information gained. For those experiments assessing CASQ “tertiary structure” in absence of Ca2+, the interpretation refers to the stability or retainment of the wild-type monomeric conformation, whereas for those experiments assessing the Ca2+ responsiveness, either conformational or in terms of binding kinetics, the interpretation is given in terms of the amplitude of the structural rearrangement or binding affinity relative to that of the wild-type protein in the same condition. ↓: lower than wt, =: identical to wt, ↑: higher than wt. References are given for each experiment. Additional details are given in the ‘Notes’ section.
A trend emerges revealing two groups with distinct biochemical behaviors, where the discriminant structural feature is the in vitro ability to dimerize or not (Table 1). Given that proper CASQ sorting and retention within the jSR is determined by the protein’s ability to multimerize, it has been hypothesized that those CASQ mutants unable to dimerize would not be targeted or be allowed to reside within the jSR, leading to haploinsufficiency when in heterozygosis, and to loss of CASQ when in homozygosis [31]. In accordance with this hypothesis, the only recessive CASQ2 mutations that have been characterized in murine models, sharing a consistent loss of CASQ2 within the jSR [92,93], fall (R33Q [94], D307H [95]) or would fall (G112 + 5X [96]) within the first non-dimerizing group of proteins (on the left side of Table 1). The second group is that of dimerization-competent mutants (on the right side of Table 1), which includes many of the dominantly inherited CASQ1 variants, the only CASQ2 mutation whose dominance has been clinically and functionally validated to date [89,91], and other mutations clinically associated with CPVT2 in heterozygosis [36]. In those cases, we hypothesize that the extremely subtle defects, undetectable at the dimer level, might be amplified and, thus, exert their pathological effect only within the polymer structure(s), which could present altered Ca2+-binding/release properties and interactome. In this regard, it is indeed interesting to note that the only mouse model carrying a mutation from the second group (CASQ2-K180R) did not show any loss in the total intra-SR calcium buffering capacity (contrary to all CPVT2 recessive models), but did show a defect in the dynamic intra-SR calcium buffering [89]. Our hypothesis points to the CASQ dimerization of pathological mutants as a critical aspect in determining the intra-SR dominant/recessive pathological effect. The altered trafficking or turnover of a non-dimerizing mutant would lead to a loss of intra-SR CASQ, which in turn affects the total Ca2+-binding capacity of the system. On the other hand, dimerization-competent mutants, residing in the jSR, can recruit the wild-type CASQ counterpart into less responsive CASQ-Ca2+ systems. Of note are two CASQ1 mutations, one correlated with malignant hyperthermia (CASQ1 M87T), and a second one involved in tubular aggregate myopathy, which have been grouped in between the non-dimerizing and the dimerizing variants because their position possibly disrupts either the dimerization or the polymerization interfaces (Table 1). We expect that our functional classification provides a starting working hypothesis for the molecular understanding of the varying penetrance of pathological CASQ variants in human diseases. From all the above, it is concluded that additional clinical in vivo and in vitro studies are necessary to assess and describe the possibly distinct pathological mechanisms between the first and the second group of mutations.
6. Polymeric CASQ Shares Similarities with Polyelectrolyte Systems
The complex physico-chemical nature of CASQ is the consequence of the multiplexed interactions between the high number of anionic groups exposed by CASQ’s monomers, dimers, and polymers, and free ions from the solution. A new possible approach to unveil CASQ complexity might consider its similarities with anionic self-assembling polymers [97]. These polyelectrolyte materials are ubiquitously employed by nature to organize complex processes such as numerous intra-cellular and extra-cellular highly charged systems, among them nuclear chromatin, and dynamically segregate delicate processes in membrane-less compartments [98,99]. To shed light on some underrated aspects of CASQ biology, some of the most interesting similarities with polyelectrolyte systems across comparable spatial scales and their relative biological meanings are discussed below.
6.1. Multiple Ionizable Groups on a Highly Flexible Substrate
Polyelectrolytes are defined by the presence of multiple charged groups on a conformationally versatile substrate. This same condition also characterizes CASQs, and it is particularly brought to the extreme at the intrinsically disordered, highly charged C-terminal tail [37]. The pervading role of this segment in determining the polymerization kinetics and the interaction with members of the CRU may lie in its ability to provide multiple weak and transient interactions in the solution at various Ca2+ concentrations [38,51].
6.2. Solvent Charges Modulate the Polymeric Architecture
Freely mobile counter-ions from the solution mask the negative charges of the polymeric phase and bridge the formation of intra- and inter-chain interactions, whose number is driven by the mobile counter-ions concentration, valence, and size [97]. The typical behavior of polyelectrolytes perfectly fits current knowledge of CASQ; its physiological polymerization is mainly triggered by Ca2+, whereas other ions promote either similar or opposite behaviors. Monovalent potassium and sodium ions support the folding of the same tertiary structure, yet they inhibit Ca2+-dependent quaternary assembly (Figure 2 and Figure 3) [4,29,51,100]. Divalent charges from transition metals (Mn2+, C02+, Ni2+, Cu2+, Zn2+) promote CASQ multimerization, probably because of their typical versatile coordination mechanisms. Divalent Mg2+ cations, abundantly present in the jSR (1 mM free) [28] have intriguing effects: the pre-incubation of CASQ with 2 mM Mg2+ before the addition of Ca2+ either facilitates the nucleation of the soluble bodies or changes their shape towards a higher radius of gyration [26,28,31].
Multiple authors have pointed out the fact that Ca2+ fluxes in and out from the jSR are coupled with cyclic variations of other cationic species. For example, Refs. [101,102] showed that net K+ countercurrent towards the SR occurs concomitantly to RyR-mediated Ca2+ release in ventricular myocytes, ensuring the neutrality of the electrochemical potential differences across the SR membrane. Likewise, in skeletal muscle SR vesicles, it was long reported that K+ (and anion) fluxes, which balance the electrical charges accumulated in the SR during Ca2+ uptake, mediate changes in SR volume by generating osmotic pressure [103]. Whether and how such cyclical variations in cation levels impact the conformation of CASQ remains obscure [25,30,36,72,104].
Protons (H+) compete for CASQ’s Ca2+-binding sites [25,27,30] and can enhance in vivo the effect of a high Ca2+ load within the jSR [30]. While we remain unaware of a demonstration of neither beat-to-beat nor single-stimulation intra-SR pH transients in living cells, we must also note that, based on observations from skeletal muscle, intra-SR pH is believed to be in equilibrium with cytosolic pH due to a large proton permeability of the SR membrane [105]. In this regard, frequency- and β-adrenergic stimulation-dependent beat-to-beat cytosolic acidifications have been measured in the cytosol of healthy ventricular myocytes (about −0.12 pH units at 0.5 Hz in rabbits [106]). Based on all the above, it is tempting to note that, if such beat-to-beat intra-SR acidifications were present (and of sufficient strength), they would decrease Ca2+ binding to CASQ2 [25], aiding in the RyR recovery from refractoriness (as free intra-SR Ca2+ would increase faster for any given SR Ca2+ uptake rate) while simultaneously helping in maintaining the assembly of CASQ. Interestingly, very low pH conditions drove the crystallization of the only polymeric structure published [31].
Finally, based on the behavior of known carboxylate-rich polyanionic gels, one final issue of “macroscopic” nature must be borne in mind: the sharp volume changes in the polyelectrolyte gel would be expected to occur as the concentration of bivalent cations and pH change [97,107]. This effect seems unlikely to occur in vivo for CASQ in short time scales (i.e., for simple polyanionic gels, the time scales required are in the order of thousands of seconds [107]). However, experiments comparing such behavior in jSR vesicles filled with either WT or mutated CASQ (or a combination of both) might provide new hypotheses about how specific mutations affect the long-term structural remodeling of the jSR (a common finding in CASQ-related diseases, e.g., [94]).
6.3. Long-Range Conductance
The high mobility of soluble charges is a key feature of polyelectrolyte matrices and accounts for their typical systemic response to changes in pH and ionic conditions [107]. Likewise, Ca2+ ions layer all over the negative surface of CASQ, where they are flexibly coordinated by both the exposed acidic residues and free water molecules. The one-dimensional diffusion of Ca2+ along the CASQ surface [16,35,72,108] is coupled with proper alignment and geometry of the polymer chains [97,108]. This “Ca2+ wire” mechanism, proposed more than two decades ago [108], might accelerate in vivo the directional Ca2+ flow from the longitudinal SR (where Ca2+ ions are continuously pumped in from the cytosol) up to the jSR release channel (Figure 5B). Two released crystal structures of a CASQ filament beautifully fit this hypothesis; in both cases, a solvent-accessible tunnel traverses the length of the linear polymer [31,109] and is lined by several Ca2+-binding carboxylates. However, the very low pH conditions of the first one, published in 2020, may pose caution on its relevance as a Ca2+-bound state [31], whereas the second one (deposited with PDB ID: 7F05) must await formal peer-reviewed publication containing experimental details on the crystallization condition and concomitant analysis [109]. Studies on the kinetics of Ca2+ dissociation from CASQ revealed that its polymeric conformation is retained during the first rapid burst of Ca2+ release, showing that a reservoir of fast-conductance, readily available Ca2+ ions can be provided without any rearrangement of the polymer [104]. This in vitro observation easily correlates with the fact that the first “hump” of the Ca2+ release curve in vivo is determined by CASQ [16]. On these bases, it is expected that the first Ca2+ ions exiting the SR are not provided by RyR-neighboring CASQ proteins as single entities, but implies the participation of the polymeric CASQ as a whole (Figure 5B–D). Further studies on the Ca2+ conductance properties of the CASQ system, including how conductance changes as release and uptake proceed, are needed to reveal important aspects of the kinetics of Ca2+ outflux from the RyR channel [109]. Further insights on the nature of the possibly multiple distinct reservoirs of Ca2+ hosted within polymerized CASQ (Figure 5) might provide interesting clues on the CASQ-driven termination of Ca2+ release [41,110] through RyR.
6.4. Ca2+-Driven Membrane-Less Compartmentalization
In the sarcoplasmic reticulum lumen, CASQ is retained within the junctional space [68,111,112,113]. The diffusion of CASQ along the longitudinal SR occurs only in response to extreme Ca2+ depletion [16]. A first explanation for this membrane-less compartmentalization lies in the fact that 30% of CASQ [16,35,72] has been found to be physically tethered to the jSR membrane through Ca2+-dependent, non-covalent bonds with the Ca2+ Release Unit [114] (Figure 1). On the other hand, it has to be taken into account that the Ca2+-dependent conformational states of CASQ feature distinct solubilities, hence they may also have a role in limiting CASQ dispersion within the SR [83]. A recent manuscript described that recombinant CASQ1 can reversibly form coacervates in vitro in response to the addition of 10 mM Ca2+ [21]. The reversible de-mixing of homogeneous solutions into liquid compartments with distinct concentrations of a solute is typical of highly charged organic polymer/salt mixtures [97], described under the common name of “liquid-liquid phase separation (LLPS)” [99,115]. Regarding CASQ, this remains an interesting hypothesis to be further tested, especially because CASQ features a seemingly Intrinsically Disordered Region (IDR) at its C-terminus (Figure 4A), a characteristic of all known proteins involved in LLPS. The multivalent, transient network of Ca2+-dependent interactions that such a domain provides may indeed seed the formation of highly CASQ populated regions of the solution, where multiple CASQ proteins would hold each other in a flexible mode within a restricted space (Figure 5A). CASQ Ca2+-dependent jSR retainment may be a sign of its physiological behavior as an organized membrane-less system. This is a hypothesis with interesting effects on SR Ca2+ release that remains to be tested.
7. Final Remarks
Since its initial discovery by MacLennan and Wong in 1971 [5], CASQ has mesmerized those involved in its study, from muscle physiologists to biochemists and medical doctors. The first three decades following its discovery were focused on biochemistry and overall characterization. Since the early 2000s, however, with the advent of genome sequencing, transgenic animals, and the discovery of CASQ-related hereditary diseases, our biochemical understanding has progressed far more slowly, as attention has focused on the link between physiopathology and associated genetic mutations. That being said, it is our belief that understanding CASQ’s pathologically relevant defects might require a renewed discovery of its structural transitions, from monomers to cross-linked polyelectrolyte-like polymers, and of the relative kinetics for the binding of Ca2+ and protein partners. Within this scenario, a hypothesis has emerged according to which the dimer-to-tetramer/polymer transition is a critical aspect of CASQ physiology and pathology. New comprehensive approaches investigating the polymeric, ionic, and dynamic nature of CASQ as a system might provide unprecedented opportunities to increase our knowledge and thus our therapeutical possibilities in the fight against striated muscular disease.
Author Contributions
All authors have read and agreed to the published version of the manuscript.
Funding
This work was partially supported by the Ricerca Corrente funding scheme of the Italian Ministry of Health. CNIC is supported by the Instituto de Salud Carlos III (ISCIII), the Ministerio de Ciencia e Innovación (MCIN) and the Pro CNIC Foundation, and is a Severo Ochoa Center of Excellence (grant CEX2020-001041-S funded by MICIN/AEI/10.13039/501100011033).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data used to compile this review work have been previously published.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Boncompagni, S.; Thomas, M.; Lopez, J.R.; Allen, P.D.; Yuan, Q.; Kranias, E.G.; Franzini-Armstrong, C.; Perez, C.F. Triadin/Junctin double null mouse reveals a differential role for Triadin and Junctin in anchoring CASQ to the jSR and regulating Ca2+ homeostasis. PLoS ONE 2012, 7, e39962. [Google Scholar] [CrossRef]
- Dulhunty, A.F.; Wei-LaPierre, L.; Casarotto, M.G.; Beard, N.A. Core skeletal muscle ryanodine receptor calcium release complex. Clin. Exp. Pharmacol. Physiol. 2016, 44, 3–12. [Google Scholar] [CrossRef]
- Lee, K.W.; Maeng, J.-S.; Choi, J.Y.; Lee, Y.R.; Hwang, C.Y.; Park, S.S.; Park, H.K.; Chung, B.H.; Lee, S.-G.; Kim, Y.-S.; et al. Role of Junctin Protein Interactions in Cellular Dynamics of Calsequestrin Polymer upon Calcium Perturbation. J. Biol. Chem. 2012, 287, 1679–1687. [Google Scholar] [CrossRef]
- Park, H.; Park, I.Y.; Kim, E.; Youn, B.; Fields, K.; Dunker, A.K.; Kang, C. Comparing skeletal and cardiac calsequestrin structures and their calcium binding: A proposed mechanism for coupled calcium binding and protein polymerization. J. Biol. Chem. 2004, 279, 18026–18033. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Wong, P.T.S. Isolation of a Calcium-Sequestering Protein from Sarcoplasmic Reticulum. Proc. Natl. Acad. Sci. USA 1971, 68, 1231–1235. [Google Scholar] [CrossRef]
- Biral, D.; Volpe, P.; Damiani, E.; Margreth, A. Coexistence of two calsequestrin isoforms in rabbit slow-twitch skeletal muscle fibers. FEBS Lett. 1992, 299, 175–178. [Google Scholar] [CrossRef]
- Paolini, C.; Quarta, M.; D’Onofrio, L.; Reggiani, C.; Protasi, F. Differential Effect of Calsequestrin Ablation on Structure and Function of Fast and Slow Skeletal Muscle Fibers. J. Biomed. Biotechnol. 2011, 2011, 634075. [Google Scholar] [CrossRef]
- Knollmann, B.C.; Chopra, N.; Hlaing, T.; Akin, B.; Yang, T.; Ettensohn, K.; Knollmann, B.E.C.; Horton, K.D.; Weissman, N.J.; Holinstat, I.; et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J. Clin. Investig. 2006, 116, 2510–2520. [Google Scholar] [CrossRef]
- Shannon, T.R.; Guo, T.; Bers, D.M. Ca2+ scraps: Local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ. Res. 2003, 93, 40–45. [Google Scholar] [CrossRef]
- Manno, C.; Sztretye, M.; Figueroa, L.; Allen, P.D.; Ríos, E. Dynamic measurement of the calcium buffering properties of the sarcoplasmic reticulum in mouse skeletal muscle. J. Physiol. 2013, 591, 423–442. [Google Scholar] [CrossRef]
- Renken, C.; Hsieh, C.-E.; Marko, M.; Rath, B.; Leith, A.; Wagenknecht, T.; Frank, J.; Mannella, C.A. Structure of frozen–hydrated triad junctions: A case study in motif searching inside tomograms. J. Struct. Biol. 2009, 165, 53–63. [Google Scholar] [CrossRef]
- Franzini-Armstrong, C.; Kenney, L.J.; Varriano-Marston, E. The structure of calsequestrin in triads of vertebrate skeletal muscle: A deep-etch study. J. Cell Biol. 1987, 105, 49–56. [Google Scholar] [CrossRef]
- Perni, S.; Close, M.; Franzini-Armstrong, C. Novel Details of Calsequestrin Gel Conformation in Situ. J. Biol. Chem. 2013, 288, 31358–31362. [Google Scholar] [CrossRef]
- Wagenknecht, T.; Hsieh, C.-E.; Rath, B.; Fleischer, S.; Marko, M. Electron Tomography of Frozen-Hydrated Isolated Triad Junctions. Biophys. J. 2002, 83, 2491–2501. [Google Scholar] [CrossRef]
- Barone, V.; Del Re, V.; Gamberucci, A.; Polverino, V.; Galli, L.; Rossi, D.; Costanzi, E.; Toniolo, L.; Berti, G.; Malandrini, A.; et al. Identification and characterization of three novel mutations in the CASQ1 gene in four patients with tubular aggregate myopathy. Hum. Mutat. 2017, 38, 1761–1773. [Google Scholar] [CrossRef]
- Manno, C.; Figueroa, L.C.; Gillespie, D.; Fitts, R.; Kang, C.; Franzini-Armstrong, C.; Rios, E. Calsequestrin depolymerizes when calcium is depleted in the sarcoplasmic reticulum of working muscle. Proc. Natl. Acad. Sci. USA 2017, 114, E638–E647. [Google Scholar] [CrossRef]
- Chen, W.; Kudryashev, M. Structure of RyR1 in native membranes. EMBO Rep. 2020, 21, e49891. [Google Scholar] [CrossRef]
- Maguire, P.B.; Briggs, F.N.; Lennon, N.J.; Ohlendieck, K. Oligomerization Is an Intrinsic Property of Calsequestrin in Normal and Transformed Skeletal Muscle. Biochem. Biophys. Res. Commun. 1997, 240, 721–727. [Google Scholar] [CrossRef]
- Glover, L.; Culligan, K.; Cala, S.; Mulvey, C.; Ohlendieck, K. Calsequestrin binds to monomeric and complexed forms of key calcium-handling proteins in native sarcoplasmic reticulum membranes from rabbit skeletal muscle. Biochim. Biophys. Acta (BBA)-Biomembr. 2001, 1515, 120–132. [Google Scholar] [CrossRef]
- O’Brian, J.J.; Ram, M.L.; Kiarash, A.; Cala, S.E. Mass Spectrometry of Cardiac Calsequestrin Characterizes Microheterogeneity Unique to Heart and Indicative of Complex Intracellular Transit. J. Biol. Chem. 2002, 277, 37154–37160. [Google Scholar] [CrossRef]
- Mayfield, J.E.; Pollak, A.J.; Worby, C.A.; Xu, J.C.; Tandon, V.; Newton, A.C.; Dixon, J.E. Ca2+-dependent liquid-liquid phase separation underlies intracellular Ca2+ stores. bioRxiv 2021. bioRxiv:2021.07.06.451223. Available online: http://biorxiv.org/content/early/2021/07/06/2021.07.06.451223.abstract (accessed on 30 September 2023).
- Beard, N.; Laver, D.; Dulhunty, A. Calsequestrin and the calcium release channel of skeletal and cardiac muscle. Prog. Biophys. Mol. Biol. 2004, 85, 33–69. [Google Scholar] [CrossRef]
- Aaron, B.M.; Oikawa, K.; A Reithmeier, R.; Sykes, B.D. Characterization of skeletal muscle calsequestrin by 1H NMR spectroscopy. J. Biol. Chem. 1984, 259, 11876–11881. [Google Scholar] [CrossRef] [PubMed]
- Ikemoto, N.; Bhatnagar, G.M.; Nagy, B.; Gergely, J. Interaction of divalent cations with the 55,000-dalton protein component of the sarcoplasmic reticulum. Studies of fluorescence and circular dichroism. J. Biol. Chem. 1972, 247, 7835–7837. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, C.; Donoso, P.; Rodriguez, P. Protons induce calsequestrin conformational changes. Biophys. J. 1996, 71, 2130–2137. [Google Scholar] [CrossRef]
- Krause, K.; Milos, M.; Luan-Rilliet, Y.; Lew, D.; Cox, J. Thermodynamics of cation binding to rabbit skeletal muscle calsequestrin. Evidence for distinct Ca2+- and Mg2+-binding sites. J. Biol. Chem. 1991, 266, 9453–9459. [Google Scholar] [CrossRef] [PubMed]
- Ostwald, T.J.; MacLennan, D.H.; Dorrington, K.J. Effects of Cation Binding on the Conformation of Calsequestrin and the High Affinity Calcium-binding Protein of Sarcoplasmic Reticulum. J. Biol. Chem. 1974, 249, 5867–5871. [Google Scholar] [CrossRef] [PubMed]
- Bal, N.C.; Jena, N.; Sopariwala, D.; Balaraju, T.; Shaikh, S.; Bal, C.; Sharon, A.; Gyorke, S.; Periasamy, M. Probing cationic selectivity of cardiac calsequestrin and its CPVT mutants. Biochem. J. 2011, 435, 391–399. [Google Scholar] [CrossRef]
- Park, H.; Wu, S.; Dunker, A.; Kang, C. Polymerization of calsequestrin. Implications for Ca2+ regulation. J. Biol. Chem. 2003, 278, 42728. [Google Scholar] [CrossRef]
- Donoso, P.; Beltrán, A.M.; Hidalgo, C. Luminal pH Regulates Calcium Release Kinetics in Sarcoplasmic Reticulum Vesicles. Biochemistry 1996, 35, 13419–13425. [Google Scholar] [CrossRef]
- Titus, E.W.; Deiter, F.H.; Shi, C.; Wojciak, J.; Scheinman, M.; Jura, N.; Deo, R.C. The structure of a calsequestrin filament reveals mechanisms of familial arrhythmia. Nat. Struct. Mol. Biol. 2020, 27, 1142–1151. [Google Scholar] [CrossRef]
- Sanchez, E.J.; Lewis, K.M.; Danna, B.R.; Kang, C. High-capacity Ca2+ Binding of Human Skeletal Calsequestrin. J. Biol. Chem. 2012, 287, 11592–11601. [Google Scholar] [CrossRef]
- Kim, E.; Youn, B.; Kemper, L.; Campbell, C.; Milting, H.; Varsanyi, M.; Kang, C. Characterization of Human Cardiac Calsequestrin and its Deleterious Mutants. J. Mol. Biol. 2007, 373, 1047–1057. [Google Scholar] [CrossRef]
- Bal, N.C.; Sharon, A.; Gupta, S.C.; Jena, N.; Shaikh, S.; Gyorke, S.; Periasamy, M. The Catecholaminergic Polymorphic Ventricular Tachycardia Mutation R33Q Disrupts the N-terminal Structural Motif That Regulates Reversible Calsequestrin Polymerization. J. Biol. Chem. 2010, 285, 17188–17196. [Google Scholar] [CrossRef]
- Lewis, K.M.; Ronish, L.A.; Ríos, E.; Kang, C. Characterization of Two Human Skeletal Calsequestrin Mutants Implicated in Malignant Hyperthermia and Vacuolar Aggregate Myopathy. J. Biol. Chem. 2015, 290, 28665–28674. [Google Scholar] [CrossRef]
- Ng, K.; Titus, E.W.; Lieve, K.V.; Roston, T.M.; Mazzanti, A.; Deiter, F.H.; Denjoy, I.; Ingles, J.; Till, J.; Robyns, T.; et al. An International Multicenter Evaluation of Inheritance Patterns, Arrhythmic Risks, and Underlying Mechanisms of CASQ2-Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2020, 142, 932–947. [Google Scholar] [CrossRef] [PubMed]
- Bal, N.C.; Jena, N.; Chakravarty, H.; Kumar, A.; Chi, M.; Balaraju, T.; Rawale, S.V.; Rawale, J.S.; Sharon, A.; Periasamy, M. The C-terminal calcium-sensitive disordered motifs regulate isoform-specific polymerization characteristics of calsequestrin. Biopolymers 2015, 103, 15–22. [Google Scholar] [CrossRef]
- Newcombe, E.A.; Fernandes, C.B.; Lundsgaard, J.E.; Brakti, I.; Lindorff-Larsen, K.; Langkilde, A.E.; Skriver, K.; Kragelund, B.B. Insight into Calcium-Binding Motifs of Intrinsically Disordered Proteins. Biomolecules 2021, 11, 1173. [Google Scholar] [CrossRef]
- Restrepo, J.G.; Weiss, J.N.; Karma, A. Calsequestrin-Mediated Mechanism for Cellular Calcium Transient Alternans. Biophys. J. 2008, 95, 3767–3789. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Valle, G.; Nani, A.; Chen, H.; Ramos-Franco, J.; Nori, A.; Volpe, P.; Fill, M. Ryanodine Receptor Luminal Ca2+ Regulation: Swapping Calsequestrin and Channel Isoforms. Biophys. J. 2009, 97, 1961–1970. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Valle, G.; Nani, A.; Nori, A.; Rizzi, N.; Priori, S.G.; Volpe, P.; Fill, M. Luminal Ca2+ Regulation of Single Cardiac Ryanodine Receptors: Insights Provided by Calsequestrin and its Mutants. J. Gen. Physiol. 2008, 131, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Terentyev, D.; Viatchenko-Karpinski, S.; Györke, I.; Volpe, P.; Williams, S.C.; Györke, S. Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: Mechanism for hereditary arrhythmia. Proc. Natl. Acad. Sci. USA 2003, 100, 11759–11764. [Google Scholar] [CrossRef]
- Terentyev, D.; Viatchenko-Karpinski, S.; Vedamoorthyrao, S.; Oduru, S.; Györke, I.; Williams, S.C.; Györke, S. Protein-protein interactions between triadin and calsequestrin are involved in modulation of sarcoplasmic reticulum calcium release in cardiac myocytes. J. Physiol. 2007, 583, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, M.; Furukawa, K.-I.; Fujimori, H.; Kuruma, A.; Kawano, S.; Hiraoka, M.; Kuniyasu, A.; Nakayama, H.; Ohizumi, Y. Dual Regulation of the Skeletal Muscle Ryanodine Receptor by Triadin and Calsequestrin. Biochemistry 1998, 37, 12987–12993. [Google Scholar] [CrossRef] [PubMed]
- Györke, I.; Hester, N.; Jones, L.R.; Györke, S. The Role of Calsequestrin, Triadin, and Junctin in Conferring Cardiac Ryanodine Receptor Responsiveness to Luminal Calcium. Biophys. J. 2004, 86, 2121–2128. [Google Scholar] [CrossRef]
- Wei, L.; Gallant, E.M.; Dulhunty, A.F.; Beard, N.A. Junctin and triadin each activate skeletal ryanodine receptors but junctin alone mediates functional interactions with calsequestrin. Int. J. Biochem. Cell Biol. 2009, 41, 2214–2224. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Lorenzini, S.; Pierantozzi, E.; Van Petegem, F.; Osamwonuyi Amadsun, D.; Sorrentino, V. Multiple regions within junctin drive its interaction with calsequestrin-1 and its localization to triads in skeletal muscle. J. Cell Sci. 2022, 135, jcs259185. [Google Scholar] [CrossRef] [PubMed]
- Chopraa, N.; Yanga, T.; Asgharib, P.; Mooreb, E.D.; Hukea, S.; Akinc, B.; Cattolicad, R.A.; Pereze, C.F.; Hlainga, T.; Knollmann-Ritschelf, B.E.; et al. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation–contraction coupling, and cardiac arrhythmias. Proc. Natl. Acad. Sci. USA 2009, 106, 7636–7641. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.M.; Alseikhan, B.A.; Jones, L.R. Localization and Characterization of the Calsequestrin-binding Domain of Triadin 1. Evidence for a charged beta-strand in mediating the protein-protein interaction. J. Biol. Chem. 2000, 275, 17639–17646. [Google Scholar] [CrossRef]
- Zhang, L.; Franzini-Armstrong, C.; Ramesh, V.; Jones, L. Structural Alterations in Cardiac Calcium Release Units Resulting from Overexpression of Junctin. J. Mol. Cell. Cardiol. 2001, 33, 233–247. [Google Scholar] [CrossRef]
- Sanchez, E.J.; Munske, G.R.; Criswell, A.; Milting, H.; Dunker, A.K.; Kang, C. Phosphorylation of human calsequestrin: Implications for calcium regulation. Mol. Cell. Biochem. 2011, 353, 195–204. [Google Scholar] [CrossRef]
- Beard, N.A.; Wei, L.; Cheung, S.N.; Kimura, T.; Varsányi, M.; Dulhunty, A.F. Phosphorylation of skeletal muscle calsequestrin enhances its Ca2+ binding capacity and promotes its association with junctin. Cell Calcium 2008, 44, 363–373. [Google Scholar] [CrossRef]
- Handhle, A.; Ormonde, C.E.; Thomas, N.L.; Bralesford, C.; Williams, A.J.; Lai, F.A.; Zissimopoulos, S. Calsequestrin interacts directly with the cardiac ryanodine receptor luminal domain. J. Cell Sci. 2016, 129, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Herzog, A.; Szegedi, C.; Jona, I.; Herberg, F.W.; Varsanyi, M. Surface plasmon resonance studies prove the interaction of skeletal muscle sarcoplasmic reticular Ca2+ release channel/ryanodine receptor with calsequestrin. FEBS Lett. 2000, 472, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Hanna, A.D.; Beard, N.A.; Dulhunty, A.F. Unique isoform-specific properties of calsequestrin in the heart and skeletal muscle. Cell Calcium 2009, 45, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Kelley, J.; Schmeisser, G.; Kobayashi, Y.M.; Jones, L.R. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J. Biol. Chem. 1997, 272, 23389–23397. [Google Scholar] [CrossRef]
- Shin, D.W.; Ma, J.; Kim, D.H. The asp-rich region at the carboxyl-terminus of calsequestrin binds to Ca2+ and interacts with triadin. FEBS Lett. 2000, 486, 178–182. [Google Scholar] [CrossRef]
- Beard, N.A.; Dulhunty, A.F. C-terminal residues of skeletal muscle calsequestrin are essential for calcium binding and for skeletal ryanodine receptor inhibition. Skelet. Muscle 2015, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, B.; Zhong, X.; Mi, T.; Guo, A.; Zhou, Q.; Tan, Z.; Wu, G.; Chen, A.W.; Fill, M.; et al. The cardiac ryanodine receptor luminal Ca2+ sensor governs Ca2+ waves, ventricular tachyarrhythmias and cardiac hypertrophy in calsequestrin-null mice. Biochem. J. 2014, 461, 99–106. [Google Scholar] [CrossRef]
- Royer, L.; Sztretye, M.; Manno, C.; Pouvreau, S.; Zhou, J.; Knollmann, B.C.; Protasi, F.; Allen, P.D.; Ríos, E. Paradoxical buffering of calcium by calsequestrin demonstrated for the calcium store of skeletal muscle. J. Gen. Physiol. 2010, 136, 325–338. [Google Scholar] [CrossRef]
- Xu, L.; Chirasani, V.R.; Carter, J.S.; Pasek, D.A.; Dokholyan, N.V.; Yamaguchi, N.; Meissner, G. Ca2+-mediated activation of the skeletal-muscle ryanodine receptor ion channel. J. Biol. Chem. 2018, 293, 19501–19509. [Google Scholar] [CrossRef]
- Picello, E.; Damiani, E.; Margreth, A. Low-affinity Ca2+-binding sites versus Zn2+-binding sites in histidine-rich Ca2+-binding protein of skeletal muscle sarcoplasmic reticulum. Biochem. Biophys. Res. Commun. 1992, 186, 659–667. [Google Scholar] [CrossRef]
- Hofmann, S.L.; Goldstein, J.L.; Orth, K.; Moomaw, C.R.; Slaughter, C.A.; Brown, M.S. Molecular cloning of a histidine-rich Ca2+-binding protein of sarcoplasmic reticulum that contains highly conserved repeated elements. J. Biol. Chem. 1989, 264, 18083–18090. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Ho, H.-T.; Brunello, L.; Unudurthi, S.D.; Lou, Q.; Belevych, A.E.; Qian, L.; Kim, D.H.; Cho, C.; Janssen, P.M.L.; et al. Ablation of HRC alleviates cardiac arrhythmia and improves abnormal Ca handling in CASQ2 knockout mice prone to CPVT. Cardiovasc. Res. 2015, 108, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Arvanitis, D.A.; Vafiadaki, E.; Sanoudou, D.; Kranias, E.G. Histidine-rich calcium binding protein: The new regulator of sarcoplasmic reticulum calcium cycling. J. Mol. Cell. Cardiol. 2011, 50, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Pollak, A.J.; Haghighi, K.; Kunduri, S.; Arvanitis, D.A.; Bidwell, P.A.; Liu, G.-S.; Singh, V.P.; Gonzalez, D.J.; Sanoudou, D.; Wiley, S.E.; et al. Phosphorylation of serine96 of histidine-rich calcium-binding protein by the Fam20C kinase functions to prevent cardiac arrhythmia. Proc. Natl. Acad. Sci. USA 2017, 114, 9098–9103. [Google Scholar] [CrossRef] [PubMed]
- Tzimas, C.; Johnson, D.M.; Santiago, D.J.; Vafiadaki, E.; Arvanitis, D.A.; Davos, C.H.; Varela, A.; Athanasiadis, N.C.; Dimitriou, C.; Katsimpoulas, M.; et al. Impaired calcium homeostasis is associated with sudden cardiac death and arrhythmias in a genetic equivalent mouse model of the human HRC-Ser96Ala variant. Cardiovasc. Res. 2017, 113, 1403–1417. [Google Scholar] [CrossRef]
- Houle, T.D.; Ram, M.L.; McMurray, W.J.; Cala, S.E. Different endoplasmic reticulum trafficking and processing pathways for calsequestrin (CSQ) and epitope-tagged CSQ. Exp. Cell Res. 2006, 312, 4150–4161. [Google Scholar] [CrossRef] [PubMed]
- Milstein, M.L.; Houle, T.D.; Cala, S.E. Calsequestrin isoforms localize to different ER subcompartments: Evidence for polymer and heteropolymer-dependent localization. Exp. Cell Res. 2009, 315, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Gatti, G.; Trifari, S.; Mesaeli, N.; Parker, J.M.; Michalak, M.; Meldolesi, J. Head-to-tail oligomerization of calsequestrin: A novel mechanism for heterogeneous distribution of endoplasmic reticulum luminal proteins. J. Cell Biol. 2001, 154, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Nori, A.; Furlan, S.; Patiri, F.; Cantini, M.; Volpe, P. Site-Directed Mutagenesis and Deletion of Three Phosphorylation Sites of Calsequestrin of Skeletal Muscle Sarcoplasmic Reticulum: Effects on Intracellular Targeting. Exp. Cell Res. 2000, 260, 40–49. [Google Scholar] [CrossRef]
- Lewis, K.M.; Munske, G.R.; Byrd, S.S.; Kang, J.; Cho, H.-J.; Ríos, E.; Kang, C. Characterization of Post-Translational Modifications to Calsequestrins of Cardiac and Skeletal Muscle. Int. J. Mol. Sci. 2016, 17, 1539. [Google Scholar] [CrossRef]
- Sato, Y.; Ferguson, D.G.; Sako, H.; Dorn, G.W.; Kadambi, V.J.; Yatani, A.; Hoit, B.D.; Walsh, R.A.; Kranias, E.G. Cardiac-specific Overexpression of Mouse Cardiac Calsequestrin Is Associated with Depressed Cardiovascular Function and Hypertrophy in Transgenic Mice. J. Biol. Chem. 1998, 273, 28470–28477. [Google Scholar] [CrossRef] [PubMed]
- Pollak, A.J.; Liu, C.; Gudlur, A.; Mayfield, J.E.; Dalton, N.D.; Gu, Y.; Chen, J.; Brown, J.H.; Hogan, P.G.; Wiley, S.E.; et al. A secretory pathway kinase regulates sarcoplasmic reticulum Ca2+ homeostasis and protects against heart failure. Elife 2018, 7, e41378. [Google Scholar] [CrossRef] [PubMed]
- Huppertz, T. Chemistry of the Caseins. In Advanced Dairy Chemistry; Springer: Boston, MA, USA, 2013; pp. 135–160. [Google Scholar]
- Mayfield, J.E.; Dixon, J.E. Emerging mechanisms of regulation for endoplasmic/sarcoplasmic reticulum Ca2+ stores by secretory pathway kinase FAM20C. Curr. Opin. Chem. Biol. 2023, 74, 102279. [Google Scholar] [CrossRef] [PubMed]
- E Cala, S.; Jones, L.R. Phosphorylation of cardiac and skeletal muscle calsequestrin isoforms by casein kinase II. Demonstration of a cluster of unique rapidly phosphorylated sites in cardiac calsequestrin. J. Biol. Chem. 1991, 266, 391–398. [Google Scholar] [CrossRef]
- McFarland, T.P.; Milstein, M.L.; Cala, S.E. Rough endoplasmic reticulum to junctional sarcoplasmic reticulum trafficking of calsequestrin in adult cardiomyocytes. J. Mol. Cell. Cardiol. 2010, 49, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.J.; Lewis, K.M.; Munske, G.R.; Nissen, M.S.; Kang, C. Glycosylation of Skeletal Calsequestrin. J. Biol. Chem. 2012, 287, 3042–3050. [Google Scholar] [CrossRef]
- Kiarash, A.; Kelly, C.E.; Phinney, B.S.; Valdivia, H.H.; Abrams, J.; Cala, S.E. Defective glycosylation of calsequestrin in heart failure. Cardiovasc. Res. 2004, 63, 264–272. [Google Scholar] [CrossRef]
- Jacob, S.; Sleiman, N.H.; Kern, S.; Jones, L.R.; Sala-Mercado, J.A.; McFarland, T.P.; Sabbah, H.H.; Cala, S.E. Altered calsequestrin glycan processing is common to diverse models of canine heart failure. Mol. Cell. Biochem. 2013, 377, 11–21. [Google Scholar] [CrossRef]
- Kirchhefer, U.; Wehrmeister, D.; Postma, A.V.; Pohlentz, G.; Mormann, M.; Kucerova, D.; Müller, F.U.; Schmitz, W.; Schulze-Bahr, E.; Wilde, A.A.; et al. The human CASQ2 mutation K206N is associated with hyperglycosylation and altered cellular calcium handling. J. Mol. Cell. Cardiol. 2010, 49, 95–105. [Google Scholar] [CrossRef]
- Rossi, D.; Vezzani, B.; Galli, L.; Paolini, C.; Toniolo, L.; Pierantozzi, E.; Spinozzi, S.; Barone, V.; Pegoraro, E.; Bello, L.; et al. A Mutation in the CASQ1 Gene Causes a Vacuolar Myopathy with Accumulation of Sarcoplasmic Reticulum Protein Aggregates. Hum. Mutat. 2014, 35, 1163–1170. [Google Scholar] [CrossRef]
- Bjorksten, A.R.; Gillies, R.L.; Hockey, B.M.; Du Sart, D. Sequencing of Genes Involved in the Movement of Calcium across Human Skeletal Muscle Sarcoplasmic Reticulum: Continuing the Search for Genes Associated with Malignant Hyperthermia. Anaesth. Intensiv. Care 2016, 44, 762–768. [Google Scholar] [CrossRef]
- Wang, Q.; Paskevicius, T.; Filbert, A.; Qin, W.; Kim, H.J.; Chen, X.-Z.; Tang, J.; Dacks, J.B.; Agellon, L.B.; Michalak, M. Phylogenetic and biochemical analysis of calsequestrin structure and association of its variants with cardiac disorders. Sci. Rep. 2020, 10, 18115. [Google Scholar] [CrossRef]
- Valle, G.; Arad, M.; Volpe, P. Molecular adaptation to calsequestrin 2 (CASQ2) point mutations leading to catecholaminergic polymorphic ventricular tachycardia (CPVT): Comparative analysis of R33Q and D307H mutants. J. Muscle Res. Cell Motil. 2020, 41, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Kalyanasundaram, A.; Viatchenko-Karpinski, S.; Belevych, A.E.; Lacombe, V.A.; Hwang, H.S.; Knollmann, B.C.; Gyorke, S.; Periasamy, M. Functional consequences of stably expressing a mutant calsequestrin (CASQ2D307H) in the CASQ2 null background. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H253–H261. [Google Scholar] [CrossRef]
- Böhm, J.; Lornage, X.; Chevessier, F.; Birck, C.; Zanotti, S.; Cudia, P.; Bulla, M.; Granger, F.; Bui, M.T.; Sartori, M.; et al. CASQ1 mutations impair calsequestrin polymerization and cause tubular aggregate myopathy. Acta Neuropathol. 2018, 135, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Wleklinski, M.J.; Kryshtal, D.O.; Kim, K.; Parikh, S.S.; Blackwell, D.J.; Marty, I.; Iyer, V.R.; Knollmann, B.C. Impaired Dynamic Sarcoplasmic Reticulum Ca Buffering in Autosomal Dominant CPVT2. Circ. Res. 2022, 131, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Valle, G.; Galla, D.; Nori, A.; Priori, S.G.; Gyorke, S.; de Filippis, V.; Volpe, P. Catecholaminergic polymorphic ventricular tachycardia-related mutations R33Q and L167H alter calcium sensitivity of human cardiac calsequestrin. Biochem. J. 2008, 413, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Gray, B.; Bagnall, R.D.; Lam, L.; Ingles, J.; Turner, C.; Haan, E.; Davis, A.; Yang, P.-C.; Clancy, C.E.; Sy, R.W.; et al. A novel heterozygous mutation in cardiac calsequestrin causes autosomal dominant catecholaminergic polymorphic ventricular tachycardia. Hear. Rhythm. 2016, 13, 1652–1660. [Google Scholar] [CrossRef]
- Györke, S.; Stevens, S.C.W.; Terentyev, D. Cardiac calsequestrin: Quest inside the SR. J. Physiol. 2009, 587 Pt 13, 3091–3094. [Google Scholar] [CrossRef]
- Knollmann, B.C. New roles of calsequestrin and triadin in cardiac muscle. J. Physiol. 2009, 587 Pt 13, 3081–3087. [Google Scholar] [CrossRef]
- Rizzi, N.; Liu, N.; Napolitano, C.; Nori, A.; Turcato, F.; Colombi, B.; Bicciato, S.; Arcelli, D.; Spedito, A.; Scelsi, M.; et al. Unexpected structural and functional consequences of the R33Q homozygous mutation in cardiac calsequestrin: A complex arrhythmogenic cascade in a knock in mouse model. Circ. Res. 2008, 103, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Kurtzwald-Josefson, E.; Yadin, D.; Harun-Khun, S.; Waldman, M.; Aravot, D.; Shainberg, A.; Eldar, M.; Hochhauser, E.; Arad, M. Viral delivered gene therapy to treat catecholaminergic polymorphic ventricular tachycardia (CPVT2) in mouse models. Hear. Rhythm. 2017, 14, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Paudel, R.; Jafri, M.S.; Ullah, A. Pacing Dynamics Determines the Arrhythmogenic Mechanism of the CPVT2-Causing CASQ2G112+5X Mutation in a Guinea Pig Ventricular Myocyte Computational Model. Genes 2022, 14, 23. [Google Scholar] [CrossRef]
- Mussel, M.; Basser, P.J.; Horkay, F. Ion-Induced Volume Transition in Gels and Its Role in Biology. Gels 2021, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Sanulli, S.; Narlikar, G.J. Generation and Biochemical Characterization of Phase-Separated Droplets Formed by Nucleic Acid Binding Proteins: Using HP1 as a Model System. Curr. Protoc. 2021, 1, e109. [Google Scholar] [CrossRef]
- Musacchio, A. On the role of phase separation in the biogenesis of membraneless compartments. EMBO J. 2022, 41, e109952. [Google Scholar] [CrossRef]
- Kang, C.; Trumble, W.R.; Dunker, A.K. Crystallization and structure-function of calsequestrin. Methods Mol. Biol. 2002, 172, 281–294. [Google Scholar]
- Gillespie, D.; Fill, M. Intracellular calcium release channels mediate their own countercurrent: The ryanodine receptor case study. Biophys. J. 2008, 95, 3706–3714. [Google Scholar] [CrossRef]
- Guo, T.; Gillespie, D.; Fill, M. Ryanodine Receptor Current Amplitude Controls Ca2+ Sparks in Cardiac Muscle. Circ. Res. 2012, 111, 28–36. [Google Scholar] [CrossRef]
- Kometani, T.; Kasai, M. Ion movement accompanied by calcium uptake of sarcoplasmic reticulum vesicles studied through the osmotic volume change by the light scattering method. J. Membr. Biol. 1980, 56, 159–168. [Google Scholar] [CrossRef]
- Beltrán, M.; Barrientos, G.; Hidalgo, C. Fast kinetics of calcium dissociation from calsequestrin. Biol. Res. 2006, 39, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Meissner, G.; Young, R. Proton permeability of sarcoplasmic reticulum vesicles. J. Biol. Chem. 1980, 255, 6814–6819. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Y.; Thai, P.N.; Ren, L.; Timofeyev, V.; Jian, Z.; Park, S.; Ginsburg, K.S.; Overton, J.; Bossuyt, J.; Bers, D.M.; et al. Beat-to-beat dynamic regulation of intracellular pH in cardiomyocytes. iScience 2021, 25, 103624. [Google Scholar] [CrossRef] [PubMed]
- Horkay, F. Polyelectrolyte Gels: A Unique Class of Soft Materials. Gels 2021, 7, 102. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Reithmeier, R.A. Ion tamers. Nat. Struct. Biol. 1998, 5, 409–411. [Google Scholar] [CrossRef]
- Fan, X.X.; Liu, Z.; Yu, H.; Huang, X.; Song, C.; Guo, X.; Wang, S.Q. A non-transmembrane channel formed by Ca2+-bound calsequestrin-2. J. Gen. Physiol. 2022, 154, e2021ecc8. [Google Scholar] [CrossRef]
- Terentyev, D.; Nori, A.; Santoro, M.; Viatchenko-Karpinski, S.; Kubalova, Z.; Gyorke, I.; Terentyeva, R.; Vedamoorthyrao, S.; Blom, N.A.; Valle, G.; et al. Abnormal Interactions of Calsequestrin with the Ryanodine Receptor Calcium Release Channel Complex Linked to Exercise-Induced Sudden Cardiac Death. Circ. Res. 2006, 98, 1151–1158. [Google Scholar] [CrossRef]
- Slupsky, J.R.; Ohnishi, M.; Carpenter, M.R.; Reithmeier, R.A.F. Characterization of cardiac calsequestrin. Biochemistry 1987, 26, 6539–6544. [Google Scholar] [CrossRef]
- Mitchell, R.D.; Simmerman, H.K.; Jones, L.R. Ca2+ binding effects on protein conformation and protein interactions of canine cardiac calsequestrin. J. Biol. Chem. 1988, 263, 1376–1381. [Google Scholar] [CrossRef]
- Gatti, G.; Podini, P.; Meldolesi, J. Overexpression of calsequestrin in L6 myoblasts: Formation of endoplasmic reticulum subdomains and their evolution into discrete vacuoles where aggregates of the protein are specifically accumulated. Mol. Biol. Cell 1997, 8, 1789–1803. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Randazzo, D.; Del Re, V.; Sorrentino, V.; Rossi, D. Organization of junctional sarcoplasmic reticulum proteins in skeletal muscle fibers. J. Muscle Res. Cell Motil. 2015, 36, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, L.; Dai, T.; Qin, Z.; Lu, H.; Zhang, L.; Zhou, F. Liquid–liquid phase separation in human health and diseases. Signal Transduct. Target. Ther. 2021, 6, 290. [Google Scholar] [CrossRef]
Figure 1.
Basic components of Ca2+ release machinery in striated muscles. Excitation–contraction coupling (ECC) is the process by which an action potential at the sarcolemma leads to a massive release of intracellular Ca2+ which, in turn, activates cell-wide contraction. ECC occurs at subcellular structures that are periodically distributed inside muscle cells, called “junctions” (when referred to in structural terms) or Ca2+ Release Units (CRUs, when referred to in functional terms). Junctions are so termed because they are composed of specialized regions of the sarcolemma and of the “junctional” sarcoplasmic reticulum (jSR), both coalescing within nanometer distances. ECC occurs via different mechanisms in skeletal and cardiac muscles, so in the figure, the sarcolemmal portion of the junctions is not decorated with tissue-specific components. At the jSR membrane, CRUs are decorated by ordered arrays of ryanodine receptors (RyR, pink), which act as the SR Ca2+ release channels. In the lumen of the jSR, CASQ (blue) buffers Ca2+ ions (yellow spheres), whereas Junctin (green) and Triadin (red) are transmembrane proteins anchoring CASQ to the RyR and acting as signaling mediators between CASQ and RyR. The actual stoichiometry for the Jnt:Trd:RyR complex is unknown.
Figure 1.
Basic components of Ca2+ release machinery in striated muscles. Excitation–contraction coupling (ECC) is the process by which an action potential at the sarcolemma leads to a massive release of intracellular Ca2+ which, in turn, activates cell-wide contraction. ECC occurs at subcellular structures that are periodically distributed inside muscle cells, called “junctions” (when referred to in structural terms) or Ca2+ Release Units (CRUs, when referred to in functional terms). Junctions are so termed because they are composed of specialized regions of the sarcolemma and of the “junctional” sarcoplasmic reticulum (jSR), both coalescing within nanometer distances. ECC occurs via different mechanisms in skeletal and cardiac muscles, so in the figure, the sarcolemmal portion of the junctions is not decorated with tissue-specific components. At the jSR membrane, CRUs are decorated by ordered arrays of ryanodine receptors (RyR, pink), which act as the SR Ca2+ release channels. In the lumen of the jSR, CASQ (blue) buffers Ca2+ ions (yellow spheres), whereas Junctin (green) and Triadin (red) are transmembrane proteins anchoring CASQ to the RyR and acting as signaling mediators between CASQ and RyR. The actual stoichiometry for the Jnt:Trd:RyR complex is unknown.

Figure 2.
Schematic diagram of CASQ structural transitions guided by increasing amounts of calcium and/or monovalent cations. Negative charges along the polypeptide backbone are represented as white spheres. At close-to-neutral pH and low ionic strength, CASQ retains an unfolded conformation. Either monovalent ions such as Na+ or K+ (blue spheres) or divalent ions such as Ca2+ (yellow spheres) mask the negative charges of the abundant exposed glutamate and aspartate residues (103 and 102 acidic residues over 362 (CASQ1) and 380 (CASQ2) amino-acid-long polypeptides, respectively). In both cases, the ionic strength increase triggers formation of the same tertiary structure. To this end, the concentrations of Ca2+ required are about three orders of magnitude lower (about 0.1 mM) than that of other monovalent cations (100 mM). Further Ca2+ addition triggers dimerization through N-terminal tail swapping. A higher Ca2+ concentration is required for tight dimerization to occur when competing Na+ and K+ ions are present. The further increase in Ca2+ ions promotes “back-to-back” tetramerization, which concomitantly leads to an enormous increase in the calcium-binding capacity of each CASQ monomer. The protein’s intrinsically disordered C-termini, involved in stabilizing the Ca2+-dependent tetramer, are represented in a fixed conformation for clarity of representation.
Figure 2.
Schematic diagram of CASQ structural transitions guided by increasing amounts of calcium and/or monovalent cations. Negative charges along the polypeptide backbone are represented as white spheres. At close-to-neutral pH and low ionic strength, CASQ retains an unfolded conformation. Either monovalent ions such as Na+ or K+ (blue spheres) or divalent ions such as Ca2+ (yellow spheres) mask the negative charges of the abundant exposed glutamate and aspartate residues (103 and 102 acidic residues over 362 (CASQ1) and 380 (CASQ2) amino-acid-long polypeptides, respectively). In both cases, the ionic strength increase triggers formation of the same tertiary structure. To this end, the concentrations of Ca2+ required are about three orders of magnitude lower (about 0.1 mM) than that of other monovalent cations (100 mM). Further Ca2+ addition triggers dimerization through N-terminal tail swapping. A higher Ca2+ concentration is required for tight dimerization to occur when competing Na+ and K+ ions are present. The further increase in Ca2+ ions promotes “back-to-back” tetramerization, which concomitantly leads to an enormous increase in the calcium-binding capacity of each CASQ monomer. The protein’s intrinsically disordered C-termini, involved in stabilizing the Ca2+-dependent tetramer, are represented in a fixed conformation for clarity of representation.

Figure 3.
CASQ monomeric structure and salt-sensitive dimerization. (A) Representation of skeletal (left, PDB ID: 5KN0) and cardiac (right, PDB ID: 1SJI) CASQ surfaces. Colors mirror the coulombic potential values according to the color legend shown. The disordered C-terminus is not present in the experimental structures. (B) Ribbon representation of CASQ monomer. The PDB model is 1SJI for cardiac CASQ2. The polypeptide is conventionally rainbow-colored starting from its N-terminus (blue) to its C-terminus (red). (C) The corresponding cardiac CASQ dimer is shown (PDB ID: 1SJI). One of the two swapped N-terminal domains within the dimeric state is colored violet for ease of identification. The N-terminal portion within the circle corresponds to the dimerization switch. (D) Left: Zoomed view of the dimerization N-terminal switch (within the circle). The most relevant pathological missense mutation residues falling close or within the dimerization interface are evidenced in gold and represented as sticks. Right: The sequences of the N-termini for both skeletal and cardiac CASQ are compared. Identical or similar residues are evidenced by an asterisk or a colon, respectively. The dimerization switch is highlighted in the violet box. (E) Surface representation of single monomers, where the dimerization interface is, is evidenced in red. For each structure, the concentrations of the cations of crystallization conditions are indicated below the relative PDB ID.
Figure 3.
CASQ monomeric structure and salt-sensitive dimerization. (A) Representation of skeletal (left, PDB ID: 5KN0) and cardiac (right, PDB ID: 1SJI) CASQ surfaces. Colors mirror the coulombic potential values according to the color legend shown. The disordered C-terminus is not present in the experimental structures. (B) Ribbon representation of CASQ monomer. The PDB model is 1SJI for cardiac CASQ2. The polypeptide is conventionally rainbow-colored starting from its N-terminus (blue) to its C-terminus (red). (C) The corresponding cardiac CASQ dimer is shown (PDB ID: 1SJI). One of the two swapped N-terminal domains within the dimeric state is colored violet for ease of identification. The N-terminal portion within the circle corresponds to the dimerization switch. (D) Left: Zoomed view of the dimerization N-terminal switch (within the circle). The most relevant pathological missense mutation residues falling close or within the dimerization interface are evidenced in gold and represented as sticks. Right: The sequences of the N-termini for both skeletal and cardiac CASQ are compared. Identical or similar residues are evidenced by an asterisk or a colon, respectively. The dimerization switch is highlighted in the violet box. (E) Surface representation of single monomers, where the dimerization interface is, is evidenced in red. For each structure, the concentrations of the cations of crystallization conditions are indicated below the relative PDB ID.

Figure 4.
Multiple sites globally design the quaternary assembly of CASQ. (A) The C-terminal Intrinsically Disordered Region is highlighted in the blue box for skeletal and cardiac CASQ isoforms. (B) Inset from the glycosylated skeletal CASQ dimer structure (PDB ID: 3TRQ), showing the relative positions and orientations of the glycosyl moieties on Asn-316, with respect to the C-terminus of the glycosyl-carrying monomer and the N-terminus of the dimeric partner. The presence of a two-mannosyl chain is sufficient to fully stabilize the N-terminus, whereas the C-terminus retains a disordered conformation. The site of the known pathological missense mutation I385T in CASQ1 is close to the glycosylation site. (C) CASQ polymeric structure is represented for a portion corresponding to five dimers (PDB ID 6OWW). Left: The structure of native glycosylated skeletal CASQ1 (PDB ID: 3TRQ) is fitted within the 6OWW polymeric structure to visualize the position of the glycosyl moieties (magenta) with respect to the linear polymer surface (transparent). Centre: For each dimer, one monomer is colored light blue, and the second monomer is colored pink. A 90° rotation from one dimer to the adjacent one is evident along the polymer axis. The inter-monomer surfaces are colored red, whereas the inter-dimer interfaces are colored blue. The empty tunnel running within the structure is colored yellow. Entry/exit spaces in continuity with the internal tunnel are visible. Right: The internal tunnel is shown in yellow, and only one monomer for each dimer (corresponding to light blue monomers in the central polymer structure) is represented as a semi-transparent surface. (D) Surface missense pathological mutations are evidenced in gold on the surface of a CASQ2 monomer (PDB ID: 6OWW). The inter-monomer surfaces are colored red, whereas the inter-dimer interfaces are colored blue. CASQ1 mutations (D44N, M87Q, G103D, D244G, and I385T) are in bold for ease of identification among the numerous CASQ2 missense mutations.
Figure 4.
Multiple sites globally design the quaternary assembly of CASQ. (A) The C-terminal Intrinsically Disordered Region is highlighted in the blue box for skeletal and cardiac CASQ isoforms. (B) Inset from the glycosylated skeletal CASQ dimer structure (PDB ID: 3TRQ), showing the relative positions and orientations of the glycosyl moieties on Asn-316, with respect to the C-terminus of the glycosyl-carrying monomer and the N-terminus of the dimeric partner. The presence of a two-mannosyl chain is sufficient to fully stabilize the N-terminus, whereas the C-terminus retains a disordered conformation. The site of the known pathological missense mutation I385T in CASQ1 is close to the glycosylation site. (C) CASQ polymeric structure is represented for a portion corresponding to five dimers (PDB ID 6OWW). Left: The structure of native glycosylated skeletal CASQ1 (PDB ID: 3TRQ) is fitted within the 6OWW polymeric structure to visualize the position of the glycosyl moieties (magenta) with respect to the linear polymer surface (transparent). Centre: For each dimer, one monomer is colored light blue, and the second monomer is colored pink. A 90° rotation from one dimer to the adjacent one is evident along the polymer axis. The inter-monomer surfaces are colored red, whereas the inter-dimer interfaces are colored blue. The empty tunnel running within the structure is colored yellow. Entry/exit spaces in continuity with the internal tunnel are visible. Right: The internal tunnel is shown in yellow, and only one monomer for each dimer (corresponding to light blue monomers in the central polymer structure) is represented as a semi-transparent surface. (D) Surface missense pathological mutations are evidenced in gold on the surface of a CASQ2 monomer (PDB ID: 6OWW). The inter-monomer surfaces are colored red, whereas the inter-dimer interfaces are colored blue. CASQ1 mutations (D44N, M87Q, G103D, D244G, and I385T) are in bold for ease of identification among the numerous CASQ2 missense mutations.

Figure 5.
The physiological consequences of CASQ complex polymerization kinetics. (A) Scheme of CASQ multimerization. Multiple factors other than the concentrations of Ca2+ ions (yellow spheres) and of CASQ monomers (blue triangles) regulate the organization of polymeric structures. The triblock design of the CASQ dimer (two negatively charged and flexible C-termini exposed from the globular dimeric architecture) allows for the formation of a cross-linked network, where the high concentration of carboxylate groups (carried by Glu and Asp residues) sequesters the free Ca2+ ions from the solution. The Ca2+ concentration sustaining the critical polymer dimensions and shape is influenced by Mg2+ ions (purple) and post-translational modifications such as phosphorylation (red) and glycosylation (pink). The changes in the dimensions and shape of CASQ-Ca2+ nM sized bodies have usually been measured as variations in the proportion of diffracted light (the Tyndall effect for colloidal solutions). Protein partners at the jSR membrane may also play a role in shaping the orientation and architecture of the CASQ-Ca2+ system. (B) Scheme of CASQ system functioning in vivo. As revealed by multiple EM images, CASQ is a high-capacity Ca2+-binding polymeric matrix within the junctional SR. Ca2+ ions are continuously pumped in the SR from the cytosol by SERCA pumps disposed along the longitudinal SR. The opening of the RyR channels allows for the rapid efflux of Ca2+ ions across a very steep electrochemical gradient. The presence of CASQ sustains the outflow of Ca2+, giving rise to a “hump” in the Ca2+ release curve [16]. This pool of Ca2+ ions may belong to a fast-conductance reservoir that is contributed by the overall polymeric architecture and geometry. (C,D) Scheme of the possible physiological scenarios for different CASQ polymer ionic properties. (C) Left panel: If a rapidly disposable reservoir of Ca2+ ions can be conducted within the CASQ polymeric network at a speed comparable with the rate of Ca2+ outflow, at least the first burst of Ca2+ efflux might be sustained by the Ca2+ conductance of the CASQ system without the need for CASQ to complete any structural transition to lower Ca2+ capacity conformations. Right panel: As more Ca2+ ions are depleted from the jSR lumen, the Ca2+-buffering role of CASQ prevails and additional ions are released as free species in the solution, in concomitance with the changes in the polymer architecture and possible rearrangements of the protein–protein interactions at multiple sites. (D) If the Ca2+ conductance rate along the CASQ- Ca2+ network is insufficient to sustain calcium outflow through the Ca2+ channel, the behavior of those CASQ polymers just beneath the RyR channels should be the main determinant of Ca2+ efflux. Only in extreme calcium depletion conditions would a second layer of CASQ lying farther from the RyR channels respond.
Figure 5.
The physiological consequences of CASQ complex polymerization kinetics. (A) Scheme of CASQ multimerization. Multiple factors other than the concentrations of Ca2+ ions (yellow spheres) and of CASQ monomers (blue triangles) regulate the organization of polymeric structures. The triblock design of the CASQ dimer (two negatively charged and flexible C-termini exposed from the globular dimeric architecture) allows for the formation of a cross-linked network, where the high concentration of carboxylate groups (carried by Glu and Asp residues) sequesters the free Ca2+ ions from the solution. The Ca2+ concentration sustaining the critical polymer dimensions and shape is influenced by Mg2+ ions (purple) and post-translational modifications such as phosphorylation (red) and glycosylation (pink). The changes in the dimensions and shape of CASQ-Ca2+ nM sized bodies have usually been measured as variations in the proportion of diffracted light (the Tyndall effect for colloidal solutions). Protein partners at the jSR membrane may also play a role in shaping the orientation and architecture of the CASQ-Ca2+ system. (B) Scheme of CASQ system functioning in vivo. As revealed by multiple EM images, CASQ is a high-capacity Ca2+-binding polymeric matrix within the junctional SR. Ca2+ ions are continuously pumped in the SR from the cytosol by SERCA pumps disposed along the longitudinal SR. The opening of the RyR channels allows for the rapid efflux of Ca2+ ions across a very steep electrochemical gradient. The presence of CASQ sustains the outflow of Ca2+, giving rise to a “hump” in the Ca2+ release curve [16]. This pool of Ca2+ ions may belong to a fast-conductance reservoir that is contributed by the overall polymeric architecture and geometry. (C,D) Scheme of the possible physiological scenarios for different CASQ polymer ionic properties. (C) Left panel: If a rapidly disposable reservoir of Ca2+ ions can be conducted within the CASQ polymeric network at a speed comparable with the rate of Ca2+ outflow, at least the first burst of Ca2+ efflux might be sustained by the Ca2+ conductance of the CASQ system without the need for CASQ to complete any structural transition to lower Ca2+ capacity conformations. Right panel: As more Ca2+ ions are depleted from the jSR lumen, the Ca2+-buffering role of CASQ prevails and additional ions are released as free species in the solution, in concomitance with the changes in the polymer architecture and possible rearrangements of the protein–protein interactions at multiple sites. (D) If the Ca2+ conductance rate along the CASQ- Ca2+ network is insufficient to sustain calcium outflow through the Ca2+ channel, the behavior of those CASQ polymers just beneath the RyR channels should be the main determinant of Ca2+ efflux. Only in extreme calcium depletion conditions would a second layer of CASQ lying farther from the RyR channels respond.


{kind=link}
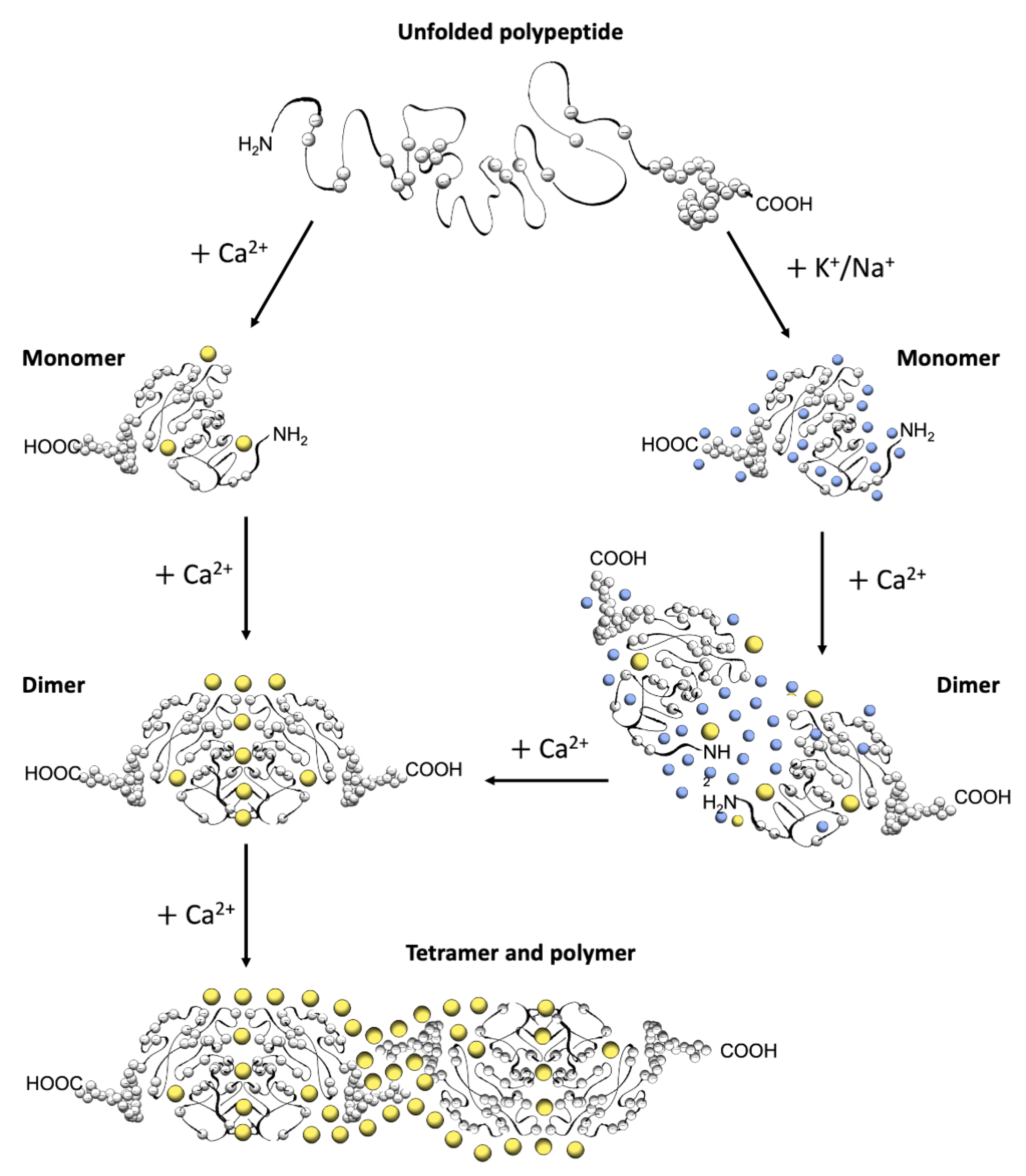{kind=link}
{kind=link}
{kind=link}
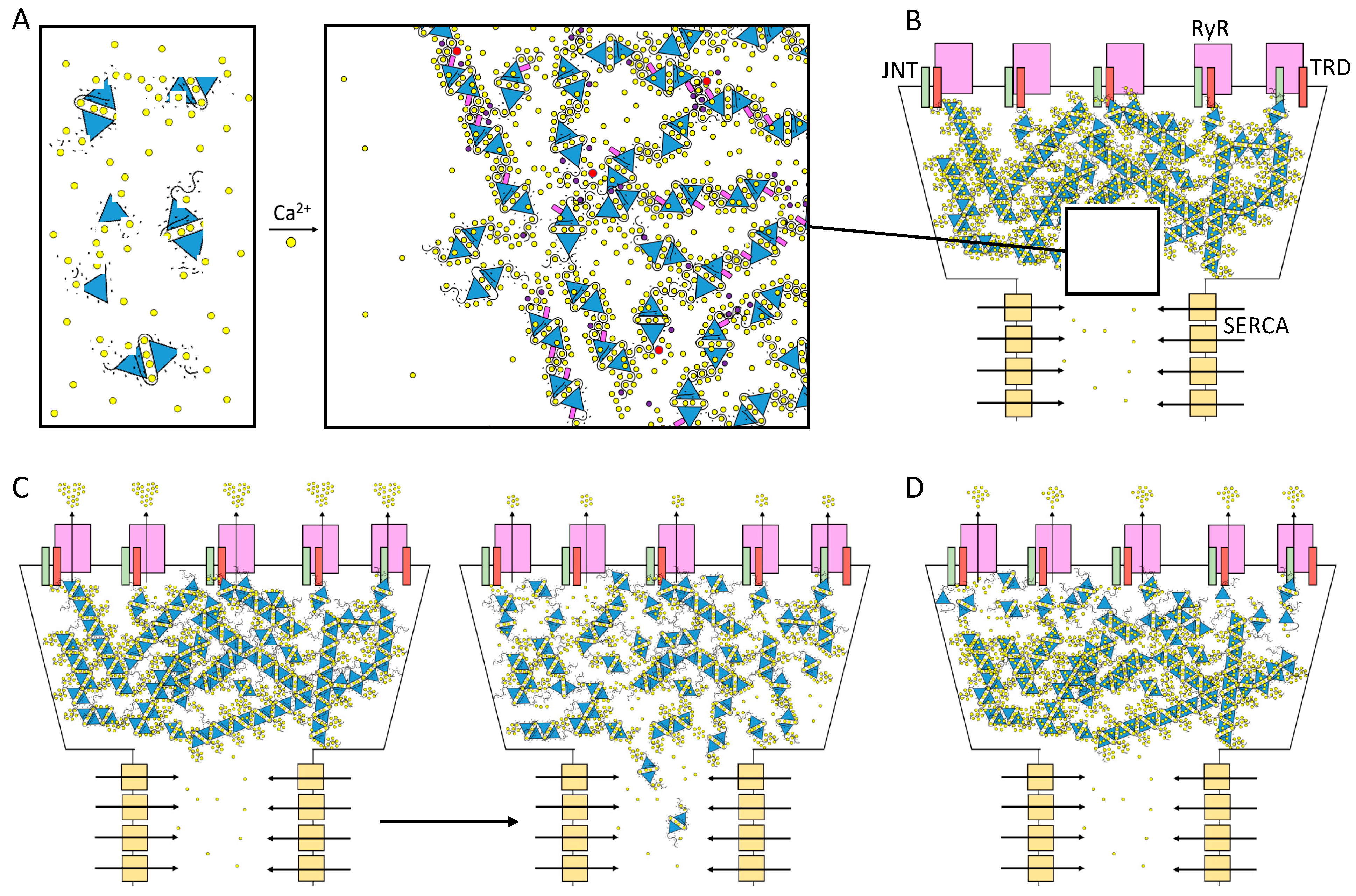{kind=link}
Table 1.
Skeletal and Cardiac CASQ pathological missense mutations.
| Altered Structure: | Monomer | Dimer | D/P | Polymer | P* | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutation | CASQ2 L167H | CASQ2 P329S | CASQ2 G332R | CASQ1 D44N | CASQ2 R33Q | CASQ2 D307H | CASQ2 P308L | CASQ1 M87T | CASQ1 G103D | CASQ2 S173I | CASQ2 K180R | CASQ1 D244G | CASQ2 D325E | CASQ2 D351G | CASQ2 R251H | CASQ1 I385T | ||
| Tertiary structure | Monomer stability | Tryptophan Fluorescence | ↓ [33] | ↓ [33] | ↓ [33] | |||||||||||||
| Circular Dichroism | ↓ [28,33,90] | = [28,33,34,90] | = [28,33] | = [28] | ||||||||||||||
| Trypsin digestion protection | = [35,85] | ↓ [85] | ↓ [85] | ↓ [85,90] | ↓ [85] = [87] | = [85] | = [85] | |||||||||||
| Thermal stability | ↓ [85] | ↓ [85] | ↓ [85] | = [34,85] | ↓ [85] | = [85] | = [85] | |||||||||||
| Mono-dispersion in SEC | = [90] | ↓ [36] | = [36] | = [35] | = [36] | = [36] | = [35] | = [36] | = [36] | |||||||||
| Ca2+-dependent properties | conformational rearrangement | Trypsin digestion protection | ↓ [90] = [28] | ↓ [28,85] | ↓ [28] | ↓ [15] | ↓ [28,85] | ↓ [28] | ↓ [15] | = [85] | = [15] | = [85] | = [15] | |||||
| Secondary structureCD, TF | ↓ [85,90] | ↓ [28] | ↓ [28] | ↓ [34,85] = [90] | ↓ [28] | = [85] | = [85] | |||||||||||
| Mass increase DLS | ↓ [15] | ↓ [15] | ↑ [15] | ↑ [15] | ||||||||||||||
| dimerization | SEC-MALS | ↓ [33] | ↓ [33] | ↓ [33] | = [35] | = [35] | ||||||||||||
| oligomerization | Turbidity | ↓ [28] | ↓ [15] | ↓ [28,34] | ↓$ [28] | ↓$ [28] | ↓ [35] | ↓ [15,88] | ↓ [31] | = [31] | ↑ [15,35] | ↑ [15,88] | ||||||
| Turbidity with Mg2+ | ↓ [36] | ↓ [31,36] | ↓ [36] | ↓ [31,36] | ↓ [31] | ↓ [36] | =£ [36] | |||||||||||
| Native PAGE | ↑§ [85] | ↑ [85] | ↑ [85] | ↓ [85] | = [85] | = [85] | = [85] | |||||||||||
| Ca2+ binding | Microscale Thermophoresis | ↓ [85] | = [85] | ↓ [85] | = [85] | ↓ [85] | = [85] | = [85] | ||||||||||
| Equilibrium dialysis | ↓ [33] | ↓ [33] | ↓ [33] | ↓ [35] | ↓ [35] | |||||||||||||
D/P: Dimer and polymer: both states could be affected. P*: Probable effect on the polymer behavior: CASQ2 R251 shapes the tunnel running within the linear polymer structure 6OWW. It may alter the kinetics of Ca2+-binding. CASQ1 I385 lies in close proximity to the glycosyl group in structure 3TRQ. It may alter the effect of glycosylation (Figure 4B). $ The defect appears only between 1.5 and 6 mM Ca2+. § Oligomers are constitutive and Ca2+-independent. £ Partial defect appears at pH 5.6.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Marabelli, C.; Santiago, D.J.; Priori, S.G. The Structural–Functional Crosstalk of the Calsequestrin System: Insights and Pathological Implications. Biomolecules 2023, 13, 1693. https://doi.org/10.3390/biom13121693
AMA Style
Marabelli C, Santiago DJ, Priori SG. The Structural–Functional Crosstalk of the Calsequestrin System: Insights and Pathological Implications. Biomolecules. 2023; 13(12):1693. https://doi.org/10.3390/biom13121693
Chicago/Turabian StyleMarabelli, Chiara, Demetrio J. Santiago, and Silvia G. Priori. 2023. "The Structural–Functional Crosstalk of the Calsequestrin System: Insights and Pathological Implications" Biomolecules 13, no. 12: 1693. https://doi.org/10.3390/biom13121693
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.