
Study of the Rv1417 and Rv2617c Membrane Proteins and Their Interactions with Nicotine Derivatives as Potential Inhibitors of Erp Virulence-Associated Factor in Mycobacterium tuberculosis: An In Silico Approach
 , , , , , , and
, , , , , , and 
Abstract
:1. Introduction
2. Computational Details
2.1. Protein–Membrane Complexes
2.2. Protein–Membrane–Drug Complexes
2.3. MD Simulations
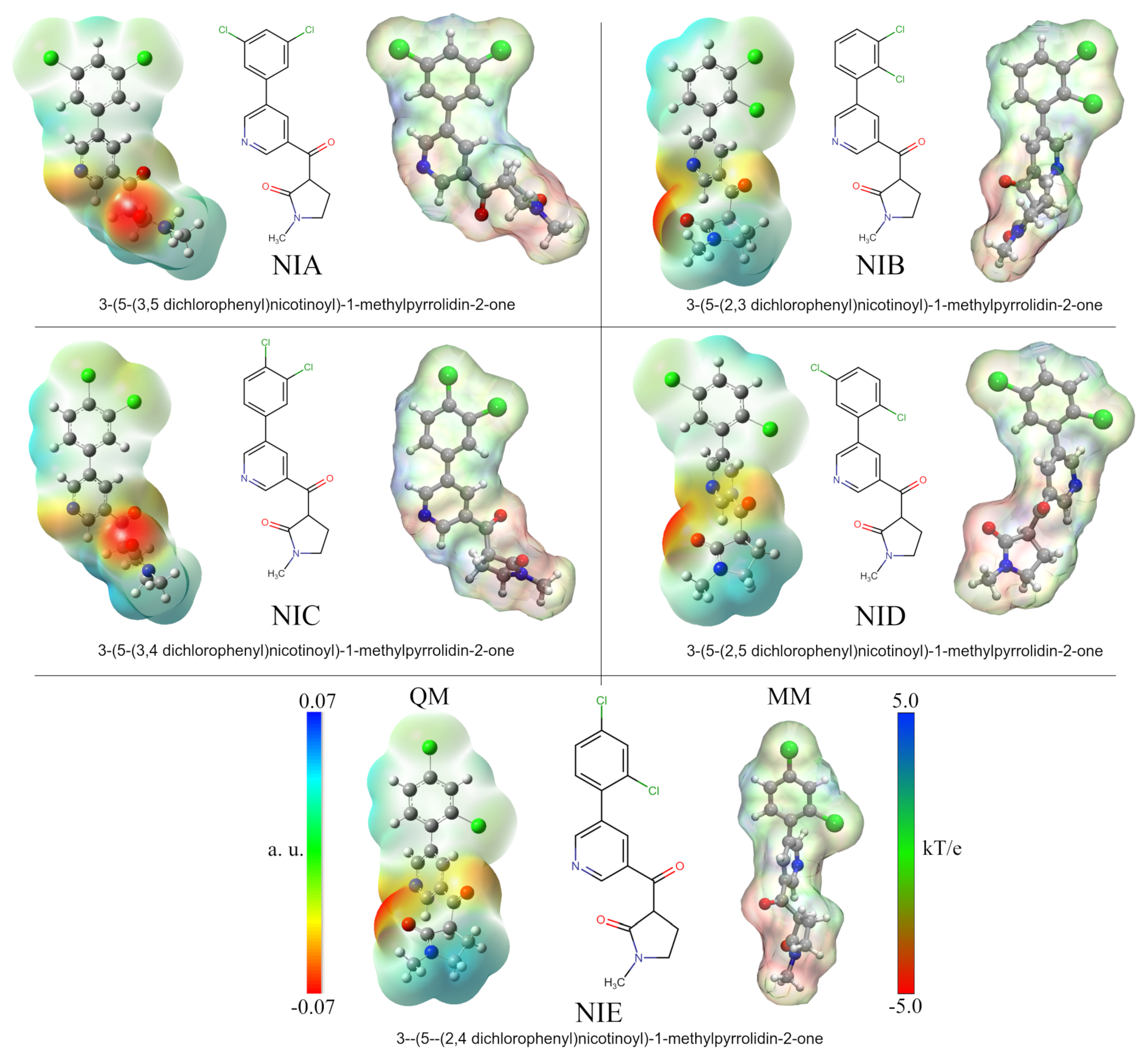
2.4. Nicotine Analogs
2.5. Molecular Docking Calculations
2.6. Computation of Binding Free Energy Using MM/PBSA Approximation
2.7. Structure and Data Analysis
3. Results and Discussion
3.1. Molecular and ADMET Analyses of the Nicotine Analogs
3.2. New MD Simulations Show High Structural Stability of the RvPs
3.3. Building the RvP–NAM Interacting Systems
3.3.1. Highly Conserved Pockets during the MD Simulations of the RvP Systems
3.3.2. Stability Descriptors Show a High Structural Affinity in the RvP–NAM Complexes
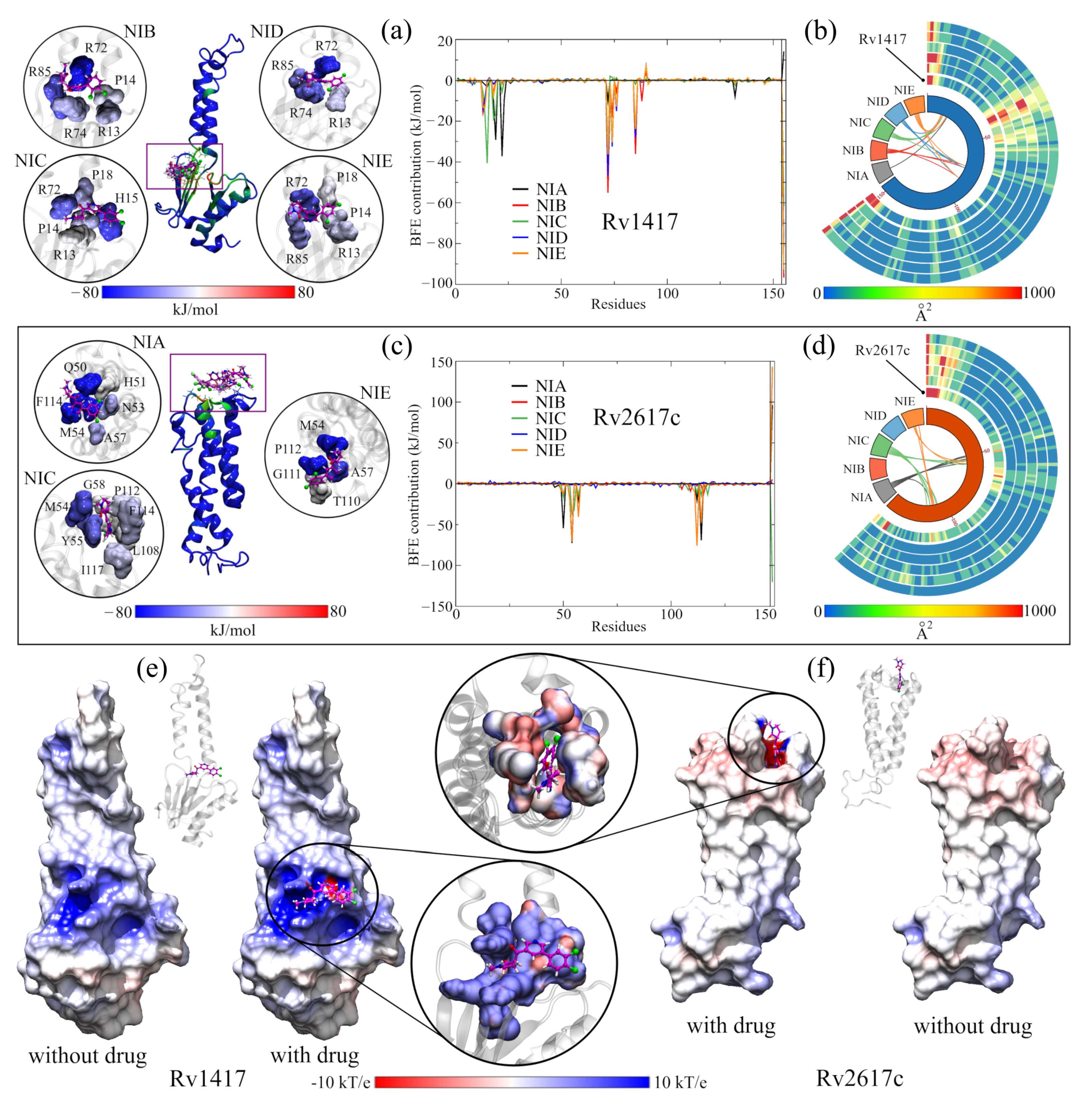
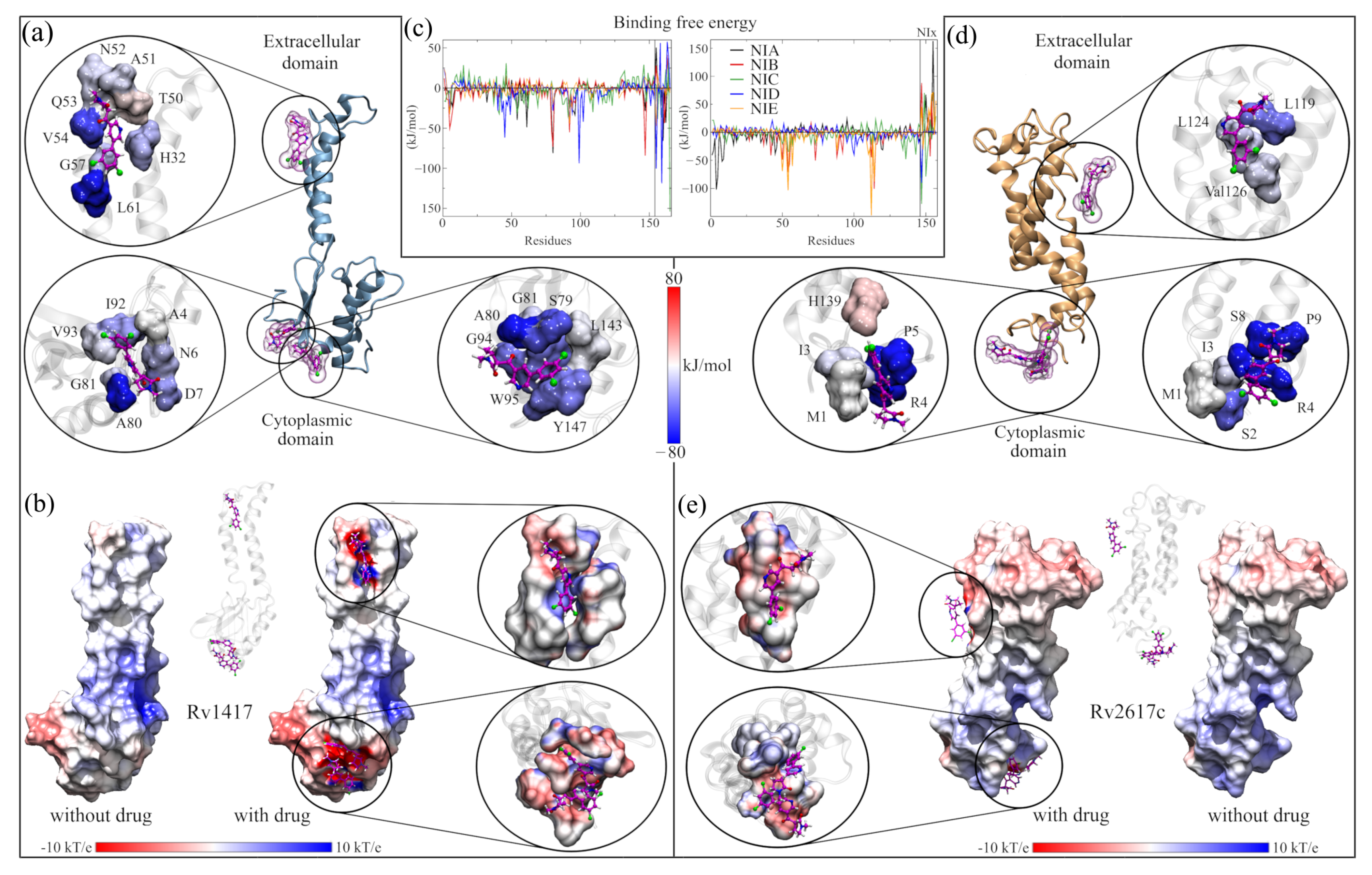
3.4. Assessment of the RvP–NAM Interactions
3.4.1. Contact Analysis between the RvPs Residues and the NAMs
3.4.2. Energy and Electrostatic Analyses Show the Affinity of NAMs with the Active Sites
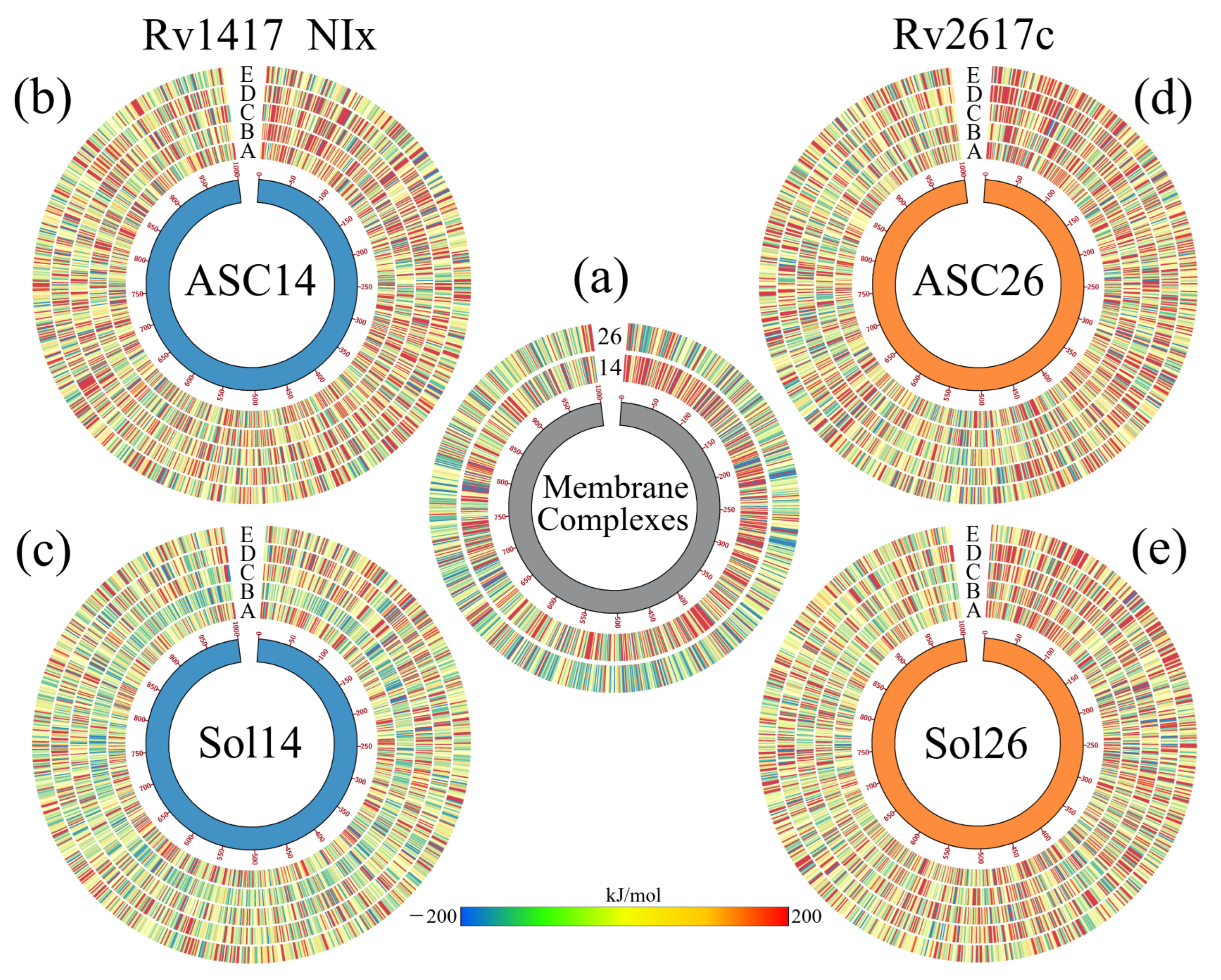
3.4.3. Solvated Systems with NAMs
3.5. Energy Analysis of RvP–Erp Heterodimers in the Presence of NAMs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Mtb | Mycobacterium tuberculosis |
| TB | Tuberculosis |
| Erp | Exported repetitive protein |
| NAMs | Nicotine analog molecules |
| RvPs | Rv1417 and Rv2617c proteins |
| DFT | Density functional theory |
| MD | Molecular dynamics |
| FEL | Free energy landscape |
| DDPC | Dipalmitoylphosphatidylcholine |
| BFE | Binding free energies |
| ESP | Electrostatic potential |
| MM-ESP | Electrostatic potential surfaces within the molecular mechanics framework |
| MM/PBSA | Molecular mechanics Poisson–Boltzmann surface area |
| ADMET | Absorption, distribution, metabolism, excretion, and toxicity) |
| ASC14–NIx | Active site complex of NAMs with Rv1417 |
| ASC26–NIx | Active site complex of NAMs with Rv2617c |
References
- McQuaid, C.F.; McCreesh, N.; Read, J.M.; Sumner, T.; Houben, R.M.; White, R.G.; Harris, R.C.; CMMID COVID-19 Working Group. The potential impact of COVID-19-related disruption on tuberculosis burden. Eur. Respir. J. 2020, 56, 2001718. [Google Scholar] [CrossRef] [PubMed]
- Antonio-Arques, V.; Franch-Nadal, J.; Caylà, J.A. Diabetes y tuberculosis: Una sindemia complicada por la COVID-19. Med. Clínica 2021, 157, 288–293. [Google Scholar] [CrossRef] [PubMed]
- W.H.O. Global Tuberculosis Report 2021; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Lee, J.; Hartman, M.; Kornfeld, H. Macrophage apoptosis in tuberculosis. Yonsei Med J. 2009, 50, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomgran, R.; Desvignes, L.; Briken, V.; Ernst, J.D. Mycobacterium tuberculosis inhibits neutrophil apoptosis, leading to delayed activation of naive CD4 T cells. Cell Host Microbe 2012, 11, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, S.; Song, Z.; Wu, Y.; Gao, Y.; Gao, M.; Liu, F.; Wang, F.; Zhang, Y. MicroRNA-27b modulates inflammatory response and apoptosis during Mycobacterium tuberculosis infection. J. Immunol. 2018, 200, 3506–3518. [Google Scholar] [CrossRef] [Green Version]
- Murray, J.F. A century of tuberculosis. Am. J. Respir. Crit. Care Med. 2004, 169, 1181–1186. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, H.C.; Feldman, W.H. Treatment of Experimental Tuberculosis: Use of Sodium P, P’-Diaminodiphenylsulfone-N, N’-Didextrose Sulfonate (Promin) with Notes on Some Toxic Effects Observed in Man. J. Am. Med Assoc. 1941, 117, 1066–1068. [Google Scholar] [CrossRef]
- Tuberculosis. Available online: https://www.who.int/es/news-room/fact-sheets/detail/tuberculosis (accessed on 25 March 2022).
- Lönnroth, K.; Migliori, G.B.; Abubakar, I.; D’Ambrosio, L.; De Vries, G.; Diel, R.; Douglas, P.; Falzon, D.; Gaudreau, M.A.; Goletti, D.; et al. Towards tuberculosis elimination: An action framework for low-incidence countries. Eur. Respir. J. 2015, 45, 928–952. [Google Scholar] [CrossRef] [Green Version]
- Horsburgh, C.R., Jr.; Barry, C.E., III; Lange, C. Treatment of tuberculosis. N. Engl. J. Med. 2015, 373, 2149–2160. [Google Scholar] [CrossRef]
- Suárez, I.; Fünger, S.M.; Kröger, S.; Rademacher, J.; Fätkenheuer, G.; Rybniker, J. The diagnosis and treatment of tuberculosis. Dtsch. Aerzteblatt Int. 2019, 116, 729–735. [Google Scholar] [CrossRef]
- Campaniço, A.; Moreira, R.; Lopes, F. Drug discovery in tuberculosis. New drug targets and antimycobacterial agents. Eur. J. Med. Chem. 2018, 150, 525–545. [Google Scholar] [CrossRef] [PubMed]
- Gibson, S.E.; Harrison, J.; Cox, J.A. Modelling a silent epidemic: A review of the in vitro models of latent tuberculosis. Pathogens 2018, 7, 88. [Google Scholar] [CrossRef] [PubMed]
- Priftakis, D.; Riaz, S.; Zumla, A.; Bomanji, J. Towards more accurate 18F-fluorodeoxyglucose positron emission tomography (18F-FDG PET) imaging in active and latent tuberculosis. Int. J. Infect. Dis. 2020, 92, S85–S90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan American Health Organization. Tuberculosis in the Americas. Regional Report 2020; Pan American Health Organization: Washington, DC, USA, 2021. [Google Scholar] [CrossRef]
- Iacobino, A.; Fattorini, L.; Giannoni, F. Drug-resistant tuberculosis 2020: Where we stand. Appl. Sci. 2020, 10, 2153. [Google Scholar] [CrossRef] [Green Version]
- Al-Mutairi, N.M.; Ahmad, S.; Mokaddas, E.; Eldeen, H.S.; Joseph, S. Occurrence of disputed rpoB mutations among Mycobacterium tuberculosis isolates phenotypically susceptible to rifampicin in a country with a low incidence of multidrug-resistant tuberculosis. BMC Infect. Dis. 2019, 19, 3. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Sun, F.; Zhang, W. Bedaquiline and delamanid in the treatment of multidrug-resistant tuberculosis: Promising but challenging. Drug Dev. Res. 2019, 80, 98–105. [Google Scholar] [CrossRef]
- Baba, I.A.; Abdulkadir, R.A.; Esmaili, P. Analysis of tuberculosis model with saturated incidence rate and optimal control. Phys. A Stat. Mech. Its Appl. 2020, 540, 123237. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report 2020; World Health Organization: Geneve, Switzerland, 2020. [Google Scholar]
- Tiberi, S.; Vjecha, M.J.; Zumla, A.; Galvin, J.; Migliori, G.B.; Zumla, A. Accelerating development of new shorter TB treatment regimens in anticipation of a resurgence of multi-drug resistant TB due to the COVID-19 pandemic. Int. J. Infect. Dis. 2021, 113, S96–S99. [Google Scholar] [CrossRef]
- Dewhare, S.S. Drug resistant tuberculosis: Current scenario and impending challenges. Indian J. Tuberc. 2021, 69, 227–233. [Google Scholar] [CrossRef]
- Grzelak, E.M.; Choules, M.P.; Gao, W.; Cai, G.; Wan, B.; Wang, Y.; McAlpine, J.B.; Cheng, J.; Jin, Y.; Lee, H.; et al. Strategies in anti-Mycobacterium tuberculosis drug discovery based on phenotypic screening. J. Antibiot. 2019, 72, 719–728. [Google Scholar] [CrossRef] [Green Version]
- Shetye, G.S.; Franzblau, S.G.; Cho, S. New tuberculosis drug targets, their inhibitors, and potential therapeutic impact. Transl. Res. 2020, 220, 68–97. [Google Scholar] [CrossRef]
- Macalino, S.J.Y.; Billones, J.B.; Organo, V.G.; Carrillo, M.C.O. In silico strategies in tuberculosis drug discovery. Molecules 2020, 25, 665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundararajan, S.; Muniyan, R. Latent tuberculosis: Interaction of virulence factors in Mycobacterium tuberculosis. Mol. Biol. Rep. 2021, 48, 6181–6196. [Google Scholar] [CrossRef]
- Forrellad, M.A.; Klepp, L.I.; Gioffré, A.; Sabio y Garcia, J.; Morbidoni, H.R.; Santangelo, M.d.l.P.; Cataldi, A.A.; Bigi, F. Virulence factors of the Mycobacterium tuberculosis complex. Virulence 2013, 4, 3–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthet, F.X.; Rauzier, J.; Lim, E.M.; Philipp, W.; Gicquel, B.; Portnoï, D. Characterization of the Mycobacterium tuberculosis erp gene encoding a potential cell surface protein with repetitive structures. Microbiology 1995, 141, 2123–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganaie, A.A.; Trivedi, G.; Kaur, A.; Jha, S.S.; Anand, S.; Rana, V.; Singh, A.; Kumar, S.; Sharma, C. Interaction of Erp protein of Mycobacterium tuberculosis with Rv2212 enhances intracellular survival of Mycobacterium smegmatis. J. Bacteriol. 2016, 198, 2841–2852. [Google Scholar] [CrossRef] [Green Version]
- Okay, S.; Çetin, R.; Karabulut, F.; Doğan, C.; Sürücüoğlu, S.; Kızıldoğan, A.K. Immune responses elicited by the recombinant Erp, HspR, LppX, MmaA4, and OmpA proteins from Mycobacterium tuberculosis in mice. Acta Microbiol. Immunol. Hung. 2019, 66, 219–234. [Google Scholar] [CrossRef] [Green Version]
- Solans, L.; Uranga, S.; Aguilo, N.; Arnal, C.; Gomez, A.B.; Monzon, M.; Badiola, J.J.; Gicquel, B.; Martin, C. Hyper-attenuated MTBVAC erp mutant protects against tuberculosis in mice. Vaccine 2014, 32, 5192–5197. [Google Scholar] [CrossRef]
- Klepp, L.I.; Soria, M.; Blanco, F.C.; Bianco, M.V.; Santangelo, M.P.; Cataldi, A.A.; Bigi, F. Identification of two proteins that interact with the Erp virulence factor from Mycobacterium tuberculosis by using the bacterial two-hybrid system. BMC Mol. Biol. 2009, 10, 3. [Google Scholar] [CrossRef]
- Paco-Chipana, M.; Febres-Molina, C.; Aguilar-Pineda, J.A.; Gómez, B. Novel In Silico Insights into Rv1417 and Rv2617c as Potential Protein Targets: The Importance of the Medium on the Structural Interactions with Exported Repetitive Protein (Erp) of Mycobacterium tuberculosis. Polymers 2022, 14, 2577. [Google Scholar] [CrossRef]
- Forrellad, M.A.; Vázquez, C.L.; Blanco, F.C.; Klepp, L.I.; García, E.A.; Rocha, R.V.; Luciana, V.; Bigi, M.M.; Gutierrez, M.G.; Bigi, F. Rv2617c and P36 are virulence factors of pathogenic mycobacteria involved in resistance to oxidative stress. Virulence 2019, 10, 1026–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosas Olvera, M.; Vivès, E.; Molle, V.; Blanc-Potard, A.B.; Gannoun-Zaki, L. Endogenous and exogenous KdpF peptide increases susceptibility of Mycobacterium bovis BCG to nitrosative stress and reduces intramacrophage replication. Front. Cell. Infect. Microbiol. 2017, 7, 115. [Google Scholar] [CrossRef] [Green Version]
- Saeed, D.K.; Ashraf, J.; Hasan, Z.; Shakoor, S.; Kanji, A.; Hasan, R. Bedaquiline resistant Mycobacterium tuberculosis clinical isolates with and without rv0678 mutations have similar growth patterns under varying BDQ drug pressure. Tuberculosis 2022, 137, 102266. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, M.C.; de Souza, M.V.; Pinheiro, A.C.; Ferreira, M.d.L.; Gonçalves, R.S.; Nogueira, T.C.M.; Peralta, M.A. Evaluation of anti-tubercular activity of nicotinic and isoniazid analogues. Arkivoc 2007, 15, 181–191. [Google Scholar] [CrossRef]
- Dang, T.; Nizamov, I.S.; Salikhov, R.Z.; Sabirzyanova, L.R.; Vorobev, V.V.; Burganova, T.I.; Shaidoullina, M.M.; Batyeva, E.S.; Cherkasov, R.A.; Abdullin, T.I. Synthesis and characterization of pyridoxine, nicotine and nicotinamide salts of dithiophosphoric acids as antibacterial agents against resistant wound infection. Bioorganic Med. Chem. 2019, 27, 100–109. [Google Scholar] [CrossRef]
- Selikoff, I.J.; Robitzek, E.H.; Ornstein, G.G. Treatment of pulmonary tuberculosis with hydrazide derivatives of isonicotinic acid. J. Am. Med. Assoc. 1952, 150, 973–980. [Google Scholar] [CrossRef]
- Jordahl, C.; Prez, R.D.; Deuschle, K.; Muschenheim, C.; McDermott, W. Ineffectiveness of nicotinamide and isoniazid in the treatment of pulmonary tuberculosis. Am. Rev. Respir. Dis. 1961, 83, 899–900. [Google Scholar]
- Murray, M.F. Nicotinamide: An oral antimicrobial agent with activity against both Mycobacterium tuberculosis and human immunodeficiency virus. Clin. Infect. Dis. 2003, 36, 453–460. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Zhang, Y.; Song, X.; Liu, Q. Research Progress in the Pharmacological Effects and Synthesis of Nicotine. ChemistrySelect 2022, 7, e202104425. [Google Scholar] [CrossRef]
- Gandhi, P.T.; Athmaram, T.N.; Arunkumar, G.R. Novel nicotine analogues with potential anti-mycobacterial activity. Bioorganic Med. Chem. 2016, 24, 1637–1647. [Google Scholar] [CrossRef]
- Mardianingrum, R.; Endah, S.R.N.; Suhardiana, E.; Ruswanto, R.; Siswandono, S. Docking and molecular dynamic study of isoniazid derivatives as anti-tuberculosis drug candidate. Chem. Data Collect. 2021, 32, 100647. [Google Scholar] [CrossRef]
- Chaudhari, K.S.; Patel, H.M.; Surana, S.J. Pyridines: Multidrug-resistant tuberculosis (MDR-TB) inhibitors. Indian J. Tuberc. 2017, 64, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Villamizar-Mogotocoro, A.F.; Vargas-Méndez, L.Y.; Kouznetsov, V.V. Pyridine and quinoline molecules as crucial protagonists in the never-stopping discovery of new agents against tuberculosis. Eur. J. Pharm. Sci. 2020, 151, 105374. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Exported Repetitive Protein, Accession Code AF-P9WIQ7-F1. Available online: https://alphafold.ebi.ac.uk/entry/P9WIQ7 (accessed on 5 June 2022).
- Uncharacterized Protein Rv1417, Accession Code AF-P9WLY1-F1. Available online: https://alphafold.ebi.ac.uk/entry/P9WLY1 (accessed on 5 June 2022).
- Probable Transmembrane Protein, Accession Code AF-I6XER9-F1. Available online: https://alphafold.ebi.ac.uk/entry/I6XER9 (accessed on 5 June 2022).
- Kandt, C.; Ash, W.L.; Tieleman, D.P. Setting up and running molecular dynamics simulations of membrane proteins. Methods 2007, 41, 475–488. [Google Scholar] [CrossRef]
- Exported Repetitive Protein, Accession Code P9WIQ7. Available online: https://www.uniprot.org/uniprot/P9WIQ7 (accessed on 20 September 2021).
- Schmidtke, P.; Le Guilloux, V.; Maupetit, J.; Tuffï?` 1/2ry, P. Fpocket: Online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010, 38, W582–W589. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2. 0: New docking methods, expanded force field, and python bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Kutzner, C.; Páll, S.; Fechner, M.; Esztermann, A.; de Groot, B.L.; Grubmüller, H. More bang for your buck: Improved use of GPU nodes for GROMACS 2018. J. Comput. Chem. 2019, 40, 2418–2431. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Tieleman, D.P.; Berendsen, H. Molecular dynamics simulations of a fully hydrated dipalmitoylphosphatidylcholine bilayer with different macroscopic boundary conditions and parameters. J. Chem. Phys. 1996, 105, 4871–4880. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView 6.0; 2016. [Google Scholar]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Ritchie, J.P.; Bachrach, S.M. Some methods and applications of electron density distribution analysis. J. Comput. Chem. 1987, 8, 499–509. [Google Scholar] [CrossRef]
- Dodda, L.S.; Cabeza de Vaca, I.; Tirado-Rives, J.; Jorgensen, W.L. LigParGen web server: An automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 2017, 45, W331–W336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duhovny, D.; Nussinov, R.; Wolfson, H.J. Efficient unbound docking of rigid molecules. In Proceedings of the International Workshop on Algorithms in Bioinformatics, Rome, Italy, 17–21 September 2002; Springer: Berlin/Heidelberg, Germany, 2002; pp. 185–200. [Google Scholar]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast interaction refinement in molecular docking. Proteins Struct. Funct. Bioinform. 2007, 69, 139–159. [Google Scholar] [CrossRef] [PubMed]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free energy calculations by the molecular mechanics Poisson- Boltzmann surface area method. Mol. Informatics 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Turner, P.; Grace, X. Center for Coastal and Land-Margin Research; Version 5.1.19; 2005. [Google Scholar]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Inc., W. Mathematica; 2020. [Google Scholar]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Pineda, J.A.; Albaghdadi, M.; Jiang, W.; Vera-Lopez, K.J.; Nieto-Montesinos, R.; Alvarez, K.L.F.; Davila Del-Carpio, G.; Gómez, B.; Lindsay, M.E.; Malhotra, R.; et al. Structural and functional analysis of female sex hormones against SARS-CoV-2 cell entry. Int. J. Mol. Sci. 2021, 22, 11508. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Pineda, J.A.; Paco-Coralla, S.G.; Febres-Molina, C.; Gamero-Begazo, P.L.; Shrivastava, P.; Vera-López, K.J.; Davila-Del-Carpio, G.; López-C, P.; Gómez, B.; Lino Cardenas, C.L. In Silico Analysis of the Antagonist Effect of Enoxaparin on the ApoE4–Amyloid-Beta (A β) Complex at Different pH Conditions. Biomolecules 2022, 12, 499. [Google Scholar] [CrossRef]
- Febres-Molina, C.; Aguilar-Pineda, J.A.; Gamero-Begazo, P.L.; Barazorda-Ccahuana, H.L.; Valencia, D.E.; Vera-López, K.J.; Davila-Del-Carpio, G.; Gómez, B. Structural and Energetic Affinity of Annocatacin B with ND1 Subunit of the Human Mitochondrial Respiratory Complex I as a Potential Inhibitor: An In Silico Comparison Study with the Known Inhibitor Rotenone. Polymers 2021, 13, 1840. [Google Scholar] [CrossRef]
- Saez, D.A.; Vöhringer-Martinez, E. A consistent S-Adenosylmethionine force field improved by dynamic Hirshfeld-I atomic charges for biomolecular simulation. J. Comput. Aided Mol. Des. 2015, 29, 951–961. [Google Scholar] [CrossRef]
- Janani, S.; Rajagopal, H.; Muthu, S.; Aayisha, S.; Raja, M.; Irfan, A. Structural, vibrational, electronic properties, hirshfeld surface analysis topological and molecular docking studies of N-[2-(diethylamino) ethyl]-2-methoxy-5-methylsulfonylbenzamide. Heliyon 2021, 7, e08186. [Google Scholar] [CrossRef]
- Uncharacterized Protein Rv1417, Accession Code P9WLY1. Available online: https://www.uniprot.org/uniprot/P9WLY1 (accessed on 20 September 2021).
- Probable Transmembrane Protein, Accession code I6XER9. Available online: https://www.uniprot.org/uniprot/I6XER9 (accessed on 20 September 2021).
- Tze-Yang Ng, J.; Tan, Y.S. Accelerated Ligand-Mapping Molecular Dynamics Simulations for the Detection of Recalcitrant Cryptic Pockets and Occluded Binding Sites. J. Chem. Theory Comput. 2022, 18, 1969–1981. [Google Scholar] [CrossRef] [PubMed]
- Giordano, D.; Langini, C.; Caflisch, A.; Marabotti, A.; Facchiano, A. Molecular dynamics analysis of the structural properties of the transglutaminases of Kutzneria albida and Streptomyces mobaraensis. Comput. Struct. Biotechnol. J. 2022, 20, 3924–3934. [Google Scholar] [CrossRef]
- Ding, T.; Karlov, D.S.; Pino-Angeles, A.; Tikhonova, I.G. Intermolecular Interactions in G Protein-Coupled Receptor Allosteric Sites at the Membrane Interface from Molecular Dynamics Simulations and Quantum Chemical Calculations. J. Chem. Inf. Model. 2022, 62, 4736–4747. [Google Scholar] [CrossRef]
- Bowman, G.R. Identifying and Exploiting Cryptic Pockets. Biophys. J. 2020, 118, 160a. [Google Scholar] [CrossRef]
- Falck, E.; Patra, M.; Karttunen, M.; Hyvönen, M.T.; Vattulainen, I. Lessons of slicing membranes: Interplay of packing, free area, and lateral diffusion in phospholipid/cholesterol bilayers. Biophys. J. 2004, 87, 1076–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leekumjorn, S.; Sum, A.K. Molecular simulation study of structural and dynamic properties of mixed DPPC/DPPE bilayers. Biophys. J. 2006, 90, 3951–3965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahane, G.; Ding, W.; Palaiokostas, M.; Orsi, M. Physical properties of model biological lipid bilayers: Insights from all-atom molecular dynamics simulations. J. Mol. Model. 2019, 25, 76. [Google Scholar] [CrossRef] [Green Version]
- Seki, T.; Mochida, J.; Okamoto, M.; Hosoya, O.; Juni, K.; Morimoto, K. Measurement of diffusion coefficients of parabens and steroids in water and 1-octanol. Chem. Pharm. Bull. 2003, 51, 734–736. [Google Scholar] [CrossRef] [Green Version]
- Bernkop-Schnürch, A.; Jalil, A. Do drug release studies from SEDDS make any sense? J. Control. Release 2018, 271, 55–59. [Google Scholar] [CrossRef]




{kind=link}
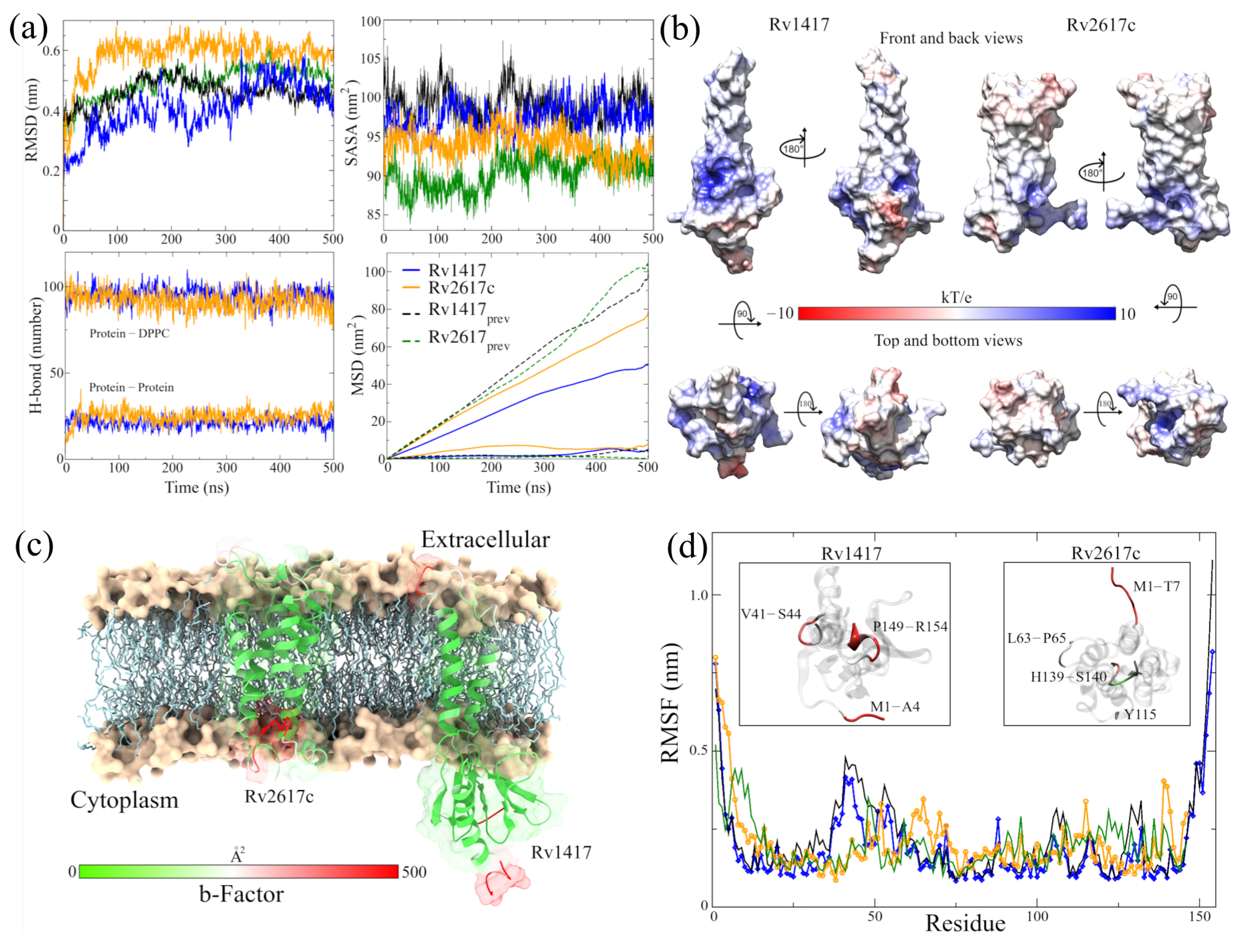{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Model Name | Predicted Value | ||||
|---|---|---|---|---|---|---|
| NIA | NIB | NIC | NID | NIE | ||
| Physicochemical | logS | −4.014 | −3.912 | −4.062 | −3.871 | −3.923 |
| logP | 3.492 | 3.254 | 3.418 | 3.271 | 3.307 | |
| logD | 2.481 | 2.191 | 2.405 | 2.257 | 2.334 | |
| Medicinal | QED | 0.627 | 0.627 | 0.627 | 0.627 | 0.627 |
| Chemistry | SAScore | 2.996 | 2.995 | 2.906 | 2.954 | 2.945 |
| NPScore | −0.866 | −0.911 | −0.962 | −0.941 | −0.979 | |
| Absorption | Caco-2 Permeability | −4.656 | −4.595 | −4.631 | −4.622 | −4.620 |
| MDCK Permeability | 1.3 × 10 | 2.9 × 10 | 2.4 × 10 | 2.0 × 10 | 2.0 × 10 | |
| Pgp inhibitor | - - - | - - - | - - - | - - | - - | |
| Pgp substrate | - - - | - - - | - - - | - - - | - - - | |
| HIA | - - - | - - - | - - - | - - - | - - - | |
| Distribution | PPB | 0.950 | 0.916 | 0.921 | 0.916 | 0.918 |
| VD | 1.14 3 | 1.280 | 1.181 | 1.275 | 1.226 | |
| Fu | 0.263 | 0.587 | 0.489 | 0.544 | 0.523 | |
| Metabolism | CYP1A2 inhibitor | +++ | +++ | +++ | +++ | +++ |
| CYP1A2 substrate | ++ | ++ | ++ | ++ | ++ | |
| CYP2C19 inhibitor | ++ | +++ | +++ | +++ | +++ | |
| CYP2C19 substrate | + | - | - | - | - | |
| CYP2C9 inhibitor | + | ++ | + | + | + | |
| CYP2C9 substrate | - - | + | - | - | - | |
| CYP2D6 inhibitor | - | - | - | + | - | |
| CYP2D6 substrate | + | + | ++ | ++ | + | |
| CYP3A4 inhibitor | +++ | +++ | +++ | +++ | +++ | |
| CYP3A4 substrate | + | + | + | + | + | |
| Excretion | CL | 3.150 | 3.568 | 3.325 | 2.804 | 2.846 |
| t | 0.260 | 0.215 | 0.176 | 0.186 | 0.171 | |
| Toxicity | hERG Blockers | + | + | ++ | + | ++ |
| H-HT | - | - | - | - | - | |
| DILI | + | ++ | ++ | ++ | ++ | |
| AMES Tox. | - - - | - - - | - - - | - - - | - - - | |
| Rat Oral Acute Tox. | - - | - | - | - - | - - | |
| FDAMDD | - | - | - | - | - | |
| Skin Sensitization | - - | - - | - - | - - | - - | |
| Carcinogenicity | - - - | - - | - - | - - | - - | |
| Eye Corrosion | - - - | - - - | - - - | - - - | - - - | |
| Eye Irritation | - - - | - - - | - - - | - - - | - - - | |
| Respiratory Tox. | - - - | - - | - - | - - - | - - - | |
| System | a RMSD | a RMSF | a RG | H Bonds | H Bonds + DPPC | |||
|---|---|---|---|---|---|---|---|---|
| Total | x-axis | y-axis | z-axis | |||||
| Membrane and Water—500 ns | ||||||||
| Rv1417 | 0.41 ± 0.08 | 0.19 ± 0.12 | 2.13 ± 0.02 | 2.00 ± 0.04 | 1.99 ± 0.03 | 1.03 ± 0.05 | 96 ± 5 | 21 ± 3 |
| Prev. | 0.46 ± 0.05 | 0.22 ± 0.14 | 2.14 ± 0.02 | 2.03 ± 0.03 | 2.01 ± 0.03 | 1.00 ± 0.05 | 93 ± 5 | 21 ± 4 |
| Rv2617c | 0.59 ± 0.08 | 0.20 ± 0.11 | 1.85 ± 0.03 | 1.70 ± 0.03 | 1.68 ± 0.03 | 1.05 ± 0.04 | 91 ± 6 | 25 ± 4 |
| Prev. | 0.50 ± 0.06 | 0.20 ± 0.07 | 1.85 ± 0.03 | 1.73 ± 0.04 | 1.71 ± 0.03 | 0.98 ± 0.04 | 100 ± 5 | 20 ± 3 |
| Erp | 0.70 ± 0.10 | 0.34 ± 0.17 | 2.01 ± 0.03 | 1.66 ± 0.15 | 1.58 ± 0.15 | 1.66 ± 0.15 | 110 ± 8 | - |
| Prev. | 0.65 ± 0.12 | 0.33 ± 0.17 | 2.04 ± 0.02 | 1.72 ± 0.12 | 1.55 ± 0.15 | 1.70 ± 0.14 | 114 ± 8 | - |
| System | RMSD a | RMSF a | RG a | H Bonds | |||
|---|---|---|---|---|---|---|---|
| Protein | Protein + lig | Intra | Inter–lig | Inter–DPPC | |||
| ASC14–NIx | |||||||
| NIA | 0.22 ± 0.03 | 0.15 ± 0.08 | 2.12 ± 0.02 | 2.11 ± 0.02 | 95 ± 5 | 0.13 ± 0.36 | 19.74 ± 2.94 |
| NIB | 0.23 ± 0.04 | 0.15 ± 0.09 | 2.14 ± 0.02 | 2.12 ± 0.02 | 94 ± 5 | 0.62 ± 0.58 | 19.54 ± 2.91 |
| NIC | 0.23 ± 0.04 | 0.16 ± 0.08 | 2.13 ± 0.02 | 2.12 ± 0.02 | 94 ± 5 | 0.43 ± 0.51 | 20.92 ± 2.98 |
| NID | 0.19 ± 0.03 | 0.14 ± 0.06 | 2.13 ± 0.02 | 2.12 ± 0.02 | 93 ± 4 | 0.39 ± 0.54 | 21.85 ± 2.91 |
| NIE | 0.22 ± 0.04 | 0.15 ± 0.06 | 2.12 ± 0.02 | 2.10 ± 0.02 | 93 ± 4 | 0.19 ± 0.42 | 21.87 ± 3.04 |
| ASC26–NIx | |||||||
| NIA | 0.30 ± 0.04 | 0.15 ± 0.06 | 1.89 ± 0.02 | 1.91 ± 0.02 | 107 ± 4 | 0.13 ± 0.42 | 19.27 ± 3.21 |
| NIB | 0.22 ± 0.03 | 0.13 ± 0.06 | 1.83 ± 0.02 | 1.86 ± 0.04 | 105 ± 5 | 0.12 ± 0.33 | 19.75 ± 2.96 |
| NIC | 0.28 ± 0.04 | 0.17 ± 0.07 | 1.90 ± 0.02 | 1.90 ± 0.02 | 100 ± 6 | 0.03 ± 0.16 | 19.27 ± 3.18 |
| NID | 0.23 ± 0.04 | 0.16 ± 0.06 | 1.83 ± 0.03 | 1.88 ± 0.04 | 104 ± 5 | 0.00 ± 0.00 | 20.17 ± 3.03 |
| NIE | 0.21 ± 0.02 | 0.11 ± 0.05 | 1.83 ± 0.02 | 1.85 ± 0.02 | 109 ± 5 | 0.00 ± 0.00 | 19.84 ± 3.20 |
| Sol14-NIx | |||||||
| NIA | 0.27 ± 0.05 | 0.17 ± 0.11 | 2.13 ± 0.02 | 2.92 ± 0.19 | 92 ± 4 | 0.49 ± 0.71 | 21.73 ± 3.74 |
| NIB | 0.27 ± 0.04 | 0.15 ± 0.08 | 2.13 ± 0.02 | 2.93 ± 0.10 | 94 ± 5 | 1.44 ± 0.92 | 19.96 ± 3.21 |
| NIC | 0.28 ± 0.05 | 0.16 ± 0.08 | 2.11 ± 0.02 | 3.14 ± 0.16 | 97 ± 5 | 0.40 ± 0.61 | 17.90 ± 3.05 |
| NID | 0.25 ± 0.04 | 0.15 ± 0.09 | 2.13 ± 0.02 | 2.88 ± 0.18 | 93 ± 5 | 1.00 ± 1.03 | 20.79 ± 3.46 |
| NIE | 0.26 ± 0.03 | 0.15 ± 0.08 | 2.14 ± 0.02 | 3.02 ± 0.26 | 94 ± 5 | 0.44 ± 0.56 | 20.17 ± 3.27 |
| Sol26-NIx | |||||||
| NIA | 0.22 ± 0.04 | 0.14 ± 0.06 | 1.88 ± 0.02 | 2.75 ± 0.20 | 104 ± 5 | 0.38 ± 0.62 | 18.09 ± 2.58 |
| NIB | 0.25 ± 0.04 | 0.14 ± 0.07 | 1.88 ± 0.02 | 2.71 ± 0.12 | 102 ± 5 | 0.40 ± 0.60 | 17.78 ± 2.94 |
| NIC | 0.23 ± 0.04 | 0.14 ± 0.07 | 1.90 ± 0.02 | 2.68 ± 0.10 | 99 ± 5 | 0.87 ± 0.77 | 16.54 ± 2.69 |
| NID | 0.37 ± 0.09 | 0.19 ± 0.14 | 1.91 ± 0.03 | 3.14 ± 0.19 | 101 ± 6 | 0.01 ± 0.06 | 19.27 ± 3.44 |
| NIE | 0.19 ± 0.02 | 0.12 ± 0.06 | 1.82 ± 0.02 | 2.30 ± 0.13 | 103 ± 5 | 0.56 ± 0.71 | 18.20 ± 2.86 |
| NAM | Active Site Complexes | Solvated Complexes | ||
|---|---|---|---|---|
| Rv1417 | Rv2617c | Rv1417 | Rv2617c | |
| NIA | L19, R74 (9.18) | Q50 (11.2), H51, N53, M54, A57, F114 | A4, N6, D7 (11.8), H32, T50, A51, D52, Q53, V54, G57, L61, S79, A80, G81, I92, V93, G94, W95 (4.0), S96 (22.9), L143, Y147 | M1, S2 (7.5), I3, R4 (7.18), P5, T7 (11.7), S8 (5.7), P9, L119, L124, V126, H139 |
| NIB | R13, P14, R72 (12.2), R74 (7.4), R85 (16.4) | - | T2, A3, P5, N6 (56.9), D7 (25.2), G78, S79, A80 (6.7), G81, L82, S83, I92, V93, G94, W95, S96, E97, L143, R146, Y147, R148 | F42, P49, Q50, H51 (6.7), N53 (6.3), M54, Y55, Q70, Y73 (8.19), A77, P112, F114 (9.9) |
| NIC | R13, P14, H15, P18, R72 (15.9) | M54, Y55, G58, N107 (25.9), L108, P112, F114, I117 | L27, A31, H32, G35, L37, V46, V47, F48, Q49 (7.0), Q53, V54, A55, F103 (12.0), G106, R108 (10.9), W109, M123, I125, A127, V128 | I3 (8.4), A19, L26, P49, Q50 (27.6), M54, Y55, N107 (10.8), L108, P112, F114, V126, G127, A130, R133, L134, S140 (17.1) I143, R145 |
| NID | R13, R72, R74 (19.9), R85 | - | W95 (12.0), S96 (14.4), I99, G100, V101, S102, D150, L151, A153 (10.6), R154 (21.2) | L36, L39, F43, L45, I60, N61, V64, A99, W100, A102, G103 |
| NIE | R13, P14, P18, R72, R74 (6.0), R85 | M54, A57, T110, G111, P112 | M1, T2 (6.8), A3 (6.6), A4, P5, N6 (14.4), A80, G81, I92, V93, G94, S96 | A10, Q50 (15.3), H51 (10.8), N53 (15.0), L63, L108, V109, T110, G111, P112, G113, F114, Y115, I129, A132, R133 |
| Complex | |||||
|---|---|---|---|---|---|
| ASC14–NIx | |||||
| NIA | −52.78 ± 2.30 | 11.53 ± 1.68 | 3.51 ± 1.57 | −7.28 ± 0.27 | −45.01 ± 4.37 |
| NIB | −100.81 ± 0.54 | −67.36 ± 0.94 | 42.12 ± 0.55 | −13.69 ± 0.05 | −139.74 ± 1.23 |
| NIC | −103.28 ± 0.64 | −15.42 ± 1.71 | 42.70 ± 0.94 | −13.27 ± 0.06 | −89.27 ± 1.32 |
| NID | −86.25 ± 0.84 | −49.19 ± 1.94 | 17.17 ± 1.56 | −12.15 ± 0.10 | −130.42 ± 2.15 |
| NIE | −100.00 ± 0.75 | −41.62 ± 1.28 | 28.09 ± 1.04 | −12.45 ± 0.07 | −125.99 ± 1.64 |
| ASC26–NIx | |||||
| NIA | −82.89 ± 0.85 | −13.30 ± 1.22 | 27.38 ± 0.98 | −10.79 ± 0.10 | −79.59 ± 2.93 |
| NIB | −12.50 ± 1.52 | 0.61 ± 0.55 | −1.53 ± 1.22 | −1.87 ± 0.21 | −15.29 ± 3.69 |
| NIC | −165.29 ± 0.83 | −7.75 ± 0.87 | 40.14 ± 0.68 | −15.34 ± 0.10 | −148.24 ± 2.00 |
| NID | −0.52 ± 0.08 | −2.15 ± 0.27 | 2.55 ± 0.98 | −0.16 ± 0.07 | −0.28 ± 2.49 |
| NIE | −76.85 ± 0.78 | −3.93 ± 0.82 | 21.05 ± 0.70 | −9.42 ± 0.10 | −69.15 ± 2.84 |
| Sol14–NIx | |||||
| NIA | −323.35 ± 2.26 | −43.97 ± 1.55 | 107.35 ± 2.13 | −32.59 ± 0.25 | −292.57 ± 6.22 |
| NIB | −281.80 ± 2.12 | −70.73 ± 1.75 | 98.53 ± 1.77 | −28.78 ± 0.24 | −282.77 ± 5.74 |
| NIC | −203.07 ± 4.48 | −30.14 ± 2.13 | 46.94 ± 1.95 | −26.86 ± 0.60 | −213.13 ± 12.23 |
| NID | −347.67 ± 3.79 | −91.54 ± 3.83 | 167.41 ± 2.64 | −40.78 ± 0.40 | −312.57 ± 8.25 |
| NIE | −138.95 ± 2.17 | −20.37 ± 1.33 | 59.84 ± 1.86 | −15.87 ± 0.27 | −115.35 ± 6.74 |
| Sol26–NIx | |||||
| NIA | −156.83 ± 2.90 | −47.89 ± 2.63 | 83.49 ± 2.14 | −21.00 ± 0.38 | −142.22 ± 7.87 |
| NIB | −313.60 ± 4.69 | −74.58 ± 3.85 | 149.28 ± 2.98 | −41.41 ± 0.53 | −280.30 ± 11.13 |
| NIC | −308.47 ± 2.46 | −30.20 ± 2.07 | 87.67 ± 1.96 | −32.08 ± 0.27 | −283.09 ± 7.56 |
| NID | −120.69 ± 2.32 | −0.75 ± 0.90 | 32.08 ± 1.16 | −14.46 ± 0.29 | −103.82 ± 6.83 |
| NIE | −290.07 ± 2.67 | −32.44 ± 2.29 | 90.30 ± 2.41 | −34.52 ± 0.31 | −266.73 ± 6.98 |
| Protein | NAM | Diffusion | Area per Lipid | ||||||
|---|---|---|---|---|---|---|---|---|---|
| H-G | G-E | A-Ch | DPPC | Protein | NIx | Up | Down | ||
| Rv1417 | NIA | 800.15 | 596.65 | 791.40 | 4.95 ± 0.91 | 2.15 ± 2.28 | 6.15 ± 2.18 | 48.61 ± 0.49 | 49.37 ± 0.40 |
| NIB | 788.40 | 588.95 | 789.35 | 5.27 ± 0.55 | 1.53 ± 0.42 | 6.30 ± 3.03 | 48.78 ± 0.48 | 49.56 ± 0.36 | |
| NIC | 791.10 | 590.20 | 786.95 | 5.24 ± 1.44 | 2.00 ± 0.44 | 7.11 ± 0.34 | 48.91 ± 0.68 | 49.64 ± 0.65 | |
| NID | 784.30 | 585.87 | 786.31 | 4.34 ± 0.49 | 0.20 ± 0.34 | 5.03 ± 2.50 | 48.87 ± 0.43 | 49.58 ± 0.42 | |
| NIE | 794.82 | 592.44 | 788.52 | 3.95 ± 0.71 | 2.01 ± 3.08 | 13.95 ± 1.7 | 48.76 ± 0.69 | 49.48 ± 0.79 | |
| Rv2617c | NIA | 837.15 | 611.15 | 780.50 | 2.41 ± 0.28 | 1.56 ± 2.46 | 2.99 ± 0.03 | 48.91 ± 0.65 | 49.46 ± 0.56 |
| NIB | 805.48 | 585.74 | 775.22 | 4.58 ± 0.33 | 1.96 ± 1.63 | 9.66 ± 10.08 | 48.96 ± 0.41 | 49.38 ± 0.60 | |
| NIC | 798.79 | 580.63 | 775.10 | 5.15 ± 2.07 | 0.21 ± 0.12 | 2.28 ± 0.39 | 48.34 ± 0.58 | 49.10 ± 0.66 | |
| NID | 822.28 | 599.22 | 779.33 | 4.48 ± 0.41 | 0.27 ± 0.14 | 5.54 ± 4.13 | 48.75 ± 0.48 | 49.00 ± 0.43 | |
| NIE | 817.93 | 594.18 | 778.69 | 4.12 ± 0.42 | 0.88 ± 0.09 | 2.16 ± 0.08 | 49.16 ± 0.32 | 49.35 ± 0.38 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguilar-Pineda, J.A.; Febres-Molina, C.; Cordova-Barrios, C.C.; Campos-Olazával, L.M.; Del-Carpio-Martinez, B.A.; Ayqui-Cueva, F.; Gamero-Begazo, P.L.; Gómez, B. Study of the Rv1417 and Rv2617c Membrane Proteins and Their Interactions with Nicotine Derivatives as Potential Inhibitors of Erp Virulence-Associated Factor in Mycobacterium tuberculosis: An In Silico Approach. Biomolecules 2023, 13, 248. https://doi.org/10.3390/biom13020248
Aguilar-Pineda JA, Febres-Molina C, Cordova-Barrios CC, Campos-Olazával LM, Del-Carpio-Martinez BA, Ayqui-Cueva F, Gamero-Begazo PL, Gómez B. Study of the Rv1417 and Rv2617c Membrane Proteins and Their Interactions with Nicotine Derivatives as Potential Inhibitors of Erp Virulence-Associated Factor in Mycobacterium tuberculosis: An In Silico Approach. Biomolecules. 2023; 13(2):248. https://doi.org/10.3390/biom13020248
Chicago/Turabian StyleAguilar-Pineda, Jorge Alberto, Camilo Febres-Molina, Cinthia C. Cordova-Barrios, Lizbeth M. Campos-Olazával, Bruno A. Del-Carpio-Martinez, Flor Ayqui-Cueva, Pamela L. Gamero-Begazo, and Badhin Gómez. 2023. "Study of the Rv1417 and Rv2617c Membrane Proteins and Their Interactions with Nicotine Derivatives as Potential Inhibitors of Erp Virulence-Associated Factor in Mycobacterium tuberculosis: An In Silico Approach" Biomolecules 13, no. 2: 248. https://doi.org/10.3390/biom13020248