
The Molecular and Genetic Mechanisms of Inherited Bone Marrow Failure Syndromes: The Role of Inflammatory Cytokines in Their Pathogenesis

Abstract
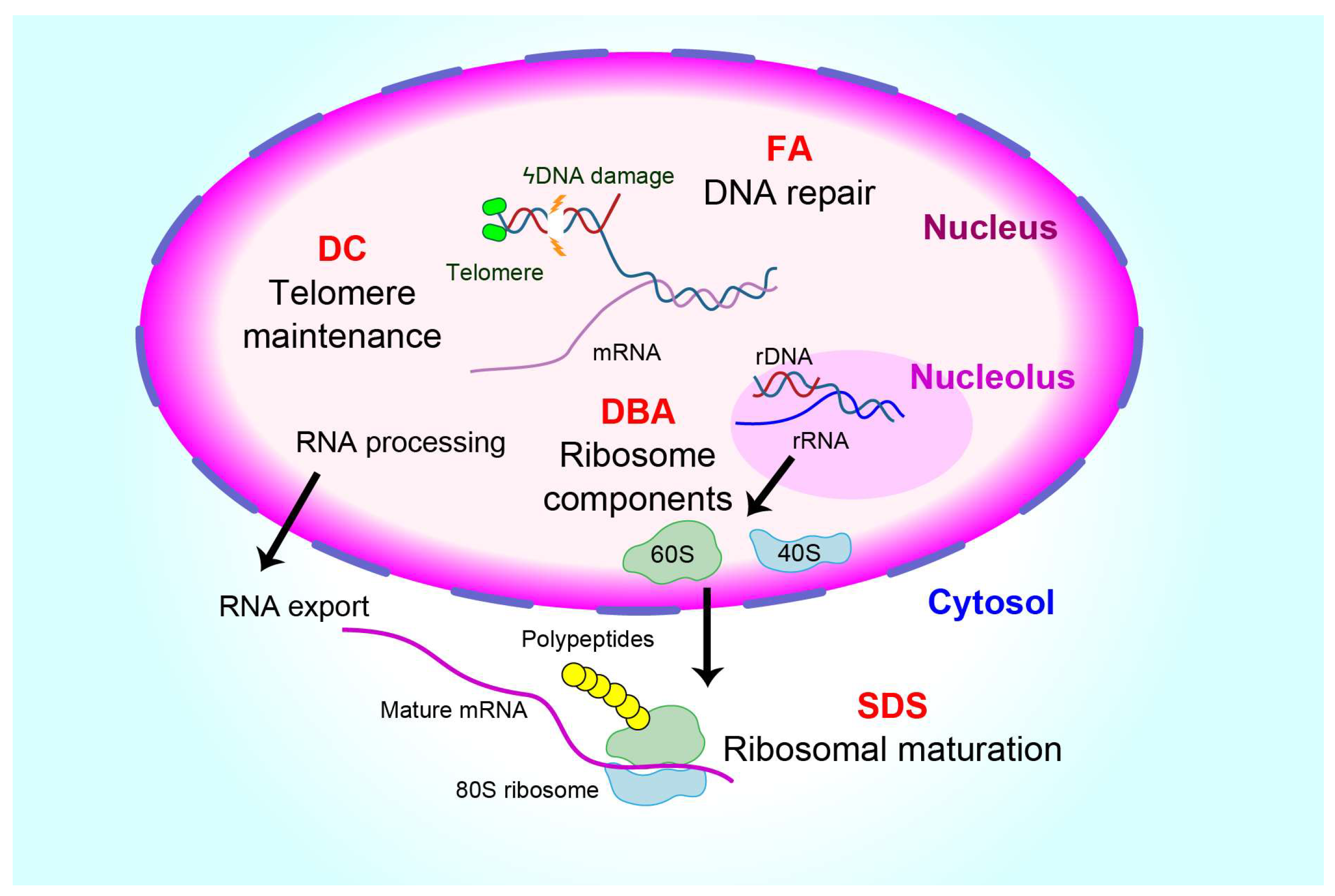
:1. Introduction
2. Common Signaling Themes
3. Common IBMFSs
3.1. Fanconi Anemia
3.1.1. Overview
3.1.2. Genetics
3.1.3. Signaling Pathway
3.1.4. Inflammatory Profile
3.2. Dyskeratosis Congenita
3.2.1. Overview
3.2.2. Genetics
3.2.3. Signaling Pathway
3.2.4. Inflammatory Profile
3.3. Diamond–Blackfan Anemia
3.3.1. Overview
3.3.2. Genetics
3.3.3. Signaling Pathway
3.3.4. Inflammatory Profile
3.4. Shwachman–Diamond Syndrome
3.4.1. Overview and Genetics
3.4.2. Signaling Pathway
3.4.3. Inflammatory Profile
4. Other Rare IBMFSs
5. The Role of Bone Vasculature in Bone Marrow Inflammation

{kind=link}
{kind=link}
| FA | DC | DBA | SDS | |
|---|---|---|---|---|
| Anti-inflammatory | ||||
| TGF-β | ↑ * | ↑ | ||
| IL-10 | ↑ | |||
| sFasL | ↑ | |||
| Pro-inflammatory | ||||
| IL-1β | ↑ * | |||
| IL-6 | ↑ * | ↑ | ||
| IL-8 | ↑ | ↑ | ||
| TNF-α | ↑ * | ↓ | ||
| IFN-γ | ↑ * | ↑ * | ↓ | |
| sCD40L | ↓ | |||
| RANTES | ↓ | ↓ | ||
| CXCL10 (IP-10) | ↑ | ↑ | ||
| CCL16 | ↑ | |||
| CCL21 | ↑ | |||
| G-CSF | ↑ | ↑ | ||
| Flt3L | ↑ | ↑ |
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsangaris, E.; Klaassen, R.; Fernandez, C.V.; Yanofsky, R.; Shereck, E.; Champagne, J.; Silva, M.; Lipton, J.H.; Brossard, J.; Michon, B.; et al. Genetic analysis of inherited bone marrow failure syndromes from one prospective, comprehensive and population-based cohort and identification of novel mutations. J. Med. Genet. 2011, 48, 618–628. [Google Scholar] [CrossRef]
- Giudice, V.; Cardamone, C.; Triggiani, M.; Selleri, C. Bone Marrow Failure Syndromes, Overlapping Diseases with a Common Cytokine Signature. Int. J. Mol. Sci. 2021, 22, 705. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Erlacher, M.; Fernandez-Orth, J. The role of inflammation in hematopoiesis and bone marrow failure: What can we learn from mouse models? Front. Immunol. 2022, 13, 951937. [Google Scholar] [CrossRef] [PubMed]
- Mazewski, C.; Perez, R.E.; Fish, E.N.; Platanias, L.C. Type I Interferon (IFN)-Regulated Activation of Canonical and Non-Canonical Signaling Pathways. Front. Immunol. 2020, 11, 606456. [Google Scholar] [CrossRef]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Jacobsen, S.E.; Jacobsen, F.W.; Fahlman, C.; Rusten, L.S. TNF-alpha, the great imitator: Role of p55 and p75 TNF receptors in hematopoiesis. Stem. Cells 1994, 12 (Suppl. S1), 111–126, discussion 126–118. [Google Scholar] [PubMed]
- Lokuta, M.A.; Huttenlocher, A. TNF-alpha promotes a stop signal that inhibits neutrophil polarization and migration via a p38 MAPK pathway. J. Leukoc. Biol. 2005, 78, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Carey, A.; Edwards, D.K.T.; Eide, C.A.; Newell, L.; Traer, E.; Medeiros, B.C.; Pollyea, D.A.; Deininger, M.W.; Collins, R.H.; Tyner, J.W.; et al. Identification of Interleukin-1 by Functional Screening as a Key Mediator of Cellular Expansion and Disease Progression in Acute Myeloid Leukemia. Cell Rep. 2017, 18, 3204–3218. [Google Scholar] [CrossRef] [PubMed]
- Tao, M.; Li, B.; Nayini, J.; Andrews, C.B.; Huang, R.W.; Devemy, E.; Song, S.; Venugopal, P.; Preisler, H.D. SCF, IL-1beta, IL-1ra and GM-CSF in the bone marrow and serum of normal individuals and of AML and CML patients. Cytokine 2000, 12, 699–707. [Google Scholar] [CrossRef]
- Hoang, T.; Haman, A.; Goncalves, O.; Letendre, F.; Mathieu, M.; Wong, G.G.; Clark, S.C. Interleukin 1 enhances growth factor-dependent proliferation of the clonogenic cells in acute myeloblastic leukemia and of normal human primitive hemopoietic precursors. J. Exp. Med. 1988, 168, 463–474. [Google Scholar] [CrossRef]
- Carter, A.; Silvian-Draxler, I.; Tatarsky, I. Effect of interleukin-1, tumor necrosis factor-alpha, and interferon-alpha on the blast cells of acute myeloblastic leukemia. Am. J. Hematol. 1992, 40, 245–251. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Estey, E.; Wen, S.; Pierce, S.; Kantarjian, H.; Albitar, M.; Kurzrock, R. The prognostic significance of cytokine levels in newly diagnosed acute myeloid leukemia and high-risk myelodysplastic syndromes. Cancer 2008, 113, 1605–1613. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, X.; Yuan, S.; Hou, S.; Guo, T.; Chu, Y.; Pang, T.; Luo, H.R.; Yuan, W.; Wang, X. Interleukin-1beta inhibits normal hematopoietic expansion and promotes acute myeloid leukemia progression via the bone marrow niche. Cytotherapy 2020, 22, 127–134. [Google Scholar] [CrossRef]
- Svensson, E.C.; Madar, A.; Campbell, C.D.; He, Y.; Sultan, M.; Healey, M.L.; Xu, H.; D’Aco, K.; Fernandez, A.; Wache-Mainier, C.; et al. TET2-Driven Clonal Hematopoiesis and Response to Canakinumab: An Exploratory Analysis of the CANTOS Randomized Clinical Trial. JAMA Cardiol. 2022, 7, 521–528. [Google Scholar] [CrossRef]
- Wong, C.C.; Baum, J.; Silvestro, A.; Beste, M.T.; Bharani-Dharan, B.; Xu, S.; Wang, Y.A.; Wang, X.; Prescott, M.F.; Krajkovich, L.; et al. Inhibition of IL1beta by Canakinumab May Be Effective against Diverse Molecular Subtypes of Lung Cancer: An Exploratory Analysis of the CANTOS Trial. Cancer Res. 2020, 80, 5597–5605. [Google Scholar] [CrossRef]
- Bagby, G.C. Multifunctional Fanconi proteins, inflammation and the Fanconi phenotype. eBioMedicine 2016, 8, 10–11. [Google Scholar] [CrossRef]
- Dufour, C.; Pierri, F. Modern management of Fanconi anemia. Hematol. Am. Soc. Hematol. Educ. Program. 2022, 2022, 649–657. [Google Scholar] [CrossRef]
- Moreno, O.M.; Paredes, A.C.; Obando, S.F.; Rojas, A. An update on Fanconi anemia: Clinical, cytogenetic and molecular approaches (Review). Biomed. Rep. 2021, 15, 74. [Google Scholar] [CrossRef]
- Soulier, J. Fanconi Anemia. Hematol. Am. Soc. Hematol. Educ. Program. 2011, 2011, 492–497. [Google Scholar] [CrossRef]
- Swuec, P.; Renault, L.; Borg, A.; Shah, F.; Murphy, V.J.; van Twest, S.; Snijders, A.P.; Deans, A.J.; Costa, A. The FA Core Complex Contains a Homo-dimeric Catalytic Module for the Symmetric Mono-ubiquitination of FANCI-FANCD2. Cell Rep. 2017, 18, 611–623. [Google Scholar] [CrossRef]
- Douwel, K.D.; Boonen, R.A.C.M.; Long, D.T.; Szypowska, A.A.; Räschle, M.; Walter, J.C.; Knipscheer, P. XPF-ERCC1 Acts in Unhooking DNA Interstrand Crosslinks in Cooperation with FANCD2 and FANCP/SLX4. Mol. Cell 2014, 54, 460–471. [Google Scholar] [CrossRef]
- Bluteau, D.; Masliah-Planchon, J.; Clairmont, C.; Rousseau, A.; Ceccaldi, R.; Dubois d’Enghien, C.; Bluteau, O.; Cuccuini, W.; Gachet, S.; Peffault de Latour, R.; et al. Biallelic inactivation of REV7 is associated with Fanconi anemia. J. Clin. Investig. 2016, 126, 3580–3584. [Google Scholar] [CrossRef]
- Zhang, H.; Kozono, D.E.; O’Connor, K.W.; Vidal-Cardenas, S.; Rousseau, A.; Hamilton, A.; Moreau, L.; Gaudiano, E.F.; Greenberger, J.; Bagby, G.; et al. TGF-β Inhibition Rescues Hematopoietic Stem Cell Defects and Bone Marrow Failure in Fanconi Anemia. Cell Stem Cell 2016, 18, 668–681. [Google Scholar] [CrossRef]
- Helbling-Leclerc, A.; Garcin, C.; Rosselli, F. Beyond DNA repair and chromosome instability—Fanconi anaemia as a cellular senescence-associated syndrome. Cell Death Differ. 2021, 28, 1159–1173. [Google Scholar] [CrossRef]
- Rickman, K.; Smogorzewska, A. Advances in understanding DNA processing and protection at stalled replication forks. J. Cell Biol. 2019, 218, 1096–1107. [Google Scholar] [CrossRef]
- Gueiderikh, A.; Maczkowiak-Chartois, F.; Rosselli, F. A new frontier in Fanconi anemia: From DNA repair to ribosome biogenesis. Blood Rev. 2022, 52, 100904. [Google Scholar] [CrossRef]
- Myers, K.C.; Bleesing, J.J.; Davies, S.M.; Zhang, X.; Martin, L.J.; Mueller, R.; Harris, R.E.; Filipovich, A.H.; Kovacic, M.B.; Wells, S.I.; et al. Impaired immune function in children with Fanconi anaemia. Br. J. Haematol. 2011, 154, 234–240. [Google Scholar] [CrossRef]
- Korthof, E.T.; Svahn, J.; de Latour, R.P.; Terranova, P.; Moins-Teisserenc, H.; Socié, G.; Soulier, J.; Kok, M.; Bredius, R.G.M.; van Tol, M.; et al. Immunological profile of Fanconi anemia: A multicentric retrospective analysis of 61 patients. Am. J. Hematol. 2013, 88, 472–476. [Google Scholar] [CrossRef]
- Justo, G.A.; Bitencourt, M.A.; Pasquini, R.; Castelo-Branco, M.T.L.; Rumjanek, V.M. Increased IL10 plasmatic levels in Fanconi anemia patients. Cytokine 2013, 64, 486–489. [Google Scholar] [CrossRef]
- Maciejewski, J.; Selleri, C.; Anderson, S.; Young, N.S. Fas Antigen Expression on CD34+ Human Marrow Cells Is Induced by Interferon γ and Tumor Necrosis Factor α and Potentiates Cytokine-Mediated Hematopoietic Suppression In Vitro. Blood 1995, 85, 3183–3190. [Google Scholar] [CrossRef]
- Dufour, C.; Corcione, A.; Svahn, J.; Haupt, R.; Poggi, V.; Békassy, A.N.; Scimè, R.; Pistorio, A.; Pistoia, V. TNF-α and IFN-γ are overexpressed in the bone marrow of Fanconi anemia patients and TNF-α suppresses erythropoiesis in vitro. Blood 2003, 102, 2053–2059. [Google Scholar] [CrossRef]
- Giri, N.; Alter, B.P.; Penrose, K.; Falk, R.T.; Pan, Y.; Savage, S.A.; Williams, M.; Kemp, T.J.; Pinto, L.A. Immune status of patients with inherited bone marrow failure syndromes. Am. J. Hematol. 2015, 90, 702–708. [Google Scholar] [CrossRef]
- Matsui, K.; Giri, N.; Alter, B.P.; Pinto, L.A. Cytokine production by bone marrow mononuclear cells in inherited bone marrow failure syndromes. Br. J. Haematol. 2013, 163, 81–92. [Google Scholar] [CrossRef]
- Ibáñez, A.; Río, P.; Casado, J.A.; Bueren, J.A.; Fernández-Luna, J.L.; Pipaón, C. Elevated levels of IL-1β in Fanconi anaemia group A patients due to a constitutively active phosphoinositide 3-kinase-Akt pathway are capable of promoting tumour cell proliferation. Biochem. J. 2009, 422, 161–170. [Google Scholar] [CrossRef]
- Epanchintsev, A.; Shyamsunder, P.; Verma, R.S.; Lyakhovich, A. IL-6, IL-8, MMP-2, MMP-9 are overexpressed in Fanconi anemia cells through a NF-κB/TNF-α dependent mechanism. Mol. Carcinog. 2015, 54, 1686–1699. [Google Scholar] [CrossRef]
- Garaycoechea, J.I.; Patel, K.J. Why does the bone marrow fail in Fanconi anemia? Blood 2014, 123, 26–34. [Google Scholar] [CrossRef]
- Fiesco-Roa, M.Ó.; García-de Teresa, B.; Leal-Anaya, P.; van ‘t Hek, R.; Wegman-Ostrosky, T.; Frías, S.; Rodríguez, A. Fanconi anemia and dyskeratosis congenita/telomere biology disorders: Two inherited bone marrow failure syndromes with genomic instability. Front. Oncol. 2022, 12, 949435. [Google Scholar] [CrossRef]
- Feld, J.; Navada, S.C.; Silverman, L.R. Myelo-deception: Luspatercept & TGF-Beta ligand traps in myeloid diseases & anemia. Leuk. Res. 2020, 97, 106430. [Google Scholar] [CrossRef]
- Tummala, H.; Walne, A.; Dokal, I. The biology and management of dyskeratosis congenita and related disorders of telomeres. Expert. Rev. Hematol. 2022, 15, 685–696. [Google Scholar] [CrossRef]
- Starace, M.; Alessandrini, A.; Piraccini, B.M. Nail Disorders in Children. Skin Appendage Disord. 2018, 4, 217–229. [Google Scholar] [CrossRef]
- Niewisch, M.R.; Savage, S.A. An update on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Rev. Hematol. 2019, 12, 1037–1052. [Google Scholar] [CrossRef]
- Dokal, I.; Vulliamy, T.; Mason, P.; Bessler, M. Clinical utility gene card for: Dyskeratosis congenita—Update 2015. Eur. J. Hum. Genet. 2015, 23, 558. [Google Scholar] [CrossRef] [PubMed]
- Karremann, M.; Neumaier-Probst, E.; Schlichtenbrede, F.; Beier, F.; Brümmendorf, T.H.; Cremer, F.W.; Bader, P.; Dürken, M. Revesz syndrome revisited. Orphanet J. Rare Dis. 2020, 15, 299. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, S.; Usta, A.M.; Urganci, N.; Kalay, N.G.; Kose, G.; Ozmen, E. Coats plus syndrome: A rare cause of severe gastrointestinal tract bleeding in children—A case report. BMC Pediatr. 2022, 22, 119. [Google Scholar] [CrossRef]
- Roake, C.M.; Artandi, S.E. Regulation of human telomerase in homeostasis and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 384–397. [Google Scholar] [CrossRef]
- Lim, C.J.; Cech, T.R. Shaping human telomeres: From shelterin and CST complexes to telomeric chromatin organization. Nat. Rev. Mol. Cell Biol. 2021, 22, 283–298. [Google Scholar] [CrossRef]
- Benyelles, M.; Episkopou, H.; O’Donohue, M.-F.; Kermasson, L.; Frange, P.; Poulain, F.; Burcu Belen, F.; Polat, M.; Bole-Feysot, C.; Langa-Vives, F.; et al. Impaired telomere integrity and rRNA biogenesis in PARN-deficient patients and knock-out models. EMBO Mol. Med. 2019, 11, e10201. [Google Scholar] [CrossRef] [PubMed]
- Ghisays, F.; Garzia, A.; Wang, H.; Canasto-Chibuque, C.; Hohl, M.; Savage, S.A.; Tuschl, T.; Petrini, J.H.J. RTEL1 influences the abundance and localization of TERRA RNA. Nat. Commun. 2021, 12, 3016. [Google Scholar] [CrossRef]
- Bernadotte, A.; Mikhelson, V.M.; Spivak, I.M. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging 2016, 8, 3–11. [Google Scholar] [CrossRef]
- Westin, E.R.; Aykin-Burns, N.; Buckingham, E.M.; Spitz, D.R.; Goldman, F.D.; Klingelhutz, A.J.; Reeves, D.; Gu, B.; Mason, P.J.; Bessler, M.; et al. The p53/p21WAF/CIP Pathway Mediates Oxidative Stress and Senescence in Dyskeratosis Congenita Cells with Telomerase Insufficiency. Antioxid. Redox Signal. 2011, 14, 985–997. [Google Scholar] [CrossRef]
- Toufektchan, E.; Lejour, V.; Durand, R.; Giri, N.; Draskovic, I.; Bardot, B.; Laplante, P.; Jaber, S.; Alter, B.P.; Londono-Vallejo, J.-A.; et al. Germline mutation of MDM4, a major p53 regulator, in a familial syndrome of defective telomere maintenance. Sci. Adv. 2020, 6, eaay3511. [Google Scholar] [CrossRef] [PubMed]
- Jyonouchi, S.; Forbes, L.; Ruchelli, E.; Sullivan, K.E. Dyskeratosis congenita: A combined immunodeficiency with broad clinical spectrum—A single-center pediatric experience. Pediatr. Allergy Immunol. 2011, 22, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Sznajer, Y.; Baumann, C.; David, A.; Journel, H.; Lacombe, D.; Perel, Y.; Blouin, P.; Segura, J.-F.; Cezard, J.-P.; Peuchmaur, M.; et al. Further delineation of the congenital form of X-linked dyskeratosis congenita (Hoyeraal-Hreidarsson syndrome). Eur. J. Pediatr. 2003, 162, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Speckmann, C.; Sahoo, S.S.; Rizzi, M.; Hirabayashi, S.; Karow, A.; Serwas, N.K.; Hoemberg, M.; Damatova, N.; Schindler, D.; Vannier, J.-B.; et al. Clinical and Molecular Heterogeneity of RTEL1 Deficiency. Front. Immunol. 2017, 8, 449. [Google Scholar] [CrossRef]
- Chu, C.-M.; Yu, H.-H.; Kao, T.-L.; Chen, Y.-H.; Lu, H.-H.; Wu, E.-T.; Yang, Y.-L.; Lin, C.-H.; Lin, S.-Y.; Tsai, M.-J.M.; et al. A missense variant in the nuclear localization signal of DKC1 causes Hoyeraal-Hreidarsson syndrome. NPJ Genom. Med. 2022, 7, 64. [Google Scholar] [CrossRef]
- Da Costa, L.; Narla, A.; Mohandas, N. An update on the pathogenesis and diagnosis of Diamond-Blackfan anemia [version 1; peer review: 2 approved]. F1000Research 2018, 7, 1350. [Google Scholar] [CrossRef]
- Giri, N.; Kang, E.; Tisdale, J.F.; Follman, D.; Rivera, M.; Schwartz, G.N.; Kim, S.; Young, N.S.; Rick, M.; Dunbar, C. Clinical and laboratory evidence for a trilineage haematopoietic defect in patients with refractory Diamond–Blackfan anaemia. Br. J. Haematol. 2000, 108, 167–175. [Google Scholar] [CrossRef]
- Aspesi, A.; Ellis, S.R. Rare ribosomopathies: Insights into mechanisms of cancer. Nat. Rev. Cancer 2019, 19, 228–238. [Google Scholar] [CrossRef]
- Sieff, C. Diamond-Blackfan Anemia (GeneReviews). Available online: https://www.ncbi.nlm.nih.gov/books/NBK7047/ (accessed on 9 June 2023).
- Gripp, K.W.; Curry, C.; Olney, A.H.; Sandoval, C.; Fisher, J.; Chong, J.X.-L.; Genomics, U.C.f.M.; Pilchman, L.; Sahraoui, R.; Stabley, D.L.; et al. Diamond–Blackfan anemia with mandibulofacial dystostosis is heterogeneous, including the novel DBA genes TSR2 and RPS28. Am. J. Med. Genet. Part A 2014, 164, 2240–2249. [Google Scholar] [CrossRef]
- Khajuria, R.K.; Munschauer, M.; Ulirsch, J.C.; Fiorini, C.; Ludwig, L.S.; McFarland, S.K.; Abdulhay, N.J.; Specht, H.; Keshishian, H.; Mani, D.R.; et al. Ribosome Levels Selectively Regulate Translation and Lineage Commitment in Human Hematopoiesis. Cell 2018, 173, 90–103.e119. [Google Scholar] [CrossRef]
- Moniz, H.; Gastou, M.; Leblanc, T.; Hurtaud, C.; Crétien, A.; Lécluse, Y.; Raslova, H.; Larghero, J.; Croisille, L.; Faubladier, M.; et al. Primary hematopoietic cells from DBA patients with mutations in RPL11 and RPS19 genes exhibit distinct erythroid phenotype in vitro. Cell Death Dis. 2012, 3, e356. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Deisenroth, C.; Zhang, Y. RP–MDM2–p53 Pathway: Linking Ribosomal Biogenesis and Tumor Surveillance. Trends Cancer 2016, 2, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Trainor, C.D.; Mas, C.; Archambault, P.; Di Lello, P.; Omichinski, J.G. GATA-1 associates with and inhibits p53. Blood 2009, 114, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Heijnen, H.F.; van Wijk, R.; Pereboom, T.C.; Goos, Y.J.; Seinen, C.W.; van Oirschot, B.A.; van Dooren, R.; Gastou, M.; Giles, R.H.; van Solinge, W.; et al. Ribosomal Protein Mutations Induce Autophagy through S6 Kinase Inhibition of the Insulin Pathway. PLoS Genet. 2014, 10, e1004371. [Google Scholar] [CrossRef]
- Hasegawa, D.; Kojima, S.; Tatsumi, E.; Hayakawa, A.; Kosaka, Y.; Nakamura, H.; Sako, M.; Osugi, Y.; Nagata, S.; Sano, K. Elevation of the Serum Fas Ligand in Patients with Hemophagocytic Syndrome and Diamond-Blackfan Anemia. Blood 1998, 91, 2793–2799. [Google Scholar] [CrossRef]
- Macečková, Z.; Kubíčková, A.; De Sanctis, J.B.; Hajdúch, M. Effect of Glucocorticosteroids in Diamond-Blackfan Anaemia: Maybe Not as Elusive as It Seems. Int. J. Mol. Sci. 2022, 23, 1886. [Google Scholar] [CrossRef]
- Wang, B.; Wang, C.; Wan, Y.; Gao, J.; Ma, Y.; Zhang, Y.; Tong, J.; Zhang, Y.; Liu, J.; Chang, L.; et al. Decoding the pathogenesis of Diamond–Blackfan anemia using single-cell RNA-seq. Cell Discov. 2022, 8, 41. [Google Scholar] [CrossRef]
- Goobie, S.; Popovic, M.; Morrison, J.; Ellis, L.; Ginzberg, H.; Boocock, G.R.B.; Ehtesham, N.; Bétard, C.; Brewer, C.G.; Roslin, N.M.; et al. Shwachman-Diamond Syndrome with Exocrine Pancreatic Dysfunction and Bone Marrow Failure Maps to the Centromeric Region of Chromosome 7. Am. J. Hum. Genet. 2001, 68, 1048–1054. [Google Scholar] [CrossRef]
- Minelli, A.; Nicolis, E.; Cannioto, Z.; Longoni, D.; Perobelli, S.; Pasquali, F.; Sainati, L.; Poli, F.; Cipolli, M.; Danesino, C. Incidence of Shwachman–Diamond syndrome. Pediatr. Blood Cancer 2012, 59, 1334–1335. [Google Scholar] [CrossRef]
- Kawashima, N.; Oyarbide, U.; Cipolli, M.; Bezzerri, V.; Corey, S.J. Shwachman-Diamond syndromes: Clinical, genetic, and biochemical insights from the rare variants. Haematologica 2023. [Google Scholar] [CrossRef]
- Menne, T.F.; Goyenechea, B.; Sánchez-Puig, N.; Wong, C.C.; Tonkin, L.M.; Ancliff, P.J.; Brost, R.L.; Costanzo, M.; Boone, C.; Warren, A.J. The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat. Genet. 2007, 39, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Weis, F.; Giudice, E.; Churcher, M.; Jin, L.; Hilcenko, C.; Wong, C.C.; Traynor, D.; Kay, R.R.; Warren, A.J. Mechanism of eIF6 release from the nascent 60S ribosomal subunit. Nat. Struct. Mol. Biol. 2015, 22, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Jomaa, A.; Chung, S.; Hwang Fu, Y.-H.; Qian, R.; Sun, X.; Hsieh, H.-H.; Chandrasekar, S.; Bi, X.; Mattei, S.; et al. Receptor compaction and GTPase rearrangement drive SRP-mediated cotranslational protein translocation into the ER. Sci. Adv. 2021, 7, eabg0942. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, A.; Cannata, E.; Perbellini, O.; Cugno, C.; Balter, R.; Zaccaron, A.; Tridello, G.; Pizzolo, G.; De Bortoli, M.; Krampera, M.; et al. Immunophenotypic analysis of hematopoiesis in patients suffering from Shwachman–Bodian–Diamond Syndrome. Eur. J. Haematol. 2015, 95, 308–315. [Google Scholar] [CrossRef]
- Dror, Y.; Freedman, M.H. Shwachman-Diamond syndrome marrow cells show abnormally increased apoptosis mediated through the Fas pathway. Blood 2001, 97, 3011–3016. [Google Scholar] [CrossRef]
- Bezzerri, V.; Vella, A.; Gennaro, G.D.; Ortolani, R.; Nicolis, E.; Cesaro, S.; Fabrizzi, B.; Bronte, V.; Corey, S.J.; Cipolli, M. Peripheral blood immunophenotyping in a large cohort of patients with Shwachman–Diamond syndrome. Pediatr. Blood Cancer 2019, 66, e27597. [Google Scholar] [CrossRef]
- Jean, D.; Odile, F.; Blandine, B.; Sandrine, B.; Florence, B.; Nizar, M.; Anne, L.; Nathalie, A.; Yves, B.; Valérie, M.; et al. Classification of and risk factors for hematologic complications in a French national cohort of 102 patients with Shwachman-Diamond syndrome. Haematologica 2012, 97, 1312–1319. [Google Scholar] [CrossRef]
- Shah, N.; Cambrook, H.; Koglmeier, J.; Mason, C.; Ancliff, P.; Lindley, K.; Smith, V.V.; Bajaj-Elliott, M.; Sebire, N.J. Enteropathic histopathological features may be associated with Shwachman–Diamond syndrome. J. Clin. Pathol. 2010, 63, 592–594. [Google Scholar] [CrossRef]
- Ambekar, C.; Das, B.; Yeger, H.; Dror, Y. SBDS-deficiency results in deregulation of reactive oxygen species leading to increased cell death and decreased cell growth. Pediatr. Blood Cancer 2010, 55, 1138–1144. [Google Scholar] [CrossRef]
- Raaijmakers, M.H.G.P.; Mukherjee, S.; Guo, S.; Zhang, S.; Kobayashi, T.; Schoonmaker, J.A.; Ebert, B.L.; Al-Shahrour, F.; Hasserjian, R.P.; Scadden, E.O.; et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010, 464, 852–857. [Google Scholar] [CrossRef]
- Zambetti, N.A.; Ping, Z.; Chen, S.; Kenswil, K.J.; Mylona, M.A.; Sanders, M.A.; Hoogenboezem, R.M.; Bindels, E.M.; Adisty, M.N.; Van Strien, P.M.; et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-leukemia. Cell Stem Cell 2016, 19, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Ball, H.L.; Zhang, B.; Riches, J.J.; Gandhi, R.; Li, J.; Rommens, J.M.; Myers, J.S. Shwachman-Bodian Diamond syndrome is a multi-functional protein implicated in cellular stress responses. Hum. Mol. Genet. 2009, 18, 3684–3695. [Google Scholar] [CrossRef]
- Tourlakis, M.E.; Zhang, S.; Ball, H.L.; Gandhi, R.; Liu, H.; Zhong, J.; Yuan, J.S.; Guidos, C.J.; Durie, P.R.; Rommens, J.M. In Vivo Senescence in the Sbds-Deficient Murine Pancreas: Cell-Type Specific Consequences of Translation Insufficiency. PLoS Genet. 2015, 11, e1005288. [Google Scholar] [CrossRef]
- Joyce, C.E.; Saadatpour, A.; Ruiz-Gutierrez, M.; Bolukbasi, O.V.; Jiang, L.; Thomas, D.D.; Young, S.; Hofmann, I.; Sieff, C.A.; Myers, K.C.; et al. TGF-β signaling underlies hematopoietic dysfunction and bone marrow failure in Shwachman-Diamond syndrome. J. Clin. Investig. 2019, 129, 3821–3826. [Google Scholar] [CrossRef] [PubMed]
- Furutani, E.; Shah, A.S.; Zhao, Y.; Andorsky, D.; Dedeoglu, F.; Geddis, A.; Zhou, Y.; Libermann, T.A.; Myers, K.C.; Shimamura, A. Inflammatory manifestations in patients with Shwachman–Diamond syndrome: A novel phenotype. Am. J. Med. Genet. A 2020, 182, 1754–1760. [Google Scholar] [CrossRef]
- Bezzerri, V.; Vella, A.; Calcaterra, E.; Finotti, A.; Gasparello, J.; Gambari, R.; Assael, B.M.; Cipolli, M.; Sorio, C. New insights into the Shwachman-Diamond Syndrome-related haematological disorder: Hyper-activation of mTOR and STAT3 in leukocytes. Sci. Rep. 2016, 6, 33165. [Google Scholar] [CrossRef] [PubMed]
- Ravera, S.; Dufour, C.; Cesaro, S.; Bottega, R.; Faleschini, M.; Cuccarolo, P.; Corsolini, F.; Usai, C.; Columbaro, M.; Cipolli, M.; et al. Evaluation of energy metabolism and calcium homeostasis in cells affected by Shwachman-Diamond syndrome. Sci. Rep. 2016, 6, 25441. [Google Scholar] [CrossRef]
- Vella, A.; D’Aversa, E.; Api, M.; Breveglieri, G.; Allegri, M.; Giacomazzi, A.; Marinelli Busilacchi, E.; Fabrizzi, B.; Cestari, T.; Sorio, C.; et al. mTOR and STAT3 Pathway Hyper-Activation is Associated with Elevated Interleukin-6 Levels in Patients with Shwachman-Diamond Syndrome: Further Evidence of Lymphoid Lineage Impairment. Cancers 2020, 12, 597. [Google Scholar] [CrossRef]
- Skokowa, J.; Dale, D.C.; Touw, I.P.; Zeidler, C.; Welte, K. Severe congenital neutropenias. Nat. Rev. Dis. Primers 2017, 3, 17032. [Google Scholar] [CrossRef]
- Ye, Y.; Carlsson, G.; Wondimu, B.; Fahlén, A.; Karlsson-Sjöberg, J.; Andersson, M.; Engstrand, L.; Yucel-Lindberg, T.; Modéer, T.; Pütsep, K. Mutations in the ELANE Gene are Associated with Development of Periodontitis in Patients with Severe Congenital Neutropenia. J. Clin. Immunol. 2011, 31, 936–945. [Google Scholar] [CrossRef]
- Acar, B.; Cagdas, D.; Tan, C.; Özbek, B.; Yaz, I.; Yıldırım, Y.D.; Özşin-Özler, C.; Karaatmaca, B.; Gür-Çetinkaya, P.; Soyak, E.; et al. Evaluation of periodontal status and cytokine/chemokine profile of GCF in patients with severe congenital neutropenia. Odontology 2021, 109, 474–482. [Google Scholar] [CrossRef]
- Iolascon, A.; Andolfo, I.; Russo, R. Congenital dyserythropoietic anemias. Blood 2020, 136, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Tamary, H.; Dgany, O. Dyserythropoietic Anemia Type I (GeneReviews). Available online: https://www.ncbi.nlm.nih.gov/books/NBK5313/ (accessed on 9 June 2023).
- Lavabre-Bertrand, T.; Blanc, P.; Navarro, R.; Saghroun, M.; Vannereau, H.; Braun, M.; Wagner, A.; Taiub, J.; Lavabre-Bertrand, C.; Navarro, M. Alpha-interferon therapy for congenital dyserythropoiesis type I. Br. J. Haematol. 1995, 89, 929–932. [Google Scholar] [CrossRef] [PubMed]
- Marwaha, R.K.; Bansal, D.; Trehan, A.; Garewal, G. Interferon Therapy in Congenital Dyserythropoietic Anemia Type I/II. Pediatr. Hematol. Oncol. 2005, 22, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, S.N. Response of CDA type I to alpha-interferon. Eur. J. Haematol. 1997, 58, 121–123. [Google Scholar] [CrossRef]
- Agrigento, V.; Barone, R.; Sclafani, S.; Di Maggio, R.; Sacco, M.; Maggio, A.; D’Alcamo, E. Response to Alpha-Interferon Treatment of the Congenital Dyserythropoietic Anemia type I in Two Sicilian Beta Thalassemia Carriers. Indian J. Hematol. Blood Transfus. 2017, 33, 621–623. [Google Scholar] [CrossRef]
- Narumi, S.; Amano, N.; Ishii, T.; Katsumata, N.; Muroya, K.; Adachi, M.; Toyoshima, K.; Tanaka, Y.; Fukuzawa, R.; Miyako, K.; et al. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat. Genet. 2016, 48, 792–797. [Google Scholar] [CrossRef]
- Chen, D.-H.; Below, J.E.; Shimamura, A.; Keel, S.B.; Matsushita, M.; Wolff, J.; Sul, Y.; Bonkowski, E.; Castella, M.; Taniguchi, T.; et al. Ataxia-Pancytopenia Syndrome Is Caused by Missense Mutations in SAMD9L. Am. J. Hum. Genet. 2016, 98, 1146–1158. [Google Scholar] [CrossRef]
- Davidsson, J.; Puschmann, A.; Tedgård, U.; Bryder, D.; Nilsson, L.; Cammenga, J. SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia 2018, 32, 1106–1115. [Google Scholar] [CrossRef]
- Nagamachi, A.; Matsui, H.; Asou, H.; Ozaki, Y.; Aki, D.; Kanai, A.; Takubo, K.; Suda, T.; Nakamura, T.; Wolff, L.; et al. Haploinsufficiency of SAMD9L, an Endosome Fusion Facilitator, Causes Myeloid Malignancies in Mice Mimicking Human Diseases with Monosomy 7. Cancer Cell 2013, 24, 305–317. [Google Scholar] [CrossRef]
- Tesi, B.; Davidsson, J.; Voss, M.; Rahikkala, E.; Holmes, T.D.; Chiang, S.C.C.; Komulainen-Ebrahim, J.; Gorcenco, S.; Rundberg Nilsson, A.; Ripperger, T.; et al. Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood 2017, 129, 2266–2279. [Google Scholar] [CrossRef] [PubMed]
- Nagamachi, A.; Kanai, A.; Nakamura, M.; Okuda, H.; Yokoyama, A.; Shinriki, S.; Matsui, H.; Inaba, T. Multiorgan failure with abnormal receptor metabolism in mice mimicking Samd9/9L syndromes. J. Clin. Investig. 2021, 131, e140147. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamed, S.; Thomas, M.E., III; Westover, T.; Umeda, M.; Xiong, E.; Rolle, C.; Walsh, M.P.; Wu, H.; Schwartz, J.R.; Valentine, V.; et al. Mutant Samd9l expression impairs hematopoiesis and induces bone marrow failure in mice. J. Clin. Investig. 2022, 132, e158869. [Google Scholar] [CrossRef] [PubMed]
- Jung, M. Inflammation fuels bone marrow exhaustion caused by Samd9l mutation. J. Clin. Investig. 2022, 132, e164136. [Google Scholar] [CrossRef] [PubMed]
- Park, M. Overview of inherited bone marrow failure syndromes. Blood Res. 2022, 57, S49–S54. [Google Scholar] [CrossRef]
- Wu, Z.; Gao, S.; Diamond, C.; Kajigaya, S.; Chen, J.; Shi, R.; Palmer, C.; Hsu, A.P.; Calvo, K.R.; Hickstein, D.D.; et al. Sequencing of RNA in single cells reveals a distinct transcriptome signature of hematopoiesis in GATA2 deficiency. Blood Adv. 2020, 4, 2702–2716. [Google Scholar] [CrossRef] [PubMed]
- Calvo, K.R.; Hickstein, D.D. The spectrum of GATA2 deficiency syndrome. Blood 2023, 141, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, R.E.; Milne, P.; Jardine, L.; Zandi, S.; Swierczek, S.I.; McGovern, N.; Cookson, S.; Ferozepurwalla, Z.; Langridge, A.; Pagan, S.; et al. The evolution of cellular deficiency in GATA2 mutation. Blood 2014, 123, 863–874. [Google Scholar] [CrossRef]
- Germeshausen, M.; Ancliff, P.; Estrada, J.; Metzler, M.; Ponstingl, E.; Rütschle, H.; Schwabe, D.; Scott, R.H.; Unal, S.; Wawer, A.; et al. MECOM-associated syndrome: A heterogeneous inherited bone marrow failure syndrome with amegakaryocytic thrombocytopenia. Blood Adv. 2018, 2, 586–596. [Google Scholar] [CrossRef]
- Voit, R.A.; Tao, L.; Yu, F.; Cato, L.D.; Cohen, B.; Fleming, T.J.; Antoszewski, M.; Liao, X.; Fiorini, C.; Nandakumar, S.K.; et al. A genetic disorder reveals a hematopoietic stem cell regulatory network co-opted in leukemia. Nat. Immunol. 2023, 24, 69–83. [Google Scholar] [CrossRef]
- Kurokawa, M.; Mitani, K.; Irie, K.; Matsuyama, T.; Takahashi, T.; Chiba, S.; Yazaki, Y.; Matsumoto, K.; Hirai, H. The oncoprotein Evi-1 represses TGF-beta signalling by inhibiting Smad3. Nature 1998, 394, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, M.; Mitani, K.; Yamagata, T.; Takahashi, T.; Izutsu, K.; Ogawa, S.; Moriguchi, T.; Nishida, E.; Yazaki, Y.; Hirai, H. The evi-1 oncoprotein inhibits c-Jun N-terminal kinase and prevents stress-induced cell death. EMBO J. 2000, 19, 2958–2968. [Google Scholar] [CrossRef] [PubMed]
- Biswas, L.; Chen, J.; De Angelis, J.; Singh, A.; Owen-Woods, C.; Ding, Z.; Pujol, J.M.; Kumar, N.; Zeng, F.; Ramasamy, S.K.; et al. Lymphatic vessels in bone support regeneration after injury. Cell 2023, 186, 382–397.e324. [Google Scholar] [CrossRef]
- Rafii, S.; Shapiro, F.; Pettengell, R.; Ferris, B.; Nachman, R.L.; Moore, M.A.; Asch, A.S. Human bone marrow microvascular endothelial cells support long-term proliferation and differentiation of myeloid and megakaryocytic progenitors. Blood 1995, 86, 3353–3363. [Google Scholar] [CrossRef]
- Sieff, C.A.; Tsai, S.; Faller, D.V. Interleukin 1 induces cultured human endothelial cell production of granulocyte-macrophage colony-stimulating factor. J. Clin. Investig. 1987, 79, 48–51. [Google Scholar] [CrossRef]
- Broudy, V.C.; Kaushansky, K.; Segal, G.M.; Harlan, J.M.; Adamson, J.W. Tumor necrosis factor type alpha stimulates human endothelial cells to produce granulocyte/macrophage colony-stimulating factor. Proc. Natl. Acad. Sci. USA 1986, 83, 7467–7471. [Google Scholar] [CrossRef]
- Fernandez, L.; Rodriguez, S.; Huang, H.; Chora, A.; Fernandes, J.; Mumaw, C.; Cruz, E.; Pollok, K.; Cristina, F.; Price, J.E.; et al. Tumor necrosis factor-alpha and endothelial cells modulate Notch signaling in the bone marrow microenvironment during inflammation. Exp. Hematol. 2008, 36, 545–558. [Google Scholar] [CrossRef]
- Bardelli, D.; Dander, E.; Bugarin, C.; Cappuzzello, C.; Pievani, A.; Fazio, G.; Pierani, P.; Corti, P.; Farruggia, P.; Dufour, C.; et al. Mesenchymal stromal cells from Shwachman-Diamond syndrome patients fail to recreate a bone marrow niche in vivo and exhibit impaired angiogenesis. Br. J. Haematol. 2018, 182, 114–124. [Google Scholar] [CrossRef]
- Leung, E.W.; Rujkijyanont, P.; Beyene, J.; Wei, K.; Abdelhaleem, M.; Freedman, M.H.; Dror, Y. Shwachman-Diamond syndrome: An inherited model of aplastic anaemia with accelerated angiogenesis. Br. J. Haematol. 2006, 133, 558–561. [Google Scholar] [CrossRef]
- Fureder, W.; Krauth, M.T.; Sperr, W.R.; Sonneck, K.; Simonitsch-Klupp, I.; Mullauer, L.; Willmann, M.; Horny, H.P.; Valent, P. Evaluation of angiogenesis and vascular endothelial growth factor expression in the bone marrow of patients with aplastic anemia. Am. J. Pathol. 2006, 168, 123–130. [Google Scholar] [CrossRef] [PubMed]


| FA | DC | DBA | SDS | SCN | |
|---|---|---|---|---|---|
| Estimated incidence (/1,000,000 births/year) * | 11.4 | 3.8 | 10.4 | 8.5 | 4.7 |
| Inheritance pattern | AR > AD, XLR | XLR > AR | AR > AD | AR | AD |
| Typical hematological findings | Pancytopenia | Pancytopenia | Anemia | Neutropenia | Neutropenia |
| Extrahematological symptoms | Growth deficiency, skeletal anomalies (radial axis), skin pigmentation, small head/eyes, genitourinary anomaly, reproductive problems | Growth deficiency, abnormal nails, leukoplakia, reticular pigmentation, pulmonary fibrosis, grey hair, cerebellar hypoplasia | Growth deficiency, head/facial anomaly, skeletal anomaly (thumb), kidney/heart anomaly | Growth deficiency, skeletal anomaly (metaphyseal dysostosis), exocrine pancreatic insufficiency | Not common |
| Short telomere | + | ++ | + | + | − |
| MDS/AML predisposition | + | + | + | + | + |
| Solid cancer predisposition | Squamous cell carcinoma (head/neck, genitourinary) | Squamous cell carcinoma (head/neck, skin) | Osteosarcoma | Not common | Not common |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawashima, N.; Bezzerri, V.; Corey, S.J. The Molecular and Genetic Mechanisms of Inherited Bone Marrow Failure Syndromes: The Role of Inflammatory Cytokines in Their Pathogenesis. Biomolecules 2023, 13, 1249. https://doi.org/10.3390/biom13081249
Kawashima N, Bezzerri V, Corey SJ. The Molecular and Genetic Mechanisms of Inherited Bone Marrow Failure Syndromes: The Role of Inflammatory Cytokines in Their Pathogenesis. Biomolecules. 2023; 13(8):1249. https://doi.org/10.3390/biom13081249
Chicago/Turabian StyleKawashima, Nozomu, Valentino Bezzerri, and Seth J. Corey. 2023. "The Molecular and Genetic Mechanisms of Inherited Bone Marrow Failure Syndromes: The Role of Inflammatory Cytokines in Their Pathogenesis" Biomolecules 13, no. 8: 1249. https://doi.org/10.3390/biom13081249