
Endometriosis-Associated Ovarian Carcinomas: How PI3K/AKT/mTOR Pathway Affects Their Pathogenesis


Abstract
:1. Introduction
2. Outline of mTOR Signaling and Its Function in Normal Endometrium
3. How mTOR Signaling Affects Endometriosis Development
4. The role of mTOR Signaling in Endometriosis-Associated Ovarian Carcinomas (EAOCs)
4.1. PI3K/AKT/mTOR Pathway Alterations in EAOC
4.2. The Role of ARID1A Gene Expression in EAOC
4.3. Synergistic Crosstalk between ARID1A and PI3K/AKT/mTOR Pathway in EAOC
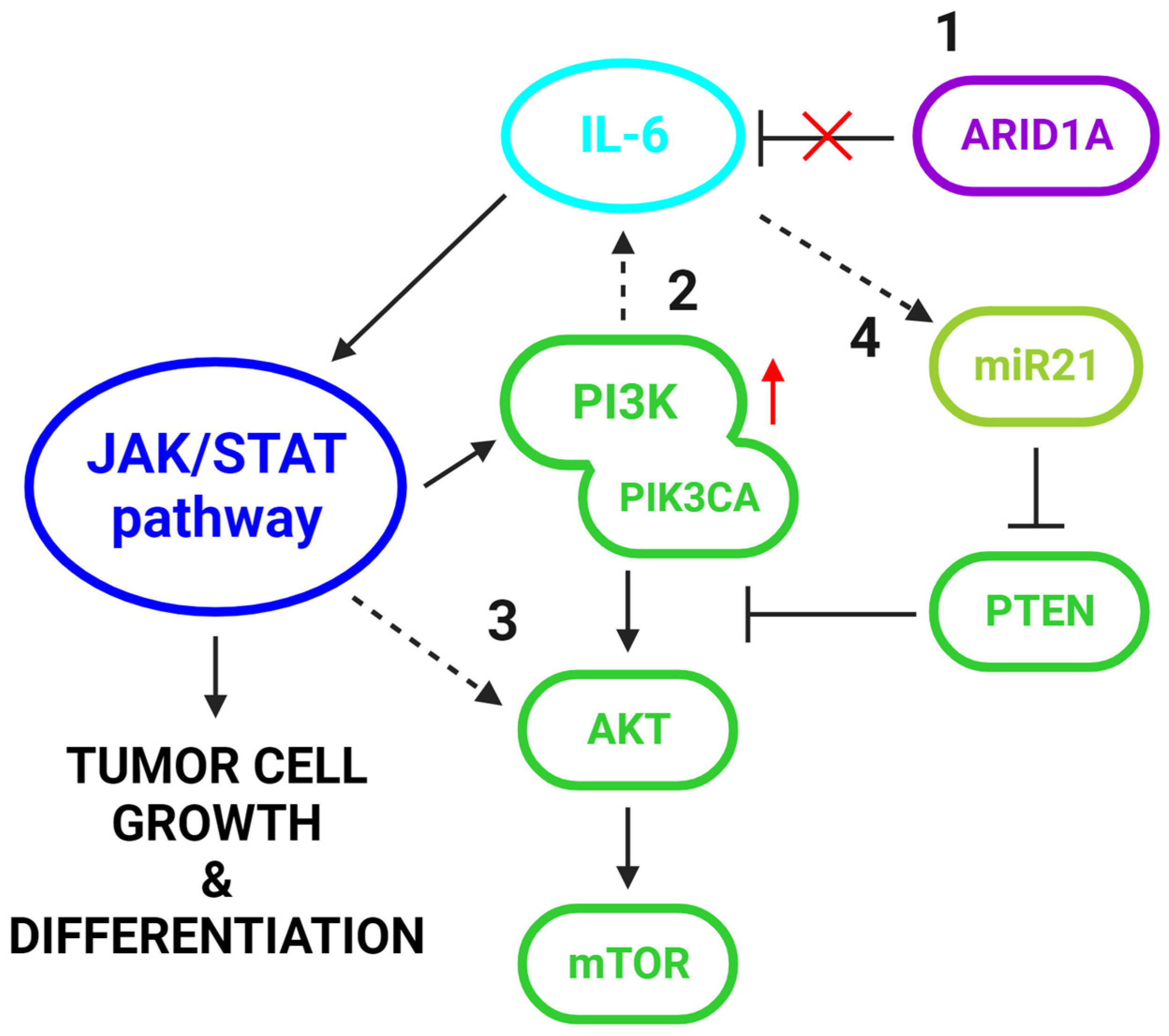
4.4. The Interplay between IL-6 and the PI3K/AKT/mTOR Pathway in EAOC
4.5. Targeting PI3K/AKT/mTOR Pathway in EAOC Treatment
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Giudice, L.C. Endometriosis. N. Engl. J. Med. 2010, 362, 2389–2398. [Google Scholar] [CrossRef]
- Králíčková, M.; Laganà, A.S.; Ghezzi, F.; Vetvicka, V. Endometriosis and Risk of Ovarian Cancer: What Do We Know? Arch. Gynecol. Obstet. 2020, 301, 1–10. [Google Scholar] [CrossRef]
- Anglesio, M.S.; Yong, P.J. Endometriosis-Associated Ovarian Cancers. Clin. Obstet. Gynecol. 2017, 60, 711–727. [Google Scholar] [CrossRef]
- Vercellini, P.; Viganò, P.; Somigliana, E.; Fedele, L. Endometriosis: Pathogenesis and Treatment. Nat. Rev. Endocrinol. 2014, 10, 261–275. [Google Scholar] [CrossRef]
- Wang, C.; Liang, Z.; Liu, X.; Zhang, Q.; Li, S. The Association between Endometriosis, Tubal Ligation, Hysterectomy and Epithelial Ovarian Cancer: Meta-Analyses. Int. J. Environ. Res. Public. Health 2016, 13, 1138. [Google Scholar] [CrossRef]
- Pearce, C.L.; Templeman, C.; Rossing, M.A.; Lee, A.; Near, A.M.; Webb, P.M.; Nagle, C.M.; Doherty, J.A.; Cushing-Haugen, K.L.; Wicklund, K.G.; et al. Association between Endometriosis and Risk of Histological Subtypes of Ovarian Cancer: A Pooled Analysis of Case–Control Studies. Lancet Oncol. 2012, 13, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Rogers-Broadway, K.R.; Kumar, J.; Sisu, C.; Wander, G.; Mazey, E.; Jeyaneethi, J.; Pados, G.; Tsolakidis, D.; Klonos, E.; Grunt, T.; et al. Differential Expression of MTOR Components in Endometriosis and Ovarian Cancer: Effects of Rapalogues and Dual Kinase Inhibitors on MTORC1 and MTORC2 Stoichiometry. Int. J. Mol. Med. 2019, 43, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Dazert, E.; Hall, M.N. MTOR Signaling in Disease. Curr. Opin. Cell Biol. 2011, 23, 744–755. [Google Scholar] [CrossRef]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Role of the PI3K/AKT/MTOR Signaling Pathway in Ovarian Cancer: Biological and Therapeutic Significance. Semin. Cancer Biol. 2019, 59, 147–160. [Google Scholar] [CrossRef]
- Driva, T.S.; Schatz, C.; Sobočan, M.; Haybaeck, J. The Role of MTOR and EIF Signaling in Benign Endometrial Diseases. Int. J. Mol. Sci. 2022, 23, 3416. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Müller, F.; Avruch, J. TOR Action in Mammalian Cells and in Caenorhabditis Elegans. Curr. Top. Microbiol. Immunol. 2004, 279, 115–138. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Luna, J.I.; Marusina, A.I.; Merleev, A.; Kundu-Raychaudhuri, S.; Fiorentino, D.; Raychaudhuri, S.P.; Maverakis, E. Dual MTOR Inhibition Is Required to Prevent TGF-β-Mediated Fibrosis: Implications for Scleroderma. J. Investig. Dermatol. 2015, 135, 2873. [Google Scholar] [CrossRef] [PubMed]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR Complexes, Only One of Which Is Rapamycin Sensitive, Have Distinct Roles in Cell Growth Control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Caron, E.; Ghosh, S.; Matsuoka, Y.; Ashton-Beaucage, D.; Therrien, M.; Lemieux, S.; Perreault, C.; Roux, P.P.; Kitano, H. A Comprehensive Map of the MTOR Signaling Network. Mol. Syst. Biol. 2010, 6, 453. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Huang, J.; Manning, B.D. The TSC1-TSC2 Complex: A Molecular Switchboard Controlling Cell Growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [PubMed]
- Ma, X.M.; Blenis, J. Molecular Mechanisms of MTOR-Mediated Translational Control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.I.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The Mammalian Target of Rapamycin (MTOR) Partner, Raptor, Binds the MTOR Substrates P70 S6 Kinase and 4E-BP1 through Their TOR Signaling (TOS) Motif. J. Biol. Chem. 2003, 278, 15461–15464. [Google Scholar] [CrossRef]
- Hwa Jung, C.; Bong Jun, C.; Ro, S.-H.; Kim, Y.-M.; Michael Otto, N.; Cao, J.; Kundu, M.; Kim, D.-H.; Schmid, S.L. ULK-Atg13-FIP200 Complexes Mediate MTOR Signaling to the Autophagy Machinery. Mol. Biol. Cell 1992, 20, 1992–2003. [Google Scholar] [CrossRef]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR Complex 2 Controls the Actin Cytoskeleton and Is Rapamycin Insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Gan, X.; Wang, J.; Wang, C.; Sommer, E.; Kozasa, T.; Srinivasula, S.; Alessi, D.; Offermanns, S.; Simon, M.I.; Wu, D. PRR5L Degradation Promotes MTORC2-Mediated PKC-δ Phosphorylation and Cell Migration Downstream of Gα 12. Nat. Cell Biol. 2012, 14, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-Induced Insulin Resistance Is Mediated by MTORC2 Loss and Uncoupled from Longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Yu, Q. Role of MTOR Signaling in Female Reproduction. Front. Endocrinol. 2019, 10, 692. [Google Scholar] [CrossRef]
- Devis-Jauregui, L.; Eritja, N.; Davis, M.L.; Matias-Guiu, X.; Llobet-Navàs, D. Autophagy in the Physiological Endometrium and Cancer. Autophagy 2021, 17, 1077–1095. [Google Scholar] [CrossRef]
- Choi, J.; Jo, M.; Lee, E.; Kim, H.J.; Choi, D. Differential Induction of Autophagy by MTOR Is Associated with Abnormal Apoptosis in Ovarian Endometriotic Cysts. Mol. Hum. Reprod. 2014, 20, 309–317. [Google Scholar] [CrossRef]
- Choi, J.; Jo, M.; Lee, E.; Oh, Y.K.; Choi, D. The Role of Autophagy in Human Endometrium. Biol. Reprod. 2012, 86, 70. [Google Scholar] [CrossRef] [PubMed]
- Gellersen, B.; Brosens, J.J. Cyclic Decidualization of the Human Endometrium in Reproductive Health and Failure. Endocr. Rev. 2014, 35, 851–905. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fu, L.-J.; Liu, X.-Q.; Hu, Z.-Y.; Jiang, Y.; Gao, R.-F.; Feng, Q.; Lan, X.; Geng, Y.-Q.; Chen, X.-M.; et al. Nm23 Regulates Decidualization through the PI3K-Akt-MTOR Signaling Pathways in Mice and Humans. Hum. Reprod. 2016, 31, 2339–2351. [Google Scholar] [CrossRef]
- Suda, K.; Nakaoka, H.; Yoshihara, K.; Ishiguro, T.; Tamura, R.; Mori, Y.; Yamawaki, K.; Adachi, S.; Takahashi, T.; Kase, H.; et al. Clonal Expansion and Diversification of Cancer-Associated Mutations in Endometriosis and Normal Endometrium. Cell Rep. 2018, 24, 1777–1789. [Google Scholar] [CrossRef]
- Lac, V.; Nazeran, T.M.; Tessier-Cloutier, B.; Aguirre-Hernandez, R.; Albert, A.; Lum, A.; Khattra, J.; Praetorius, T.; Mason, M.; Chiu, D.; et al. Oncogenic Mutations in Histologically Normal Endometrium: The New Normal? J. Pathol. 2019, 249, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.; Leongamornlert, D.; Coorens, T.H.H.; Sanders, M.A.; Ellis, P.; Dentro, S.C.; Dawson, K.J.; Butler, T.; Rahbari, R.; Mitchell, T.J.; et al. The Mutational Landscape of Normal Human Endometrial Epithelium. Nature 2020, 580, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Kyo, S.; Sato, S.; Nakayama, K. Cancer-Associated Mutations in Normal Human Endometrium: Surprise or Expected? Cancer Sci. 2020, 111, 3458–3467. [Google Scholar] [CrossRef]
- Lac, V.; Verhoef, L.; Aguirre-Hernandez, R.; Nazeran, T.M.; Tessier-Cloutier, B.; Praetorius, T.; Orr, N.L.; Noga, H.; Lum, A.; Khattra, J.; et al. Iatrogenic Endometriosis Harbors Somatic Cancer-Driver Mutations. Hum. Reprod. 2019, 34, 69–78. [Google Scholar] [CrossRef]
- Yachida, N.; Yoshihara, K.; Yamaguchi, M.; Suda, K.; Tamura, R.; Enomoto, T. How Does Endometriosis Lead to Ovarian Cancer? The Molecular Mechanism of Endometriosis-Associated Ovarian Cancer Development. Cancers 2021, 13, 1439. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Pavone, M.E.; Lu, Z.; Wei, J.J.; Kim, J.J. Increased Activation of the PI3K/AKT Pathway Compromises Decidualization of Stromal Cells from Endometriosis. J. Clin. Endocrinol. Metab. 2012, 97, E35–E43. [Google Scholar] [CrossRef]
- Makker, A.; Goel, M.M.; Das, V.; Agarwal, A. PI3K-Akt-MTOR and MAPK Signaling Pathways in Polycystic Ovarian Syndrome, Uterine Leiomyomas and Endometriosis: An Update. Gynecol. Endocrinol. 2012, 28, 175–181. [Google Scholar] [CrossRef]
- Cinar, O.; Seval, Y.; Uz, Y.H.; Cakmak, H.; Ulukus, M.; Kayisli, U.A.; Arici, A. Differential Regulation of Akt Phosphorylation in Endometriosis. Reprod. Biomed. Online 2009, 19, 864–871. [Google Scholar] [CrossRef]
- Laudanski, P.; Szamatowicz, J.; Kowalczuk, O.; Kuźmicki, M.; Grabowicz, M.; Chyczewski, L. Expression of Selected Tumor Suppressor and Oncogenes in Endometrium of Women with Endometriosis. Hum. Reprod. 2009, 24, 1880–1890. [Google Scholar] [CrossRef]
- Honda, H.; Barrueto, F.F.; Gogusev, J.; Im, D.D.; Morin, P.J. Serial Analysis of Gene Expression Reveals Differential Expression between Endometriosis and Normal Endometrium. Possible Roles for AXL and SHC1 in the Pathogenesis of Endometriosis. Reprod. Biol. Endocrinol. 2008, 6, 59. [Google Scholar] [CrossRef]
- Zhang, H.; Li, M.; Zheng, X.; Sun, Y.; Wen, Z.; Zhao, X. Endometriotic Stromal Cells Lose the Ability to Regulate Cell-Survival Signaling in Endometrial Epithelial Cells In Vitro. Mol. Hum. Reprod. 2009, 15, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Madanes, D.; Bilotas, M.A.; Bastón, J.I.; Singla, J.J.; Meresman, G.F.; Barañao, R.I.; Ricci, A.G. PI3K/AKT Pathway Is Altered in the Endometriosis Patient’s Endometrium and Presents Differences According to Severity Stage. Gynecol. Endocrinol. 2020, 36, 436–440. [Google Scholar] [CrossRef]
- Kim, T.H.; Yu, Y.; Luo, L.; Lydon, J.P.; Jeong, J.W.; Kim, J.J. Activated AKT Pathway Promotes Establishment of Endometriosis. Endocrinology 2014, 155, 1921–1930. [Google Scholar] [CrossRef]
- Anglesio, M.S.; Papadopoulos, N.; Ayhan, A.; Nazeran, T.M.; Noë, M.; Horlings, H.M.; Lum, A.; Jones, S.; Senz, J.; Seckin, T.; et al. Cancer-Associated Mutations in Endometriosis without Cancer. N. Engl. J. Med. 2017, 376, 1835–1848. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, B.; Mueller, M.; Montgomery, G. Progesterone Resistance in Endometriosis: An Acquired Property? Trends Endocrinol. Metab. 2018, 29, 535–548. [Google Scholar] [CrossRef]
- Marquardt, R.M.; Kim, T.H.; Shin, J.H.; Jeong, J.W. Progesterone and Estrogen Signaling in the Endometrium: What Goes Wrong in Endometriosis? Int. J. Mol. Sci. 2019, 20, 3822. [Google Scholar] [CrossRef]
- Li, M.; Peng, J.; Shi, Y.; Sun, P. MiR-92a Promotes Progesterone Resistance in Endometriosis through PTEN/AKT Pathway. Life Sci. 2020, 242, 117190. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Han, B.; Zhang, Y.; Su, K.; Wang, C.; Hai, P.; Bian, A.; Guo, R. Effect of miR-194-5p regulating STAT1/mTOR signaling pathway on the biological characteristics of ectopic endometrial cells from mice. Am. J. Transl. Res. 2020, 2, 6136–6148. [Google Scholar]
- Zhou, X.; Chen, Z.; Pei, L.; Sun, J. MicroRNA MiR-106a-5p Targets Forkhead Box Transcription Factor FOXC1 to Suppress the Cell Proliferation, Migration, and Invasion of Ectopic Endometrial Stromal Cells via the PI3K/Akt/MTOR Signaling Pathway. Bioengineered 2021, 12, 2203–2213. [Google Scholar] [CrossRef]
- Choi, J.; Jo, M.; Lee, E.; Lee, D.Y.; Choi, D. Dienogest Enhances Autophagy Induction in Endometriotic Cells by Impairing Activation of AKT, ERK1/2, and MTOR. Fertil. Steril. 2015, 104, 655–664.e1. [Google Scholar] [CrossRef]
- Leconte, M.; Nicco, C.; Ng, C.; Chéreau, C.; Chouzenoux, S.; Marut, W.; Guibourdenche, J.; Arkwright, S.; Weill, B.; Chapron, C.; et al. The MTOR/AKT Inhibitor Temsirolimus Prevents Deep Infiltrating Endometriosis in Mice. Am. J. Pathol. 2011, 179, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Wang, Y.; Xu, G.; Dai, L. Effect of Rapamycin on Endometriosis in Mice. Exp. Ther. Med. 2016, 12, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Ye, Q.; Zhuang, M.; Xie, S.; Zhong, R.; Cui, J.; Zhou, J.; Zhu, Y.; Zhang, T.; Cao, L. Ginsenoside Rg3 Inhibits Angiogenesis in a Rat Model of Endometriosis through the VEGFR-2-Mediated PI3K/Akt/MTOR Signaling Pathway. PLoS ONE 2017, 12, e0186520. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, J.B.; Kjær, S.K.; Mellemkjær, L.; Jensen, A. Endometriosis and Risks for Ovarian, Endometrial and Breast Cancers: A Nationwide Cohort Study. Gynecol. Oncol. 2016, 143, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Murakami, R.; Matsumura, N.; Brown, J.B.; Higasa, K.; Tsutsumi, T.; Kamada, M.; Abou-Taleb, H.; Hosoe, Y.; Kitamura, S.; Yamaguchi, K.; et al. Exome Sequencing Landscape Analysis in Ovarian Clear Cell Carcinoma Shed Light on Key Chromosomal Regions and Mutation Gene Networks. Am. J. Pathol. 2017, 187, 2246–2258. [Google Scholar] [CrossRef]
- Anglesio, M.S.; Bashashati, A.; Wang, Y.K.; Senz, J.; Ha, G.; Yang, W.; Aniba, M.R.; Prentice, L.M.; Farahani, H.; Li Chang, H.; et al. Multifocal Endometriotic Lesions Associated with Cancer Are Clonal and Carry a High Mutation Burden. J. Pathol. 2015, 236, 201–209. [Google Scholar] [CrossRef]
- Worley, M.; Welch, W.; Berkowitz, R.; Ng, S.-W. Endometriosis-Associated Ovarian Cancer: A Review of Pathogenesis. Int. J. Mol. Sci. 2013, 14, 5367–5379. [Google Scholar] [CrossRef]
- Watanabe, T.; Nanamiya, H.; Endo, Y.; Kojima, M.; Nomura, S.; Furukawa, S.; Soeda, S.; Tamura, H.; Ryufuku, M.; Tanaka, D.; et al. Identification and Clinical Significance of Somatic Oncogenic Mutations in Epithelial Ovarian Cancer. J. Ovarian Res. 2021, 14, 129. [Google Scholar] [CrossRef]
- Cybulska, P.; Paula, A.D.C.; Tseng, J.; Leitao, M.M., Jr.; Bashashati, A.; Huntsman, D.G.; Nazeran, T.M.; Aghajanian, C.; Abu-Rustum, N.R.; DeLair, D.F.; et al. Molecular Profiling and Molecular Classification of Endometrioid Ovarian Carcinomas. Gynecol. Oncol. 2019, 154, 516–523. [Google Scholar] [CrossRef]
- Hollis, R.L.; Thomson, J.P.; Stanley, B.; Churchman, M.; Meynert, A.M.; Rye, T.; Bartos, C.; Iida, Y.; Croy, I.; Mackean, M.; et al. Molecular Stratification of Endometrioid Ovarian Carcinoma Predicts Clinical Outcome. Nat. Commun. 2020, 11, 4995. [Google Scholar] [CrossRef]
- Pierson, W.E.; Peters, P.N.; Chang, M.T.; Chen, L.; Quigley, D.A.; Ashworth, A.; Chapman, J.S. An Integrated Molecular Profile of Endometrioid Ovarian Cancer. Gynecol. Oncol. 2020, 157, 55–61. [Google Scholar] [CrossRef]
- Samartzis, E.P.; Noske, A.; Dedes, K.J.; Fink, D.; Imesch, P. ARID1A Mutations and PI3K/AKT Pathway Alterations in Endometriosis and Endometriosis-Associated Ovarian Carcinomas. Int. J. Mol. Sci. 2013, 14, 18824–18849. [Google Scholar] [CrossRef]
- Pavlidou, E.N.; Balis, V. Diagnostic Significance and Prognostic Role of the ARID1A Gene in Cancer Outcomes (Review). World Acad. Sci. J. 2020, 2, 49–64. [Google Scholar] [CrossRef]
- Wu, R.-C.; Wang, T.-L.; Shih, I.-M. The Emerging Roles of ARID1A in Tumor Suppression. Cancer Biol. Ther. 2014, 15, 655–664. [Google Scholar] [CrossRef]
- Samartzis, E.P.; Labidi-Galy, S.I.; Moschetta, M.; Uccello, M.; Kalaitzopoulos, D.R.; Perez-Fidalgo, J.A.; Boussios, S. Endometriosis-Associated Ovarian Carcinomas: Insights into Pathogenesis, Diagnostics, and Therapeutic Targets—A Narrative Review. Ann. Transl. Med. 2020, 8, 1712. [Google Scholar] [CrossRef]
- Stamp, J.P.; Gilks, C.B.; Wesseling, M.; Eshragh, S.; Ceballos, K.; Anglesio, M.S.; Kwon, J.S.; Tone, A.; Huntsman, D.G.; Carey, M.S. BAF250a Expression in Atypical Endometriosis and Endometriosis-Associated Ovarian Cancer. Int. J. Gynecol. Cancer 2016, 26, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Zhou, J.Y.; Guo, J.B.; Wang, L.Q.; Luo, Y.; Zhang, Z.Y.; Liu, F.Y.; Tan, J.; Wang, F.; Huang, O.P. The Presence of KRAS, PPP2R1A and ARID1A Mutations in 101 Chinese Samples with Ovarian Endometriosis. Mutat. Res.-Fundam. Mol. Mech. Mutagen. 2018, 809, 1–5. [Google Scholar] [CrossRef]
- Xie, H.; Chen, P.; Huang, H.-W.; Liu, L.-P.; Zhao, F. Reactive Oxygen Species Downregulate ARID1A Expression via Its Promoter Methylation during the Pathogenesis of Endometriosis. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4509–4515. [Google Scholar]
- Xiao, W.; Awadallah, A.; Xin, W. Loss of ARID1A/BAF250a expression in ovarian endometriosis and clear cell carcinoma. Int. J. Clin. Exp. Pathol. 2012, 5, 642–650. [Google Scholar] [PubMed]
- Yamamoto, S.; Tsuda, H.; Takano, M.; Tamai, S.; Matsubara, O. Loss of ARID1A Protein Expression Occurs as an Early Event in Ovarian Clear-Cell Carcinoma Development and Frequently Coexists with PIK3CA Mutations. Mod. Pathol. 2012, 25, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Worley, M.J.; Liu, S.; Hua, Y.; Kwok, J.S.L.; Samuel, A.; Hou, L.; Shoni, M.; Lu, S.; Sandberg, E.M.; Keryan, A.; et al. Molecular Changes in Endometriosis-Associated Ovarian Clear Cell Carcinoma. Eur. J. Cancer 2015, 51, 1831–1842. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A Mutations in Endometriosis-Associated Ovarian Carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Ayhan, A.; Mao, T.L.; Seckin, T.; Wu, C.H.; Guan, B.; Ogawa, H.; Futagami, M.; Mizukami, H.; Yokoyama, Y.; Kurman, R.J.; et al. Loss of ARID1A Expression Is an Early Molecular Event in Tumor Progression from Ovarian Endometriotic Cyst to Clear Cell and Endometrioid Carcinoma. Int. J. Gynecol. Cancer 2012, 22, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Yachida, N.; Yoshihara, K.; Suda, K.; Nakaoka, H.; Ueda, H.; Sugino, K.; Yamaguchi, M.; Mori, Y.; Yamawaki, K.; Tamura, R.; et al. ARID1A Protein Expression Is Retained in Ovarian Endometriosis with ARID1A Loss-of-Function Mutations: Implication for the Two-Hit Hypothesis. Sci. Rep. 2020, 10, 14260. [Google Scholar] [CrossRef]
- Chandler, R.L.; Damrauer, J.S.; Raab, J.R.; Schisler, J.C.; Wilkerson, M.D.; Didion, J.P.; Starmer, J.; Serber, D.; Yee, D.; Xiong, J.; et al. Coexistent ARID1A-PIK3CA Mutations Promote Ovarian Clear-Cell Tumorigenesis through pro-Tumorigenic Inflammatory Cytokine Signalling. Nat. Commun. 2015, 6, 6118. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.N.; Lin, M.C.; Huang, W.C.; Chiang, Y.C.; Kuo, K.T. Loss of ARID1A Expression and Its Relationship with PI3K-Akt Pathway Alterations and ZNF217 Amplification in Ovarian Clear Cell Carcinoma. Mod. Pathol. 2014, 27, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, K.C.; Hennessy, B.T.; Leung, S.; Wang, Y.; Ju, Z.; McGahren, M.; Kalloger, S.E.; Finlayson, S.; Stemke-Hale, K.; Lu, Y.; et al. A Functional Proteogenomic Analysis of Endometrioid and Clear Cell Carcinomas Using Reverse Phase Protein Array and Mutation Analysis: Protein Expression Is Histotype-Specific and Loss of ARID1A/BAF250a Is Associated with AKT Phosphorylation. BMC Cancer 2014, 14, 120. [Google Scholar] [CrossRef]
- Chene, G.; Ouellet, V.; Rahimi, K.; Barres, V.; Provencher, D.; Mes-Masson, A.M. The ARID1A Pathway in Ovarian Clear Cell and Endometrioid Carcinoma, Contiguous Endometriosis, and Benign Endometriosis. Int. J. Gynecol. Obstet. 2015, 130, 27–30. [Google Scholar] [CrossRef]
- Jones, S.; Wang, T.L.; Shih, I.M.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A.; Vogelstein, B.; et al. Frequent Mutations of Chromatin Remodeling Gene ARID1A in Ovarian Clear Cell Carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef]
- Krig, S.R.; Miller, J.K.; Frietze, S.; Beckett, L.A.; Neve, R.M.; Farnham, P.J.; Yaswen, P.I.; Sweeney, C.A. ZNF217, a Candidate Breast Cancer Oncogene Amplified at 20q13, Regulates Expression of the ErbB3 Receptor Tyrosine Kinase in Breast Cancer Cells. Oncogene 2010, 29, 5500–5510. [Google Scholar] [CrossRef]
- McConechy, M.K.; Ding, J.; Senz, J.; Yang, W.; Melnyk, N.; Tone, A.A.; Prentice, L.M.; Wiegand, K.C.; McAlpine, J.N.; Shah, S.P.; et al. Ovarian and Endometrial Endometrioid Carcinomas Have Distinct CTNNB1 and PTEN Mutation Profiles. Mod. Pathol. 2014, 27, 128–134. [Google Scholar] [CrossRef]
- Shibuya, Y.; Tokunaga, H.; Saito, S.; Shimokawa, K.; Katsuoka, F.; Bin, L.; Kojima, K.; Nagasaki, M.; Yamamoto, M.; Yaegashi, N.; et al. Identification of Somatic Genetic Alterations in Ovarian Clear Cell Carcinoma with next Generation Sequencing. Genes. Chromosomes Cancer 2018, 57, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Nakayama, K.; Nakamura, K.; Ono, R.; Sanuki, K.; Yamashita, H.; Ishibashi, T.; Minamoto, T.; Iida, K.; Razia, S.; et al. Affinity-Purified DNA-Based Mutation Profiles of Endometriosis-Related Ovarian Neoplasms in Japanese Patients. Oncotarget 2018, 9, 14754–14763. [Google Scholar] [CrossRef] [PubMed]
- Maru, Y.; Tanaka, N.; Ohira, M.; Itami, M.; Hippo, Y.; Nagase, H. Identification of Novel Mutations in Japanese Ovarian Clear Cell Carcinoma Patients Using Optimized Targeted NGS for Clinical Diagnosis. Gynecol. Oncol. 2017, 144, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Itamochi, H.; Oishi, T.; Oumi, N.; Takeuchi, S.; Yoshihara, K.; Mikami, M.; Yaegashi, N.; Terao, Y.; Takehara, K.; Ushijima, K.; et al. Whole-Genome Sequencing Revealed Novel Prognostic Biomarkers and Promising Targets for Therapy of Ovarian Clear Cell Carcinoma. Br. J. Cancer 2017, 117, 717–724. [Google Scholar] [CrossRef]
- Rahman, M.; Nakayama, K.; Rahman, M.T.; Nakayama, N.; Ishikawa, M.; Katagiri, A.; Iida, K.; Nakayama, S.; Otsuki, Y.; Shih, I.M.; et al. Clinicopathologic and Biological Analysis of PIK3CA Mutation in Ovarian Clear Cell Carcinoma. Hum. Pathol. 2012, 43, 2197–2206. [Google Scholar] [CrossRef]
- Er, T.K.; Su, Y.F.; Wu, C.C.; Chen, C.C.; Wang, J.; Hsieh, T.H.; Herreros-Villanueva, M.; Chen, W.T.; Chen, Y.T.; Liu, T.C.; et al. Targeted Next-Generation Sequencing for Molecular Diagnosis of Endometriosis-Associated Ovarian Cancer. J. Mol. Med. 2016, 94, 835–847. [Google Scholar] [CrossRef]
- Su, Y.F.; Tsai, E.M.; Chen, C.C.; Wu, C.C.; Er, T.K. Targeted Sequencing of a Specific Gene Panel Detects a High Frequency of ARID1A and PIK3CA Mutations in Ovarian Clear Cell Carcinoma. Clin. Chim. Acta 2019, 494, 1–7. [Google Scholar] [CrossRef]
- Sa, J.K.; Kim, J.; Kang, S.; Kim, S.W.; Song, T.; Shim, S.H.; Choi, M.C.; No, J.H.; Song, J.Y.; Kim, D.; et al. Somatic Genomic Landscape of East Asian Epithelial Ovarian Carcinoma and Its Clinical Implications from Prospective Clinical Sequencing: A Korean Gynecologic Oncology Group Study (KGOG 3047). Int. J. Cancer 2022, 151, 1086–1097. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, C.; Ren, Y.; Yi, H.; Luo, T.; Xing, F.; Bai, X.; Cui, L.; Zhu, L.; Ouyang, J.; et al. Genomic Characterization of Chinese Ovarian Clear Cell Carcinoma Identifies Driver Genes by Whole Exome Sequencing. Neoplasia 2020, 22, 399–430. [Google Scholar] [CrossRef]
- Wang, Y.K.; Bashashati, A.; Anglesio, M.S.; Cochrane, D.R.; Grewal, D.S.; Ha, G.; McPherson, A.; Horlings, H.M.; Senz, J.; Prentice, L.M.; et al. Genomic Consequences of Aberrant DNA Repair Mechanisms Stratify Ovarian Cancer Histotypes. Nat. Genet. 2017, 49, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Lee, J.W.; Lee, M.; Kim, H.S.; Chung, H.H.; Kim, J.W.; Park, N.H.; Song, Y.S.; Seo, J.S. Genomic Landscape of Ovarian Clear Cell Carcinoma via Whole Exome Sequencing. Gynecol. Oncol. 2018, 148, 375–382. [Google Scholar] [CrossRef]
- Lapke, N.; Chen, C.H.; Chang, T.C.; Chao, A.; Lu, Y.J.; Lai, C.H.; Tan, K.T.; Chen, H.C.; Lu, H.Y.; Chen, S.J. Genetic Alterations and Their Therapeutic Implications in Epithelial Ovarian Cancer. BMC Cancer 2021, 21, 499. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, D.V.N.P.; Schnack, T.H.; Poulsen, T.S.; Christiansen, A.P.; Høgdall, C.K.; Høgdall, E.V. Genomic Sub-Classification of Ovarian Clear Cell Carcinoma Revealed by Distinct Mutational Signatures. Cancers 2021, 13, 5242. [Google Scholar] [CrossRef] [PubMed]
- Bolton, K.L.; Chen, D.; de la Fuente, R.C.; Fu, Z.; Murali, R.; Köbel, M.; Tazi, Y.; Cunningham, J.M.; Chan, I.C.C.; Wiley, B.J.; et al. Molecular Subclasses of Clear Cell Ovarian Carcinoma and Their Impact on Disease Behavior and Outcomes. Clin. Cancer Res. 2022, 28, 4947–4956. [Google Scholar] [CrossRef]
- Yanaihara, N.; Anglesio, M.S.; Ochiai, K.; Hirata, Y.; Saito, M.; Nagata, C.; Iida, Y.; Takakura, S.; Yamada, K.; Tanaka, T.; et al. Cytokine Gene Expression Signature in Ovarian Clear Cell Carcinoma. Int. J. Oncol. 2012, 41, 1094–1100. [Google Scholar] [CrossRef]
- Michaud, D.S.; Daugherty, S.E.; Berndt, S.I.; Platz, E.A.; Yeager, M.; Crawford, E.D.; Hsing, A.; Huang, W.-Y.; Hayes, R.B. Genetic Polymorphisms of Interleukin-1B (IL-1B), IL-6, IL-8, and IL-10 and Risk of Prostate Cancer. Cancer Res. 2006, 66, 4525–4530. [Google Scholar] [CrossRef]
- Martínez-Pérez, C.; Leung, J.; Kay, C.; Meehan, J.; Gray, M.; Dixon, J.M.; Turnbull, A.K. The Signal Transducer Il6st (Gp130) as a Predictive and Prognostic Biomarker in Breast Cancer. J. Pers. Med. 2021, 11, 618. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, F.; Lu, T.; Duan, Z.; Zhang, Z. Interleukin-6 Signaling Pathway in Targeted Therapy for Cancer. Cancer Treat. Rev. 2012, 38, 904–910. [Google Scholar] [CrossRef]
- Naugler, W.E.; Karin, M. The Wolf in Sheep’s Clothing: The Role of Interleukin-6 in Immunity, Inflammation and Cancer. Trends Mol. Med. 2008, 14, 109–119. [Google Scholar] [CrossRef]
- Chang, Q.; Daly, L.; Bromberg, J. The IL-6 Feed-Forward Loop: A Driver of Tumorigenesis. Semin. Immunol. 2014, 26, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT Signaling Pathway: From Bench to Clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Rädler, P.D.; Wehde, B.L.; Wagner, K.-U. Crosstalk between STAT5 Activation and PI3K/AKT Functions in Normal and Transformed Mammary Epithelial Cells. Mol. Cell Endocrinol. 2017, 451, 31–39. [Google Scholar] [CrossRef]
- Tasian, S.K.; Doral, M.Y.; Borowitz, M.J.; Wood, B.L.; Chen, I.-M.; Harvey, R.C.; Gastier-Foster, J.M.; Willman, C.L.; Hunger, S.P.; Mullighan, C.G.; et al. Aberrant STAT5 and PI3K/MTOR Pathway Signaling Occurs in Human CRLF2-Rearranged B-Precursor Acute Lymphoblastic Leukemia. Blood 2012, 120, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Jaeger, S.A.; Hirsch, H.A.; Bulyk, M.L.; Struhl, K. STAT3 Activation of MiR-21 and MiR-181b-1 via PTEN and CYLD Are Part of the Epigenetic Switch Linking Inflammation to Cancer. Mol. Cell 2010, 39, 493–506. [Google Scholar] [CrossRef]
- Hirata, Y.; Murai, N.; Yanaihara, N.; Saito, M.; Saito, M.; Urashima, M.; Murakami, Y.; Matsufuji, S.; Okamoto, A. MicroRNA-21 Is a Candidate Driver Gene for 17q23-25 Amplification in Ovarian Clear Cell Carcinoma. BMC Cancer 2014, 14, 799. [Google Scholar] [CrossRef]
- Suryawanshi, S.; Vlad, A.M.; Lin, H.M.; Mantia-Smaldone, G.; Laskey, R.; Lee, M.; Lin, Y.; Donnellan, N.; Klein-Patel, M.; Lee, T.; et al. Plasma MicroRNAs as Novel Biomarkers for Endometriosis and Endometriosis-Associated Ovarian Cancer. Clin. Cancer Res. 2013, 19, 1213–1224. [Google Scholar] [CrossRef]
- Ohlsson Teague, E.M.C.; Print, C.G.; Hull, M.L. The Role of MicroRNAs in Endometriosis and Associated Reproductive Conditions. Hum. Reprod. Update 2009, 16, 142–165. [Google Scholar] [CrossRef]
- Nagaraja, A.K.; Creighton, C.J.; Yu, Z.; Zhu, H.; Gunaratne, P.H.; Reid, J.G.; Olokpa, E.; Itamochi, H.; Ueno, N.T.; Hawkins, S.M.; et al. A Link between Mir-100 and FRAP1/MTOR in Clear Cell Ovarian Cancer. Mol. Endocrinol. 2010, 24, 447–463. [Google Scholar] [CrossRef]
- Colombo, N.; Sessa, C.; du Bois, A.; Ledermann, J.; McCluggage, W.G.; McNeish, I.; Morice, P.; Pignata, S.; Ray-Coquard, I.; Vergote, I.; et al. ESMO–ESGO Consensus Conference Recommendations on Ovarian Cancer: Pathology and Molecular Biology, Early and Advanced Stages, Borderline Tumours and Recurrent Disease. Ann. Oncol. 2019, 30, 672–705. [Google Scholar] [CrossRef]
- Shoji, T.; Tatsuki, S.; Abe, M.; Tomabechi, H.; Takatori, E.; Kaido, Y.; Nagasawa, T.; Kagabu, M.; Baba, T.; Itamochi, H. Novel Therapeutic Strategies for Refractory Ovarian Cancers: Clear Cell and Mucinous Carcinomas. Cancers 2021, 13, 6120. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, S.; Kawase, C.; Altomare, D.A.; Morishige, K.; Sawada, K.; Hayashi, M.; Tsujimoto, M.; Yamoto, M.; Klein-Szanto, A.J.; Schilder, R.J.; et al. MTOR Is a Promising Therapeutic Target Both in Cisplatin-Sensitive and Cisplatin-Resistant Clear Cell Carcinoma of the Ovary. Clin. Cancer Res. 2009, 15, 5404–5413. [Google Scholar] [CrossRef] [PubMed]
- Oishi, T.; Itamochi, H.; Kudoh, A.; Nonaka, M.; Kato, M.; Nishimura, M.; Oumi, N.; Sato, S.; Naniwa, J.; Sato, S.; et al. The PI3K/MTOR Dual Inhibitor NVP-BEZ235 Reduces the Growth of Ovarian Clear Cell Carcinoma. Oncol. Rep. 2014, 32, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Caumanns, J.J.; van Wijngaarden, A.; Kol, A.; Meersma, G.J.; Jalving, M.; Bernards, R.; van der Zee, A.G.J.; Wisman, G.B.A.; de Jong, S. Low-Dose Triple Drug Combination Targeting the PI3K/AKT/MTOR Pathway and the MAPK Pathway Is an Effective Approach in Ovarian Clear Cell Carcinoma. Cancer Lett. 2019, 461, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Papp, E.; Hallberg, D.; Konecny, G.E.; Bruhm, D.C.; Adleff, V.; Noë, M.; Kagiampakis, I.; Palsgrove, D.; Conklin, D.; Kinose, Y.; et al. Integrated Genomic, Epigenomic, and Expression Analyses of Ovarian Cancer Cell Lines. Cell Rep. 2018, 25, 2617–2633. [Google Scholar] [CrossRef]
- Basu, S. PP2A in the Regulation of Cell Motility and Invasion. Curr. Protein Pept. Sci. 2011, 12, 3–11. [Google Scholar] [CrossRef]
- Herreros-Villanueva, M.; Chen, C.C.; Tsai, E.M.; Er, T.K. Endometriosis-Associated Ovarian Cancer: What Have We Learned so Far? Clin. Chim. Acta 2019, 493, 63–72. [Google Scholar] [CrossRef]
- Samartzis, E.P.; Gutsche, K.; Dedes, K.J.; Fink, D.; Stucki, M.; Imesch, P. Loss of ARID1A expression sensitizes cancer cells to PI3K- and AKT-inhibition. Oncotarget 2014, 5, 5295–5303. [Google Scholar] [CrossRef]
- Chien, W.; Tyner, J.W.; Gery, S.; Zheng, Y.; Li, L.-Y.; Gopinatha Pillai, M.S.; Nam, C.; Bhowmick, N.A.; Lin, D.-C.; Koeffler, H.P. Treatment for Ovarian Clear Cell Carcinoma with Combined Inhibition of WEE1 and ATR. J. Ovarian Res. 2023, 16, 80. [Google Scholar] [CrossRef]
- Berns, K.; Sonnenblick, A.; Gennissen, A.; Brohée, S.; Hijmans, E.M.; Evers, B.; Fumagalli, D.; Desmedt, C.; Loibl, S.; Denkert, C.; et al. Loss of ARID1A Activates ANXA1, Which Serves as a Predictive Biomarker for Trastuzumab Resistance. Clin. Cancer Res. 2016, 22, 5238–5248. [Google Scholar] [CrossRef]
- Farley, J.H.; Brady, W.E.; O’Malley, D.; Fujiwara, K.; Yonemori, K.; Bonebrake, A.; Secord, A.A.; Stephan, J.-M.; Walker, J.L.; Nam, J.-H.; et al. A Phase II Evaluation of Temsirolimus with Carboplatin and Paclitaxel Followed by Temsirolimus Consolidation in Clear Cell Ovarian Cancer: An NRG Oncology Trial. Gynecol. Oncol. 2022, 167, 423–428. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Authors [Ref] | HISTOLOGICAL Type | No of Samples | ARID1A | PIK3CA | PTEN | ARID1A-PIK3CA Co-Mutations |
|---|---|---|---|---|---|---|
| Wang et al., 2017 [91] | OCCC | 35 | 54% (19/35) | 54% (19/35) | 6% (2/35) | 40% (14/35) |
| Itamochi et al., 2017 [85] | OCCC | 55 | 42% (23/55) | 35% (19/55) | 2% (1/55) | 25% (14/55) |
| Murakami et al., 2017 [55] | OCCC | 39 | 62% (24/39) | 51% (20/39) | 5% (2/39) | NA |
| Shibuya et al., 2018 [82] | OCCC | 48 | 67% (32/48) | 50% (24/48) | 2% (1/48) | 46% (22/48) |
| Kim et al., 2018 [92] | OCCC | 15 | 4% (6/15) | 40% (6/15) | 13% (2/15) | 20% (3/15) |
| Yang et al., 2020 [90] | OCCC | 42 | 64% (27/42) | 29% (12/42) | 7% (3/42) | 26%(11/42) |
| Lapke et al., 2021 [93] | OCCC | 23 | 39% (9/23) | 43% (10/23) | 0% (0/23) | 22% (5/23) |
| Oliveira et al., 2021 [94] | OCCC | 55 | 49% (27/55) | 42% (23/55) | NA | 36% (13/36) |
| Bolton et al., 2022 [95] | OCCC | 421 | 49% (205/421) | 45% (188/421) | NA | ≤40% (≤167/421) * |
| Wang et al., 2017 [91] | EnOC | 29 | 41% (12/29) | 52% (15/29) | 41% (12/29) | 3% (1/36) |
| Cybulska et al., 2019 [59] | EnOC | 36 | 19% (7/36) | 39% (14/36) | 33% (12/36) | 3% (1/36) |
| Pierson et al., 2020 [61] | EnOC | 26 | 19% (5/26) | 27% (7/26) | 46% (12/26) | 12% (3/26) |
| Hollis et al., 2020 [60] | EnOC | 112 | 36% (40/112) | 43% (48/112) | 29% (32/112) | 21% (23/112) |
| Lapke et al., 2021 [93] | EnOC | 22 | 32% (7/22) | 32% (7/22) | 27% (6/22) | 23% (5/22) |
| Su et al., 2019 [88] | OCCC and EnOC | 16 | 56% (9/16) | 50% (8/16) | NA | 44%(7/16) |
| Total of frequencies | 974 | 47% (462/974) | 43% (420/974) | 18% (85/482) | 24% (122/514) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Driva, T.S.; Schatz, C.; Haybaeck, J. Endometriosis-Associated Ovarian Carcinomas: How PI3K/AKT/mTOR Pathway Affects Their Pathogenesis. Biomolecules 2023, 13, 1253. https://doi.org/10.3390/biom13081253
Driva TS, Schatz C, Haybaeck J. Endometriosis-Associated Ovarian Carcinomas: How PI3K/AKT/mTOR Pathway Affects Their Pathogenesis. Biomolecules. 2023; 13(8):1253. https://doi.org/10.3390/biom13081253
Chicago/Turabian StyleDriva, Tatiana S., Christoph Schatz, and Johannes Haybaeck. 2023. "Endometriosis-Associated Ovarian Carcinomas: How PI3K/AKT/mTOR Pathway Affects Their Pathogenesis" Biomolecules 13, no. 8: 1253. https://doi.org/10.3390/biom13081253