
The Ca2+ Sensor STIM in Human Diseases
 , , , ,
, , , ,  , and
, and 
Abstract
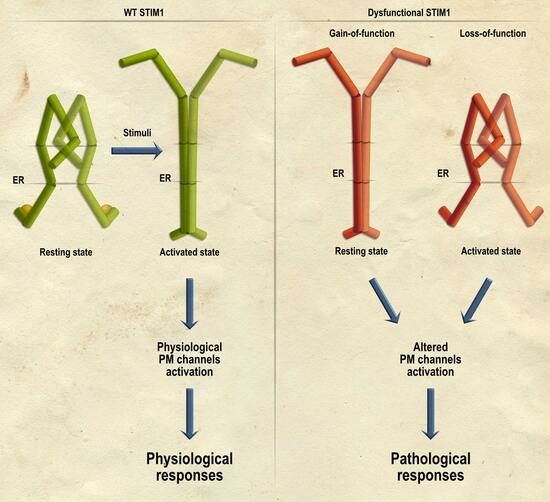
:
1. Introduction
The STIM Family
2. Disorders Associated with Gain-of-Function STIM1 Mutations
2.1. Stormorken Syndrome and Tubular Aggregate Myopathy (TAM)
2.2. York Platelet Syndrome (YPS)
3. Disorders Associated with Loss-of-Function STIM1 Mutations
4. STIM2 in Human Diseases
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W., Jr. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Feske, S. CRAC channels and disease—From human CRAC channelopathies and animal models to novel drugs. Cell Calcium 2019, 80, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Rosado, J.A. STIM and calcium channel complexes in cancer. Biochim. Biophys. Acta 2016, 1863, 1418–1426. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Calcium signalling remodelling and disease. Biochem. Soc. Trans. 2012, 40, 297–309. [Google Scholar] [CrossRef]
- Putney, J.W. Origins of the concept of store-operated calcium entry. Front. Biosci. Schol. Ed. 2011, 3, 980–984. [Google Scholar] [CrossRef]
- Gericke, M.; Oike, M.; Droogmans, G.; Nilius, B. Inhibition of capacitative Ca2+ entry by a Cl- channel blocker in human endothelial cells. Eur. J. Pharmacol. 1994, 269, 381–384. [Google Scholar] [CrossRef]
- Putney, J.W. Store-Operated Calcium Entry: An Historical Overview. Adv. Exp. Med. Biol. 2017, 981, 205–214. [Google Scholar] [CrossRef]
- Rosado, J.A.; Redondo, P.C.; Sage, S.O.; Pariente, J.A.; Salido, G.M. Store-operated Ca2+ entry: Vesicle fusion or reversible trafficking and de novo conformational coupling? J. Cell Physiol. 2005, 205, 262–269. [Google Scholar] [CrossRef]
- Bolotina, V.M.; Csutora, P. CIF and other mysteries of the store-operated Ca2+-entry pathway. Trends Biochem. Sci. 2005, 30, 378–387. [Google Scholar] [CrossRef]
- Jaconi, M.; Pyle, J.; Bortolon, R.; Ou, J.; Clapham, D. Calcium release and influx colocalize to the endoplasmic reticulum. Curr. Biol. 1997, 7, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef] [PubMed]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Luik, R.M.; Wu, M.M.; Buchanan, J.; Lewis, R.S. The elementary unit of store-operated Ca2+ entry: Local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J. Cell Biol. 2006, 174, 815–825. [Google Scholar] [CrossRef]
- Butorac, C.; Krizova, A.; Derler, I. Review: Structure and Activation Mechanisms of CRAC Channels. Adv. Exp. Med. Biol. 2020, 1131, 547–604. [Google Scholar] [CrossRef]
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.J.; Worley, P.F.; Muallem, S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 2009, 11, 337–343. [Google Scholar] [CrossRef]
- Muik, M.; Fahrner, M.; Derler, I.; Schindl, R.; Bergsmann, J.; Frischauf, I.; Groschner, K.; Romanin, C. A Cytosolic Homomerization and a Modulatory Domain within STIM1 C Terminus Determine Coupling to ORAI1 Channels. J. Biol. Chem. 2009, 284, 8421–8426. [Google Scholar] [CrossRef]
- Derler, I.; Jardin, I.; Romanin, C. Molecular mechanisms of STIM/Orai communication. Am. J. Physiol. Cell Physiol. 2016, 310, C643–C662. [Google Scholar] [CrossRef]
- Emrich, S.M.; Yoast, R.E.; Trebak, M. Physiological Functions of CRAC Channels. Annu. Rev. Physiol. 2022, 84, 355–379. [Google Scholar] [CrossRef] [PubMed]
- Sabbioni, S.; Veronese, A.; Trubia, M.; Taramelli, R.; Barbanti-Brodano, G.; Croce, C.M.; Negrini, M. Exon structure and promoter identification of STIM1 (alias GOK), a human gene causing growth arrest of the human tumor cell lines G401 and RD. Cytogenet. Cell Genet. 1999, 86, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Dionisio, N.; Frischauf, I.; Berna-Erro, A.; Woodard, G.E.; Lopez, J.J.; Salido, G.M.; Rosado, J.A. The polybasic lysine-rich domain of plasma membrane-resident STIM1 is essential for the modulation of store-operated divalent cation entry by extracellular calcium. Cell. Signal. 2013, 25, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Zbidi, H.; Jardin, I.; Woodard, G.E.; Lopez, J.J.; Berna-Erro, A.; Salido, G.M.; Rosado, J.A. STIM1 and STIM2 are located in the acidic Ca2+ stores and associates with Orai1 upon depletion of the acidic stores in human platelets. J. Biol. Chem. 2011, 286, 12257–12270. [Google Scholar] [CrossRef] [PubMed]
- Spassova, M.A.; Soboloff, J.; He, L.P.; Xu, W.; Dziadek, M.A.; Gill, D.L. STIM1 has a plasma membrane role in the activation of store-operated Ca(2+) channels. Proc. Natl. Acad. Sci. USA 2006, 103, 4040–4045. [Google Scholar] [CrossRef]
- Fahrner, M.; Schindl, R.; Muik, M.; Derler, I.; Romanin, C. The STIM-Orai Pathway: The Interactions Between STIM and Orai. Adv. Exp. Med. Biol. 2017, 993, 59–81. [Google Scholar] [CrossRef]
- Stathopulos, P.B.; Zheng, L.; Li, G.Y.; Plevin, M.J.; Ikura, M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell 2008, 135, 110–122. [Google Scholar] [CrossRef]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef]
- Luik, R.M.; Wang, B.; Prakriya, M.; Wu, M.M.; Lewis, R.S. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature 2008, 454, 538–542. [Google Scholar] [CrossRef]
- Zheng, L.; Stathopulos, P.B.; Schindl, R.; Li, G.Y.; Romanin, C.; Ikura, M. Auto-inhibitory role of the EF-SAM domain of STIM proteins in store-operated calcium entry. Proc. Natl. Acad. Sci. USA 2011, 108, 1337–1342. [Google Scholar] [CrossRef]
- Stathopulos, P.B.; Li, G.Y.; Plevin, M.J.; Ames, J.B.; Ikura, M. Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: An initiation mechanism for capacitive Ca2+ entry. J. Biol. Chem. 2006, 281, 35855–35862. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Wei, M.; He, L.; Liu, C.; Wu, B.; Zhang, S.L.; Jing, J.; Liang, X.; Senes, A.; Tan, P.; et al. Inside-out Ca(2+) signalling prompted by STIM1 conformational switch. Nat. Commun. 2015, 6, 7826. [Google Scholar] [CrossRef] [PubMed]
- Van Dorp, S.; Qiu, R.; Choi, U.B.; Wu, M.M.; Yen, M.; Kirmiz, M.; Brunger, A.T.; Lewis, R.S. Conformational dynamics of auto-inhibition in the ER calcium sensor STIM1. Elife 2021, 10, e66194. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, M.; Muik, M.; Schindl, R.; Butorac, C.; Stathopulos, P.; Zheng, L.; Jardin, I.; Ikura, M.; Romanin, C. A coiled-coil clamp controls both conformation and clustering of stromal interaction molecule 1 (STIM1). J. Biol. Chem. 2014, 289, 33231–33244. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, M.; Stadlbauer, M.; Muik, M.; Rathner, P.; Stathopulos, P.; Ikura, M.; Muller, N.; Romanin, C. A dual mechanism promotes switching of the Stormorken STIM1 R304W mutant into the activated state. Nat. Commun. 2018, 9, 825. [Google Scholar] [CrossRef]
- Rathner, P.; Fahrner, M.; Cerofolini, L.; Grabmayr, H.; Horvath, F.; Krobath, H.; Gupta, A.; Ravera, E.; Fragai, M.; Bechmann, M.; et al. Interhelical interactions within the STIM1 CC1 domain modulate CRAC channel activation. Nat. Chem. Biol. 2021, 17, 196–204. [Google Scholar] [CrossRef]
- Stathopulos, P.B.; Schindl, R.; Fahrner, M.; Zheng, L.; Gasmi-Seabrook, G.M.; Muik, M.; Romanin, C.; Ikura, M. STIM1/Orai1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat. Commun. 2013, 4, 2963. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Zhou, Y.; Hendron, E.; Mancarella, S.; Andrake, M.D.; Rothberg, B.S.; Soboloff, J.; Gill, D.L. Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat. Commun. 2014, 5, 3183. [Google Scholar] [CrossRef]
- Yang, X.; Jin, H.; Cai, X.; Li, S.; Shen, Y. Structural and mechanistic insights into the activation of Stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. USA 2012, 109, 5657–5662. [Google Scholar] [CrossRef]
- Misceo, D.; Holmgren, A.; Louch, W.E.; Holme, P.A.; Mizobuchi, M.; Morales, R.J.; De Paula, A.M.; Stray-Pedersen, A.; Lyle, R.; Dalhus, B.; et al. A dominant STIM1 mutation causes Stormorken syndrome. Hum. Mutat. 2014, 35, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Lee, M.H.; Jeong, S.J.; Qin, X.; Lee, A.R.; Park, H.; Park, C.Y. The inactivation domain of STIM1 acts through intramolecular binding to the coiled-coil domain in the resting state. J. Cell Sci. 2020, 133, jcs237354. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.L.; Chen, Y.J.; Quintanilla, C.G.; Hsieh, T.S.; Liou, J. EB1 binding restricts STIM1 translocation to ER-PM junctions and regulates store-operated Ca(2+) entry. J. Cell Biol. 2018, 217, 2047–2058. [Google Scholar] [CrossRef] [PubMed]
- Honnappa, S.; Gouveia, S.M.; Weisbrich, A.; Damberger, F.F.; Bhavesh, N.S.; Jawhari, H.; Grigoriev, I.; van Rijssel, F.J.; Buey, R.M.; Lawera, A.; et al. An EB1-binding motif acts as a microtubule tip localization signal. Cell 2009, 138, 366–376. [Google Scholar] [CrossRef]
- Sallinger, M.; Grabmayr, H.; Humer, C.; Bonhenry, D.; Romanin, C.; Schindl, R.; Derler, I. Activation mechanisms and structural dynamics of STIM proteins. J. Physiol. 2023. ahead of print. [Google Scholar] [CrossRef]
- Stormorken, H.; Sjaastad, O.; Langslet, A.; Sulg, I.; Egge, K.; Diderichsen, J. A new syndrome: Thrombocytopathia, muscle fatigue, asplenia, miosis, migraine, dyslexia and ichthyosis. Clin. Genet. 1985, 28, 367–374. [Google Scholar] [CrossRef]
- Lopez, J.J.; Salido, G.M.; Pariente, J.A.; Rosado, J.A. Thrombin induces activation and translocation of Bid, Bax and Bak to the mitochondria in human platelets. J. Thromb. Haemost. 2008, 6, 1780–1788. [Google Scholar] [CrossRef]
- Rosado, J.A.; Meijer, E.M.; Hamulyak, K.; Novakova, I.; Heemskerk, J.W.; Sage, S.O. Fibrinogen binding to the integrin alpha(IIb)beta(3) modulates store-mediated calcium entry in human platelets. Blood 2001, 97, 2648–2656. [Google Scholar] [CrossRef]
- Morin, G.; Bruechle, N.O.; Singh, A.R.; Knopp, C.; Jedraszak, G.; Elbracht, M.; Bremond-Gignac, D.; Hartmann, K.; Sevestre, H.; Deutz, P.; et al. Gain-of-Function Mutation in STIM1 (P.R304W) Is Associated with Stormorken Syndrome. Hum. Mutat. 2014, 35, 1221–1232. [Google Scholar] [CrossRef]
- Stormorken, H.; Holmsen, H.; Sund, R.; Sakariassen, K.S.; Hovig, T.; Jellum, E.; Solum, O. Studies on the haemostatic defect in a complicated syndrome. An inverse Scott syndrome platelet membrane abnormality? Thromb. Haemost. 1995, 74, 1244–1251. [Google Scholar]
- Grosse, J.; Braun, A.; Varga-Szabo, D.; Beyersdorf, N.; Schneider, B.; Zeitlmann, L.; Hanke, P.; Schropp, P.; Muhlstedt, S.; Zorn, C.; et al. An EF hand mutation in Stim1 causes premature platelet activation and bleeding in mice. J. Clin. Investig. 2007, 117, 3540–3550. [Google Scholar] [CrossRef] [PubMed]
- Nesin, V.; Wiley, G.; Kousi, M.; Ong, E.C.; Lehmann, T.; Nicholl, D.J.; Suri, M.; Shahrizaila, N.; Katsanis, N.; Gaffney, P.M.; et al. Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc. Natl. Acad. Sci. USA 2014, 111, 4197–4202. [Google Scholar] [CrossRef] [PubMed]
- Muik, M.; Fahrner, M.; Schindl, R.; Stathopulos, P.; Frischauf, I.; Derler, I.; Plenk, P.; Lackner, B.; Groschner, K.; Ikura, M.; et al. STIM1 couples to ORAI1 via an intramolecular transition into an extended conformation. EMBO J. 2011, 30, 1678–1689. [Google Scholar] [CrossRef] [PubMed]
- Gamage, T.H.; Gunnes, G.; Lee, R.H.; Louch, W.E.; Holmgren, A.; Bruton, J.D.; Lengle, E.; Kolstad, T.R.S.; Revold, T.; Amundsen, S.S.; et al. STIM1 R304W causes muscle degeneration and impaired platelet activation in mice. Cell Calcium 2018, 76, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.J.; Zhao, X.; Dou, Z.Y.; Su, Q.X.; Rong, Z.H. Stormorken Syndrome Caused by a Novel STIM1 Mutation: A Case Report. Front. Neurol. 2021, 12, 522513. [Google Scholar] [CrossRef]
- Bohm, J.; Laporte, J. Gain-of-function mutations in STIM1 and ORAI1 causing tubular aggregate myopathy and Stormorken syndrome. Cell Calcium 2018, 76, 1–9. [Google Scholar] [CrossRef]
- Protasi, F.; Girolami, B.; Roccabianca, S.; Rossi, D. Store-operated calcium entry: From physiology to tubular aggregate myopathy. Curr. Opin. Pharmacol. 2023, 68, 102347. [Google Scholar] [CrossRef]
- Lee, J.M.; Noguchi, S. Calcium Dyshomeostasis in Tubular Aggregate Myopathy. Int. J. Mol. Sci. 2016, 17, 1952. [Google Scholar] [CrossRef]
- Lacruz, R.S.; Feske, S. Diseases caused by mutations in ORAI1 and STIM1. Ann. N. Y. Acad. Sci. 2015, 1356, 45–79. [Google Scholar] [CrossRef]
- Bohm, J.; Chevessier, F.; Maues De Paula, A.; Koch, C.; Attarian, S.; Feger, C.; Hantai, D.; Laforet, P.; Ghorab, K.; Vallat, J.M.; et al. Constitutive activation of the calcium sensor STIM1 causes tubular-aggregate myopathy. Am. J. Hum. Genet. 2013, 92, 271–278. [Google Scholar] [CrossRef]
- Bohm, J.; Chevessier, F.; Koch, C.; Peche, G.A.; Mora, M.; Morandi, L.; Pasanisi, B.; Moroni, I.; Tasca, G.; Fattori, F.; et al. Clinical, histological and genetic characterisation of patients with tubular aggregate myopathy caused by mutations in STIM1. J. Med. Genet. 2014, 51, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Hedberg, C.; Niceta, M.; Fattori, F.; Lindvall, B.; Ciolfi, A.; D’Amico, A.; Tasca, G.; Petrini, S.; Tulinius, M.; Tartaglia, M.; et al. Childhood onset tubular aggregate myopathy associated with de novo STIM1 mutations. J. Neurol. 2014, 261, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.C.; Rossius, M.; Zitzelsberger, M.; Vorgerd, M.; Muller-Felber, W.; Ertl-Wagner, B.; Zhang, Y.; Brinkmeier, H.; Senderek, J.; Schoser, B. 50 years to diagnosis: Autosomal dominant tubular aggregate myopathy caused by a novel STIM1 mutation. Neuromuscul. Disord. 2015, 25, 577–584. [Google Scholar] [CrossRef]
- Barone, V.; Del Re, V.; Gamberucci, A.; Polverino, V.; Galli, L.; Rossi, D.; Costanzi, E.; Toniolo, L.; Berti, G.; Malandrini, A.; et al. Identification and characterization of three novel mutations in the CASQ1 gene in four patients with tubular aggregate myopathy. Hum. Mutat. 2017, 38, 1761–1773. [Google Scholar] [CrossRef] [PubMed]
- White, J.G. Giant electron-dense chains, clusters and granules in megakaryocytes and platelets with normal dense bodies: An inherited thrombocytopenic disorder. Platelets 2003, 14, 109–121. [Google Scholar] [CrossRef]
- White, J.G. Giant electron-dense chains, clusters and granules in megakaryocytes and platelets with normal dense bodies: An inherited thrombocytopenic disorder I. Megakaryocytes. Platelets 2003, 14, 53–60. [Google Scholar] [CrossRef]
- White, J.G.; Ahlstrand, G.G. Giant electron dense chains, clusters and granules in megakaryocytes and platelets with normal dense bodies: An inherited thrombocytopenic disorder IV. Ultrastructural cytochemistry and analytical electron microscopy. Platelets 2003, 14, 313–324. [Google Scholar] [CrossRef]
- White, J.G.; Ahlstrand, G.G. Giant electron dense chains, clusters and granules in megakaryocytes and platelets with normal dense bodies: An inherited thrombocytopenic disorder III. Platelet analytical electron microscopy. Platelets 2003, 14, 305–312. [Google Scholar] [CrossRef]
- White, J.G.; Gunay-Aygun, M. The York Platelet Syndrome: A third case. Platelets 2011, 22, 117–134. [Google Scholar] [CrossRef]
- Markello, T.; Chen, D.; Kwan, J.Y.; Horkayne-Szakaly, I.; Morrison, A.; Simakova, O.; Maric, I.; Lozier, J.; Cullinane, A.R.; Kilo, T.; et al. York platelet syndrome is a CRAC channelopathy due to gain-of-function mutations in STIM1. Mol. Genet. Metab. 2015, 114, 474–482. [Google Scholar] [CrossRef]
- Roman, J.; Palmer, M.I.; Palmer, C.A.; Johnson, N.E.; Butterfield, R.J. Myopathy in the York Platelet Syndrome: An Underrecognized Complication. Case Rep. Pathol. 2018, 2018, 5130143. [Google Scholar] [CrossRef] [PubMed]
- Ticci, C.; Cassandrini, D.; Rubegni, A.; Riva, B.; Vattemi, G.; Mata, S.; Ricci, G.; Baldacci, J.; Guglielmi, V.; Di Muzio, A.; et al. Expanding the clinical and genetic spectrum of pathogenic variants in STIM1. Muscle Nerve 2021, 64, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Darbellay, B.; Arnaudeau, S.; Bader, C.R.; Konig, S.; Bernheim, L. STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J. Cell Biol. 2011, 194, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Riva, B.; Pessolano, E.; Quaglia, E.; Cordero-Sanchez, C.; Bhela, I.P.; Topf, A.; Serafini, M.; Cox, D.; Harris, E.; Garibaldi, M.; et al. STIM1 and ORAI1 mutations leading to tubular aggregate myopathies are sensitive to the Store-operated Ca(2+)-entry modulators CIC-37 and CIC-39. Cell Calcium 2022, 105, 102605. [Google Scholar] [CrossRef] [PubMed]
- Barde, P.J.; Viswanadha, S.; Veeraraghavan, S.; Vakkalanka, S.V.; Nair, A. A first-in-human study to evaluate the safety, tolerability and pharmacokinetics of RP3128, an oral calcium release-activated calcium (CRAC) channel modulator in healthy volunteers. J. Clin. Pharm. Ther. 2021, 46, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Bruen, C.; Miller, J.; Wilburn, J.; Mackey, C.; Bollen, T.L.; Stauderman, K.; Hebbar, S. Auxora for the Treatment of Patients With Acute Pancreatitis and Accompanying Systemic Inflammatory Response Syndrome: Clinical Development of a Calcium Release-Activated Calcium Channel Inhibitor. Pancreas 2021, 50, 537–543. [Google Scholar] [CrossRef]
- Feske, S. CRAC channelopathies. Pflugers Arch. 2010, 460, 417–435. [Google Scholar] [CrossRef]
- Picard, C.; McCarl, C.A.; Papolos, A.; Khalil, S.; Luthy, K.; Hivroz, C.; LeDeist, F.; Rieux-Laucat, F.; Rechavi, G.; Rao, A.; et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N. Engl. J. Med. 2009, 360, 1971–1980. [Google Scholar] [CrossRef]
- Byun, M.; Abhyankar, A.; Lelarge, V.; Plancoulaine, S.; Palanduz, A.; Telhan, L.; Boisson, B.; Picard, C.; Dewell, S.; Zhao, C.; et al. Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J. Exp. Med. 2010, 207, 2307–2312. [Google Scholar] [CrossRef]
- Mesri, E.A.; Cesarman, E.; Boshoff, C. Kaposi’s sarcoma and its associated herpesvirus. Nat. Rev. Cancer 2010, 10, 707–719. [Google Scholar] [CrossRef]
- Schaballie, H.; Rodriguez, R.; Martin, E.; Moens, L.; Frans, G.; Lenoir, C.; Dutre, J.; Canioni, D.; Bossuyt, X.; Fischer, A.; et al. A novel hypomorphic mutation in STIM1 results in a late-onset immunodeficiency. J. Allergy Clin. Immunol. 2015, 136, 816–819.e814. [Google Scholar] [CrossRef]
- Fuchs, S.; Rensing-Ehl, A.; Speckmann, C.; Bengsch, B.; Schmitt-Graeff, A.; Bondzio, I.; Maul-Pavicic, A.; Bass, T.; Vraetz, T.; Strahm, B.; et al. Antiviral and regulatory T cell immunity in a patient with stromal interaction molecule 1 deficiency. J. Immunol. 2012, 188, 1523–1533. [Google Scholar] [CrossRef]
- Maus, M.; Jairaman, A.; Stathopulos, P.B.; Muik, M.; Fahrner, M.; Weidinger, C.; Benson, M.; Fuchs, S.; Ehl, S.; Romanin, C.; et al. Missense mutation in immunodeficient patients shows the multifunctional roles of coiled-coil domain 3 (CC3) in STIM1 activation. Proc. Natl. Acad. Sci. USA 2015, 112, 6206–6211. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Choi, M.; Richardson, A.S.; Reid, B.M.; Seymen, F.; Yildirim, M.; Tuna, E.; Gencay, K.; Simmer, J.P.; Hu, J.C. STIM1 and SLC24A4 Are Critical for Enamel Maturation. J. Dent. Res. 2014, 93, 94S–100S. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, N.; Hye-Ryong Shim, A.; Maneshi, M.M.; See-Wai Yeung, P.; Yamashita, M.; Prakriya, M. Mapping interactions between the CRAC activation domain and CC1 regulating the activity of the ER Ca(2+) sensor STIM1. J. Biol. Chem. 2022, 298, 102157. [Google Scholar] [CrossRef] [PubMed]
- Salvi, A.; Skrypnyk, C.; Da Silva, N.; Urtizberea, J.A.; Bakhiet, M.; Robert, C.; Levy, N.; Megarbane, A.; Delague, V.; Bartoli, M. A novel bi-allelic loss-of-function mutation in STIM1 expands the phenotype of STIM1-related diseases. Clin. Genet. 2021, 100, 84–89. [Google Scholar] [CrossRef]
- Di, J.; Yenwongfai, L.; Rieger, H.T.; Zhang, S.; Wei, S. Familial 4p Interstitial Deletion Provides New Insights and Candidate Genes Underlying This Rare Condition. Genes 2023, 14, 635. [Google Scholar] [CrossRef]
- Chen, C.P.; Lee, M.J.; Chern, S.R.; Wu, P.S.; Su, J.W.; Chen, Y.T.; Lee, M.S.; Wang, W. Prenatal diagnosis and molecular cytogenetic characterization of a de novo proximal interstitial deletion of chromosome 4p (4p15.2-->p14). Gene 2013, 529, 351–356. [Google Scholar] [CrossRef]
- Crippa, M.; Malatesta, P.; Bonati, M.T.; Trapasso, F.; Fortunato, F.; Annesi, G.; Larizza, L.; Labate, A.; Finelli, P.; Perrotti, N.; et al. A familial t(4;8) translocation segregates with epilepsy and migraine with aura. Ann. Clin. Transl. Neurol. 2020, 7, 855–859. [Google Scholar] [CrossRef]
- Pchitskaya, E.; Kraskovskaya, N.; Chernyuk, D.; Popugaeva, E.; Zhang, H.; Vlasova, O.; Bezprozvanny, I. Stim2-Eb3 Association and Morphology of Dendritic Spines in Hippocampal Neurons. Sci. Rep. 2017, 7, 17625. [Google Scholar] [CrossRef]
- Berna-Erro, A.; Jardin, I.; Salido, G.M.; Rosado, J.A. Role of STIM2 in cell function and physiopathology. J. Physiol. 2017, 595, 3111–3128. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Xie, Y.; Shen, Y.; Yin, Q.; Yuan, H. A Rare de novo Interstitial Duplication at 4p15.2 in a Boy with Severe Congenital Heart Defects, Limb Anomalies, Hypogonadism, and Global Developmental Delay. Cytogenet. Genome Res. 2016, 150, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Chitayat, D.; Ruvalcaba, R.H.; Babul, R.; Teshima, I.E.; Posnick, J.C.; Vekemans, M.J.; Scarpelli, H.; Thuline, H. Syndrome of proximal interstitial deletion 4p15: Report of three cases and review of the literature. Am. J. Med. Genet. 1995, 55, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Berna-Erro, A.; Braun, A.; Kraft, R.; Kleinschnitz, C.; Schuhmann, M.K.; Stegner, D.; Wultsch, T.; Eilers, J.; Meuth, S.G.; Stoll, G.; et al. STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci. Signal 2009, 2, ra67. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Point Mutation | Disease | Patient Symptoms |
|---|---|---|
| Gain-of-function STIM1 mutations | ||
| STIM1-R304W/Q | Stormorken syndrome | Bleeding diathesis, thrombocytopenia, miosis, mild anemia, asplenia, headache, ichthyosis, and proximal muscle weakness. |
| STIM1-R304W | YPS | Enlarged platelets with giant organelles called “opaque organelles”, target organelles, and reduced number of alpha granules. Myopathy with tubular aggregates and asplenia/hyposplenia. |
| STIM1-K365N | Stormorken syndrome | Bleeding diathesis, headache, stroke-like episodes, anemia, thrombocytopenia, and mild myopathy. |
| STIM1-H72Q | TAM | Muscle weakness, cramps, myalgia, elevated levels of creatine kinase, accumulation of packed membrane tubules highly ordered and aligned in parallel, large variations in muscle fiber size, type 1 fiber predominance, and increased number of fibers with internalized nuclei. |
| STIM1-N80T | TAM | |
| STIM1-G81D | TAM | |
| STIM1-D84G | TAM | |
| STIM1-L96V | TAM | |
| STIM1-F108I/L | TAM | |
| STIM1-H109N/R | TAM | |
| STIM1-I115F | TAM | |
| YPS | Enlarged platelets with giant organelles called “opaque organelles”, target organelles, and reduced number of alpha granules. Myopathy with tubular aggregates and asplenia/hyposplenia. | |
| STIM1-K104N | Muscle disorders | Muscle tubular aggregates. |
| STIM1-V138L | Muscle disorders | Congenial fiber type size disproportion. |
| STIM1L-S630F | Muscle disorders | Type I fiber atrophy. |
| STIM1L-R749H | Muscle disorders | Severe muscle dystrophy with inflammatory infiltrates. |
| STIM1L-H632fs | Muscle disorders | Mild variation in fiber size. |
| Loss-of-function STIM1 mutations | ||
| STIM1-E128R | Not classified | Autoimmune hemolytic anemia and immune thrombocytopenia, hepatosplenomegaly, lymphadenopathy, and nonprogressive muscular hypotonia. |
| STIM1 1538-1G>A | Not classified | T-cell deficiency. |
| STIM1 P165Q | Not classified | Immune and inflammatory disorders. |
| STIM1-R429C | Not classified | Dental enamel defect, anhidrosis, generalized eczema, mild muscular hypotonia, light-insensitive pupils, primary enuresis, and combined immunodeficiency. |
| STIM1-R426C | Not classified | Enamel defects with dental attrition, nail dysplasia, and frequent throat infections. |
| STIM1-F229L | Not classified | Mild immune deficiency, muscle weakness, dysmorphic facies, hypoplastic patellae, dental abnormalities, and hyperelasticity. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berna-Erro, A.; Sanchez-Collado, J.; Nieto-Felipe, J.; Macias-Diaz, A.; Redondo, P.C.; Smani, T.; Lopez, J.J.; Jardin, I.; Rosado, J.A. The Ca2+ Sensor STIM in Human Diseases. Biomolecules 2023, 13, 1284. https://doi.org/10.3390/biom13091284
Berna-Erro A, Sanchez-Collado J, Nieto-Felipe J, Macias-Diaz A, Redondo PC, Smani T, Lopez JJ, Jardin I, Rosado JA. The Ca2+ Sensor STIM in Human Diseases. Biomolecules. 2023; 13(9):1284. https://doi.org/10.3390/biom13091284
Chicago/Turabian StyleBerna-Erro, Alejandro, Jose Sanchez-Collado, Joel Nieto-Felipe, Alvaro Macias-Diaz, Pedro C. Redondo, Tarik Smani, Jose J. Lopez, Isaac Jardin, and Juan A. Rosado. 2023. "The Ca2+ Sensor STIM in Human Diseases" Biomolecules 13, no. 9: 1284. https://doi.org/10.3390/biom13091284
APA StyleBerna-Erro, A., Sanchez-Collado, J., Nieto-Felipe, J., Macias-Diaz, A., Redondo, P. C., Smani, T., Lopez, J. J., Jardin, I., & Rosado, J. A. (2023). The Ca2+ Sensor STIM in Human Diseases. Biomolecules, 13(9), 1284. https://doi.org/10.3390/biom13091284