
Pegylated Liposomal Alendronate Biodistribution, Immune Modulation, and Tumor Growth Inhibition in a Murine Melanoma Model
 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Key Chemicals and Formulations
2.2. Cell Culture
2.3. In Vitro Cytotoxicity
2.4. RT-qPCR
2.5. Animals
2.6. Alendronate Quantitation
2.7. Fluorescence Microscopy
2.8. Flow Cytometric Immunophenotyping
2.9. Statistical Analyses
3. Results
3.1. Characterization of Liposomal Formulations
3.2. In Vitro Cytotoxicity
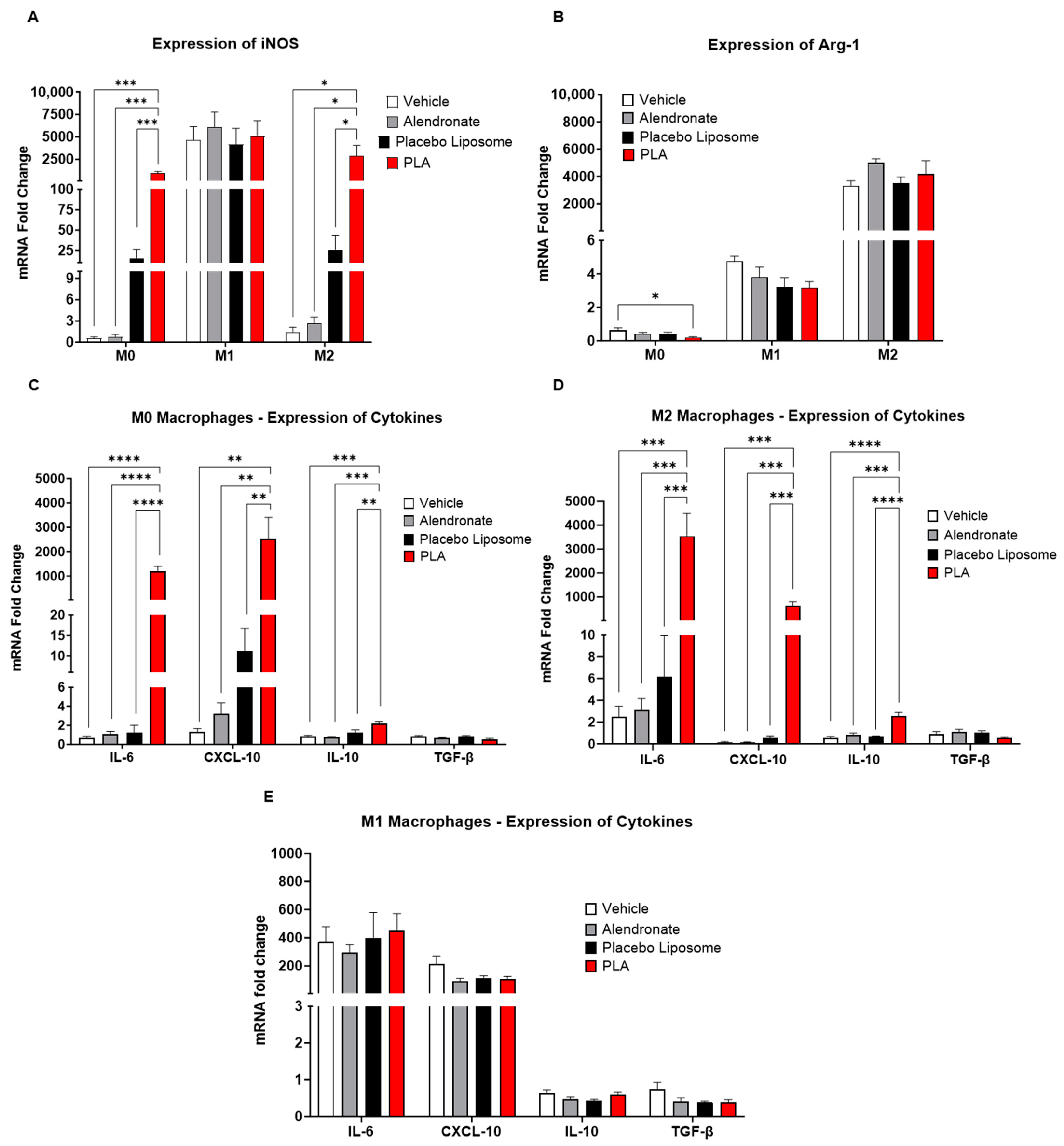
3.3. Modulation of Immunological Pathways in Macrophages
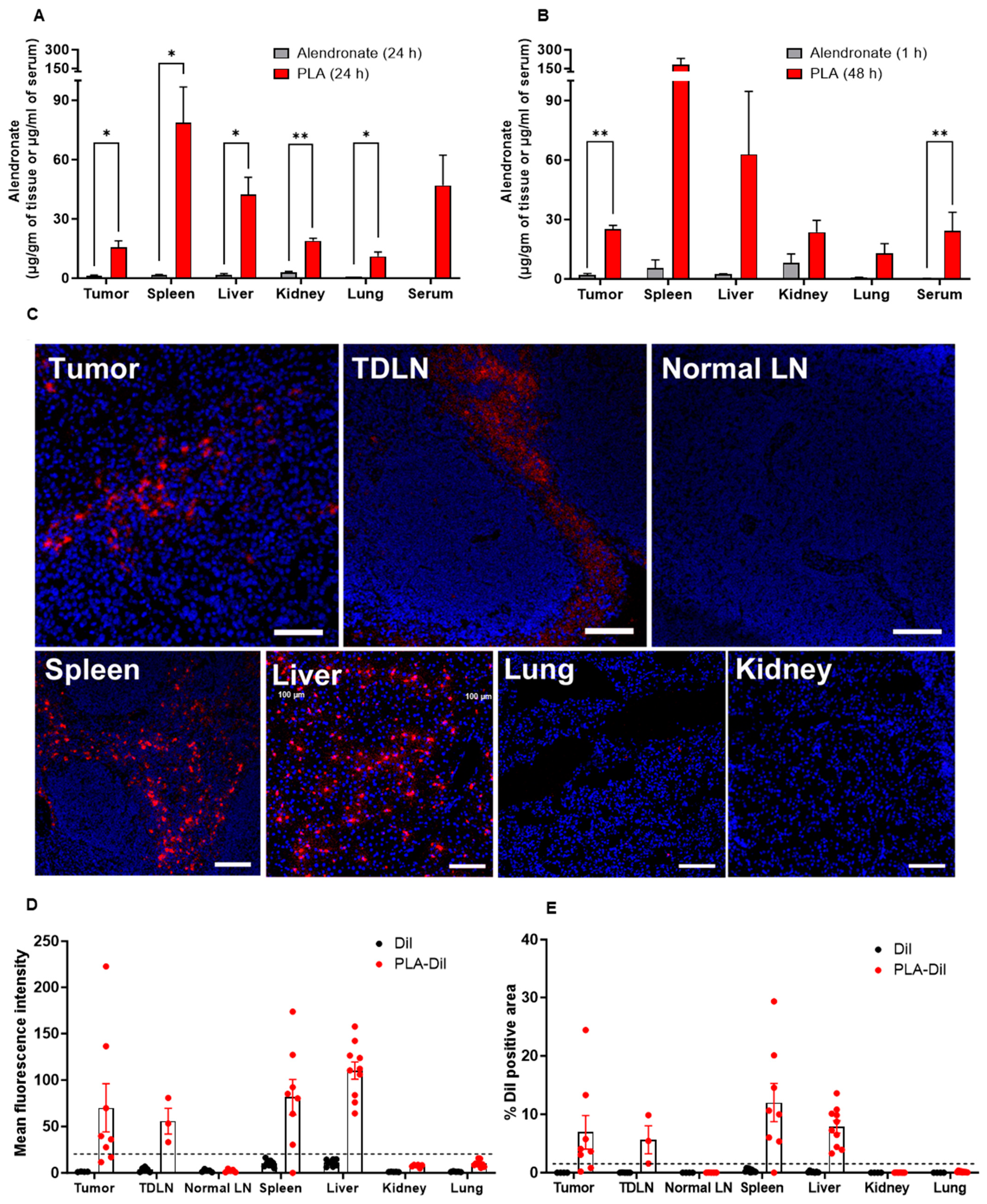
3.4. PLA Is Stable In Vivo and Accumulates in Tumors and Tumor-Draining Lymph Nodes
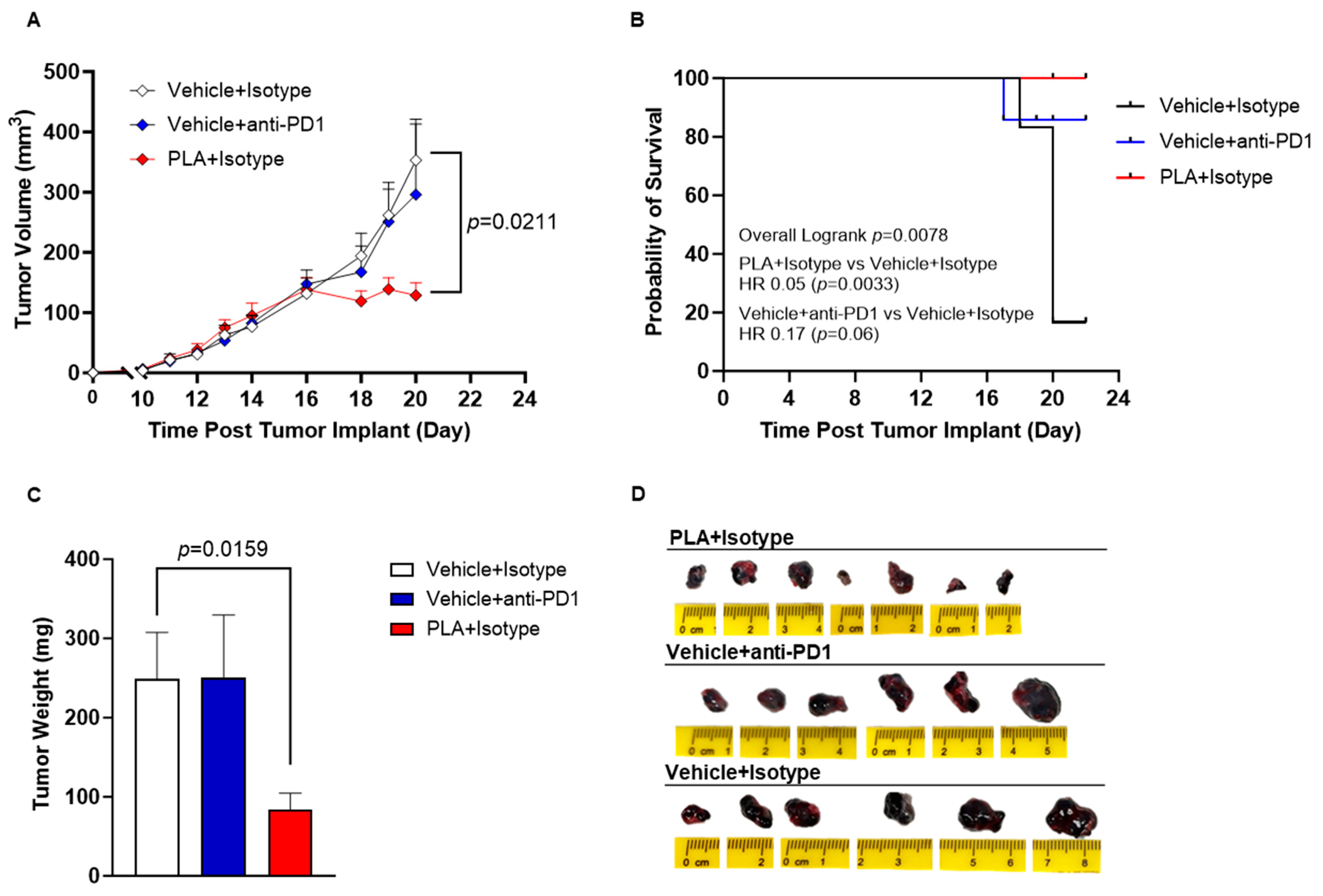
3.5. PLA Inhibits Tumor Growth
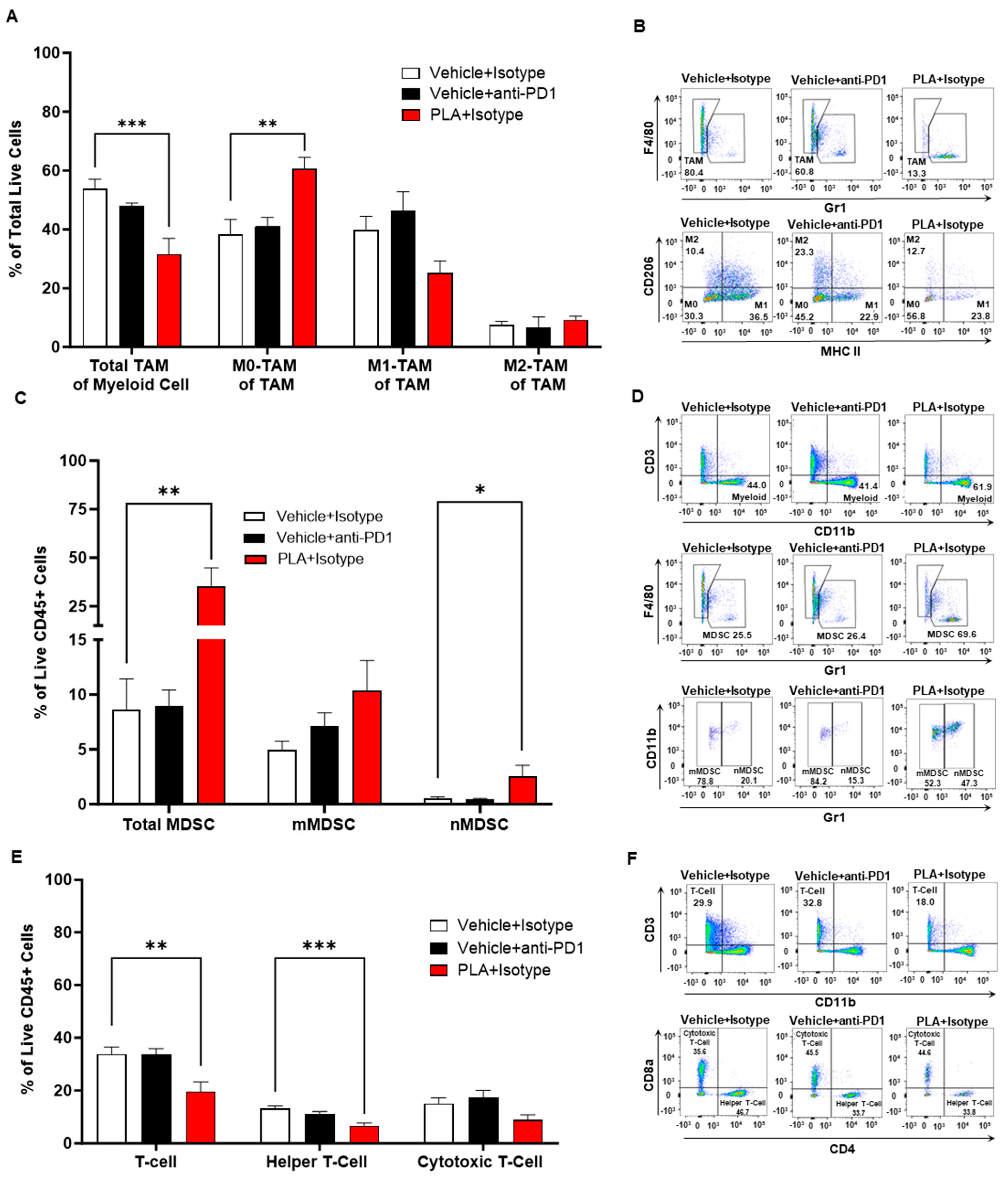
3.6. PLA Remodels the Tumor Immunologic Milieu
3.7. PLA Is Tolerable in Mouse Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sato, M.; Grasser, W.; Endo, N.; Akins, R.; Simmons, H.; Thompson, D.D.; Golub, E.; Rodan, G.A. Bisphosphonate action. Alendronate localization in rat bone and effects on osteoclast ultrastructure. J. Clin. Investig. 1991, 88, 2095–2105. [Google Scholar] [CrossRef] [PubMed]
- Rodan, G.A.; Fleisch, H.A. Bisphosphonates: Mechanisms of action. J. Clin. Investig. 1996, 97, 2692–2696. [Google Scholar] [CrossRef]
- Roelofs, A.J.; Thompson, K.; Gordon, S.; Rogers, M.J. Molecular Mechanisms of Action of Bisphosphonates: Current Status. Clin. Cancer Res. 2006, 12, 6222s–6230s. [Google Scholar] [CrossRef]
- Luckman, S.P.; Hughes, D.E.; Coxon, F.P.; Russell, R.G.G.; Rogers, M.J. Nitrogen-Containing Bisphosphonates Inhibit the Mevalonate Pathway and Prevent Post-Translational Prenylation of GTP-Binding Proteins, Including Ras. J. Bone Miner. Res. 1998, 13, 581–589. [Google Scholar] [CrossRef]
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Adjuvant bisphosphonate treatment in early breast cancer: Meta-analyses of individual patient data from randomised trials. Lancet 2015, 386, 1353–1361. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Van Poznak, C.; Harker, W.G.; Gradishar, W.J.; Chew, H.; Dakhil, S.R.; Haley, B.B.; Sauter, N.; Mohanlal, R.; Zheng, M.; et al. Continued Treatment Effect of Zoledronic Acid Dosing Every 12 vs. 4 Weeks in Women with Breast Cancer Metastatic to Bone: The OPTIMIZE-2 randomized clinical trial. JAMA Oncol. 2017, 3, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Himelstein, A.L.; Foster, J.C.; Khatcheressian, J.L.; Roberts, J.D.; Seisler, D.K.; Novotny, P.J.; Qin, R.; Go, R.S.; Grubbs, S.S.; O’connor, T.; et al. Effect of Longer-Interval vs Standard Dosing of Zoledronic Acid on Skeletal Events in Patients with Bone Metastases: A Randomized Clinical Trial. JAMA 2017, 317, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.; Gnant, M.; Morgan, G.; Clezardin, P. Effects of Bone-Targeted Agents on Cancer Progression and Mortality. J. Natl. Cancer Inst. 2012, 104, 1059–1067. [Google Scholar] [CrossRef]
- Coleman, R.; Cameron, D.; Dodwell, D.; Bell, R.; Wilson, C.; Rathbone, E.; Keane, M.; Gil, M.; Burkinshaw, R.; Grieve, R.; et al. Adjuvant zoledronic acid in patients with early breast cancer: Final efficacy analysis of the AZURE (BIG 01/04) randomised open-label phase 3 trial. Lancet Oncol. 2014, 15, 997–1006. [Google Scholar] [CrossRef]
- van Rooijen, N.; van Nieuwmegen, R. Elimination of phagocytic cells in the spleen after intravenous injection of liposome-encapsulated dichloromethylene diphosphonate: An enzyme-histochemical study. Cell Tissue Res. 1984, 238, 355–358. [Google Scholar] [CrossRef]
- Zeisberger, S.M.; Odermatt, B.; Marty, C.; Zehnder-Fjällman, A.H.M.; Ballmer-Hofer, K.; Schwendener, R.A. Clodronate-liposome-mediated depletion of tumour-associated macrophages: A new and highly effective antiangiogenic therapy approach. Br. J. Cancer 2006, 95, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Llorente, C.; Schnabl, B. Fast-Track Clearance of Bacteria from the Liver. Cell Host Microbe 2016, 20, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Hoh, C.H.; Tan, Y.H.; Gan, Y.-H. Protective Role of Kupffer Cells and Macrophages in Klebsiella pneumoniae-Induced Liver Abscess Disease. Infect. Immun. 2019, 87, 10–128. [Google Scholar] [CrossRef]
- O’sullivan, T.; Saddawi-Konefka, R.; Vermi, W.; Koebel, C.M.; Arthur, C.; White, J.M.; Uppaluri, R.; Andrews, D.M.; Ngiow, S.F.; Teng, M.W.; et al. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J. Exp. Med. 2012, 209, 1869–1882. [Google Scholar] [CrossRef]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy against Pancreatic Carcinoma in Mice and Humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef]
- La-Beck, N.M.; Liu, X.; Shmeeda, H.; Shudde, C.; Gabizon, A.A. Repurposing amino-bisphosphonates by liposome formulation for a new role in cancer treatment. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2021; Volume 68, pp. 175–185. [Google Scholar] [CrossRef]
- Bone, H.G.; Hosking, D.; Devogelaer, J.-P.; Tucci, J.R.; Emkey, R.D.; Tonino, R.P.; Rodriguez-Portales, J.A.; Downs, R.W.; Gupta, J.; Santora, A.C.; et al. Ten Years’ Experience with Alendronate for Osteoporosis in Postmenopausal Women. N. Engl. J. Med. 2004, 350, 1189–1199. [Google Scholar] [CrossRef]
- Hodgins, N.O.; Al-Jamal, W.T.; Wang, J.T.-W.; Parente-Pereira, A.C.; Liu, M.; Maher, J.; Al-Jamal, K.T. In vitro potency, in vitro and in vivo efficacy of liposomal alendronate in combination with γδ T cell immunotherapy in mice. J. Control. Release 2016, 241, 229–241. [Google Scholar] [CrossRef]
- Parente-Pereira, A.C.; Shmeeda, H.; Whilding, L.M.; Zambirinis, C.P.; Foster, J.; van der Stegen, S.J.C.; Beatson, R.; Zabinski, T.; Brewig, N.; Sosabowski, J.K.; et al. Adoptive Immunotherapy of Epithelial Ovarian Cancer with Vγ9Vδ2 T Cells, Potentiated by Liposomal Alendronic Acid. J. Immunol. 2014, 193, 5557–5566. [Google Scholar] [CrossRef]
- Guo, R.; Schreyer, M.; Chang, J.C.; Rothenberg, S.M.; Henry, D.; Cotzia, P.; Kris, M.G.; Rekhtman, N.; Young, R.J.; Hyman, D.M.; et al. Response to Selective RET Inhibition with LOXO-292 in a Patient with RET Fusion-Positive Lung Cancer with Leptomeningeal Metastases. JCO Precis. Oncol. 2019, 3, PO.19.00021. [Google Scholar] [CrossRef]
- Wang, S.; Zhou, Z.; Hu, R.; Dong, M.; Zhou, X.; Ren, S.; Zhang, Y.; Chen, C.; Huang, R.; Zhu, M.; et al. Metabolic Intervention Liposome Boosted Lung Cancer Radio-Immunotherapy via Hypoxia Amelioration and PD-L1 Restraint. Adv. Sci. 2023, 10, e2207608. [Google Scholar] [CrossRef]
- Golombek, S.K.; May, J.-N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.J.; Mohammadtaghi, S.; Uster, P.S.; Glass, D.; Peters, A.M.; Vile, R.G.; Stewart, J.S. Effective targeting of solid tumors in patients with locally advanced cancers by radiolabeled pegylated liposomes. Clin. Cancer Res. 2001, 7, 243–254. [Google Scholar]
- Lee, H.; Shields, A.F.; Siegel, B.A.; Miller, K.D.; Krop, I.; Ma, C.X.; LoRusso, P.M.; Munster, P.N.; Campbell, K.; Gaddy, D.F.; et al. 64Cu-MM-302 Positron Emission Tomography Quantifies Variability of Enhanced Permeability and Retention of Nanoparticles in Relation to Treatment Response in Patients with Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 4190–4202. [Google Scholar] [CrossRef]
- Shmeeda, H.; Amitay, Y.; Gorin, J.; Tzemach, D.; Mak, L.; Stern, S.T.; Barenholz, Y.; Gabizon, A. Coencapsulation of alendronate and doxorubicin in pegylated liposomes: A novel formulation for chemoimmunotherapy of cancer. J. Drug Target. 2016, 24, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, G.R. Phosphorus Assay in Column Chromatography. J. Biol. Chem. 1959, 234, 466–468. [Google Scholar] [CrossRef]
- Breil, C.; Vian, M.A.; Zemb, T.; Kunz, W.; Chemat, F. “Bligh and Dyer” and Folch Methods for Solid–Liquid–Liquid Extraction of Lipids from Microorganisms. Comprehension of Solvatation Mechanisms and towards Substitution with Alternative Solvents. Int. J. Mol. Sci. 2017, 18, 708. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, X.; Tang, Y.; Lu, Y.; Guo, L.; Zhong, D. Bioequivalence of generic alendronate sodium tablets (70 mg) to Fosamax® tablets (70 mg) in fasting, healthy volunteers: A randomized, open-label, three-way, reference-replicated crossover study. Drug Des. Dev. Ther. 2017, ume 11, 2109–2119. [Google Scholar] [CrossRef]
- Kedl, R.M.; Jordan, M.; Potter, T.; Kappler, J.; Marrack, P.; Dow, S. CD40 stimulation accelerates deletion of tumor-specific CD8+T cells in the absence of tumor-antigen vaccination. Proc. Natl. Acad. Sci. USA 2001, 98, 10811–10816. [Google Scholar] [CrossRef]
- Gabizon, A.; Shmeeda, H.; Barenholz, Y. Pharmacokinetics of Pegylated Liposomal Doxorubicin: Review of animal and human studies. Clin. Pharmacokinet. 2003, 42, 419–436. [Google Scholar] [CrossRef]
- Zhuang, H.; Dai, X.; Zhang, X.; Mao, Z.; Huang, H. Sophoridine suppresses macrophage-mediated immunosuppression through TLR4/IRF3 pathway and subsequently upregulates CD8+ T cytotoxic function against gastric cancer. Biomed. Pharmacother. 2019, 121, 109636. [Google Scholar] [CrossRef]
- Olmsted-Davis, E.; Mejia, J.; Salisbury, E.; Gugala, Z.; Davis, A.R. A Population of M2 Macrophages Associated with Bone Formation. Front. Immunol. 2021, 12, 686769. [Google Scholar] [CrossRef]
- Bieghs, V.; Wouters, K.; van Gorp, P.J.; Gijbels, M.J.; de Winther, M.P.; Binder, C.J.; Lütjohann, D.; Febbraio, M.; Moore, K.J.; van Bilsen, M.; et al. Role of Scavenger Receptor A and CD36 in Diet-Induced Nonalcoholic Steatohepatitis in Hyperlipidemic Mice. Gastroenterology 2010, 138, 2477–2486.e3. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.P.; Lin, F.; Yi, D.; Xie, Y.; Dinh, J.; Xue, P.; Sul, H.S. Aging-dependent regulatory cells emerge in subcutaneous fat to inhibit adipogenesis. Dev. Cell 2021, 56, 1437–1451.e3. [Google Scholar] [CrossRef] [PubMed]
- Surette, F.A.; Guthmiller, J.J.; Li, L.; Sturtz, A.J.; Vijay, R.; Pope, R.L.; McClellan, B.L.; Pack, A.D.; Zander, R.A.; Shao, P.; et al. Extrafollicular CD4 T cell-derived IL-10 functions rapidly and transiently to support anti-Plasmodium humoral immunity. PLoS Pathog. 2021, 17, e1009288. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.; Sun, T.; Lu, W.; Smith, A.O.; Bao, Y.; Adzraku, S.Y.; Qi, K.; Xu, K.; Qiao, J.; Zeng, L. Reference gene selection and validation for mRNA expression analysis by RT-qPCR in murine M1- and M2-polarized macrophage. Mol. Biol. Rep. 2020, 47, 2735–2748. [Google Scholar] [CrossRef]
- Chen, M.; Liu, K.; Zhong, D.; Chen, X. Trimethylsilyldiazomethane derivatization coupled with solid-phase extraction for the determination of alendronate in human plasma by LC-MS/MS. Anal. Bioanal. Chem. 2011, 402, 791–798. [Google Scholar] [CrossRef]
- Rajan, R.; Sabnani, M.K.; Mavinkurve, V.; Shmeeda, H.; Mansouri, H.; Bonkoungou, S.; Le, A.D.; Wood, L.M.; Gabizon, A.A.; La-Beck, N.M. Liposome-induced immunosuppression and tumor growth is mediated by macrophages and mitigated by liposome-encapsulated alendronate. J. Control. Release 2017, 271, 139–148. [Google Scholar] [CrossRef]
- Sabnani, M.K.; Rajan, R.; Rowland, B.; Mavinkurve, V.; Wood, L.M.; Gabizon, A.A.; La-Beck, N.M. Liposome promotion of tumor growth is associated with angiogenesis and inhibition of antitumor immune responses. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 259–262. [Google Scholar] [CrossRef]
- La-Beck, N.M.; Gabizon, A.A. Nanoparticle Interactions with the Immune System: Clinical Implications for Liposome-Based Cancer Chemotherapy. Front. Immunol. 2017, 8, 416. [Google Scholar] [CrossRef]
- Edmonds, S.; Volpe, A.; Shmeeda, H.; Parente-Pereira, A.C.; Radia, R.; Baguña-Torres, J.; Szanda, I.; Severin, G.W.; Livieratos, L.; Blower, P.J.; et al. Exploiting the Metal-Chelating Properties of the Drug Cargo for in Vivo Positron Emission Tomography Imaging of Liposomal Nanomedicines. ACS Nano 2016, 10, 10294–10307. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Mantovani, A. Immunology in the clinic review series; focus on cancer: Tumour-associated macrophages: Undisputed stars of the inflammatory tumour microenvironment. Clin. Exp. Immunol. 2012, 167, 195–205. [Google Scholar] [CrossRef]
- Gazzaniga, S.; Bravo, A.I.; Guglielmotti, A.; van Rooijen, N.; Maschi, F.; Vecchi, A.; Mantovani, A.; Mordoh, J.; Wainstok, R. Targeting Tumor-Associated Macrophages and Inhibition of MCP-1 Reduce Angiogenesis and Tumor Growth in a Human Melanoma Xenograft. J. Investig. Dermatol. 2007, 127, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Bottazzi, B.; Colotta, F.; Sozzani, S.; Ruco, L. The origin and function of tumor-associated macrophages. Immunol. Today 1992, 13, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Tcyganov, E.; Mastio, J.; Chen, E.; Gabrilovich, D.I. Plasticity of myeloid-derived suppressor cells in cancer. Curr. Opin. Immunol. 2018, 51, 76–82. [Google Scholar] [CrossRef]
- Davis, R.J.; Moore, E.C.; Clavijo, P.E.; Friedman, J.; Cash, H.; Chen, Z.; Silvin, C.; Van Waes, C.; Allen, C. Anti-PD-L1 Efficacy Can Be Enhanced by Inhibition of Myeloid-Derived Suppressor Cells with a Selective Inhibitor of PI3Kδ/γ. Cancer Res. 2017, 77, 2607–2619. [Google Scholar] [CrossRef]
- Li, J.; Kim, S.Y.; Lainez, N.M.; Coss, D.; Nair, M.G. Macrophage-Regulatory T Cell Interactions Promote Type 2 Immune Homeostasis Through Resistin-Like Molecule α. Front. Immunol. 2021, 12, 710406. [Google Scholar] [CrossRef]
- Wang, S.; Liu, G.; Li, Y.; Pan, Y. Metabolic Reprogramming Induces Macrophage Polarization in the Tumor Microenvironment. Front. Immunol. 2022, 13, 840029. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, Y.; Song, W.; Jiang, X.; Deng, Z.; Xiong, W.; Shen, J. Metabolic reprogramming mediated PD-L1 depression and hypoxia reversion to reactivate tumor therapy. J. Control. Release 2022, 352, 793–812. [Google Scholar] [CrossRef]
- Gu, S.; Sachleben, J.R.; Boughter, C.T.; Nawrocka, W.I.; Borowska, M.T.; Tarrasch, J.T.; Skiniotis, G.; Roux, B.; Adams, E.J. Phosphoantigen-induced conformational change of butyrophilin 3A1 (BTN3A1) and its implication on Vγ9Vδ2 T cell activation. Proc. Natl. Acad. Sci. USA 2017, 114, E7311–E7320. [Google Scholar] [CrossRef]
- Roelofs, A.J.; Jauhiainen, M.; Mönkkönen, H.; Rogers, M.J.; Mönkkönen, J.; Thompson, K. Peripheral blood monocytes are responsible for γδ T cell activation induced by zoledronic acid through accumulation of IPP/DMAPP. Br. J. Haematol. 2008, 144, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Märten, A.; von Lilienfeld-Toal, M.; Büchler, M.W.; Schmidt, J. Zoledronic Acid has Direct Antiproliferative and Antimetastatic Effect on Pancreatic Carcinoma Cells and Acts as an Antigen for δ2 γ/δ T Cells. J. Immunother. 2007, 30, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Benzaïd, I.; Mönkkönen, H.; Stresing, V.; Bonnelye, E.; Green, J.; Mönkkönen, J.; Touraine, J.-L.; Clézardin, P. High Phosphoantigen Levels in Bisphosphonate-Treated Human Breast Tumors Promote Vγ9Vδ2 T-Cell Chemotaxis and Cytotoxicity in Vivo. Cancer Res. 2011, 71, 4562–4572. [Google Scholar] [CrossRef]
- Kunzmann, V.; Jomaa, H.; Feurle, J.; Bauer, E.; Herderich, M.; Wihelm, M. Stimulation of γδ T cells by aminobisphosphonates used in treatment of bone disorders. Bood 1997, 90, 575. [Google Scholar]
- Kunzmann, V.; Bauer, E.; Wilhelm, M. γ/δ T-Cell Stimulation by Pamidronate. N. Engl. J. Med. 1999, 340, 737–738. [Google Scholar] [CrossRef]
- Ding, Y.; Zeng, J.-C.; Yin, F.; Zhang, C.-L.; Zhang, Y.; Li, S.-X.; Liu, X.; Zhang, C.; Xue, Q.-Y.; Lin, H.; et al. Multicenter Study on Observation of Acute-phase Responses after Infusion of Zoledronic Acid 5 mg in Chinese Women with Postmenopausal Osteoporosis. Orthop. Surg. 2017, 9, 284–289. [Google Scholar] [CrossRef]
- Anastasilakis, A.D.; Polyzos, S.A.; Makras, P.; Sakellariou, G.T.; Bisbinas, I.; Gkiomisi, A.; Delaroudis, S.; Gerou, S.; Ballaouri, I.; Oikonomou, D.; et al. Acute phase response following intravenous zoledronate in postmenopausal women with low bone mass. Bone 2012, 50, 1130–1134. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, S.-H.; Chen, S.-C.; Chen, C.-Y.; Lin, T.-M. Zoledronic acid blocks the interaction between breast cancer cells and regulatory T-cells. BMC Cancer 2019, 19, 176. [Google Scholar] [CrossRef]
- Hodgins, N.O.; Al-Jamal, W.T.; Wang, J.T.-W.; Klippstein, R.; Costa, P.M.; Sosabowski, J.K.; Marshall, J.F.; Maher, J.; Al-Jamal, K.T. Investigating in vitro and in vivo αvβ6 integrin receptor-targeting liposomal alendronate for combinatory γδ T cell immunotherapy. J. Control. Release 2017, 256, 141–152. [Google Scholar] [CrossRef]
- Man, F.; Lim, L.; Volpe, A.; Gabizon, A.; Shmeeda, H.; Draper, B.; Parente-Pereira, A.C.; Maher, J.; Blower, P.J.; Fruhwirth, G.O.; et al. In Vivo PET Tracking of 89Zr-Labeled Vγ9Vδ2 T Cells to Mouse Xenograft Breast Tumors Activated with Liposomal Alendronate. Mol. Ther. 2018, 27, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Trinh, S.; Le, A.; Gowani, S.; La-Beck, N.M. Management of Immune-Related Adverse Events Associated with Immune Checkpoint Inhibitor Therapy: A Minireview of Current Clinical Guidelines. Asia-Pac. J. Oncol. Nurs. 2019, 6, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Chulkina, M.; Beswick, E.J.; Pinchuk, I.V. Role of PD-L1 in Gut Mucosa Tolerance and Chronic Inflammation. Int. J. Mol. Sci. 2020, 21, 9165. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, M.R.; Patel, J.; Back, P.I.; Shmeeda, H.; Kallem, R.R.; Shudde, C.; Markiewski, M.; Putnam, W.C.; Gabizon, A.A.; La-Beck, N.M. Pegylated Liposomal Alendronate Biodistribution, Immune Modulation, and Tumor Growth Inhibition in a Murine Melanoma Model. Biomolecules 2023, 13, 1309. https://doi.org/10.3390/biom13091309
Islam MR, Patel J, Back PI, Shmeeda H, Kallem RR, Shudde C, Markiewski M, Putnam WC, Gabizon AA, La-Beck NM. Pegylated Liposomal Alendronate Biodistribution, Immune Modulation, and Tumor Growth Inhibition in a Murine Melanoma Model. Biomolecules. 2023; 13(9):1309. https://doi.org/10.3390/biom13091309
Chicago/Turabian StyleIslam, Md. Rakibul, Jalpa Patel, Patricia Ines Back, Hilary Shmeeda, Raja Reddy Kallem, Claire Shudde, Maciej Markiewski, William C. Putnam, Alberto A. Gabizon, and Ninh M. La-Beck. 2023. "Pegylated Liposomal Alendronate Biodistribution, Immune Modulation, and Tumor Growth Inhibition in a Murine Melanoma Model" Biomolecules 13, no. 9: 1309. https://doi.org/10.3390/biom13091309