
Immunologic Aspects in Fibrodysplasia Ossificans Progressiva

Division of Endocrinology and Metabolism, Department of Medicine, the Institute for Human Genetics, the Program in Craniofacial Biology, University of California, San Francisco, CA 94143, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Biomolecules 2024, 14(3), 357; https://doi.org/10.3390/biom14030357
Submission received: 3 February 2024
/
Revised: 2 March 2024
/
Accepted: 11 March 2024
/
Published: 16 March 2024
(This article belongs to the Special Issue Fibrodysplasia Ossificans Progressiva (FOP): From Molecular Mechanisms to Therapeutic Strategies)
Abstract
:Background: Inflammation is a major driver of heterotopic ossification (HO), a condition of abnormal bone growth in a site that is not normally mineralized. Purpose of review: This review will examine recent findings on the roles of inflammation and the immune system in fibrodysplasia ossificans progressiva (FOP). FOP is a genetic condition of aggressive and progressive HO formation. We also examine how inflammation may be a valuable target for the treatment of HO. Rationale/Recent findings: Multiple lines of evidence indicate a key role for the immune system in driving FOP pathogenesis. Critical cell types include macrophages, mast cells, and adaptive immune cells, working through hypoxia signaling pathways, stem cell differentiation signaling pathways, vascular regulatory pathways, and inflammatory cytokines. In addition, recent clinical reports suggest a potential role for immune modulators in the management of FOP. Future perspectives: The central role of inflammatory mediators in HO suggests that the immune system may be a common target for blocking HO in both FOP and non-genetic forms of HO. Future research focusing on the identification of novel inflammatory targets will help support the testing of potential therapies for FOP and other related conditions.
1. Introduction
Bone homeostasis is closely linked with immune function. The key cell types responsible for maintaining the balance between bone formation and resorption are osteoblasts and osteoclasts. Osteoblasts originate from mesenchymal stem cells (MSCs) in the long bones, or MSC-like cells in the craniofacial bones, specifically in the neural crest lineage [1]. Conversely, osteoclasts originate from hematopoietic progenitors from the monocyte/macrophage lineage [2]. Osteoclast formation is driven by the binding of the receptor activator of Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) ligand (RANKL) to the RANK receptor [3,4]. Additionally, macrophages appear to promote osteoblast formation, as evidenced by studies showing that macrophage-deficient mice exhibit reduced bone density and impaired MSC differentiation into osteoblasts [5]. Osteal macrophages (also known as “osteomacs”, a specific subtype of macrophages residing in bone tissues) collaborate with pro-inflammatory macrophages to stimulate anabolic bone formation during fracture repair [6]. Furthermore, various diseases affecting the immune system or causing autoimmunity are associated with bone loss [7]. Together, these findings suggest that innate immune cells, including macrophages, may play a significant role in the development of bone disorders and could potentially serve as therapeutic targets for the treatment of a spectrum of bone diseases.
While inflammation is essential for normal tissue repair, an uncontrolled inflammatory response can lead to pathological conditions [8]. The connections between the immune system and conditions of abnormal bone formation, such as heterotopic ossification (HO), have been investigated for several decades. HO is a severe and debilitating process of abnormal bone formation that occurs at non-mineralized sites [9,10]. HO can be triggered by trauma or burn injuries, or in conditions of severe inflammation such as in rheumatologic diseases [10,11,12,13,14].
2. Inflammatory Responses in the Context of Bone
Inflammatory cytokines and inflammatory cells are considered major drivers of HO formation after injuries or in pathologies prone to HO [15,16,17,18,19,20]. Pro-inflammatory cytokines, such as interleukin 3 (IL-3), are elevated in patients with combat wounds [18]. Additionally, in murine models, interleukin (IL) 1β (IL-1β) secreted by the inflammasome is elevated in bone marrow-derived macrophages (BMDMs), while suppression of IL-1β secretion can cause recovery of bone mass in ovariectomized mice [21,22]. Interestingly, in human MSCS, inflammasome stimulation results in enhanced adipogenesis and decreased osteogenesis [21,23]. Another member of the interleukin family, interleukin 6 (IL-6), activates bone degradation, suggesting potential involvement in osteoporosis [24]. The natural antagonist of the pro-inflammatory cytokine IL-18, IL-18BP, has been found to be low in osteoporotic women; animal studies indicate a stimulatory role of IL-18BP in osteoblast differentiation, preservation of cortical bone, and restoration of trabecular microarchitecture, as well as an inhibitory role in inflammasome activation and osteoclastogenesis [25]. Further, loss of the anti-inflammatory cytokine IL-10 worsens the bone loss in type 1 diabetes [26]. Another bone-protecting cytokine is IL-33, which may hold therapeutic potential against bone resorption [21]. Additionally, TNF-α is known to repress osteoblast function during differentiation, while triggering osteoclast proliferation and differentiation [27]. Non-genetic HO after high energy trauma in penetrating war injuries show elevations in IL-6, IL-10, and monocyte chemoattractant protein 1 (MCP-1) [17]. Based on the large number of inflammatory cytokines found in HO, a number of anti-inflammatory therapies are used clinically in an attempt to decrease the risk of HO after major hip procedures, although the blockade is not complete [16,19,28].
Because of the heterogeneity of triggers in non-genetic HO, it is useful to examine the signaling pathways and mechanisms active in genetic forms of HO. Much has changed since our last review of this area in 2019 [29]. This review updates the current state of knowledge regarding macrophages’ involvement in fibrodysplasia ossificans progressiva (FOP), a genetic disorder of severe progressive HO formation. We also emphasize the impact of the causative activin A type 1 receptor (ACVR1) genetic mutations on the inflammatory microenvironment, fibrotic tissue transformation, and unique interactions with other signaling pathways. Additionally, we discuss potential therapeutic strategies targeting the immune system being studied for FOP management.
3. Molecular Aspects of Osteogenic and Chondrogenic Differentiation
Bone remodeling comprises a series of sequential events [30]. The first is activation, where elusive stimulatory triggers determine the starting point of bone formation by activating osteoblasts and bone lining cells. This is the location to which osteoclasts are then summoned and where they transform into their mature form [31]. Next, the previously developed mature osteoclasts enzymatically degrade the inorganic matrix during the resorption phase. After bone resorption is complete, osteoclast apoptosis follows, and macrophage-like cells drift to the resorbed area to remove any remaining osteoclast-generated debris in a step called reversal. During the end stages of the reversal process, when the osteoclast-mediated bone resorption wanes, osteoclasts secrete sphingosine 1-phosphate, which incites osteoblast recruitment [32]. Next, more osteoblasts are recruited to the resorbed site, where they assemble a new inorganic matrix, and the subsequent mineralization fills up the resorption lacuna during formation. After formation, the bone remodeling cycle concludes with the termination phase, whereby osteoblasts have three possible fates, meaning that they undergo apoptosis, transform into bone lining cells, or become trapped into the bone matrix, where they end up differentiating into osteocytes [30,32]. The bone matrix-embedded osteocytes will control and coordinate future events of bone remodeling [32].
Bone tissue turnover and regeneration require crucial, yet complex and not fully elucidated, interactions with the immune system. Disruption can result in a wide range of inflammatory disorders in the skeleton [30,33]. Intriguingly, there is a crosstalk between cells of the immune system and cytokines, with bone cells (i.e., osteoblasts, osteoclasts, and osteocytes) implicated in the modulation of both bone homeostasis and immune cell proliferation, differentiation, and function in target cells. This bidirectional crosstalk has been termed ‘osteoimmunology’ [30,32,34,35]. Immune cells involved in bone regeneration include neutrophils, macrophages, dendritic cells, innate lymphoid cells, T cells, Th1/Th2 and Th17 cells, T regulatory cells (Tregs), and B cells [30,35]. Additionally, bone cells and immune cells are not merely co-present in the bone marrow milieu, but also appear to have overlapping progenitor cells and modulatory molecules, which renders their two-way communication more intricate [30].
Immune cytokines that play an essential role in bone regeneration include TNF-α, IL-1, IL-6, IL-17, and IFN-γ, as well as numerous other cytokines (e.g., IL-12, IL-23, IL-27, IL-35) [30,33,36]. More specifically, the immune-related cytokines IL-1β, ΙL-6, IL-17, and TNF-α are thought to be the primary inducers of osteoclastogenesis by enhancing the expression of factors involved in osteoclast differentiation, such as RANKL and the macrophage-colony-stimulating factor (M-CSF), favoring bone resorption [33,37]. Another class of inflammatory molecules promoting osteoclastogenesis is that of the chemokines, involving several C-C motif chemokine ligands (CCLs) (e.g., CCL3, CCL5, CCL9, etc.), the C-X3-C motif chemokine ligand (CX3CL1), as well as the X-C motif ligand 1 (XCL1), all of which enhance osteoclast differentiation [33,37]. Some of these inflammatory molecules exert adverse effects on osteoblasts by inhibiting their differentiation and stimulating osteocyte apoptosis, leading to decreased formation, mineralization, and density in osseous tissue [33]. In contrast, other immune- and bone-cell-secreted factors, such as osteoprotegerin (OPG), which functions as a decoy receptor for RANKL, impede osteoclast differentiation [31,33], while immune-related molecules, such as IL-4, IL-10, IL-27, interferons, annexin A1, and cardiotrophin-like cytokine factor 1 (CLCF1), constrain osteoclastogenesis and osteoclast differentiation [33].
Bone remodeling is primarily maintained by the absorptive properties of osteoclasts and the bone formative properties of osteoblasts in a finely tuned spatiotemporal manner, with the additional contribution of osteocytes that sense mechanical stressors, and bone lining cells that instigate skeletal turnover, by degrading the bone matrix [30,31,32,35,36], and capillary blood supply [32]. The bone forming osteoblasts arise from multi-potent MSCs and their main actions are to generate bone matrix proteins and mineralize the bone, but which also express factors involved in osteoclastogenesis [32]. Together, these different types of cells form an intricate complex termed a ‘basic multicellular unit’ (BMU), which is essential for efficient bone remodeling [32]. The transcription factor runt-related transcription factor 2 (RUNX2) is indispensable for osteoblast development and differentiation [32,35]. Separately, osteoclasts emanate from the monocyte/macrophage hematopoietic lineage and hold the capacity to degrade mineralized bone, cartilage, and dentine [31,32]. Osteoclast maturation and function are also necessary, and can be followed by osteoclast markers (e.g., integrin receptors, cathepsin K, calcitonin receptor, tartrate-resistant acid phosphatase), RANKL and M-CSF [31,32,35].
4. Inflammatory Responses and Inflammation-Associated Molecules and Immune Cells
The contribution of the immune and cytokine microenvironment to skeleton turnover and regeneration is critical for bone homeostasis and remodeling across the lifespan. For example, activated macrophages help direct the differentiation of osteoblasts, possibly through the secretion of oncostatin M (OSM) [38]. Additionally, the anabolic properties of parathyroid hormone on bone (i.e., IL-6 and IL-6 receptor (IL-6R)) may be mediated by T cells [39]. Interestingly, defects in the calcineurin (CN)/nuclear factor of activated T cells (NFAT) signaling pathway have been implicated in the enhancement of lipopolysaccharide (LPS) tolerance in macrophages, activating T cells and natural killer cells, and regulating B cell development—all of which may affect osteoclastogenesis regulation [32]. The wingless-related integration site 1 (Wnt1) has recently been shown to be produced by B-cells within the bone marrow niche, and to induce bone formation [30,35,40]. Injury of bone tissue stimulates an early innate immune response consisting of neutrophils, mast cells, monocytes, and macrophages, whereas a later adaptive immune response also occurs, consisting of T and B cells [30].
Infection is another trigger for the initiation of complex cell–cell communication between osteoclasts, osteoblasts, and immune cells [32]. Bone resorption during infection is modulated by both the innate and adaptive immune system, with the involvement of T cells, Tregs (i.e., specialized CD4 T cell subgroup), Th17 cells (i.e., IL-17-producing helper T cell), B cells, neutrophils, dendritic cells, and macrophages [32]. Immune cells serve as both anabolic and catabolic mediators in bone tissue: in the early stages of bone fracture healing, acute inflammatory response leads to chemokine secretion, which triggers the proliferation of MSCs and their downstream differentiation into osteoblasts, contributing to bone regeneration (i.e., anabolic function) [30]. In contrast, chronic inflammatory response results in bone resorption (i.e., catabolic function) through the repression of osteoblast-stimulated bone formation and the induction of osteolysis [30].
5. Fibrodysplasia Ossificans Progressiva Can Be Considered a Genetic Disease of Inflammation-Driven Heterotopic Ossification
FOP is a rare genetic disorder affecting approximately one in 1.36 million to 2 million individuals [41,42]. Clinically, FOP is characterized by large amounts of abnormal bone formation in tissues that are not normally mineralized, such as skeletal muscle, tendons, and connective tissues. Over time, the new HO leads to immobility and severe pain from nerve compression and stretching. Treatments remain largely symptomatic with only one approved directed therapy available.
The main cause of FOP is the presence of an activating mutation in the ACVR1, which is part of the bone morphogenetic protein (BMP) pathway [43]. The identified mutations in FOP are primarily located in the glycine-serine (GS) region of ACVR1, with the most common being the single-point mutation ACVR1 c. G617A/p.R206H [43]. This mutation reduces the stability of the GS region, resulting in continuous activation of ACVR1 and neoligand activity to Activin A, leading to HO and eventual joint fusion in patients with FOP (Figure 1). Other rare missense mutations in the GS or protein kinase (PK) domain of ACVR1 have also been reported in patients with FOP [44,45,46,47]. All of these gene mutations in FOP disrupt normal homeostasis and cell differentiation processes, triggering the abnormal endochondral ossification that is a hallmark of FOP [48].
Activin A serves as a competitive inhibitor of the BMP signaling pathway, as indicated by various studies [52,53,54,55]. Under normal conditions, activin A binds to the wild-type ACVR1 receptor and activates the signaling cascade through the SMAD2/3 pathway. The presence of the ACVR1R206H mutation changes the ACVR1 response to activin A, instead allowing signaling through the SMAD1/5/9 pathway—effectively misinterpreting activin A as a BMP. This is discussed in more detail below.
A striking and pathognomonic clinical feature of FOP is that patients can develop massive inflammatory lesions that ultimately lead to HO formation. These can occur with severe traumatic events but have been reported even after mild trauma [41]. These inflammatory lesions, often called “flare-ups,” are accompanied by classical symptoms of inflammation such as induration, erythema, and pain [56]. Tissue injury exacerbates the progression of FOP development. Histology shows the presence of many cell types in these developing FOP lesions, including macrophages, mast cells, MSCs, osteocytes, chondrocytes, and fibroblasts. Cytokines are reportedly elevated in the connective tissues, and within blood vessels and skeletal muscles [57,58,59,60]. 20% or more of these flare-ups result in significant bone formation. The mainstays of current FOP therapy are anti-inflammatory agents, including nonsteroidal anti-inflammatory drugs (NSAIDs) and corticosteroids. These can help reduce FOP HO formation [28], and are also used in the treatment of non-genetic HO [29]. However, these strategies still show poor efficacy when blocking new HO formation.
6. The Role of the Immune System in Fibrodysplasia Ossificans Progressiva
The role of the immune system in FOP has garnered significant interest. Hematopoietic-lineage cells have long been recognized as contributors to the early inflammatory phases [61]. A mouse model with an ACVR1R206H mutation showed the strong presence of macrophages and mast cells at the site of HO formation [62]. Suppression of the transforming growth factor-beta (TGF-β) in FOP mouse models attenuates HO [63], sugesting that macrophages have a critical role in the early phase of inflammation in developing FOP lesions. Our own research has revealed a significant increase in different cytokine levels, such as IL-3, IL-7, and IL-8 in the blood of patients with FOP [20] and has also shown that NF-κB activation is a key factor for inflammation in FOP. Additionally, we have found that blood samples obtained from FOP individuals have high levels of proinflammatory interleukins even in the absence of flare-up symptoms [29,64]. We also found increased monocyte production of TGF-β in patients with FOP, consistent with findings in mouse models [63]. However, the mechanism by which the ACVR1 mutation induces macrophage hyper-responsiveness remains largely unknown.
7. Macrophages in Fibrodysplasia Ossificans Progressiva: A Key Player in Disease Pathogenesis
Most forms of HO formation occur through a process of endochondral ossification [65,66,67]. This process involves four stages: inflammation, cartilage formation, osteogenesis, and ectopic bone maturation. In the inflammatory phase, immune cells infiltrate the injury site, as has been well demonstrated in FOP [68]. As the HO formation process progresses, local MSCs differentiate into chondrocytes [66], followed by chondrocyte hypertrophy and calcification during osteogenesis [67]. In the final stages of HO formation, the bone matures to form cancellous bone [65,66,67].
Although the specific influence of individual macrophage subtypes on chondrogenic formation in HO patients is not yet fully understood, insights can be drawn from other diseases that affect cartilage. Inflammatory factors like TGF-β can promote chondrogenesis and osteogenesis [63]. Studies on osteoarthritis patients suggest that M1 (inflammatory) macrophages can also induce cartilage apoptosis, while M2 (tissue repair) macrophages can promote cartilage hypertrophy and ectopic bone formation [69,70].
Notably, the osteogenic and chondrogenic differentiation of MSCs or MSC-like cells is a critical process in the development of HO [71]. Different subtypes of macrophages are able to regulate the osteogenic differentiation of MSCs [72], and the depletion of macrophages can decrease the osteogenic potential of MSCs [5]. Conversely, MSCs release signaling factors, such as prostaglandin E-2 (PGE-2), that control the polarization of macrophages [73]. Thus, the control induced by different subtypes of macrophages via production of factors like TGF-β1, BMP, activin A, OSM, substance P (SP), and neurotrophin 3 (NT-3), play vital roles in the development of HO by helping to regulate the inflammatory process, bone repair process, and potential progression of HO [57,74,75,76,77,78]
Macrophage depletion can mitigate new HO formation in mice [57]. To overcome the barrier of obtaining large numbers of characterized human cells for understanding the tissue effects of macrophages, several protocols with which to generate macrophages from mouse- [79,80] and human- [51,80,81,82,83,84] induced pluripotent stem cells (iPSCs) have been described. Using methods to create human iPSC-derived macrophages (iMACs) from FOP and non-FOP [51], we have previously shown the heightened production of pro-inflammatory cytokines in unstimulated FOP-M1-like iMACs. This aligns with our earlier observations of a pro-inflammatory state as identified in FOP serum and in primary monocyte-derived M1-like macrophages [20]. Furthermore, the significantly elevated IP-10 (CXCL10) and RANTES (CCL5) in FOP-M2-like iMACs mirrored our observations in FOP-patient-derived primary monocytes. Unexpectedly, cytokine production in M1-like-iMACs reached saturation upon stimulation, with the lowest concentration of LPS that we tested. In contrast, FOP-M1-like iMACs exhibited enhanced inflammatory cytokine production, with stronger differences emerging after the M1-like polarization steps. This disparity in timing indicates that FOP-iMACs may retain a pro-inflammatory phenotype after LPS stimulation, for an extended duration, as compared with control cells. These findings align with our previous experiments involving primary FOP samples, indicating that LPS stimulation of naïve FOP monocytes exhibits prolonged NF-κB activation, compared with control monocytes.
Surprisingly, we found no significant differences in cytokine production responses to the damage-associated molecular patterns (DAMPs) that may be released after tissue injury, suggesting that the initiation process of the macrophages by tissue injury is not changed, but that the magnitude and/or duration is prolonged in FOP [51]. FOP-M1-like macrophages also exhibited increased activin A production, potentially acting as a source of the neo-ligand for the ACVR1R206H receptor. This FOP iMA̧C model provides important opportunities for detailed studies on the way in which ACVR1 regulates inflammation in the human FOP disease state and highlights the crucial role of macrophages as a driver of HO formation in FOP patients.
8. Other Immune Cell Types in Fibrodysplasia Ossificans Progressiva
In addition to myeloid cells, other types of immune cells, such as mast cells and lymphoid cells, are thought to contribute to the inflammation of FOP.
Mast Cells: Mast cells are immune cells known for their involvement in allergic and inflammatory responses [85,86]. These mast cells release pro-inflammatory mediators in patients with FOP and contribute to the inflammatory micro-environment. Depletion of mast cells can reduce HO volume by about 50% in the conditional knock-in mouse ACVR1R206H model. Additionally, the combined depletion of mast cells and macrophages, together with the use of clodronate, reduced HO volume by about 75%, suggesting that these two cell types are contributors to HO formation in FOP [57]. Our own prior study provides additional support for this concept, as serum isolated from patients with FOP had high levels of interleukin 9 (IL-9), a cytokine produced by mast cells [20].
Lymphocytes: Both B and T lymphocytes have been identified as potential contributors to the development of FOP. Perivascular lymphocytic infiltration has been reported in the skeletal muscle in a 2-year-old child with FOP, despite normal histology [87]. The presence of perivascular lymphocytic infiltrates in FOP lesions suggests that lymphocytes play a role in the inflammatory process. Studies undertaken on animal models have shown that the adaptive immune system may impact the advancement of HO. RAG1 null mice in an Nse-BMP4 background. These mice lack mature B or T lymphocytes, and form HO after injury without any temporal delay. Notably, the loss of adaptive cells decreased the rate of spreading and the overall amount of HO [88]. These findings suggest that adaptive immunity may not be necessary for the initiation of HO but might be a critical regulator for the expansion of HO lesions [88,89]. Prior studies have also shown that HO expansion after a burn injury is attenuated when mature B- and T-lymphocytes are not present [90]. Finally, histology on early FOP lesions has shown extensive perivascular inflammatory infiltrates with a significant expression of hypoxia-inducible factor 1 alpha (HIF1α) [91]. Taken together, these human and mouse data indicate that a complex inflammatory response involving lymphocytes, mast cells, macrophages, and likely other immune cell types are key drivers of FOP HO formation.
9. Activin A—A Complex Contributor to Fibrodysplasia Ossificans Progressiva Inflammation
Activin A has an obligatory role in the initiation of FOP HO, through aberrant signaling via the neo-ligand activity of the ACVR1R206H receptor [52,54,92]. However, there are still questions about the precise origin of activin A in FOP patients.
Activin A as an inflammatory mediator: Activin A is a member of the TGF-β/BMP family of ligands [93]. Recently, de Ruiter et al. discovered that TGF-β1 stimulates the generation of activin A in dermal fibroblasts obtained from FOP patients, as evidenced by an increase in the inhibin subunit beta A (INHBA) gene and protein expression levels, functioning as an upstream activator of activin A generation, specifically in FOP [94]. These findings suggest that TGF-β may be an additional target along with activin A, as its blockade could hinder, at least in part, the signaling cascade concluding in the HO formation arbitrated by activin A in FOP [94]. This is in agreement with previous reports implicating the TGF-β pathway as a strong driver of HO, as HO has been shown to be abated following the circulating repression of TGF-β in FOP mouse models [63]. Increased inflammatory responses and canonical BMP receptor (BMPR) signaling all occur during the early stages of trauma-induced HO formation in FOP, and it has been proposed that the crosstalk of several inflammatory signaling pathways may exert a synergistic effect when enhancing inducement of Smad1/5/8 activity in various cell types mediated by BMP [95].
Receptors downstream of Activin A: Many of the inflammatory signals induced early after injury are mediated through toll-like receptors (TLRs) and IL-1 receptors via MyD88-dependent pathways [95]. Despite the reported synergistic effect of IL-1β and LPS when the phosphorylation of Smad1/5 is enhanced by activin A in ACVR1R206H-expressing fibroadipo-progenitors (FAPs), the adapter protein MyD88 has been indicated to be dispensable for trauma-induced HO formation in FOP [95]. Further, it has been demonstrated that activin A promotes early differential gene expression in ligament fibroblasts from FOP patients, with the activin, TGFβ, and BMP signaling being significantly enriched, and upregulated transcript levels in genes involved in bone metabolism (SHOC2 Leucine Rich Repeat Scaffold Protein (SHOC2), Tetratricopeptide Repeat Domain 1 (TTC1), Tetratricopeptide Repeat Domain 1 (PAPSS2), Dedicator Of Cytokinesis 7 (DOCK7), and Lysyl Oxidase (LOX)) [96]. Activation of ACVR1R206H by activin A can also occur by receptor-dependent clustering, thus inducing its auto-activation without the need for upstream kinases that are otherwise required for WT ACVR1 activation. This appeared to employ the type II receptors ACVR2A/B instead [97].
Surprisingly, ACVR1 is the only subtype of the BMP type I receptors capable of interceding in the activin A-induced BMP signaling that is consequent to the disengagement of FKBP12, while FKBP12 inhibition results in failure of BMP4 to cross-activate the TGF-β pathway [98]. Additionally, type II BMP receptors can enhance activin A-induced BMP signaling via their kinase activity [98]. Recently, polypeptide substrate accessibility has been proposed as another potential mechanism, where the ACVR1 gain-of-function R206H mutation allosterically changes the ACVR1 kinase activity [99] and results in altered responsiveness to activin A.
Pathways potentially involved in FOP heterotopic bone formation: Intriguingly, FOP HO development has been shown to be closely correlated with spatiotemporal patterns of human body infrared thermographs, suggesting that temperature responsive FOP connective tissue progenitor cells (CTPCs) may act as potent determinants of the anatomic distribution of HO in FOP [100]. Recently, vitamin D3 has been raised as another potential modulator of BMP signaling, chondrogenesis, and possibly enhanced activin A expression. This is an intriguing area of study as it may highlight the importance of lifestyle factors, such as diet, as potential disease modifying factors in the context of FOP [101].
Another pathway likely involved in aberrant chondrogenesis in FOP implicates the mammalian target of rapamycin (mTOR) signaling [102]. By employing a high-throughput screening (HTS) using FOP-patient-derived iPSCs, Hino and colleagues found that the lysophosphatidic acid-producing enzyme ectonucleotide pyrophosphatase/phosphodiesterase 2 (ENPP2) may link ACVR1R206H and mTOR signaling in chondrogenesis via an activin A/ACVR1R206H/ENPP2/mTOR axis [102].
As noted previously, the pathways involved in inflammation in FOP show significant roles for IL-1 and p38 [20,102]. Elevated IL-1β plasma levels have been found in a case study of FOP patient with repeated and extremely active flare-ups, which were mitigated with anti-IL-1 treatment, as evidenced by reported flare-up occurrence and IL-1β levels [103]. In marked contrast, no differences in systemic activin A levels have been detected between patients with FOP and healthy individuals, even when a patient with FOP has an active flare-up [104]. This finding has led to the hypothesis that activin A levels may be regulated at the local level, highlighting the pivotal role of the local environment in driving HO formation in FOP. Nonetheless, further research is warranted in order to elucidate the source of these pro-inflammatory cytokines, as well as their spatiotemporal interplay, and the microenvironment conformation where they drive HO in FOP.
Schoenmaker and colleagues have shown that activin A promotes the development of fewer, albeit more expanded and active, osteoclasts, independent of the FOP mutation, resulting in osteoclastogenesis induction of unknown underlying mechanism [105]. More recently, they have demonstrated that the presence of activin A instigates transcriptional changes in osteoclast formation both in healthy individuals and patients with FOP, likely through the upregulation of genes involved in the differentiation and function of osteoclasts, cell fusion, and inflammation [106]. However, Ye et al. reported no significant differences in serum levels of activin A, BMP4 or BMP6 between healthy individuals and FOP patients, nor between FOP patients with active flare-ups or in remission, suggesting that activin A, BMP4 and BMP6 may be instigators for flare-ups in FOP, but not biomarkers for FOP disease activity [107]. This finding is consistent with previous reports that indicate that, although activin A intensifies dysregulated BMP signaling in human FOP primary CTPCs via stimulation of the pSmad1/5/8 pathway to induce chondro-osseous differentiation, basal overstimulated pSmad1/5/8 signaling in FOP can be independent of activin A and BMP4 [108].
Despite post-injury activin A production in both traumatic HO (tHO) and FOP HO, single cell RNA sequencing (scRNA-seq) data reveal that activin A is produced by different cell types, suggesting that the precursor signals for HO formation may be distinct between FOP and traumatic HO [104]. Activin A inhibition can effectively block HO in FOP, highlighting the importance of the pro-osteogenic signal induced and sustained by activin A as a critical antecedent event in genetic HO [104]. Findings stemming from the phase 2 LUMINA-1 clinical trial using garetosmab, an activin A-blocking antibody, showed suppressed development of new HO lesions, as well as a lower volume of new abnormal bone formation lesions [109]. In contrast, antibodies targeting ACVR1 activation by its ligands have been shown to stimulate HO and activate the signaling of FOP-mutant ACVR1 in some circumstances [110].
Additionally, a newly developed adeno-associated virus (AAV)-based gene therapy approach that carries the combination of a codon-optimized human ACVR1 and engineered miRNAs targeting activin A and its receptor ACVR1R206H has been demonstrated to be effective for downregulating the BMP-Smad1/5 signaling pathway and the osteogenic differentiation of heterozygous ACVR1R206H/+ skeletal progenitors, leading to protection against trauma-induced HO in FOP mice [111]. Another AAV-compatible artificial miRNA gene therapy strategy, ablating the activin A signal and suppressing chondrogenic and osteogenic differentiation, has been proven efficacious in the prevention of HO in FOP mice [112]. Interestingly, constitutive overexpression of WT ACVR1 in FOP mice has been shown to rescue the murine perinatal lethality associated with the disease and inhibit spontaneous abnormal bone formation and injury-induced HO in FOP mice [113]. Considering the highly variable nature of FOP disease presentation, and the fact that genetic deficiency of the WT Acvr1 allele aggravates HO, it has been recently suggested that the ratio of WT and mutant receptors may impact FOP severity [113]. It has also been proposed that overexpression of WT ACVR1 may protect against abnormal skeletogenesis by increasing the levels of activin A-bound non-signaling complexes (NSCs) with WT ACVR1, thus decreasing osteogenic signaling in response to activin A [113]. Further corroboration of the placating effects of activin A-bound NSCs on FOP HO can be found from the finding that ‘agonist-only’ activin A muteins that activate ACVR1B but are unable to form NSCs with WT ACVR1 lead to more aggravated disease pathology, highlighting the finger two tip loop (F2TL) region of activin A as a crucial component of the NSC formation [114].
10. Inflammation as a Therapeutic Target in Fibrodysplasia Ossificans Progressiva
Because inflammation is a major contributor to FOP and other types of HO, a major avenue for treating HO has focused on disrupting the inflammatory drivers that lead to abnormal bone formation (Figure 2). The mainstay for FOP therapy remains glucocorticoids, such as prednisone, in order to decrease the inflammation in an FOP flare [28]. However, detailed study of the benefits of steroids remains sparse. Retinoid signaling is a key regulator of chondrogenesis. Downregulation is required for HO formation in order to trigger the endochondral ossification pathway [115]. Therefore, retinoid agonists hold potential as a therapy by which to block chondrogenesis [116]. Palovarotene is a potent retinoic acid receptor gamma (RARγ) agonist that can reduce HO in FOP mouse models [117,118] and in a blast-related HO rat model [119]. Palovarotene dampened systemic and local inflammatory responses and specifically reduced levels of IL-6, TNF-α, and IFN-γ [119]. Palovarotene has also been shown to downregulate the inflammatory microenvironment and decrease HO formation of tendon stem cells [120]. Clinical trials for the treatment of FOP patients using palovarotene (NCT03312634, NCT02190747, and NCT02279095) showed 50–60% efficacy in reducing new HO formation [121], although detailed analyses of the immune response in patients receiving palovarotene have yet to be undertaken. In addition, direct inhibition of activin A by the neutralizing antibody garetosmab is being tested in clinical trials as a potential target for blocking HO in FOP (NCT03188666) and has shown efficacy in decreasing the flare activity in patients with FOP [109].
Several other strategies that are commonly tried in patients with non-genetic HO have also been tried in patients with FOP. Rapamycin affects the mTOR signaling pathway. The mTOR pathway is recognized as a critical driver of hypoxia and inflammation—two factors that occur in traumatic injuries [122] and in FOP [102,123]. Rapamycin can prevent HO after blast-related injury in rats; this is thought to occur via the suppression of the expression of inflammatory genes like Cxcl5, C-X-C motif chemokine ligand 10 (Cxcl10), IL-6, and Ccl2 [124]. This background supports the testing of rapamycin in clinical trials as a potential therapy for FOP [123,125,126].
The NSAID celecoxib is a potent cyclooxygenase-2 inhibitor that appears to significantly reduce HO formation after surgical trauma in mouse models [127]. Clinical studies have shown some positive results for patients with non-genetic HO [128]. Surprisingly, other types of NSAIDs do not appear to have similar reductions in HO formation; for example, indomethacin was seen to fail to prevent HO in a blast-related HO rat model [129]. More research is needed to better understand how and which NSAIDs may benefit patients with FOP and how these responses differ from non-genetic HO.
Targeting inflammatory pathways directly has shown some promise for FOP and other forms of HO. A case report of anti-IL1 therapy with anakinra or canakinumab suggests a potential benefit for patients with severe intractable FOP disease activity [103]. IL1 may also have a more general role, as it has also been implicated in a mouse model of neurogenic HO [130]. Targeting the Janus kinase 1 and 2 (JAK1/2) tyrosine kinase is another strategy for reducing inflammation. One factor produced by the pro-inflammatory cytokine OSM, which is normally produced by myeloid cells [76], is phosphorylation of signal transducer and activator of transcription 3 (Stat3). Stat3 is a part of the JAK-STAT signaling pathway and is a critical regulator of cytokine production. In a neurogenic HO mouse model induced by spinal cord injury, ruxolitinib reduced phosphorylation of Stat3 and decreased the formation of new HO [76]. Recently, the JAK inhibitor tofacitinib was reported to decrease inflammatory flare-ups in a cohort of patients with FOP [131]. Saracatinib, which has shown anti-inflammatory activity in atherosclerosis [132], is being investigated as a possible therapy for FOP [133]. Imatinib, another kinase inhibitor with anti-fibrotic and anti-inflammatory activity [134,135,136], has also been described for use in severe cases of FOP [137,138]. While most of these strategies are off-label and do not yet have full studies showing an ability to reduce new HO formation, they provide an important foundation upon which to study the role of inflammatory mediators in the prevention of new HO.
11. Future Research Directions and Conclusions
Inflammation and trauma are key factors influencing the development of HO, yet the precise mechanisms connecting these processes, as well as potential therapeutic targets within, largely remain a mystery. It is clear that these mechanisms involve macrophages, mast cells, and adaptive immune cells, each playing distinct roles throughout the initiation and progression of HO (Table 1). There are shared characteristics between inflammation in FOP and other types of HO that provide both translational relevance for understanding the inflammatory drivers and also for finding therapies that may be effective for both forms of HO. These include the engagement of hypoxia signaling pathways, the initiation of MSC differentiation, the targeting of inflammatory pathways such as IL1, and the activation of vascular signaling pathways.
There are still many critical questions yet to be answered about HO formation. For instance, some rheumatic diseases, such as systemic scleroderma, have been reported to be complicated by HO [139]. However, we still do not understand why these inflammatory disorders result in HO, yet other chronic inflammatory and rheumatic diseases result in bone loss [7]. These cellular interactions between the inflammatory triggers and tissue resident skeletal stem cell lineages remain an area of active research.
Pharmacologic modulation of the immune system appears to be a promising strategy for the mitigation of HO. However, our understanding of exactly how pharmacologic modulation of immunity reduces (or potentially enhances) HO formation remains largely empirical. Thus, a systematic understanding of the immune process is necessary for us to develop effective treatment strategies that likely combine targets in multiple pathways. Continued research into these mechanisms holds high potential for new knowledge that will guide future treatments to prevent and reverse HO.
Funding
ECH received support from 1R01AR073015 and the UCSF Robert L Kroc Chair in Rheumatic and Connective Tissue Diseases III.
Conflicts of Interest
ECH serves in a volunteer capacity on the Medical Registry Advisory Board of the International Fibrodysplasia Ossificans Progressiva Association, the International Clinical Council on FOP, and on the Fibrous Dysplasia Foundation Medical Advisory Board. ECH receives clinical trial research support from Ipsen Pharmaceuticals Inc. to support clinical trials in FOP, and from Ascendis Bio for a clinical trial in hypoparathyroidism. These pose no conflicts of interest.
References
- Long, F. Building strong bones: Molecular regulation of the osteoblast lineage. Nat. Rev. Mol. Cell Biol. 2011, 13, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, N. The mechanism of osteoclast differentiation from macrophages: Possible roles of T lymphocytes in osteoclastogenesis. J. Bone Miner. Metab. 2003, 21, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; Yasuda, H.; Yano, K.; Morinaga, T.; Higashio, K. RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem. Biophys. Res. Commun. 1998, 253, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA 1998, 95, 3597–3602. [Google Scholar] [CrossRef] [PubMed]
- Vi, L.; Baht, G.S.; Whetstone, H.; Ng, A.; Wei, Q.; Poon, R.; Mylvaganam, S.; Grynpas, M.; Alman, B.A. Macrophages promote osteoblastic differentiation in-vivo: Implications in fracture repair and bone homeostasis. J. Bone Miner. Res. 2015, 30, 1090–1102. [Google Scholar] [CrossRef] [PubMed]
- Raggatt, L.J.; Wullschleger, M.E.; Alexander, K.A.; Wu, A.C.; Millard, S.M.; Kaur, S.; Maugham, M.L.; Gregory, L.S.; Steck, R.; Pettit, A.R. Fracture healing via periosteal callus formation requires macrophages for both initiation and progression of early endochondral ossification. Am. J. Pathol. 2014, 184, 3192–3204. [Google Scholar] [CrossRef] [PubMed]
- Negishi-Koga, T.; Gober, H.J.; Sumiya, E.; Komatsu, N.; Okamoto, K.; Sawa, S.; Suematsu, A.; Suda, T.; Sato, K.; Takai, T. Immune complexes regulate bone metabolism through FcRgamma signalling. Nat. Commun. 2015, 6, 6637. [Google Scholar] [CrossRef]
- Wynn, T.A.; Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Chakkalakal, S.A.; Shore, E.M. Fibrodysplasia ossificans progressiva: Mechanisms and models of skeletal metamorphosis. Dis. Models Mech. 2012, 5, 756–762. [Google Scholar] [CrossRef]
- McCarthy, E.F.; Sundaram, M. Heterotopic ossification: A review. Skelet. Radiol. 2005, 34, 609–619. [Google Scholar] [CrossRef]
- Kocic, M.; Lazovic, M.; Mitkovic, M.; Djokic, B. Clinical significance of the heterotopic ossification after total hip arthroplasty. Orthopedics 2010, 33, 16. [Google Scholar] [CrossRef]
- Leung, C.; Casey, A.T.; Goffin, J.; Kehr, P.; Liebig, K.; Lind, B.; Logroscino, C.; Pointillart, V. Clinical significance of heterotopic ossification in cervical disc replacement: A prospective multicenter clinical trial. Neurosurgery 2005, 57, 759–763, discussion 63. [Google Scholar] [CrossRef]
- Hurvitz, E.A.; Mandac, B.R.; Davidoff, G.; Johnson, J.H.; Nelson, V.S. Risk factors for heterotopic ossification in children and adolescents with severe traumatic brain injury. Arch. Phys. Med. Rehabil. 1992, 73, 459–462. [Google Scholar] [PubMed]
- Potter, B.K.; Burns, T.C.; Lacap, A.P.; Granville, R.R.; Gajewski, D.A. Heterotopic ossification following traumatic and combat-related amputations. Prevalence, risk factors, and preliminary results of excision. J. Bone Jt. Surg. Am. 2007, 89, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Schett, G. Effects of inflammatory and anti-inflammatory cytokines on the bone. Eur. J. Clin. Investig. 2011, 41, 1361–1366. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, J.T.; Wylie, J.D.; Potter, M.Q.; Maak, T.G.; Greene, T.H.; Aoki, S.K. Effect of Naproxen Prophylaxis on Heterotopic Ossification Following Hip Arthroscopy: A Double-Blind Randomized Placebo-Controlled Trial. J. Bone Jt. Surg. Am. 2015, 97, 2032–2037. [Google Scholar] [CrossRef] [PubMed]
- Evans, K.N.; Forsberg, J.A.; Potter, B.K.; Hawksworth, J.S.; Brown, T.S.; Andersen, R.; Dunne, J.R.; Tadaki, D.; Elster, E.A. Inflammatory cytokine and chemokine expression is associated with heterotopic ossification in high-energy penetrating war injuries. J. Orthop. Trauma 2012, 26, e204–e213. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, J.A.; Potter, B.K.; Polfer, E.M.; Safford, S.D.; Elster, E.A. Do inflammatory markers portend heterotopic ossification and wound failure in combat wounds? Clin. Orthop. Relat. Res. 2014, 472, 2845–2854. [Google Scholar] [CrossRef]
- Pakos, E.E.; Ioannidis, J.P. Radiotherapy vs. nonsteroidal anti-inflammatory drugs for the prevention of heterotopic ossification after major hip procedures: A meta-analysis of randomized trials. Int. J. Radiat. Oncol. Biol. Phys. 2004, 60, 888–895. [Google Scholar] [CrossRef]
- Barruet, E.; Morales, B.M.; Cain, C.J.; Ton, A.N.; Wentworth, K.L.; Chan, T.V.; Moody, T.A.; Haks, M.C.; Ottenhoff, T.H.; Hellman, J. NF-kappaB/MAPK activation underlies ACVR1-mediated inflammation in human heterotopic ossification. JCI Insight 2018, 3, e122958. [Google Scholar] [CrossRef]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Kim, K.S.; Kim, M.J.; Kim, H.S.; Lee, K.Y.; Kang, K.W. Auranofin Inhibits RANKL-Induced Osteoclastogenesis by Suppressing Inhibitors of kappaB Kinase and Inflammasome-Mediated Interleukin-1beta Secretion. Oxid. Med. Cell Longev. 2019, 2019, 3503912. [Google Scholar] [CrossRef]
- Wang, L.; Chen, K.; Wan, X.; Wang, F.; Guo, Z.; Mo, Z. NLRP3 inflammasome activation in mesenchymal stem cells inhibits osteogenic differentiation and enhances adipogenic differentiation. Biochem. Biophys. Res. Commun. 2017, 484, 871–877. [Google Scholar] [CrossRef]
- Yan, L.; Hu, R.; Tu, S.; Cheng, W.J.; Zheng, Q.; Wang, J.W.; Kan, W.S.; Ren, Y.J. Meta-analysis of association between IL-6 -634C/G polymorphism and osteoporosis. Genet. Mol. Res. 2015, 14, 19225–19232. [Google Scholar] [CrossRef]
- Mansoori, M.N.; Shukla, P.; Kakaji, M.; Tyagi, A.M.; Srivastava, K.; Shukla, M.; Dixit, M.; Kureel, J.; Gupta, S.; Singh, D. IL-18BP is decreased in osteoporotic women: Prevents Inflammasome mediated IL-18 activation and reduces Th17 differentiation. Sci. Rep. 2016, 6, 33680. [Google Scholar] [CrossRef]
- Rios-Arce, N.D.; Dagenais, A.; Feenstra, D.; Coughlin, B.; Kang, H.J.; Mohr, S.; McCabe, L.R.; Parameswaran, N. Loss of interleukin-10 exacerbates early Type-1 diabetes-induced bone loss. J. Cell. Physiol. 2020, 235, 2350–2365. [Google Scholar] [CrossRef]
- Wang, T.; He, C. TNF-alpha and IL-6: The Link between Immune and Bone System. Curr. Drug Targets 2020, 21, 213–227. [Google Scholar]
- The International Clinical Consortium on FOP The Medical Management of Fibrodysplasia Ossificans Progressiva: Current Treatment Considerations. Proc. Int. Clin. Consort. FOP 2022, 2, 1–127. [CrossRef] [PubMed]
- Matsuo, K.; Chavez, R.D.; Barruet, E.; Hsiao, E.C. Inflammation in Fibrodysplasia Ossificans Progressiva and Other Forms of Heterotopic Ossification. Curr. Osteoporos. Rep. 2019, 17, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Liu, Y. The Role of the Immune Microenvironment in Bone Regeneration. Int. J. Med. Sci. 2021, 18, 3697–3707. [Google Scholar] [CrossRef] [PubMed]
- Epsley, S.; Tadros, S.; Farid, A.; Kargilis, D.; Mehta, S.; Rajapakse, C.S. The Effect of Inflammation on Bone. Front. Physiol. 2021, 11, 511799. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Roger, P.-M. From Crosstalk between Immune and Bone Cells to Bone Erosion in Infection. Int. J. Mol. Sci. 2019, 20, 5154. [Google Scholar] [CrossRef]
- Terkawi, M.A.; Matsumae, G.; Shimizu, T.; Takahashi, D.; Kadoya, K.; Iwasaki, N. Interplay between Inflammation and Pathological Bone Resorption: Insights into Recent Mechanisms and Pathways in Related Diseases for Future Perspectives. Int. J. Mol. Sci. 2022, 23, 1786. [Google Scholar] [CrossRef] [PubMed]
- Arron, J.R.; Choi, Y. Bone versus immune system. Nature 2000, 408, 535–536. [Google Scholar] [CrossRef] [PubMed]
- Guder, C.; Gravius, S.; Burger, C.; Wirtz, D.C.; Schildberg, F.A. Osteoimmunology: A Current Update of the Interplay Between Bone and the Immune System. Front. Immunol. 2020, 11, 00058. [Google Scholar] [CrossRef]
- Cai, L.; Lv, Y.; Yan, Q.; Guo, W. Cytokines: The links between bone and the immune system. Injury 2024, 55, 111203. [Google Scholar] [CrossRef]
- Kushioka, J.; Chow, S.K.-H.; Toya, M.; Tsubosaka, M.; Shen, H.; Gao, Q.; Li, X.; Zhang, N.; Goodman, S.B. Bone regeneration in inflammation with aging and cell-based immunomodulatory therapy. Inflamm. Regen. 2023, 43, 29. [Google Scholar] [CrossRef]
- Guihard, P.; Danger, Y.; Brounais, B.; David, E.; Brion, R.; Delecrin, J.; Richards, C.D.; Chevalier, S.; Redini, F.; Heymann, D. Induction of osteogenesis in mesenchymal stem cells by activated monocytes/macrophages depends on oncostatin M signaling. Stem Cells 2012, 30, 762–772. [Google Scholar] [CrossRef]
- Mirosavljevic, D.; Quinn, J.M.; Elliott, J.; Horwood, N.J.; Martin, T.J.; Gillespie, M.T. T-cells mediate an inhibitory effect of interleukin-4 on osteoclastogenesis. J. Bone Miner. Res. 2003, 18, 984–993. [Google Scholar] [CrossRef]
- Laine, C.M.; Joeng, K.S.; Campeau, P.M.; Kiviranta, R.; Tarkkonen, K.; Grover, M.; Lu, J.T.; Pekkinen, M.; Wessman, M.; Heino, T.J. WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N. Engl. J. Med. 2013, 368, 1809–1816. [Google Scholar] [CrossRef]
- Baujat, G.; Choquet, R.; Bouee, S.; Jeanbat, V.; Courouve, L.; Ruel, A.; Michot, C.; Le Quan Sang, K.H.; Lapidus, D.; Messiaen, C. Prevalence of fibrodysplasia ossificans progressiva (FOP) in France: An estimate based on a record linkage of two national databases. Orphanet J. Rare Dis. 2017, 12, 123. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Hsiao, E.C.; Baujat, G.; Lapidus, D.; Sherman, A.; Kaplan, F.S. Prevalence of fibrodysplasia ossificans progressiva (FOP) in the United States: Estimate from three treatment centers and a patient organization. Orphanet J. Rare Dis. 2021, 16, 350. [Google Scholar] [CrossRef]
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Cho, T.J.; Choi, I.H.; Connor, J.M.; Delai, P.; Glaser, D.L.; LeMerrer, M. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Furuya, H.; Ikezoe, K.; Wang, L.; Ohyagi, Y.; Motomura, K.; Fujii, N.; Kira, J.; Fukumaki, Y. A unique case of fibrodysplasia ossificans progressiva with an ACVR1 mutation, G356D, other than the common mutation (R206H). Am. J. Med. Genet. A 2008, 146A, 459–463. [Google Scholar] [CrossRef]
- Petrie, K.A.; Lee, W.H.; Bullock, A.N.; Pointon, J.J.; Smith, R.; Russell, R.G.; Brown, M.A.; Wordsworth, B.P.; Triffitt, J.T. Novel mutations in ACVR1 result in atypical features in two fibrodysplasia ossificans progressiva patients. PLoS ONE 2009, 4, e5005. [Google Scholar] [CrossRef]
- Kaliya-Perumal, A.K.; Carney, T.J.; Ingham, P.W. Fibrodysplasia ossificans progressiva: Current concepts from bench to bedside. Dis. Models Mech. 2020, 13, dmm046441. [Google Scholar] [CrossRef]
- Lin, G.T.; Chang, H.W.; Liu, C.S.; Huang, P.J.; Wang, H.C.; Cheng, Y.M. De novo 617G-A nucleotide mutation in the ACVR1 gene in a Taiwanese patient with fibrodysplasia ossificans progressiva. J. Hum. Genet. 2006, 51, 1083–1086. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Hahn, G.V.; Zasloff, M.A. Heterotopic Ossification: Two Rare Forms and What They Can Teach Us. J. Am. Acad. Orthop. Surg. 1994, 2, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Hsiao, E.C.; Sami, S.; Lancero, M.; Schlieve, C.R.; Nguyen, T.; Yano, K.; Nagahashi, A.; Ikeya, M.; Matsumoto, Y. BMP-SMAD-ID promotes reprogramming to pluripotency by inhibiting p16/INK4A-dependent senescence. Proc. Natl. Acad. Sci. USA 2016, 113, 13057–13062. [Google Scholar] [CrossRef] [PubMed]
- Haupt, J.; Stanley, A.; McLeod, C.M.; Cosgrove, B.D.; Culbert, A.L.; Wang, L.; Mourkioti, F.; Mauck, R.L.; Shore, E.M. ACVR1(R206H) FOP mutation alters mechanosensing and tissue stiffness during heterotopic ossification. Mol. Biol. Cell 2019, 30, 17–29. [Google Scholar] [CrossRef]
- Matsuo, K.; Lepinski, A.; Chavez, R.D.; Barruet, E.; Pereira, A.; Moody, T.A.; Ton, A.N.; Sharma, A.; Hellman, J.; Tomoda, K. ACVR1(R206H) extends inflammatory responses in human induced pluripotent stem cell-derived macrophages. Bone 2021, 153, 116129. [Google Scholar] [CrossRef]
- Hatsell, S.J.; Idone, V.; Wolken, D.M.; Huang, L.; Kim, H.J.; Wang, L.; Wen, X.; Nannuru, K.C.; Jimenez, J.; Xie, L. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 2015, 7, 303ra137. [Google Scholar] [CrossRef]
- Aykul, S.; Martinez-Hackert, E. Transforming Growth Factor-beta Family Ligands Can Function as Antagonists by Competing for Type II Receptor Binding. J. Biol. Chem. 2016, 291, 10792–10804. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Ikeya, M.; Horigome, K.; Matsumoto, Y.; Ebise, H.; Nishio, M.; Sekiguchi, K.; Shibata, M.; Nagata, S.; Matsuda, S. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 2015, 112, 15438–15443. [Google Scholar] [CrossRef]
- Olsen, O.E.; Wader, K.F.; Hella, H.; Mylin, A.K.; Turesson, I.; Nesthus, I.; Waage, A.; Sundan, A.; Holien, T. Activin A inhibits BMP-signaling by binding ACVR2A and ACVR2B. Cell Commun. Signal. 2015, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Pignolo, R.J.; Bedford-Gay, C.; Liljesthrom, M.; Durbin-Johnson, B.P.; Shore, E.M.; Rocke, D.M.; Kaplan, F.S. The Natural History of Flare-Ups in Fibrodysplasia Ossificans Progressiva (FOP): A Comprehensive Global Assessment. J. Bone Miner. Res. 2016, 31, 650–656. [Google Scholar] [CrossRef]
- Convente, M.R.; Chakkalakal, S.A.; Yang, E.; Caron, R.J.; Zhang, D.; Kambayashi, T.; Kaplan, F.S.; Shore, E.M. Depletion of Mast Cells and Macrophages Impairs Heterotopic Ossification in an Acvr1(R206H) Mouse Model of Fibrodysplasia Ossificans Progressiva. J. Bone Miner. Res. 2018, 33, 269–282. [Google Scholar] [CrossRef]
- Loder, S.J.; Agarwal, S.; Chung, M.T.; Cholok, D.; Hwang, C.; Visser, N.; Vasquez, K.; Sorkin, M.; Habbouche, J.; Sung, H.H. Characterizing the Circulating Cell Populations in Traumatic Heterotopic Ossification. Am. J. Pathol. 2018, 188, 2464–2473. [Google Scholar] [CrossRef]
- Grgurevic, L.; Novak, R.; Trkulja, V.; Hamzic, L.F.; Hrkac, S.; Grazio, S.; Santini, M. Elevated plasma RANTES in fibrodysplasia ossificans progressiva—A novel therapeutic target? Med. Hypotheses 2019, 131, 109313. [Google Scholar] [CrossRef]
- Kan, L.; Lounev, V.Y.; Pignolo, R.J.; Duan, L.; Liu, Y.; Stock, S.R.; McGuire, T.L.; Lu, B.; Gerard, N.P.; Shore, E.M. Substance P signaling mediates BMP-dependent heterotopic ossification. J. Cell. Biochem. 2011, 112, 2759–2772. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Glaser, D.L.; Shore, E.M.; Pignolo, R.J.; Xu, M.; Zhang, Y.; Senitzer, D.; Forman, S.J.; Emerson, S.G. Hematopoietic stem-cell contribution to ectopic skeletogenesis. J. Bone Jt. Surg. Am. 2007, 89, 347–357. [Google Scholar] [CrossRef]
- Chakkalakal, S.A.; Zhang, D.; Culbert, A.L.; Convente, M.R.; Caron, R.J.; Wright, A.C.; Maidment, A.D.; Kaplan, F.S.; Shore, E.M. An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J. Bone Miner. Res. 2012, 27, 1746–1756. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, F.; Xie, L.; Crane, J.; Zhen, G.; Mishina, Y.; Deng, R.; Gao, B.; Chen, H.; Liu, S.; et al. Inhibition of overactive TGF-beta attenuates progression of heterotopic ossification in mice. Nat. Commun. 2018, 9, 551. [Google Scholar] [CrossRef] [PubMed]
- Pignolo, R.J.; McCarrick-Walmsley, R.; Wang, H.; Qiu, S.; Hunter, J.; Barr, S.; He, K.; Zhang, H.; Kaplan, F.S. Plasma-Soluble Biomarkers for Fibrodysplasia Ossificans Progressiva (FOP) Reflect Acute and Chronic Inflammatory States. J. Bone Miner. Res. 2022, 37, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Hu, J.; Zhou, X.; Yang, Y. Heterotopic ossification: Mechanistic insights and clinical challenges. Bone 2018, 109, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Dilling, C.F.; Wada, A.M.; Lazard, Z.W.; Salisbury, E.A.; Gannon, F.H.; Vadakkan, T.J.; Gao, L.; Hirschi, K.; Dickinson, M.E.; Davis, A.R. Vessel formation is induced prior to the appearance of cartilage in BMP-2-mediated heterotopic ossification. J. Bone Miner. Res. 2010, 25, 1147–1156. [Google Scholar] [CrossRef]
- O’Brien, E.J.; Frank, C.B.; Shrive, N.G.; Hallgrimsson, B.; Hart, D.A. Heterotopic mineralization (ossification or calcification) in tendinopathy or following surgical tendon trauma. Int. J. Exp. Pathol. 2012, 93, 319–331. [Google Scholar] [CrossRef]
- Dey, D.; Bagarova, J.; Hatsell, S.J.; Armstrong, K.A.; Huang, L.; Ermann, J.; Vonner, A.J.; Shen, Y.; Mohedas, A.H.; Lee, A. Two tissue-resident progenitor lineages drive distinct phenotypes of heterotopic ossification. Sci. Transl. Med. 2016, 8, 366ra163. [Google Scholar] [CrossRef]
- Fernandes, T.L.; Gomoll, A.H.; Lattermann, C.; Hernandez, A.J.; Bueno, D.F.; Amano, M.T. Macrophage: A Potential Target on Cartilage Regeneration. Front. Immunol. 2020, 11, 111. [Google Scholar] [CrossRef]
- Dai, M.; Sui, B.; Xue, Y.; Liu, X.; Sun, J. Cartilage repair in degenerative osteoarthritis mediated by squid type II collagen via immunomodulating activation of M2 macrophages, inhibiting apoptosis and hypertrophy of chondrocytes. Biomaterials 2018, 180, 91–103. [Google Scholar] [CrossRef]
- Peterson, J.R.; Eboda, O.N.; Brownley, R.C.; Cilwa, K.E.; Pratt, L.E.; De La Rosa, S.; Agarwal, S.; Buchman, S.R.; Cederna, P.S.; Morris, M.D.; et al. Effects of aging on osteogenic response and heterotopic ossification following burn injury in mice. Stem Cells Dev. 2015, 24, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bose, T.; Unger, R.E.; Jansen, J.A.; Kirkpatrick, C.J.; van den Beucken, J. Macrophage type modulates osteogenic differentiation of adipose tissue MSCs. Cell Tissue Res. 2017, 369, 273–286. [Google Scholar] [CrossRef]
- Maggini, J.; Mirkin, G.; Bognanni, I.; Holmberg, J.; Piazzon, I.M.; Nepomnaschy, I.; Costa, H.; Canones, C.; Raiden, S.; Vermeulen, M.; et al. Mouse bone marrow-derived mesenchymal stromal cells turn activated macrophages into a regulatory-like profile. PLoS ONE 2010, 5, e9252. [Google Scholar] [CrossRef]
- Horwood, N.J. Macrophage Polarization and Bone Formation: A review. Clin. Rev. Allergy Immunol. 2016, 51, 79–86. [Google Scholar] [CrossRef]
- Sorkin, M.; Huber, A.K.; Hwang, C.; Carson, W.F.t.; Menon, R.; Li, J.; Vasquez, K.; Pagani, C.; Patel, N.; Li, S.; et al. Regulation of heterotopic ossification by monocytes in a mouse model of aberrant wound healing. Nat. Commun. 2020, 11, 722. [Google Scholar] [CrossRef]
- Alexander, K.A.; Tseng, H.W.; Fleming, W.; Jose, B.; Salga, M.; Kulina, I.; Millard, S.M.; Pettit, A.R.; Genet, F.; Levesque, J.P. Inhibition of JAK1/2 Tyrosine Kinases Reduces Neurogenic Heterotopic Ossification After Spinal Cord Injury. Front. Immunol. 2019, 10, 377. [Google Scholar] [CrossRef] [PubMed]
- Tuzmen, C.; Verdelis, K.; Weiss, L.; Campbell, P. Crosstalk between substance P and calcitonin gene-related peptide during heterotopic ossification in murine Achilles tendon. J. Orthop. Res. 2018, 36, 1444–1455. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, L.; Chu, J.; Ao, X.; Jiang, T.; Bin, Y.; Huang, M.; Zhang, Z. Macrophage-derived neurotrophin-3 promotes heterotopic ossification in rats. Lab. Investig. 2020, 100, 762–776. [Google Scholar] [CrossRef] [PubMed]
- Senju, S.; Haruta, M.; Matsunaga, Y.; Fukushima, S.; Ikeda, T.; Takahashi, K.; Okita, K.; Yamanaka, S.; Nishimura, Y. Characterization of dendritic cells and macrophages generated by directed differentiation from mouse induced pluripotent stem cells. Stem Cells 2009, 27, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Takata, K.; Kozaki, T.; Lee, C.Z.W.; Thion, M.S.; Otsuka, M.; Lim, S.; Utami, K.H.; Fidan, K.; Park, D.S.; Malleret, B.; et al. Induced-Pluripotent-Stem-Cell-Derived Primitive Macrophages Provide a Platform for Modeling Tissue-Resident Macrophage Differentiation and Function. Immunity 2017, 47, 183–198.e6. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, N.; Ackermann, M.; Frenzel, E.; Liebhaber, S.; Brennig, S.; Happle, C.; Hoffmann, D.; Klimenkova, O.; Luttge, D.; Buchegger, T.; et al. Large-scale hematopoietic differentiation of human induced pluripotent stem cells provides granulocytes or macrophages for cell replacement therapies. Stem Cell Rep. 2015, 4, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.H.; Aubourg, P.; Ransohoff, R.M.; et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef]
- Nenasheva, T.; Gerasimova, T.; Serdyuk, Y.; Grigor’eva, E.; Kosmiadi, G.; Nikolaev, A.; Dashinimaev, E.; Lyadova, I. Macrophages Derived From Human Induced Pluripotent Stem Cells Are Low-Activated “Naive-Like” Cells Capable of Restricting Mycobacteria Growth. Front. Immunol. 2020, 11, 1016. [Google Scholar] [CrossRef]
- van Wilgenburg, B.; Browne, C.; Vowles, J.; Cowley, S.A. Efficient, long term production of monocyte-derived macrophages from human pluripotent stem cells under partly-defined and fully-defined conditions. PLoS ONE 2013, 8, e71098. [Google Scholar] [CrossRef]
- Gilfillan, A.M.; Austin, S.J.; Metcalfe, D.D. Mast cell biology: Introduction and overview. Adv. Exp. Med. Biol. 2011, 716, 2–12. [Google Scholar]
- Gannon, F.H.; Glaser, D.; Caron, R.; Thompson, L.D.; Shore, E.M.; Kaplan, F.S. Mast cell involvement in fibrodysplasia ossificans progressiva. Hum. Pathol. 2001, 32, 842–848. [Google Scholar] [CrossRef]
- Gannon, F.H.; Valentine, B.A.; Shore, E.M.; Zasloff, M.A.; Kaplan, F.S. Acute lymphocytic infiltration in an extremely early lesion of fibrodysplasia ossificans progressiva. Clin. Orthop. Relat. Res. 1998, 19–25. [Google Scholar] [CrossRef]
- Kan, L.; Liu, Y.; McGuire, T.L.; Berger, D.M.; Awatramani, R.B.; Dymecki, S.M.; Kessler, J.A. Dysregulation of local stem/progenitor cells as a common cellular mechanism for heterotopic ossification. Stem Cells 2009, 27, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Kan, L.; Hu, M.; Gomes, W.A.; Kessler, J.A. Transgenic mice overexpressing BMP4 develop a fibrodysplasia ossificans progressiva (FOP)-like phenotype. Am. J. Pathol. 2004, 165, 1107–1115. [Google Scholar] [CrossRef]
- Ranganathan, K.; Agarwal, S.; Cholok, D.; Loder, S.; Li, J.; Sung Hsieh, H.H.; Wang, S.C.; Buchman, S.R.; Levi, B. The role of the adaptive immune system in burn-induced heterotopic ossification and mesenchymal cell osteogenic differentiation. J. Surg. Res. 2016, 206, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lindborg, C.; Lounev, V.; Kim, J.H.; McCarrick-Walmsley, R.; Xu, M.; Mangiavini, L.; Groppe, J.C.; Shore, E.M.; Schipani, E.; et al. Cellular Hypoxia Promotes Heterotopic Ossification by Amplifying BMP Signaling. J. Bone Miner. Res. 2016, 31, 1652–1665. [Google Scholar] [CrossRef] [PubMed]
- Alessi Wolken, D.M.; Idone, V.; Hatsell, S.J.; Yu, P.B.; Economides, A.N. The obligatory role of Activin A in the formation of heterotopic bone in Fibrodysplasia Ossificans Progressiva. Bone 2018, 109, 210–217. [Google Scholar] [CrossRef]
- Hinck, A.P.; Mueller, T.D.; Springer, T.A. Structural Biology and Evolution of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2016, 8, a022103. [Google Scholar] [CrossRef]
- de Ruiter, R.D.; Wisse, L.E.; Schoenmaker, T.; Yaqub, M.; Sánchez-Duffhues, G.; Eekhoff, E.M.W.; Micha, D. TGF-Beta Induces Activin A Production in Dermal Fibroblasts Derived from Patients with Fibrodysplasia Ossificans Progressiva. Int. J. Mol. Sci. 2023, 24, 2299. [Google Scholar] [CrossRef]
- Lyu, H.; Elkins, C.M.; Pierce, J.L.; Serezani, C.H.; Perrien, D.S. MyD88 Is Not Required for Muscle Injury-Induced Endochondral Heterotopic Ossification in a Mouse Model of Fibrodysplasia Ossificans Progressiva. Biomedicines 2021, 9, 630. [Google Scholar] [CrossRef]
- Schoenmaker, T.; Mokry, M.; Micha, D.; Netelenbos, C.; Bravenboer, N.; Gilijamse, M.; Eekhoff, E.M.W.; de Vries, T.J. Activin-A Induces Early Differential Gene Expression Exclusively in Periodontal Ligament Fibroblasts from Fibrodysplasia Ossificans Progressiva Patients. Biomedicines 2021, 9, 629. [Google Scholar] [CrossRef]
- Ramachandran, A.; Mehić, M.; Wasim, L.; Malinova, D.; Gori, I.; Blaszczyk, B.K.; Carvalho, D.M.; Shore, E.M.; Jones, C.; Hyvönen, M.; et al. Pathogenic ACVR1(R206H) activation by Activin A-induced receptor clustering and autophosphorylation. EMBO J. 2021, 40, e106317. [Google Scholar] [CrossRef]
- Xie, C.; Jiang, W.; Lacroix, J.J.; Luo, Y.; Hao, J. Insight into Molecular Mechanism for Activin A-Induced Bone Morphogenetic Protein Signaling. Int. J. Mol. Sci. 2020, 21, 6498. [Google Scholar] [CrossRef] [PubMed]
- Groppe, J.C.; Lu, G.; Tandang-Silvas, M.R.; Pathi, A.; Konda, S.; Wu, J.; Le, V.Q.; Culbert, A.L.; Shore, E.M.; Wharton, K.A.; et al. Polypeptide Substrate Accessibility Hypothesis: Gain-of-Function R206H Mutation Allosterically Affects Activin Receptor-like Protein Kinase Activity. Biomolecules 2023, 13, 1129. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; De Cunto, C.L.; Pignolo, R.J.; Kaplan, F.S. Spatial patterns of heterotopic ossification in fibrodysplasia ossificans progressiva correlate with anatomic temperature gradients. Bone 2021, 149, 115978. [Google Scholar] [CrossRef] [PubMed]
- Pierce, J.L.; Perrien, D.S. Do Interactions of Vitamin D(3) and BMP Signaling Hold Implications in the Pathogenesis of Fibrodysplasia Ossificans Progressiva? Curr. Osteoporos. Rep. 2021, 19, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Horigome, K.; Nishio, M.; Komura, S.; Nagata, S.; Zhao, C.; Jin, Y.; Kawakami, K.; Yamada, Y.; Ohta, A.; et al. Activin-A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressiva. J. Clin. Investig. 2017, 127, 3339–3352. [Google Scholar] [CrossRef]
- Haviv, R.; Moshe, V.; De Benedetti, F.; Prencipe, G.; Rabinowicz, N.; Uziel, Y. Is fibrodysplasia ossificans progressiva an interleukin-1 driven auto-inflammatory syndrome? Pediatr. Rheumatol. 2019, 17, 84. [Google Scholar] [CrossRef]
- Hwang, C.; Pagani, C.A.; Das, N.; Marini, S.; Huber, A.K.; Xie, L.; Jimenez, J.; Brydges, S.; Lim, W.K.; Nannuru, K.C.; et al. Activin A does not drive post-traumatic heterotopic ossification. Bone 2020, 138, 115473. [Google Scholar] [CrossRef]
- Schoenmaker, T.; Botman, E.; Sariyildiz, M.; Micha, D.; Netelenbos, C.; Bravenboer, N.; Kelder, A.; Eekhoff, E.M.W.; De Vries, T.J. Activin-A Induces Fewer, but Larger Osteoclasts From Monocytes in Both Healthy Controls and Fibrodysplasia Ossificans Progressiva Patients. Front. Endocrinol. 2020, 11, 501. [Google Scholar] [CrossRef]
- Schoenmaker, T.; Zwaak, J.; Loos, B.G.; Volckmann, R.; Koster, J.; Eekhoff, E.M.W.; de Vries, T.J. Transcriptomic Differences Underlying the Activin-A Induced Large Osteoclast Formation in Both Healthy Control and Fibrodysplasia Ossificans Progressiva Osteoclasts. Int. J. Mol. Sci. 2023, 24, 6822. [Google Scholar] [CrossRef]
- Ye, Z.; Wang, S.; Shan, C.; Zhu, Q.; Xue, Y.; Zhang, K. The serum levels of activin A and bone morphogenetic protein-4 and -6 in patients with fibrodysplasia ossificans progressiva. Orphanet J. Rare Dis. 2023, 18, 111. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shore, E.M.; Pignolo, R.J.; Kaplan, F.S. Activin A amplifies dysregulated BMP signaling and induces chondro-osseous differentiation of primary connective tissue progenitor cells in patients with fibrodysplasia ossificans progressiva (FOP). Bone 2018, 109, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Di Rocco, M.; Forleo-Neto, E.; Pignolo, R.J.; Keen, R.; Orcel, P.; Funck-Brentano, T.; Roux, C.; Kolta, S.; Madeo, A.; Bubbear, J.S.; et al. Garetosmab in fibrodysplasia ossificans progressiva: A randomized, double-blind, placebo-controlled phase 2 trial. Nat. Med. 2023, 29, 2615–2624. [Google Scholar] [CrossRef] [PubMed]
- Aykul, S.; Huang, L.; Wang, L.; Das, N.M.; Reisman, S.; Ray, Y.; Zhang, Q.; Rothman, N.; Nannuru, K.C.; Kamat, V.; et al. Anti-ACVR1 antibodies exacerbate heterotopic ossification in fibrodysplasia ossificans progressiva (FOP) by activating FOP-mutant ACVR1. J. Clin. Investig. 2022, 132, e153792. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.S.; Lin, C.; Ma, H.; Xie, J.; Kaplan, F.S.; Gao, G.; Shim, J.H. AAV-Mediated Targeting of the Activin A-ACVR1(R206H) Signaling in Fibrodysplasia Ossificans Progressiva. Biomolecules 2023, 13, 1364. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-S.; Kim, J.-M.; Xie, J.; Chaugule, S.; Lin, C.; Ma, H.; Hsiao, E.; Hong, J.; Chun, H.; Shore, E.M.; et al. Suppression of heterotopic ossification in fibrodysplasia ossificans progressiva using AAV gene delivery. Nat. Commun. 2022, 13, 6175. [Google Scholar] [CrossRef]
- Yamamoto, M.; Stoessel, S.J.; Yamamoto, S.; Goldhamer, D.J. Overexpression of Wild-Type ACVR1 in Fibrodysplasia Ossificans Progressiva Mice Rescues Perinatal Lethality and Inhibits Heterotopic Ossification. J. Bone Miner. Res. 2022, 37, 2077–2093. [Google Scholar] [CrossRef] [PubMed]
- Aykul, S.; Corpina, R.A.; Goebel, E.J.; Cunanan, C.J.; Dimitriou, A.; Kim, H.J.; Zhang, Q.; Rafique, A.; Leidich, R.; Wang, X.; et al. Activin A forms a non-signaling complex with ACVR1 and type II Activin/BMP receptors via its finger 2 tip loop. eLife 2020, 9, e54582. [Google Scholar] [CrossRef]
- Weston, A.D.; Rosen, V.; Chandraratna, R.A.; Underhill, T.M. Regulation of skeletal progenitor differentiation by the BMP and retinoid signaling pathways. J. Cell Biol. 2000, 148, 679–690. [Google Scholar] [CrossRef]
- Pacifici, M.; Cossu, G.; Molinaro, M.; Tato, F. Vitamin A inhibits chondrogenesis but not myogenesis. Exp. Cell Res. 1980, 129, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Chakkalakal, S.A.; Uchibe, K.; Convente, M.R.; Zhang, D.; Economides, A.N.; Kaplan, F.S.; Pacifici, M.; Iwamoto, M.; Shore, E.M. Palovarotene Inhibits Heterotopic Ossification and Maintains Limb Mobility and Growth in Mice With the Human ACVR1(R206H) Fibrodysplasia Ossificans Progressiva (FOP) Mutation. J. Bone Miner. Res. 2016, 31, 1666–1675. [Google Scholar] [CrossRef] [PubMed]
- Lees-Shepard, J.B.; Nicholas, S.E.; Stoessel, S.J.; Devarakonda, P.M.; Schneider, M.J.; Yamamoto, M.; Goldhamer, D.J. Palovarotene reduces heterotopic ossification in juvenile FOP mice but exhibits pronounced skeletal toxicity. eLife 2018, 7, e40814. [Google Scholar] [CrossRef]
- Wheatley, B.M.; Cilwa, K.E.; Dey, D.; Qureshi, A.T.; Seavey, J.G.; Tomasino, A.M.; Sanders, E.M.; Bova, W.; Boehm, C.A.; Iwamoto, M.; et al. Palovarotene inhibits connective tissue progenitor cell proliferation in a rat model of combat-related heterotopic ossification. J. Orthop. Res. 2018, 36, 1135–1144. [Google Scholar] [CrossRef]
- Huang, J.; Lin, J.; Li, C.; Tang, B.; Xiao, H. Palovarotene Can Attenuate Heterotopic Ossification Induced by Tendon Stem Cells by Downregulating the Synergistic Effects of Smad and NF-kappaB Signaling Pathway following Stimulation of the Inflammatory Microenvironment. Stem Cells Int. 2022, 2022, 1560943. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Hsiao, E.C.; Al Mukaddam, M.; Baujat, G.; Berglund, S.K.; Brown, M.A.; Cheung, A.M.; De Cunto, C.; Delai, P.; Haga, N.; et al. Reduction of New Heterotopic Ossification (HO) in the Open-Label, Phase 3 MOVE Trial of Palovarotene for Fibrodysplasia Ossificans Progressiva (FOP). J. Bone Miner. Res. 2023, 38, 381–394. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Zhao, C.; Horigome, K.; Nishio, M.; Okanishi, Y.; Nagata, S.; Komura, S.; Yamada, Y.; Toguchida, J.; Ohta, A.; et al. An mTOR Signaling Modulator Suppressed Heterotopic Ossification of Fibrodysplasia Ossificans Progressiva. Stem Cell Rep. 2018, 11, 1106–1119. [Google Scholar] [CrossRef]
- Qureshi, A.T.; Dey, D.; Sanders, E.M.; Seavey, J.G.; Tomasino, A.M.; Moss, K.; Wheatley, B.; Cholok, D.; Loder, S.; Li, J.; et al. Inhibition of Mammalian Target of Rapamycin Signaling with Rapamycin Prevents Trauma-Induced Heterotopic Ossification. Am. J. Pathol. 2017, 187, 2536–2545. [Google Scholar] [CrossRef]
- IFOPA. Rapamycin Clinical Trial for FOP 2019. Available online: https://www.ifopa.org/rapamycin (accessed on 1 March 2024).
- Kaplan, F.S.; Zeitlin, L.; Dunn, S.P.; Benor, S.; Hagin, D.; Al Mukaddam, M.; Pignolo, R.J. Acute and chronic rapamycin use in patients with Fibrodysplasia Ossificans Progressiva: A report of two cases. Bone 2018, 109, 281–284. [Google Scholar] [CrossRef]
- Peterson, J.R.; De La Rosa, S.; Eboda, O.; Cilwa, K.E.; Agarwal, S.; Buchman, S.R.; Cederna, P.S.; Xi, C.; Morris, M.D.; Herndon, D.N.; et al. Treatment of heterotopic ossification through remote ATP hydrolysis. Sci. Transl. Med. 2014, 6, 255ra132. [Google Scholar] [CrossRef] [PubMed]
- Lavernia, C.J.; Contreras, J.S.; Villa, J.M.; Rossi, M.D. Celecoxib and heterotopic bone formation after total hip arthroplasty. J. Arthroplast. 2014, 29, 390–392. [Google Scholar] [CrossRef]
- Robertson, A.D.; Chiaramonti, A.M.; Nguyen, T.P.; Jaffe, D.E.; Holmes, R.E.; Hanna, E.L.; Rhee, J.G.; Barfield, W.R.; Fourney, W.B.; Stains, J.P.; et al. Failure of Indomethacin and Radiation to Prevent Blast-induced Heterotopic Ossification in a Sprague-Dawley Rat Model. Clin. Orthop. Relat. Res. 2019, 477, 644–654. [Google Scholar] [CrossRef]
- Tseng, H.W.; Kulina, I.; Girard, D.; Gueguen, J.; Vaquette, C.; Salga, M.; Fleming, W.; Jose, B.; Millard, S.M.; Pettit, A.R.; et al. Interleukin-1 Is Overexpressed in Injured Muscles Following Spinal Cord Injury and Promotes Neurogenic Heterotopic Ossification. J. Bone Miner. Res. 2022, 37, 531–546. [Google Scholar] [CrossRef]
- Nikishina, I.P.; Arsenyeva, S.V.; Matkava, V.G.; Arefieva, A.N.; Kaleda, M.I.; Smirnov, A.V.; Blank, L.M.; Kostik, M.M. Successful experience of tofacitinib treatment in patients with Fibrodysplasia Ossificans Progressiva. Pediatr. Rheumatol. 2023, 21, 92. [Google Scholar] [CrossRef]
- Amadori, L.; Calcagno, C.; Fernandez, D.M.; Koplev, S.; Fernandez, N.; Kaur, R.; Mury, P.; Khan, N.S.; Sajja, S.; Shamailova, R.; et al. Systems immunology-based drug repurposing framework to target inflammation in atherosclerosis. Nat. Cardiovasc. Res. 2023, 2, 550–571. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.; Bagarova, J.; Kerr, G.; Xia, D.D.; Place, E.S.; Dey, D.; Shen, Y.; Bocobo, G.A.; Mohedas, A.H.; Huang, X.; et al. Saracatinib is an efficacious clinical candidate for fibrodysplasia ossificans progressiva. JCI Insight 2021, 6, e95042. [Google Scholar] [CrossRef] [PubMed]
- Daniels, C.E.; Wilkes, M.C.; Edens, M.; Kottom, T.J.; Murphy, S.J.; Limper, A.H.; Leof, E.B. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J. Clin. Investig. 2004, 114, 1308–1316. [Google Scholar] [CrossRef]
- Juurikivi, A.; Sandler, C.; Lindstedt, K.A.; Kovanen, P.T.; Juutilainen, T.; Leskinen, M.J.; Maki, T.; Eklund, K.K. Inhibition of c-kit tyrosine kinase by imatinib mesylate induces apoptosis in mast cells in rheumatoid synovia: A potential approach to the treatment of arthritis. Ann. Rheum. Dis. 2005, 64, 1126–1131. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.M.; Wolf, D.; Rumpold, H.; Ludwiczek, S.; Enrich, B.; Gastl, G.; Weiss, G.; Tilg, H. The kinase inhibitor imatinib mesylate inhibits TNF-alpha production in vitro and prevents TNF-dependent acute hepatic inflammation. Proc. Natl. Acad. Sci. USA 2005, 102, 13622–13627. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Andolina, J.R.; Adamson, P.C.; Teachey, D.T.; Finklestein, J.Z.; Ebb, D.H.; Whitehead, B.; Jacobs, B.; Siegel, D.M.; Keen, R.; et al. Early clinical observations on the use of imatinib mesylate in FOP: A report of seven cases. Bone 2018, 109, 276–280. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Teachey, D.T.; Andolina, J.R.; Siegel, D.M.; Mancilla, E.E.; Hsiao, E.C.; Al Mukaddam, M.; Rocke, D.M.; Pignolo, R.J. Off-on-off-on use of imatinib in three children with fibrodysplasia ossificans progressiva. Bone 2021, 150, 116016. [Google Scholar] [CrossRef]
- Botzoris, V.G.; Argyropoulou, M.I.; Voulgari, P.V.; Zikou, A.K.; Drosos, A.A. Heterotopic ossification in systemic sclerosis. Scand. J. Rheumatol. 2009, 38, 317–319. [Google Scholar] [CrossRef]
Figure 1.
Potential inflammatory contributors to the pathogenesis of FOP. The ACVR1R206H genetic variant causes osteoprogenitors, such as fibroadipocyte progenitor cells (FAPs) to have an increased ability to enter the osteogenic pathway, possibly due to cell fate instability [49] and the abnormal mechanosensation [50] seen in other cell types. The ACVR1R206H variant also causes innate immune cells to be in a “primed” inflammatory state [20], where a subsequent trigger results in immune hyperactivation. Macrophages appear to be a potential source of activin A [51], which then leads to neo-ligand activation of ACVR1R206H [52], with subsequent tissue inflammation, fibrosis, and endochondral ossification leading to the formation of heterotopic ossification. Figure created with Biorender.
Figure 1.
Potential inflammatory contributors to the pathogenesis of FOP. The ACVR1R206H genetic variant causes osteoprogenitors, such as fibroadipocyte progenitor cells (FAPs) to have an increased ability to enter the osteogenic pathway, possibly due to cell fate instability [49] and the abnormal mechanosensation [50] seen in other cell types. The ACVR1R206H variant also causes innate immune cells to be in a “primed” inflammatory state [20], where a subsequent trigger results in immune hyperactivation. Macrophages appear to be a potential source of activin A [51], which then leads to neo-ligand activation of ACVR1R206H [52], with subsequent tissue inflammation, fibrosis, and endochondral ossification leading to the formation of heterotopic ossification. Figure created with Biorender.

Figure 2.
Potential therapies and investigational compounds for FOP and their relation to inflammation. Multiple therapies are being used or developed to treat FOP. These include investigational agents (green), and current medications used for standard of care. Palovarotene is approved by the US Food and Drug Administration and by Health Canada. Therapies that directly target the ACVR1 receptor or signaling pathway will also likely mitigate the inflammatory response induced by the FOP mutation. The long-term goal is to eventually develop strategies by which to reverse or allow for surgical resection of HO in patients with FOP.
Figure 2.
Potential therapies and investigational compounds for FOP and their relation to inflammation. Multiple therapies are being used or developed to treat FOP. These include investigational agents (green), and current medications used for standard of care. Palovarotene is approved by the US Food and Drug Administration and by Health Canada. Therapies that directly target the ACVR1 receptor or signaling pathway will also likely mitigate the inflammatory response induced by the FOP mutation. The long-term goal is to eventually develop strategies by which to reverse or allow for surgical resection of HO in patients with FOP.


{kind=link}
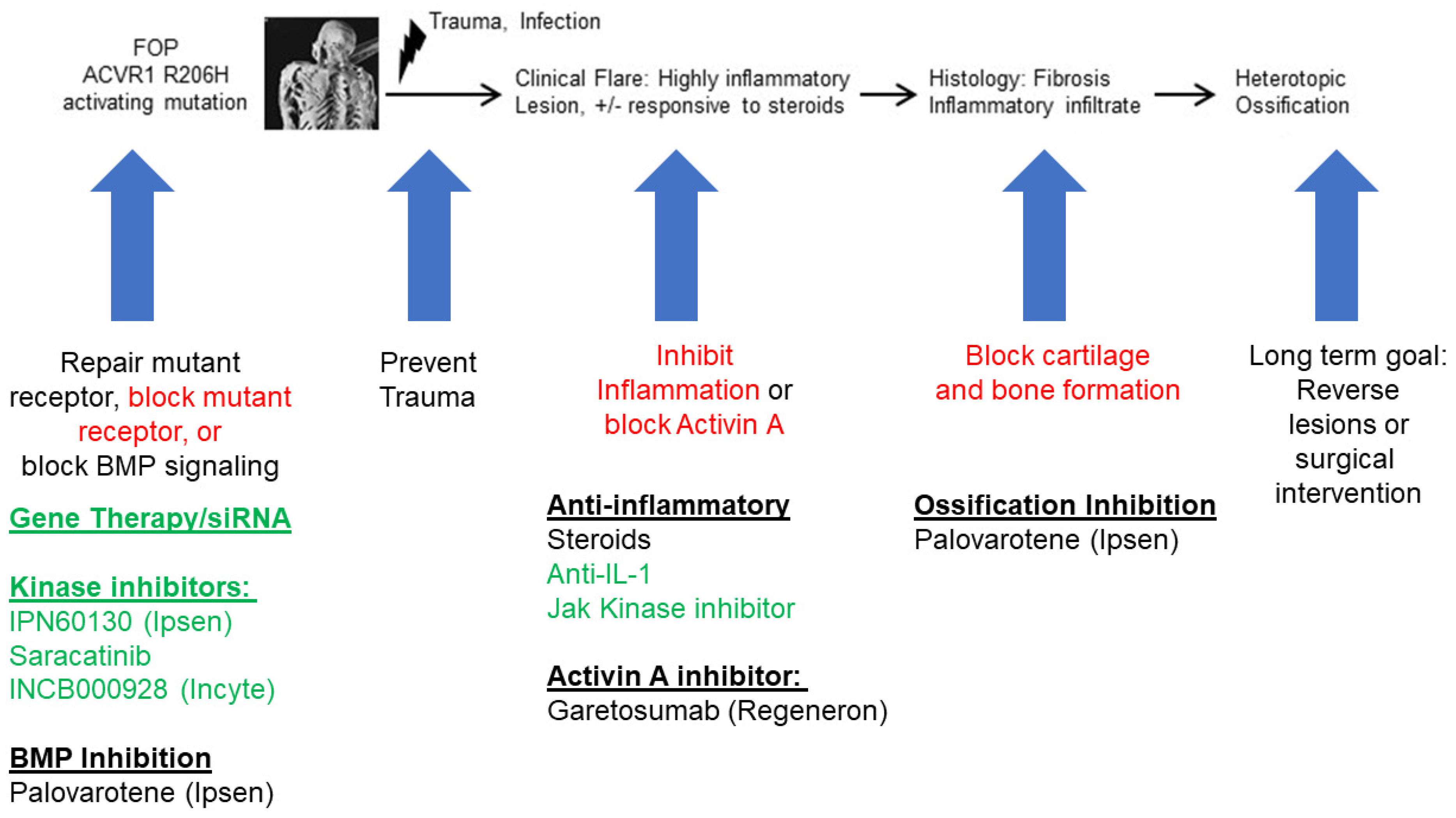{kind=link}
Table 1.
High-level summary of immune cells, including macrophage, mast cells, and adaptive immune cells, and related molecular target and intervention for FOP.
Table 1.
High-level summary of immune cells, including macrophage, mast cells, and adaptive immune cells, and related molecular target and intervention for FOP.
| Immune Cell Type | Secreted Cytokines | Molecular Targets | Involvement in FOP | Intervention Strategies | References | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Macrophages | IL-1β, IL-6, TGF-β, TNF-α, BMPs, activin A, OSM, SP, NT-3 | BMP signaling pathway | Elevated levels of pro-inflammatory cytokines observed in FOP lesions, contributing to inflammation and aberrant tissue repair. | Targeting BMP signaling with inhibitors. Mitigating inflammation and aberrant bone formation using anti-inflammatory medications. | [16,19,62,63] | |||||
| Mast Cells | IL-6, IL-9, TNF-α | Histamine receptors | Increased mast cell infiltration observed in FOP lesion, potentially contributing to inflammatory responses and tissue remodeling. | Blocking histamine receptors with antagonists to reduce mast cell activation and inflammation. | [20,62,86,87] | |||||
| Adaptive Immune Cells (T cell and B cell) | IL-2, IL-4, IL-10, IL-17, IFN-γ | T cell receptors, B cell receptors | T cells may modulate inflammatory responses in FOP through cytokine production and regulation of immune cell activity. Cytokine production by B cells may contribute to inflammation and immune dysregulation. | Immune suppressive therapies targeting T and B cell activity to reduce inflammation and tissue damage. | [88,89,90] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Diolintzi, A.; Pervin, M.S.; Hsiao, E.C. Immunologic Aspects in Fibrodysplasia Ossificans Progressiva. Biomolecules 2024, 14, 357. https://doi.org/10.3390/biom14030357
AMA Style
Diolintzi A, Pervin MS, Hsiao EC. Immunologic Aspects in Fibrodysplasia Ossificans Progressiva. Biomolecules. 2024; 14(3):357. https://doi.org/10.3390/biom14030357
Chicago/Turabian StyleDiolintzi, Anastasia, Mst Shaela Pervin, and Edward C. Hsiao. 2024. "Immunologic Aspects in Fibrodysplasia Ossificans Progressiva" Biomolecules 14, no. 3: 357. https://doi.org/10.3390/biom14030357
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.