Preserving Yeast Genetic Heritage through DNA Damage Checkpoint Regulation and Telomere Maintenance

Abstract
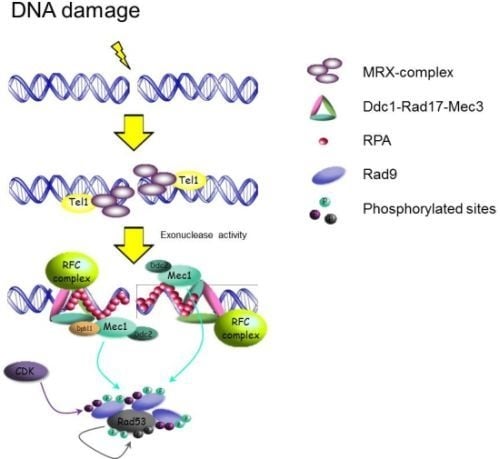
:
1. Introduction: The Importance of Genome Stability

2. The DNA Damage Checkpoint in S. cerevisiae
2.1. Activation of the DNA Damage Checkpoint in Yeast
2.1.1. Sensors: How the Signal Transduction Cascade Starts

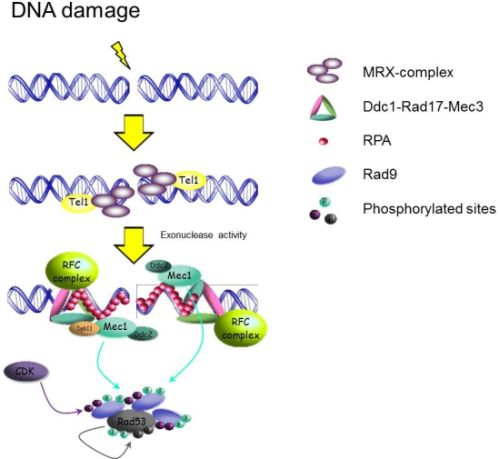{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Function | S. cerevisiae | H.sapiens | S.pombe |
|---|---|---|---|
| Sensors | |||
| 9-1-1 complex | Ddc1 | hRad9 | Rad9 |
| Mec3 | hHus1 | Hus1 | |
| Rad17 | hRad1 | Rad1 | |
| RFC-like clamp loader | Rad24 | hRad17 | Rad17 |
| Rfc2-5 | hRfc2-5 | Rfc2-5 | |
| MRX complex | Mre11 | hMre11 | Mre11 |
| Rad50 | hRad50 | Rad50 | |
| Xrs2 | hNbs1 | Nbs1 | |
| BRCT-containing | Dpb11 | TopBP1 | Cut5 |
| BRCA1 | |||
| hMdc1 | |||
| ss-DNA binding | RPA | RPA | RPA |
| Transducers | |||
| PI3K-like kinases | Mec1-Ddc2 | ATR-ATRIP | Rad3-Rad26 |
| Tel1 | ATM | Tel1 | |
| Adaptors | |||
| Rad9 | 53BP1 | Crb2 | |
| Mrc1 | CLSPN | Mrc1 | |
| Effectors | |||
| Checkpoint kinases | Chk1 | CHK1 | Chk1 |
| Rad53 | CHK2 | Cds1 |

2.1.2. Adaptors and Activation of Downstream Effector kinases
2.2. Inactivation of the DNA Damage Checkpoint: How to Silence the Checkpoint
2.2.1. Checkpoint Recovery: When It Is Safe to Switch the Checkpoint Off
2.2.2. Checkpoint Adaptation: Escaping the Checkpoint in the Presence of Damage
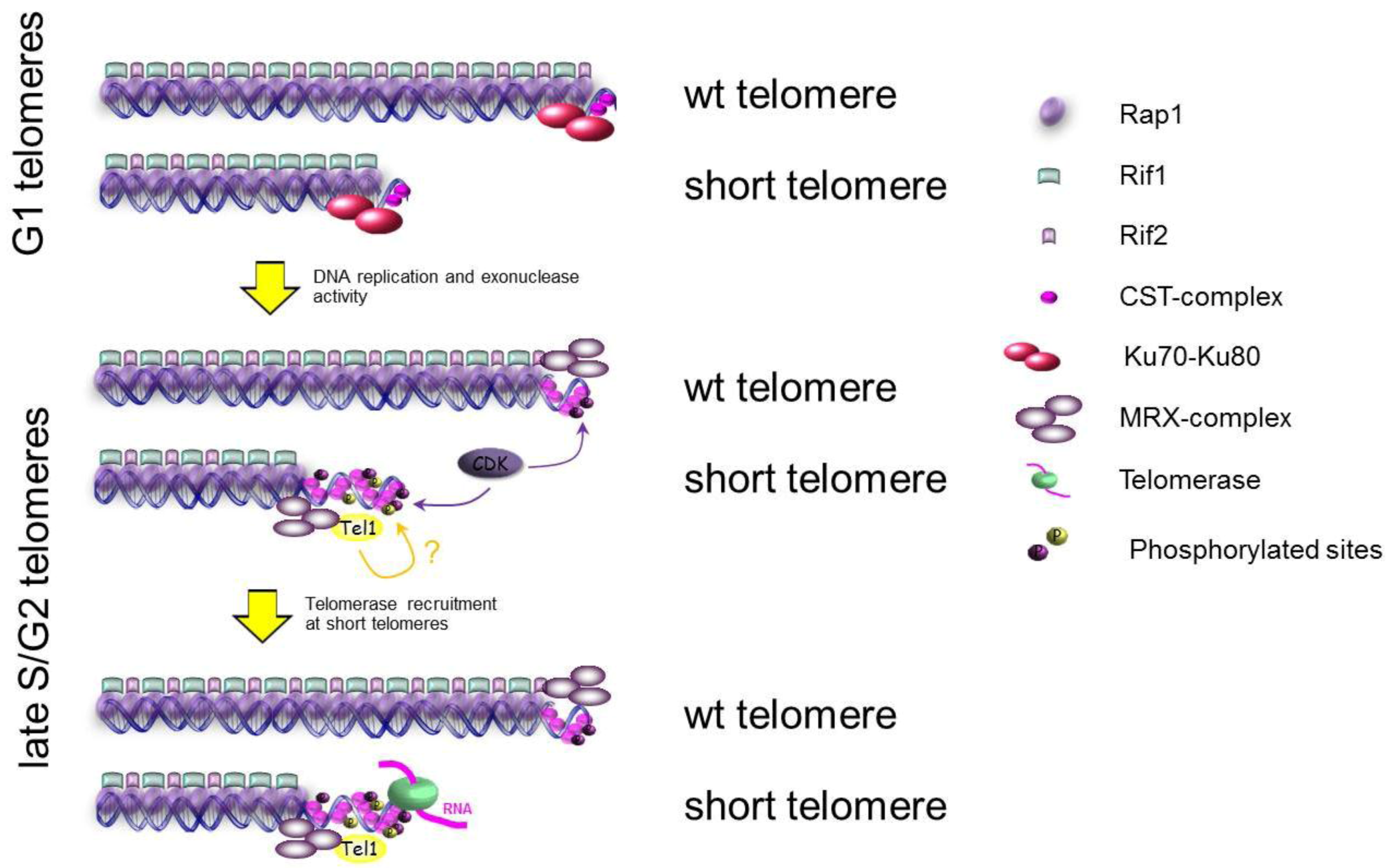
3. Genome Maintenance: Focusing on Telomere
3.1. Protection of Chromosome Ends: Telomeres
3.2. Replication of Telomeres: When and Where?
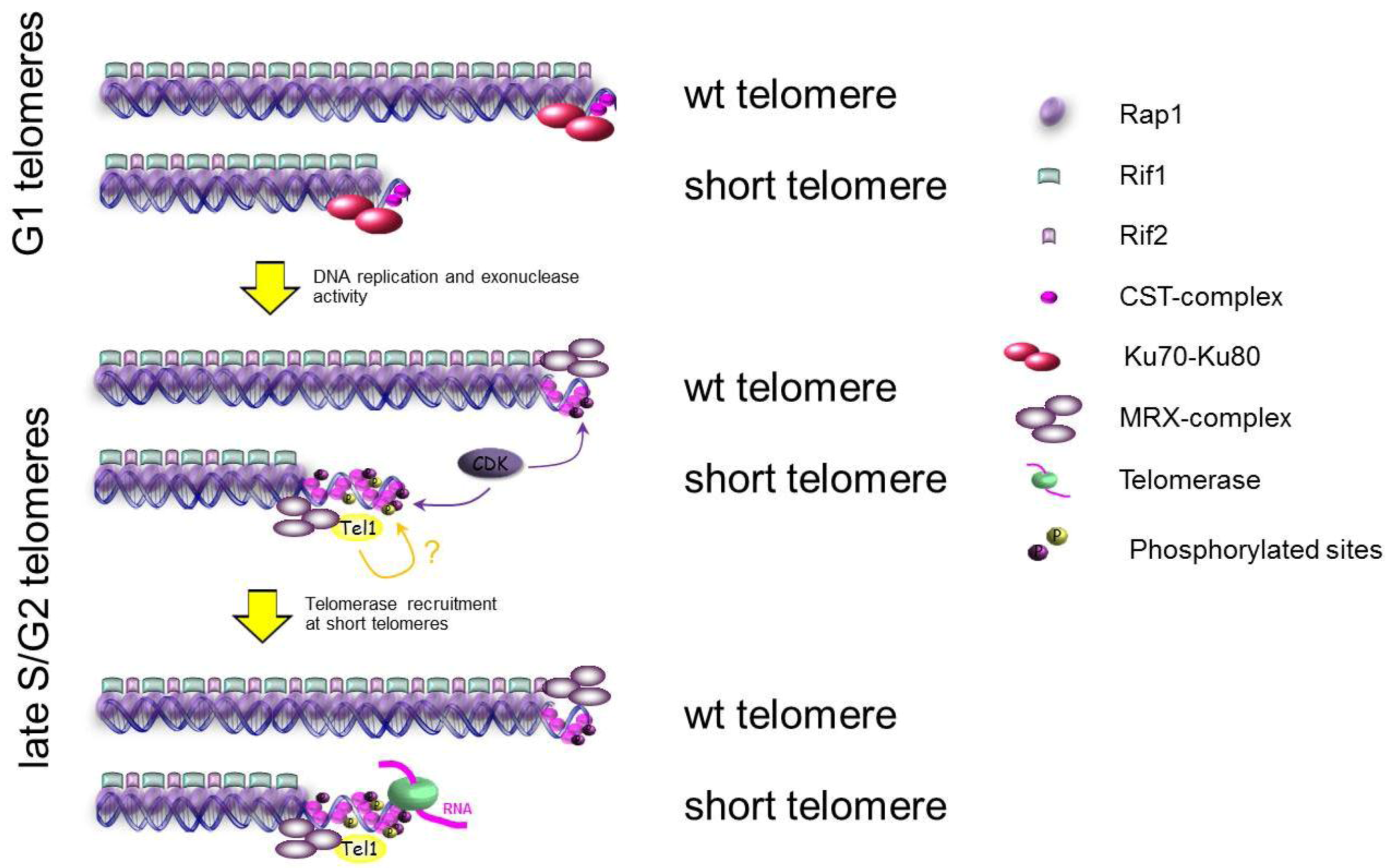
3.3. The Ends of Chromosomes Are Not DSBs
4. Conclusions
Acknowledgements
References
- Ciccia, A.; Elledge, S.J. The DNA damage response: making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef]
- Shiloh, Y. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef]
- Langerak, P.; Russell, P. Regulatory networks integrating cell cycle control with DNA damage checkpoints and double-strand break repair. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 3562–3571. [Google Scholar] [CrossRef]
- Flynn, R.L.; Zou, L. ATR: a master conductor of cellular responses to DNA replication stress. Trends Biochem. Sci. 2011, 36, 133–140. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef]
- Kolodner, R.D.; Putnam, C.D.; Myung, K. Maintenance of genome stability in Saccharomyces cerevisiae. Science 2002, 297, 552–557. [Google Scholar] [CrossRef]
- Vessey, C.J.; Norbury, C.J.; Hickson, I.D. Genetic disorders associated with cancer predisposition and genomic instability. Prog. Nucleic Acid Res. Mol. Biol. 1999, 63, 189–221. [Google Scholar] [CrossRef]
- Bayani, J.; Squire, J.A. Advances in the detection of chromosomal aberrations using spectral karyotyping. Clin. Genet. 2001, 59, 65–73. [Google Scholar] [CrossRef]
- Deininger, P.L.; Batzer, M.A. Alu repeats and human disease. Mol. Genet. MeTable 1999, 67, 183–193. [Google Scholar] [CrossRef]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Weinert, T.A.; Hartwell, L.H. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science 1988, 241, 317–322. [Google Scholar]
- Hartwell, L.H.; Weinert, T.A. Checkpoints: controls that ensure the order of cell cycle events. Science 1989, 246, 629–634. [Google Scholar]
- Lowndes, N.F.; Murguia, J.R. Sensing and responding to DNA damage. Curr. Opin. Genet. Dev. 2000, 10, 17–25. [Google Scholar] [CrossRef]
- Bartek, J.; Lukas, J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr. Opin. Cell Biol. 2007, 19, 238–245. [Google Scholar] [CrossRef]
- Longhese, M.P.; Foiani, M.; Muzi-Falconi, M.; Lucchini, G.; Plevani, P. DNA damage checkpoint in budding yeast. EMBO J. 1998, 17, 5525–5528. [Google Scholar] [CrossRef]
- Foiani, M.; Pellicioli, A.; Lopes, M.; Lucca, C.; Ferrari, M.; Liberi, G.; Muzi-Falconi, M.; Plevani1, P. DNA damage checkpoints and DNA replication controls in Saccharomyces cerevisiae. Mutat. Res. 2000, 451, 187–196. [Google Scholar] [CrossRef]
- Finn, K.; Lowndes, N.F.; Grenon, M. Eukaryotic DNA damage checkpoint activation in response to double-strand breaks. Cell Mol. Life Sci. 2012, 69, 1447–1473. [Google Scholar] [CrossRef]
- Harper, J.W.; Elledge, S.J. The DNA damage response: ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef]
- Vinella, D.; D'Ari, R. Overview of controls in the Escherichia coli cell cycle. Bioessays. 1995, 17, 527–536. [Google Scholar] [CrossRef]
- Lisby, M.; Barlow, J.H.; Burgess, R.C.; Rothstein, R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 2004, 118, 699–713. [Google Scholar] [CrossRef]
- Mantiero, D.; Clerici, M.; Lucchini, G.; Longhese, M.P. Dual role for Saccharomyces cerevisiae Tel1 in the checkpoint response to double-strand breaks. EMBO Rep. 2007, 8, 380–387. [Google Scholar] [CrossRef]
- Lee, J.-H.; Paull, T.T. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 2004, 304, 93–96. [Google Scholar] [CrossRef]
- Lee, J.-H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef]
- Fukunaga, K.; Kwon, Y.; Sung, P.; Sugimoto, K. Activation of protein kinase Tel1 through recognition of protein-bound DNA ends. Mol. Cell Biol. 2011, 31, 1959–1971. [Google Scholar] [CrossRef]
- Shiotani, B.; Zou, L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol. Cell 2009, 33, 547–558. [Google Scholar] [CrossRef]
- You, Z.; Chahwan, C.; Bailis, J.; Hunter, T.; Russell, P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol. Cell Biol. 2005, 25, 5363–5379. [Google Scholar] [CrossRef]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C. M.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef]
- Kondo, T.; Matsumoto, K.; Sugimoto, K. Role of a complex containing Rad17, Mec3, and Ddc1 in the yeast DNA damage checkpoint pathway. Mol. Cell Biol. 1999, 19, 1136–1143. [Google Scholar]
- Majka, J.; Burgers, P.M. J. The PCNA-RFC families of DNA clamps and clamp loaders. Prog. Nucleic Acid Res. Mol. Biol. 2004, 78, 227–260. [Google Scholar] [CrossRef]
- Lydall, D.; Weinert, T. G2/M checkpoint genes of Saccharomyces cerevisiae: Further evidence for roles in DNA replication and/or repair. Mol. Gen. Genet. 1997, 256, 638–651. [Google Scholar] [CrossRef]
- de, M.A.; Green, C.M.; Lowndes, N.F. RAD9 and RAD24 define two additive, interacting branches of the DNA damage checkpoint pathway in budding yeast normally required for Rad53 modification and activation. EMBO J. 1998, 17, 2687–2698. [Google Scholar] [CrossRef]
- Green, C.M.; Erdjument-Bromage, H.; Tempst, P.; Lowndes, N.F. A novel Rad24 checkpoint protein complex closely related to replication factor C. Curr. Biol. 2000, 10, 39–42. [Google Scholar] [CrossRef]
- Crabbé, L.; Thomas, A.; Pantesco, V.; De Vos, J.; Pasero, P.; Lengronne, A. Analysis of replication profiles reveals key role of RFC-Ctf18 in yeast replication stress response. Nat. Struct. Mol. Biol. 2010, 17, 1391–1397. [Google Scholar] [CrossRef]
- Bellaoui, M.; Chang, M.; Ou, J.; Xu, H.; Boone, C.; Brown, G.W. Elg1 forms an alternative RFC complex important for DNA replication and genome integrity. EMBO J. 2003, 22, 4304–4313. [Google Scholar] [CrossRef]
- Ben-Aroya, S.; Koren, A.; Liefshitz, B.; Steinlauf, R.; Kupiec, M. ELG1, a yeast gene required for genome stability, forms a complex related to replication factor C. Proc. Natl. Acad. Sci. USA 2003, 100, 9906–9911. [Google Scholar] [CrossRef]
- Aroya, S.B.; Kupiec, M. The Elg1 replication factor C-like complex: A novel guardian of genome stability. DNA Repair (Amst.) 2005, 4, 409–417. [Google Scholar] [CrossRef]
- Kanellis, P.; Agyei, R.; Durocher, D. Elg1 forms an alternative PCNA-interacting RFC complex required to maintain genome stability. Curr. Biol. 2003, 13, 1583–1595. [Google Scholar] [CrossRef]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef]
- Mordes, D.A.; Nam, E.A.; Cortez, D. Dpb11 activates the Mec1-Ddc2 complex. Proc. Natl. Acad. Sci. USA 2008, 105, 18730–18734. [Google Scholar]
- Navadgi-Patil, V.M.; Burgers, P.M. Yeast DNA replication protein Dpb11 activates the Mec1/ATR checkpoint kinase. J. Biol. Chem. 2008, 283, 35853–35859. [Google Scholar]
- Pfander, B.; Diffley, J.F. X. Dpb11 coordinates Mec1 kinase activation with cell cycle-regulated Rad9 recruitment. EMBO J. 2011.
- Wang, H.; Elledge, S.J. Genetic and physical interactions between DPB11 and DDC1 in the yeast DNA damage response pathway. Genetics 2002, 160, 1295–1304. [Google Scholar]
- Puddu, F.; Granata, M.; Di Nola, L.; Balestrini, A.; Piergiovanni, G.; Lazzaro, F.; Giannattasio, M.; Plevani, P.; Muzi-Falconi, M. Phosphorylation of the budding yeast 9-1-1 complex is required for Dpb11 function in the full activation of the UV-induced DNA damage checkpoint. Mol. Cell Biol. 2008, 28, 4782–4793. [Google Scholar] [CrossRef]
- Longhese, M.P.; Paciotti, V.; Fraschini, R.; Zaccarini, R.; Plevani, P.; Lucchini, G. The novel DNA damage checkpoint protein Ddc1p is phosphorylated periodically during the cell cycle and in response to DNA damage in budding yeast. EMBO J. 1997, 16, 5216–5226. [Google Scholar] [CrossRef]
- Navadgi-Patil, V.M.; Burgers, P.M. The unstructured C-terminal tail of the 9-1-1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Mol. Cell 2009, 36, 743–753. [Google Scholar]
- Tanaka, K.; Russell, P. Mrc1 channels the DNA replication arrest signal to checkpoint kinase Cds1. Nat. Cell Biol. 2001, 3, 966–972. [Google Scholar]
- Alcasabas, A.A.; Osborn, A.J.; Bachant, J.; Hu, F.; Werler, P.J.; Bousset, K.; Furuya, K.; Diffley, J.F.; Carr, A.M.; Elledge, S.J. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat. Cell Biol. 2001, 3, 958–965. [Google Scholar] [CrossRef]
- Schwartz, M.F.; Duong, J.K.; Sun, Z.; Morrow, J.S.; Pradhan, D.; Stern, D.F. Rad9 phosphorylation sites couple Rad53 to the Saccharomyces cerevisiae DNA damage checkpoint. Mol. Cell 2002, 9, 1055–1065. [Google Scholar] [CrossRef]
- Bando, M.; Katou, Y.; Komata, M.; Tanaka, H.; Itoh, T.; Sutani, T.; Shirahige, K. Csm3, Tof1, and Mrc1 form a heterotrimeric mediator complex that associates with DNA replication forks. J. Biol. Chem. 2009, 284, 34355–34365. [Google Scholar] [CrossRef]
- Tourrière, H.; Versini, G.; Cordón-Preciado, V.; Alabert, C.; Pasero, P. Mrc1 and Tof1 promote replication fork progression and recovery independently of Rad53. Mol. Cell 2005, 19, 699–706. [Google Scholar] [CrossRef]
- Szyjka, S.J.; Viggiani, C.J.; Aparicio, O.M. Mrc1 is required for normal progression of replication forks throughout chromatin in S. cerevisiae. Mol. Cell 2005, 19, 691–697. [Google Scholar] [CrossRef]
- Chen, S.-H.; Zhou, H. Reconstitution of Rad53 activation by Mec1 through adaptor protein Mrc1. J. Biol. Chem. 2009, 284, 18593–18604. [Google Scholar] [CrossRef]
- Berens, T.J.; Toczyski, D.P. Colocalization of Mec1 and Mrc1 is sufficient for Rad53 phosphorylation in vivo. Mol. Biol. Cell 2012, 23, 1058–1067. [Google Scholar] [CrossRef]
- Du, L.-L.; Nakamura, T.M.; Russell, P. Histone modification-dependent and -independent pathways for recruitment of checkpoint protein Crb2 to double-strand breaks. Genes Dev. 2006, 20, 1583–1596. [Google Scholar] [CrossRef]
- Huyen, Y.; Zgheib, O.; Ditullio, R.A.; Gorgoulis, V.G.; Zacharatos, P.; Petty, T.J.; Sheston, E.A.; Mellert, H.S.; Stavridi, E.S.; Halazonetis, T.D. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 2004, 432, 406–411. [Google Scholar] [CrossRef]
- Manke, I.A.; Lowery, D.M.; Nguyen, A.; Yaffe, M.B. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 2003, 302, 636–639. [Google Scholar] [CrossRef]
- Yu, X.; Chini, C.C. S.; He, M.; Mer, G.; Chen, J. The BRCT domain is a phospho-protein binding domain. Science 2003, 302, 639–642. [Google Scholar] [CrossRef]
- Hammet, A.; Magill, C.; Heierhorst, J.; Jackson, S.P. Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep. 2007, 8, 851–857. [Google Scholar] [CrossRef]
- Bonilla, C.Y.; Melo, J.A.; Toczyski, D.P. Colocalization of sensors is sufficient to activate the DNA damage checkpoint in the absence of damage. Mol. Cell 2008, 30, 267–276. [Google Scholar] [CrossRef]
- Wang, G.; Tong, X.; Weng, S.; Zhou, H. Multiple phosphorylation of Rad9 by CDK is required for DNA damage checkpoint activation. Cell Cycle 2012, 11, 3792–3800. [Google Scholar]
- Lee, S.J.; Schwartz, M.F.; Duong, J.K.; Stern, D.F. Rad53 phosphorylation site clusters are important for Rad53 regulation and signaling. Mol. Cell Biol. 2003, 23, 6300–6314. [Google Scholar] [CrossRef]
- Chen, S.-H.; Smolka, M.B.; Zhou, H. Mechanism of Dun1 activation by Rad53 phosphorylation in Saccharomyces cerevisiae. J. Biol. Chem. 2007, 282, 986–995. [Google Scholar]
- Bashkirov, V.I.; Bashkirova, E.V.; Haghnazari, E.; Heyer, W.D. Direct kinase-to-kinase signaling mediated by the FHA phosphoprotein recognition domain of the Dun1 DNA damage checkpoint kinase. Mol. Cell Biol. 2003, 23, 1441–1452. [Google Scholar] [CrossRef]
- Lee, H.; Yuan, C.; Hammet, A.; Mahajan, A.; Chen, E.S.-W.; Wu, M.-R.; Su, M.-I.; Heierhorst, J.; Tsai, M.-D. Diphosphothreonine-specific interaction between an SQ/TQ cluster and an FHA domain in the Rad53-Dun1 kinase cascade. Mol. Cell 2008, 30, 767–778. [Google Scholar] [CrossRef]
- Lee, S.E.; Pellicioli, A.; Demeter, J.; Vaze, M.P.; Gasch, A.P.; Malkova, A.; Brown, P.O.; Botstein, D.; Stearns, T.; Foiani, M.; Haber, J.E. Arrest, adaptation, and recovery following a chromosome double-strand break in Saccharomyces cerevisia. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 303–314. [Google Scholar] [CrossRef]
- Clémenson, C.; Marsolier-Kergoat, M.C. DNA damage checkpoint inactivation: adaptation and recovery. DNA Repair 2009, 8, 1101–1109. [Google Scholar] [CrossRef]
- Vaze, M.B.; Pellicioli, A.; Lee, S.E.; Ira, G.; Liberi, G.; Arbel-Eden, A.; Foiani, M.; Haber, J.E. Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol. Cell 2002, 10, 373–385. [Google Scholar] [CrossRef]
- Yeung, M.; Durocher, D. Srs2 enables checkpoint recovery by promoting disassembly of DNA damage foci from chromatin. DNA Repair 2011, 10, 1213–1222. [Google Scholar] [CrossRef]
- Keogh, M.-C.; Kim, J.-A.; Downey, M.; Fillingham, J.; Chowdhury, D.; Harrison, J.C.; Onishi, M.; Datta, N.; Galicia, S.; Emili, A.; Lieberman, J.; Shen, X.; Buratowski, S.; Haber, J.E.; Durocher, D.; Greenblatt, J.F.; Krogan, N.J. A phosphatase complex that dephosphorylates gammaH2AX regulates DNA damage checkpoint recovery. Nature 2006, 439, 497–501. [Google Scholar]
- O'Neill, B.M.; Szyjka, S.J.; Lis, E.T.; Bailey, A.O.; Yates, J.R.; Aparicio, O.M.; Romesberg, F.E. Pph3-Psy2 is a phosphatase complex required for Rad53 dephosphorylation and replication fork restart during recovery from DNA damage. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 9290–9295. [Google Scholar]
- Woolstencroft, R.N.; Beilharz, T.H.; Cook, M.A.; Preiss, T.; Durocher, D.; Tyers, M. Ccr4 contributes to tolerance of replication stress through control of CRT1 mRNA poly(A) tail length. J. Cell Sci. 2006, 119, 5178–5192. [Google Scholar] [CrossRef]
- Clerici, M.; Mantiero, D.; Lucchini, G.; Longhese, M.P. The Saccharomyces cerevisiae Sae2 protein promotes resection and bridging of double strand break ends. J. Biol. Chem. 2005, 280, 38631–38638. [Google Scholar]
- Baroni, E.; Viscardi, V.; Cartagena-Lirola, H.; Lucchini, G.; Longhese, M.P. The functions of budding yeast Sae2 in the DNA damage response require Mec1- and Tel1-dependent phosphorylation. Mol. Cell Biol. 2004, 24, 4151–4165. [Google Scholar] [CrossRef]
- Clerici, M.; Mantiero, D.; Lucchini, G.; Longhese, M.P. The Saccharomyces cerevisiae Sae2 protein negatively regulates DNA damage checkpoint signalling. EMBO Rep. 2006, 7, 212–218. [Google Scholar] [CrossRef]
- Leroy, C.; Lee, S.E.; Vaze, M.B.; Ochsenbein, F.; Ochsenbien, F.; Guérois, R.; Haber, J.E.; Marsolier-Kergoat, M.-C. PP2C phosphatases Ptc2 and Ptc3 are required for DNA checkpoint inactivation after a double-strand break. Mol. Cell 2003, 11, 827–835. [Google Scholar] [CrossRef]
- Smolka, M.B.; Chen, S.H.; Maddox, P.S.; Enserink, J.M.; Albuquerque, C.P.; Wei, X.X.; Desai, A.; Kolodner, R.D.; Zhou, H. An FHA domain-mediated protein interaction network of Rad53 reveals its role in polarized cell growth. J. Cell Biol. 2006, 175, 743–753. [Google Scholar] [CrossRef]
- Guillemain, G.; Ma, E.; Mauger, S.; Miron, S.; Thai, R.; Guérois, R.; Ochsenbein, F.; Marsolier-Kergoat, M.-C. Mechanisms of checkpoint kinase Rad53 inactivation after a double-strand break in Saccharomyces cerevisiae. Mol. Cell Biol. 2007, 27, 3378–3389. [Google Scholar] [CrossRef]
- Bazzi, M.; Mantiero, D.; Trovesi, C.; Lucchini, G.; Longhese, M.P. Dephosphorylation of gamma H2A by Glc7/protein phosphatase 1 promotes recovery from inhibition of DNA replication. Mol. Cell Biol. 2010, 30, 131–145. [Google Scholar] [CrossRef]
- Travesa, A.; Duch, A.; Quintana, D.G. Distinct phosphatases mediate the deactivation of the DNA damage checkpoint kinase Rad53. J. Biol. Chem. 2008, 283, 17123–17130. [Google Scholar] [CrossRef]
- Kim, J.-A.; Hicks, W.M.; Li, J.; Tay, S.Y.; Haber, J.E. Protein phosphatases Pph3, Ptc2, and Ptc3 play redundant roles in DNA double-strand break repair by homologous recombination. Mol. Cell Biol. 2011, 31, 507–516. [Google Scholar] [CrossRef]
- Toczyski, D.P.; Galgoczy, D.J.; Hartwell, L.H. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 1997, 90, 1097–1106. [Google Scholar] [CrossRef]
- Vidanes, G.M.; Sweeney, F.D.; Galicia, S.; Cheung, S.; Doyle, J.P.; Durocher, D.; Toczyski, D.P. CDC5 inhibits the hyperphosphorylation of the checkpoint kinase Rad53, leading to checkpoint adaptation. PLoS Biol. 2010, 8, e1000286. [Google Scholar] [CrossRef]
- Tercero, J.A.; Longhese, M.P.; Diffley, J.F. X. A central role for DNA replication forks in checkpoint activation and response. Mol. Cell 2003, 11, 1323–1336. [Google Scholar] [CrossRef]
- Pellicioli, A.; Lee, S.E.; Lucca, C.; Foiani, M.; Haber, J.E. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol. Cell 2001, 7, 293–300. [Google Scholar] [CrossRef]
- Myung, K.; Datta, A.; Kolodner, R.D. Suppression of spontaneous chromosomal rearrangements by S phase checkpoint functions in Saccharomyces cerevisiae. Cell 2001, 104, 397–408. [Google Scholar] [CrossRef]
- Craven, R.J.; Greenwell, P.W.; Dominska, M.; Petes, T.D. Regulation of genome stability by TEL1 and MEC1, yeast homologs of the mammalian ATM and ATR genes. Genetics 2002, 161, 493–507. [Google Scholar]
- Mieczkowski, P.A.; Mieczkowska, J.O.; Dominska, M.; Petes, T.D. Genetic regulation of telomere-telomere fusions in the yeast Saccharomyces cerevisae. Proc. Natl. Acad. Sci. USA 2003, 100, 10854–10859. [Google Scholar] [CrossRef]
- Smogorzewska, A.; de Lange, T. Regulation of telomerase by telomeric proteins. Annu. Rev. Biochem. 2004, 73, 177–208. [Google Scholar] [CrossRef]
- Vega, L.R.; Mateyak, M.K.; Zakian, V.A. Getting to the end: telomerase access in yeast and humans. Nat. Rev. Mol. Cell Biol. 2003, 4, 948–959. [Google Scholar]
- Verdun, R.E.; Karlseder, J. Replication and protection of telomeres. Nature 2007, 447, 924–931. [Google Scholar] [CrossRef]
- Nugent, C.I.; Hughes, T.R.; Lue, N.F.; Lundblad, V. Cdc13p: a single-strand telomeric DNA-binding protein with a dual role in yeast telomere maintenance. Science 1996, 274, 249–252. [Google Scholar] [CrossRef]
- Lin, J.J.; Zakian, V.A. The Saccharomyces CDC13 protein is a single-strand TG1-3 telomeric DNA-binding protein in vitro that affects telomere behavior in vivo. Proc. Natl. Acad. Sci. USA 1996, 93, 13760–13765. [Google Scholar] [CrossRef]
- Grandin, N.; Damon, C.; Charbonneau, M. Ten1 functions in telomere end protection and length regulation in association with Stn1 and Cdc13. EMBO J. 2001, 20, 1173–1183. [Google Scholar] [CrossRef]
- Grandin, N.; Reed, S.I.; Charbonneau, M. Stn1, a new Saccharomyces cerevisiae protein, is implicated in telomere size regulation in association with Cdc13. Genes Dev. 1997, 11, 512–527. [Google Scholar] [CrossRef]
- Sun, J.; Yu, E.Y.; Yang, Y.; Confer, L.A.; Sun, S.H.; Wan, K.; Lue, N.F.; Lei, M. Stn1-Ten1 is an Rpa2-Rpa3-like complex at telomeres. Genes Dev. 2009, 23, 2900–2914. [Google Scholar] [CrossRef]
- Gao, H.; Cervantes, R.B.; Mandell, E.K.; Otero, J.H.; Lundblad, V. RPA-like proteins mediate yeast telomere function. Nat. Struct. Mol. Biol. 2007, 14, 208–214. [Google Scholar] [CrossRef]
- Pennock, E.; Buckley, K.; Lundblad, V. Cdc13 delivers separate complexes to the telomere for end protection and replication. Cell 2001, 104, 387–396. [Google Scholar] [CrossRef]
- Taggart, A.K. P.; Teng, S.-C.; Zakian, V.A. Est1p as a cell cycle-regulated activator of telomere-bound telomerase. Science 2002, 297, 1023–1026. [Google Scholar] [CrossRef]
- Bonetti, D.; Martina, M.; Clerici, M.; Lucchini, G.; Longhese, M.P. Multiple pathways regulate 3' overhang generation at S. cerevisiae telomeres. Mol. Cell 2009, 35, 70–81. [Google Scholar] [CrossRef]
- Zhu, Z.; Chung, W.-H.; Shim, E.Y.; Lee, S.E.; Ira, G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 2008, 134, 981–994. [Google Scholar] [CrossRef]
- Mimitou, E.P.; Symington, L.S. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 2008, 455, 770–774. [Google Scholar] [CrossRef]
- Ira, G.; Pellicioli, A.; Balijja, A.; Wang, X.; Fiorani, S.; Carotenuto, W.; Liberi, G.; Bressan, D.; Wan, L.; Hollingsworth, N.M.; Haber, J.E.; Foiani, M. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004, 431, 1011–1017. [Google Scholar] [CrossRef]
- Chen, X.; Niu, H.; Chung, W.-H.; Zhu, Z.; Papusha, A.; Shim, E.Y.; Lee, S.E.; Sung, P.; Ira, G. Cell cycle regulation of DNA double-strand break end resection by Cdk1-dependent Dna2 phosphorylation. Nat. Struct. Mol. Biol. 2011, 18, 1015–1019. [Google Scholar] [CrossRef]
- Huertas, P.; Cortés-Ledesma, F.; Sartori, A.A.; Aguilera, A.; Jackson, S.P. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 2008, 455, 689–692. [Google Scholar] [CrossRef]
- Bonetti, D.; Clerici, M.; Anbalagan, S.; Martina, M.; Lucchini, G.; Longhese, M.P. Shelterin-like proteins and Yku inhibit nucleolytic processing of Saccharomyces cerevisiae telomeres. PLoS Genet. 2010, 6, e1000966. [Google Scholar] [CrossRef]
- Marcand, S.; Wotton, D.; Gilson, E.; Shore, D. Rap1p and telomere length regulation in yeast. Ciba Found. Symp. 1997, 211, 76–93, discussion 93-103. [Google Scholar]
- Vodenicharov, M.D.; Laterreur, N.; Wellinger, R.J. Telomere capping in non-dividing yeast cells requires Yku and Rap1. EMBO J. 2010, 29, 3007–3019. [Google Scholar] [CrossRef]
- Fisher, T.S.; Taggart, A.K. P.; Zakian, V.A. Cell cycle-dependent regulation of yeast telomerase by Ku. Nat. Struct. Mol. Biol. 2004, 11, 1198–1205. [Google Scholar] [CrossRef]
- Tuzon, C.T.; Wu, Y.; Chan, A.; Zakian, V.A. The Saccharomyces cerevisiae telomerase subunit Est3 binds telomeres in a cell cycle- and Est1-dependent manner and interacts directly with Est1 in vitro. PLoS Genet. 2011, 7. [Google Scholar] [CrossRef]
- Chan, A.; Boulé, J.-B.; Zakian, V.A. Two pathways recruit telomerase to Saccharomyces cerevisiae telomeres. PLoS Genet. 2008, 4. [Google Scholar] [CrossRef]
- Qi, H.; Zakian, V.A. The Saccharomyces telomere-binding protein Cdc13p interacts with both the catalytic subunit of DNA polymerase alpha and the telomerase-associated est1 protein. Genes Dev. 2000, 14, 1777–1788. [Google Scholar]
- Li, S.; Makovets, S.; Matsuguchi, T.; Blethrow, J.D.; Shokat, K.M.; Blackburn, E.H. Cdk1-dependent phosphorylation of Cdc13 coordinates telomere elongation during cell-cycle progression. Cell 2009, 136, 50–61. [Google Scholar] [CrossRef]
- Gao, H.; Toro, T.B.; Paschini, M.; Braunstein-Ballew, B.; Cervantes, R.B.; Lundblad, V. Telomerase recruitment in Saccharomyces cerevisiae is not dependent on Tel1-mediated phosphorylation of Cdc13. Genetics 2010, 186, 1147–1159. [Google Scholar] [CrossRef]
- Teixeira, M.T.; Arneric, M.; Sperisen, P.; Lingner, J. Telomere length homeostasis is achieved via a switch between telomerase- extendible and -nonextendible states. Cell 2004, 117, 323–335. [Google Scholar] [CrossRef]
- Levy, D.L.; Blackburn, E.H. Counting of Rif1p and Rif2p on Saccharomyces cerevisiae telomeres regulates telomere length. Mol. Cell Biol. 2004, 24, 10857–10867. [Google Scholar] [CrossRef]
- McGee, J.S.; Phillips, J.A.; Chan, A.; Sabourin, M.; Paeschke, K.; Zakian, V.A. Reduced Rif2 and lack of Mec1 target short telomeres for elongation rather than double-strand break repair. Nat. Struct. Mol. Biol. 2010, 17, 1438–1445. [Google Scholar] [CrossRef]
- Wotton, D.; Shore, D. A novel Rap1p-interacting factor, Rif2p, cooperates with Rif1p to regulate telomere length in Saccharomyces cerevisia. Genes Dev. 1997, 11, 748–760. [Google Scholar] [CrossRef]
- Sabourin, M.; Tuzon, C.T.; Zakian, V.A. Telomerase and Tel1p preferentially associate with short telomeres in S. cerevisiae. Mol. Cell 2007, 27, 550–561. [Google Scholar] [CrossRef]
- Hector, R.E.; Shtofman, R.L.; Ray, A.; Chen, B.-R.; Nyun, T.; Berkner, K.L.; Runge, K.W. Tel1p preferentially associates with short telomeres to stimulate their elongation. Mol. Cell 2007, 27, 851–858. [Google Scholar] [CrossRef]
- Bianchi, A.; Shore, D. Increased association of telomerase with short telomeres in yeast. Genes Dev. 2007, 21, 1726–1730. [Google Scholar] [CrossRef]
- Hirano, Y.; Fukunaga, K.; Sugimoto, K. Rif1 and Rif2 inhibit localization of tel1 to DNA ends. Mol. Cell 2009, 33, 312–322. [Google Scholar] [CrossRef]
- Tseng, S.-F.; Lin, J.-J.; Teng, S.-C. The telomerase-recruitment domain of the telomere binding protein Cdc13 is regulated by Mec1p/Tel1p-dependent phosphorylation. Nucleic Acids Res. 2006, 34, 6327–6336. [Google Scholar]
- Wu, Y.; Zakian, V.A. The telomeric Cdc13 protein interacts directly with the telomerase subunit Est1 to bring it to telomeric DNA ends in vitro. Proc. Natl. Acad. Sci. USA 2011, 108, 20362–20369. [Google Scholar] [CrossRef]
- Smolka, M.B.; Albuquerque, C.P.; Chen, S.H.; Zhou, H. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc. Natl. Acad. Sci. USA. 2007, 104, 10364–10369. [Google Scholar] [CrossRef]
- Longhese, M.P. DNA damage response at functional and dysfunctional telomeres. Genes Dev. 2008, 22, 125–140. [Google Scholar] [CrossRef]
- Ribeyre, C.; Shore, D. Anticheckpoint pathways at telomeres in yeast. Nat. Struct. Mol. Biol. 2012, 19, 307–313. [Google Scholar]
- Michelson, R.J.; Rosenstein, S.; Weinert, T. A telomeric repeat sequence adjacent to a DNA double-stranded break produces an anticheckpoint. Genes Dev. 2005, 19, 2546–2559. [Google Scholar] [CrossRef]
- Xue, Y.; Rushton, M.D.; Maringele, L. A novel checkpoint and RPA inhibitory pathway regulated by Rif1. PLoS Genet. 2011, 7. [Google Scholar] [CrossRef]
- Chen, S.H.; Albuquerque, C.P.; Liang, J.; Suhandynata, R.T.; Zhou, H. A proteome-wide analysis of kinase-substrate network in the DNA damage response. J. Biol. Chem. 2010, 285, 12803–12812. [Google Scholar]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; Shiloh, Y.; Gygi, S.P.; Elledge, S.J. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Baldo, V.; Liang, J.; Wang, G.; Zhou, H. Preserving Yeast Genetic Heritage through DNA Damage Checkpoint Regulation and Telomere Maintenance. Biomolecules 2012, 2, 505-523. https://doi.org/10.3390/biom2040505
Baldo V, Liang J, Wang G, Zhou H. Preserving Yeast Genetic Heritage through DNA Damage Checkpoint Regulation and Telomere Maintenance. Biomolecules. 2012; 2(4):505-523. https://doi.org/10.3390/biom2040505
Chicago/Turabian StyleBaldo, Veronica, Jason Liang, Guoliang Wang, and Huilin Zhou. 2012. "Preserving Yeast Genetic Heritage through DNA Damage Checkpoint Regulation and Telomere Maintenance" Biomolecules 2, no. 4: 505-523. https://doi.org/10.3390/biom2040505