Recent Advances in Cellular Glycomic Analyses

Laboratory of Medical and Functional Glycomics, Graduate School of Advanced Life Science and Frontier Research Center for Post-Genome Science and Technology, Hokkaido University, Sapporo 001-0021, Japan
*
Author to whom correspondence should be addressed.
Biomolecules 2013, 3(1), 198-225; https://doi.org/10.3390/biom3010198
Submission received: 2 December 2012
/
Revised: 28 January 2013
/
Accepted: 14 February 2013
/
Published: 21 February 2013
(This article belongs to the Special Issue Challenges in Glycan, Glycoprotein and Proteoglycan Research)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:A large variety of glycans is intricately located on the cell surface, and the overall profile (the glycome, given the entire repertoire of glycoconjugate-associated sugars in cells and tissues) is believed to be crucial for the diverse roles of glycans, which are mediated by specific interactions that control cell-cell adhesion, immune response, microbial pathogenesis and other cellular events. The glycomic profile also reflects cellular alterations, such as development, differentiation and cancerous change. A glycoconjugate-based approach would therefore be expected to streamline discovery of novel cellular biomarkers. Development of such an approach has proven challenging, due to the technical difficulties associated with the analysis of various types of cellular glycomes; however, recent progress in the development of analytical methodologies and strategies has begun to clarify the cellular glycomics of various classes of glycoconjugates. This review focuses on recent advances in the technical aspects of cellular glycomic analyses of major classes of glycoconjugates, including N- and O-linked glycans, derived from glycoproteins, proteoglycans and glycosphingolipids. Articles that unveil the glycomics of various biologically important cells, including embryonic and somatic stem cells, induced pluripotent stem (iPS) cells and cancer cells, are discussed.
1. Introduction
Human embryonic stem (ES) cells derived from the inner cell mass of the blastocyst and induced pluripotent stem (iPS) cells reprogrammed from somatic cells display indefinite growth, while maintaining pluripotency [1,2]. Extensive research has been conducted with these cells to help develop disease models, methods for drug screening and regenerative therapies. As progress using stem-cell-driven therapy has evolved dramatically, effective quality control methods and standardization of human iPS cells have become important requirements for clinical application of these therapies. The significance of identifying highly validated cellular biomarkers that enable prediction of health conditions and screening of patients is also evident. The Critical Path Initiative of the US Food and Drug Administration (FDA) highlights the importance of biomarkers that can predict or monitor responses to therapy for in vitro diagnostics, imaging and preclinical toxicogenomics [3]. Cells are currently defined by a combination of physical, phenotypic and functional properties; the identification of novel cell surface markers is highly advantageous to the rapid detection, characterization and isolation of particular cell populations. In fact, many studies aimed at discovering novel cellular biomarkers based on gene expression, proteomics and metabolite analyses are currently in progress [4].
Glycan expression analysis is an attractive option for the development of novel cellular biomarkers, because many of the frequently employed biomarkers, such as stage-specific embryonic antigens (SSEA-3/4/5) and tumor-rejection antigens (Tra-1-60 and Tra-1-81), are glycoconjugates [5,6,7,8,9]. Most FDA-approved tumor markers are either glycans or glycoproteins (e.g., AFP, CA10-9, CA125, CEA, PSA and HER2/NEU) [10]. These glycomarkers were identified following the rather fortuitous development of specific anti-glycoconjugate antibodies. The glycome is indirectly related to the genome through the specificity of glycosyltransferases that perform non-template mediated biosynthesis of glycans. By regulating the expression of glycan degradation enzymes and supply of nucleotide-activated sugars, a cell can produce glycan structures that are distinct from those of neighboring cell types. The glycans produced by each cell are a highly heterogeneous nested set of related structures that result from alternative branching patterns, incomplete glycosylation and post-glycosylational modifications, such as sulfation and acetylation [11]. These variations produce an enormous number of biosynthetically permissible glycan structures. The size of the cellular glycome is a matter of debate, but the number of glycan structures is estimated to be in excess of 100,000 to 500,000 [12]. Due to these complexities, direct analysis of glycans is highly valuable for accurate clarification of the spectra of cellular glycomics. Additional research into the cellular glycome may help to identify a potential goldmine of biomarkers.
Contrary to linear DNA and protein sequences, glycans have heterogeneous structures that differ in composition, branching, linkage and anomericity. These differences have caused glycomic research to lag far behind DNA- and protein-based research. However, rapid growth and interest in glycomic research, along with improved mass spectrometric (MS) analytical methodologies, have begun to aid development of powerful analytical procedures that meet the demand for cellular glycomics.
Cell surfaces are coated with a variety of intricately arranged glycoconjugates. Major components of the cellular glycome include N- and O-linked glycans derived from glycoproteins, glycosaminoglycans and glycosphingolipids. It is widely appreciated that the relative positioning of glycans, which can be regulated by their display on glycoprotein scaffolds with three-dimensional geometries or the spatial arrangement of glycoconjugates, can profoundly influence the avidity and specificity of glycan-oligomerized receptor interactions [13]. In addition, some carbohydrate epitopes (e.g., Lewisx, also known as SSEA-1) are known to be constituents of different glycoconjugates [14]. Therefore, it is important to clarify the distribution of particular epitopes in a cross-glycomics manner. Transgenic and knockout mice in which various glycogenes were removed or altered had no apparent phenotype [15], suggesting that compensation by alternate glycogene(s) may occur. These results indicate the importance of understanding the role of intra- and inter-glycomic correlations (correlations within and between the individual glycomes of various glycoconjugates) in maintaining cellular homeostasis and response to changes. Therefore, it is important to delineate a comprehensive cellular glycome by analysis of all major glycoconjugates, rather than individual glycomes. In this review, we will discuss structurally intensive methods that focus on the glycan moiety and enable analysis of the elemental glycomics of N- and O-linked glycans derived from glycoproteins, proteoglycans and glycosphingolipids, which are the major components of cell surface glycoconjugates.
2. Recent Advances in Elementary Glycomic Analysis
2.1. N-Glycans
Protein N-glycosylations play important roles in folding, oligomerization, sorting and transport of proteins [16,17]. De-N-glycosylation by peptide N-glycosidase F (PNGase F), and subsequent purification and detection protocols are well established; therefore, the cellular N-glycome is the most routinely analyzed glycome among various classes of glycoconjugates. In the Human Proteome Organization’s human disease glycomics/proteomics initiative project, the relative abundance of various N-glycans in a number of glycoprotein samples was analyzed in 20 laboratories, and the results of chromatographic and MS analyses were evaluated [18]. Although it is generally accepted that MS is not capable of accurately quantifying oligosaccharides unless stable isotope-labeled analogs are incorporated as internal standards, the results of this multi-institutional study indicated that matrix-assisted laser desorption/ionization (MALDI) time-of-flight MS and liquid chromatography (LC)/electrospray ionization (ESI) MS yielded quite compatible results with chromatography in the quantitation of oligosaccharides. A comparable study of the signal intensities generated from a mixture of equimolar amounts of various underivatized N-glycans in the MALDI mass spectrum did not show any significance for compounds with molecular masses greater than approximately 1 kDa [19]. It was also reported that methyl esterification of sialic acid renders sialylated oligosaccharides that are chemically equivalent to neutral oligosaccharides and allows simultaneous analysis of neutral and sialylated oligosaccharides by MALDI-TOF MS [20,21]. The inherently rapid and accurate nature of detection by MALDI-TOF MS gives this approach unique and significant advantages for high-throughput glycomic analysis [22].
Considering that quantitative cellular N-glycomics relies on highly efficient and reproducible liberation of oligosaccharides from cellular glycoproteins, it is important to maximize the efficiency of N-glycan liberation. Glycoproteins differ widely in their susceptibility to enzymatic digestion, since glycosylated sites are often obstructed by secondary and tertiary protein structures. Therefore, differences in the conditions of de-N-glycosylation by PNGase F may cause alterations in liberation efficiency. For example, preparation of tryptic glycopeptides and reductive alkylation facilitate the release of N-glycans via PNGase F digestion [23].
Upon liberation, N-glycans are analyzed in their intact form, reduced form, permethylated form or other derivatives used for reducing end modifications [24,25]. Permethylation of glycan hydroxyl groups has been used to facilitate sample clean-up; increase glycan hydrophobicity and volatility, which translates into higher sensitivity during MS analysis; allow simultaneous analysis of neutral and sialylated oligosaccharides; and facilitate MS/MS analysis by leading to predictable fragmentation. Miniaturized approaches using micro-spin columns packed with NaOH that permit effective derivatization with methyl iodide in less than one minute have also been described [26]. High-throughput quantitative analysis of N-glycans by MALDI-TOF MS has been demonstrated by using prior solid-phase permethylation in a 96-well plate configuration [27]. The lack of hydroxyl groups prevents the cleavage of other glycosidic bonds, rendering the permethylated oligosaccharides resistant to in-source fragmentation [28]. To confirm glycan composition, as well as obtain detailed structural information, selected ions are often subjected to tandem mass spectrometry (e.g., TOF/TOF). To define monosaccharides and their anomeric configurations and to confirm tentative sequences, glycans are often treated with exoglycosidases. These enzymes are specific to the stereochemical and anomeric configurations of the monosaccharides, including α2-6-, α2-3- and α2-8-bound sialic acid; α1-2-, α1-3/4- and α1-6-bound fucose; and β1-3- and β1-4-bound galactose.
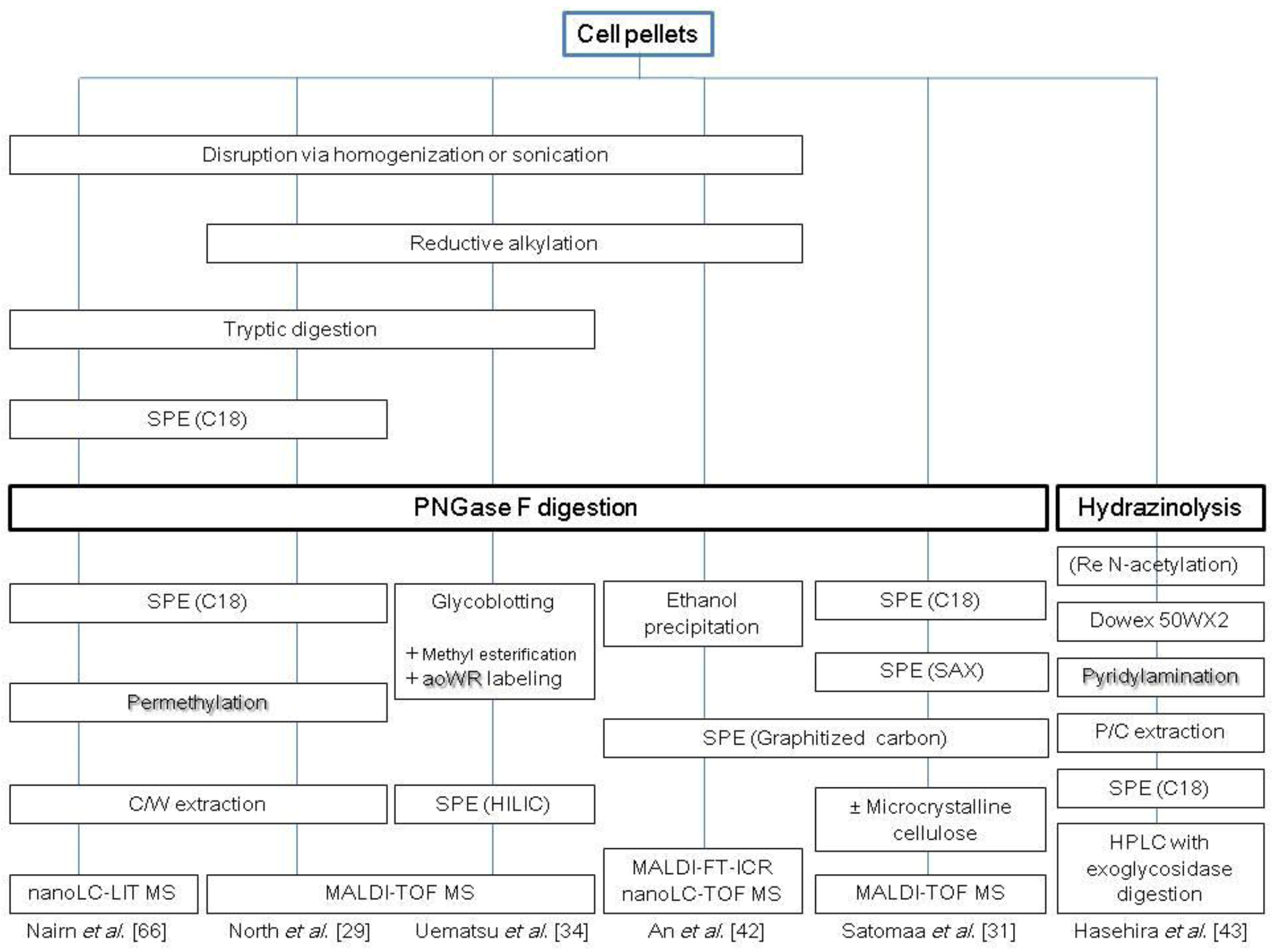
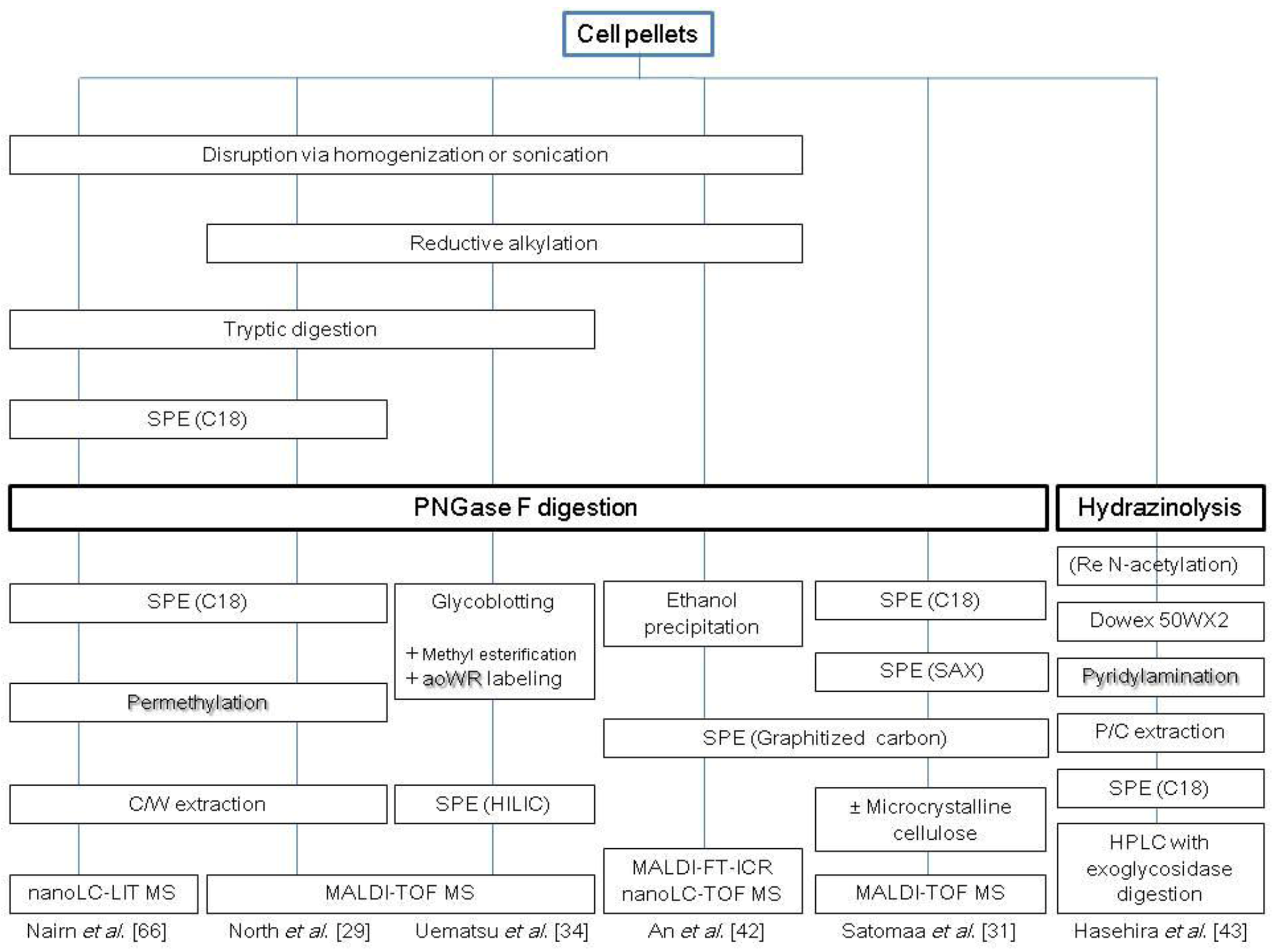
With the advent of MALDI-TOF MS technology and the basic research described above, it has become possible to define the structures of the multitude of individual glycans in various cell lines. A number of glycomic studies clarifying cellular N-glycomics have been performed. North et al. reported the N-glycomic profiles of parental Chinese Hamster Ovary (CHO Pro-5) and glycosylation mutant (CHO Lec1, CHO Lec2, etc.) cells [29]. In this study, cell pellets were sonicated, reduced and carboxymethylated and then digested with trypsin. Permethylated purified glycans were subjected to MALDI-TOF MS. This analysis revealed an unexpectedly large complexity of N-glycans in CHO cell mutants. The N-glycosylation profile of mouse and human immune cells, assessed by the same approach, has also been reviewed recently [30]. Satomaa et al. were the first to report the N-glycomic profile of human ES cells and their differentiated progeny [31]. In this study, cell pellets were subjected to PNGase F digestion and then purified by sequential precipitation/extraction using C18 silica chromatography columns with strong cation-exchange resin, porous graphitized carbon and microcrystalline cellulose (for the sialylated glycans). The purified N-glycans were directly subjected to MALDI-TOF MS analysis without any derivatization. This analysis revealed statistical significance for the human ES cells-associated N-glycan signals, e.g., large high-mannose type N-glycan signals, Man7, Man8 and Man9, as well as complex fucosylated N-glycan signals, (Hex)5(HexNAc)4(Fuc)2, (NeuAc)1(Hex)5(HexNAc)4(Fuc)2 and (NeuAc)1(Hex)5(HexNAc)4(Fuc)3. This study also demonstrated that the N-glycan phenotype of human ESs reflected their differentiation stage. Notably, the authors of the study analyzed the glycans by 800 MHz 1H nuclear magnetic resonance (NMR) using cryo-probes.
Furukawa et al. captured glycans with a solid-phase hydrazide-functionalized glycoblotting polymer (BlotGlyco H), followed by washing to remove the non-bound material [32]. Unreacted hydrazide groups were blocked with acetic anhydride, and sialic acids were stabilized by methyl ester formation using 3-methyl-p-tolyltriazine [21]. The blotted glycans were recovered in their reducing terminal-derivative form by adding the aminooxy-containing compound aoWR, which substantially improves detection sensitivity with MALDI-TOF MS analysis [33]. This procedure can be performed in a 96-well plate format and can be used with MALDI-TOF MS for N-glycomic analysis of murine dermis and epidermis [34], as well as murine ES cells and their differentiated progenies (i.e., cardiomyocytes or neural cells) [35].
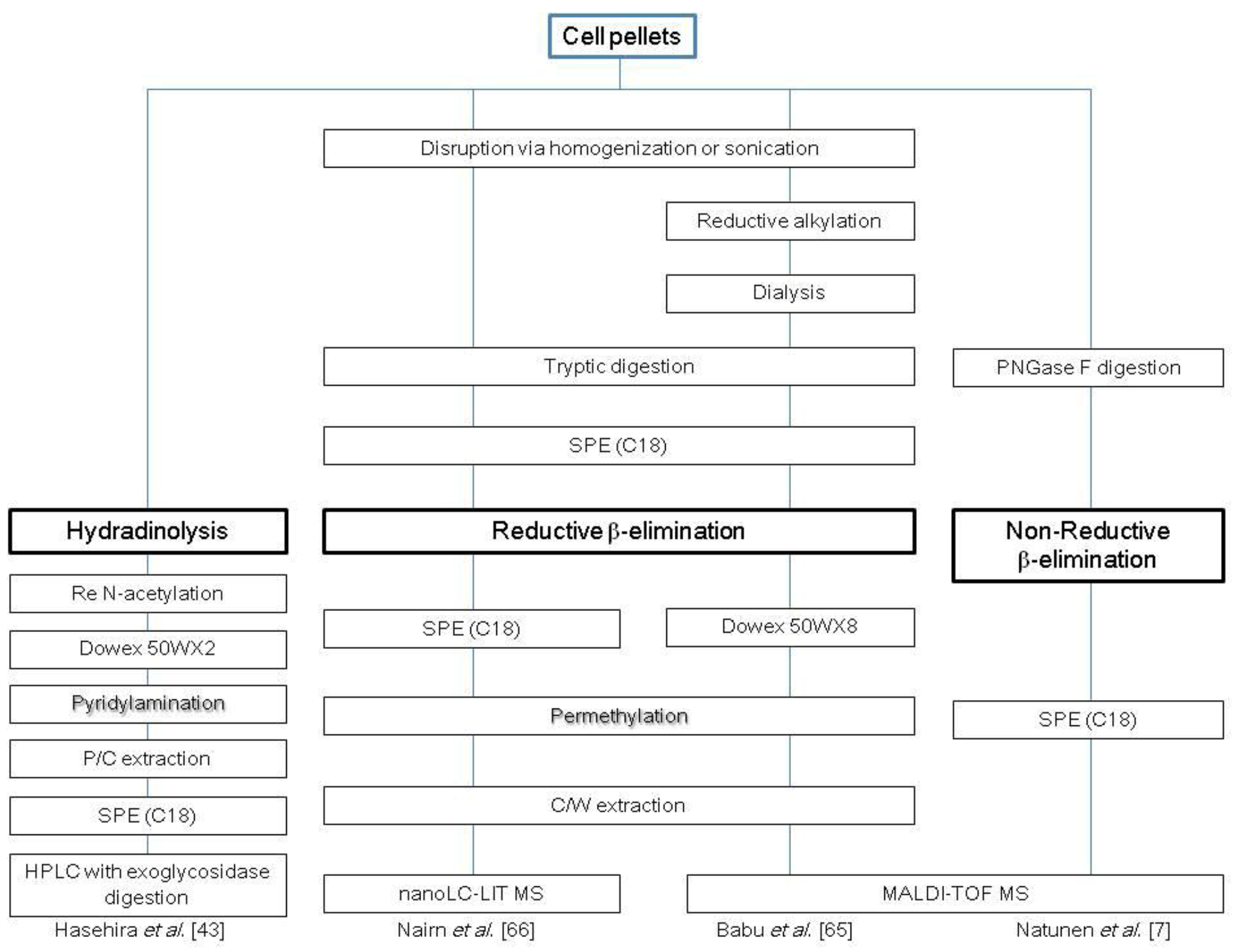
Chromatographic profiling of glycans enables not only the discovery of structure-specific markers, but also unprecedented structural analysis of the glycome. Coupling of chromatographic profiling with MS complements the strategy of glycan mass profiling by MALDI-TOF MS, since MALDI-TOF MS profiling alone is often unable to discriminate isomers and detect low abundant glycan species due to its limited dynamic range (typically in the order of 102 to 103). Separation modes for N-glycan analysis include reversed-phase (RP) chromatography [36,37], normal-phase chromatography/hydrophilic interaction chromatography (HILIC)[38,39] and graphitized carbon chromatography [40], all of which display good compatibility with MS and high resolution power. MS analyzers, such as ion trap, quadrupole, FTICR orbitrap and ion mobility with electrospray ionization have been used for glycomic and glycoproteomic analysis [41]. An et al. reported the N-glycomic profile of enriched plasma membranes from human ES cells [42]. In combination with MALDI-TOF MS analysis, this study analyzed N-glycans using a microfluidic HPLC-ChiP-TOF MS system to discriminate isomers. The microfluidic HPLC-Ch consisted of an enrichment column, an LC separation column packed with porous graphitized carbon and a nanoelectrospray tip. The microchip was interfaced with a TOF mass analyzer that routinely provides a mass measurement accuracy of less than 5 ppm. The isolated membrane fraction was reduced with dithiothreitol (DTT) and then subjected to PNGase F digestion. Following ethanol precipitation, N-glycans were purified by solid-phase extraction (SPE) using a graphitized carbon cartridge. This study identified an average of 170 distinct features, which included anomers and other isomers, arising from an average of 69 glycan compositions. Hasehira et al. reported the N-glycan profiles of a human iPS cell line and human dermal fibroblasts [43]. In this study, the glycans were liberated by gas-phase hydrazinolysis, fluorescently tagged with 2-aminopyridine (PA) at their reducing terminus, and then, the derived PA-glycans were purified by multiple-mode (anion-exchange, size-fractionation and RP) high-performance liquid chromatography (HPLC). The glycan structures were determined and quantified by HPLC mapping with MALDI-TOF-MS and exoglycosidase digestion analyses. It was found that the type of linkage of sialic acid (Sia) on N-linked glycans was dramatically changed from α-2-3 to α-2-6, and the expression of α-1-2 fucose and type 1 LacNAc structures became clearly and apparently increased upon induction of pluripotency. Several representative strategies for the analysis of cellular N-glycans are summarized in Scheme 1.
Analysis of extensive N-glycosylation sites can be performed using hydrazide-capture methods, as described previously [44]. Briefly, glycoproteins are chemically oxidized with sodium peroxidate and captured on hydrazide beads. The unbound non-glycosylated proteins are removed by rinsing, and the captured glycoproteins are subjected to on-bead reduction, alkylation, denaturation and tryptic digestion. The bound peptides are then labeled with stable isotope reagents for quantification, and the N-glycosylated peptides are released with PNGase F, purified by RP-SPE and analyzed by LC-MS/MS. This method has been utilized to elucidate the N-glycosylation site of multiple cell types, including CHO cells [45], human bronchial epithelial cells [46] and a human pancreatic carcinoma cell line [47]. Kurogochi et al. described a strategy for analysis of sialic acid-containing glycopeptides that involves selective oxidation of sialic acid residues to elaborate terminal aldehyde groups and subsequent enrichment by chemical ligation with hydrazide beads [48].
Scheme 1.
Representative protocols for cellular N-glycomic analysis. C, chloroform; M, methanol; W, water; P, phenol; SPE, solid-phase extraction.
Scheme 1.
Representative protocols for cellular N-glycomic analysis. C, chloroform; M, methanol; W, water; P, phenol; SPE, solid-phase extraction.
Recent progress in MS techniques, including electron capture dissociation (ECD) and electron transfer dissociation (ETD) fragmentation, have enabled analysis of both glycan and peptide moieties, thus providing a powerful measurement tool for N-glycoproteomics [49]. However, extensive direct analysis of the cellular N-glycoproteome that unveils both glycan and peptide moieties is still a challenging task, due to the tremendous heterogeneity generated by both glycan and peptide moieties. Despite this difficulty, once target glycopeptides are identified, multiple reaction monitoring allows quantification of glycopeptides from complex biological samples [48,50].
2.2. O-Glycans
O-glycosylation represents a very diverse group of modifications, including the O-GalNAc type typical of mucin and the O-GlcNAc, O-Fuc, O-Man and O-Xyl types [51]. These modifications not only activate proteins, but also serve as markers indicating binding to other proteins. O-GlcNAcylation involves a single O-GlcNAc residue, while other modifications subsequently form more complicated oligosaccharide structures through the actions of various glycosyltransferases. Site-specific O-glycosylation is an important regulator of protein function; the most recently discovered function of O-glycosylation is co-regulation of protein processing by proprotein convertase [52,53]. The dynamic cell- and protein-specific regulation of site-directed glycosylation can be essential for development and health [52,54,55]. The initiating event of O-glycan biosynthesis that is classified into the mucin type is transfer of monosaccharidic GalNAc (from UDP-GalNAc) to serine and threonine residues via catalysis by a polypeptide GalNAc transferase. The resultant protein-linked GalNAc can be extended into different structural core classes (core 1 to core 8) by various glycosyl transferases. Mucins are high molecular weight glycoproteins characterized by a high density of glycans that form complex macromolecular structures [56,57]. Changes in mucin O-glycosylation appear to be a general phenomenon of malignancies [58,59]. The O-glycopeptide of MUC1 has been targeted in cancer and may be of diagnostic value [60].
Although the N-glycans in glycoproteins can be released by specific enzymes, such as PNGase F and PNGase A [14,61], O-glycan release is limited because an analogous endoglycosidase is currently unavailable. The commercially available endo-α-N-acetylgalactosaminidase (O-glycanase) has a high substrate specificity and is used only for the removal of core 1 disaccharides (Galβ1-3GalNAc) from mucin glycoproteins [62]. Therefore, most O-glycans are released from the core proteins by chemical procedures. Various chemical digestion approaches are often accompanied by significant loss of the intact O-glycans, due to serious glycan degradation (referred to as peeling reactions). Over the last two decades, several chemical release procedures have been studied. In the Human Proteome Organization’s human disease glycomics/proteomics initiative, O-glycans in a number of glycoprotein samples were analyzed in 15 laboratories worldwide [63]. Unlike the same analysis of N-glycans, where fairly consistent results were obtained among different laboratories and techniques, the results obtained for O-glycans differed substantially among the laboratories and techniques, suggesting that there continues to be a strong and growing need for sensitive, rapid and highly quantitative analysis of O-glycans. However, recent rapid progress in O-glycan analysis has begun to allow clarification of cellular O-glycomics.
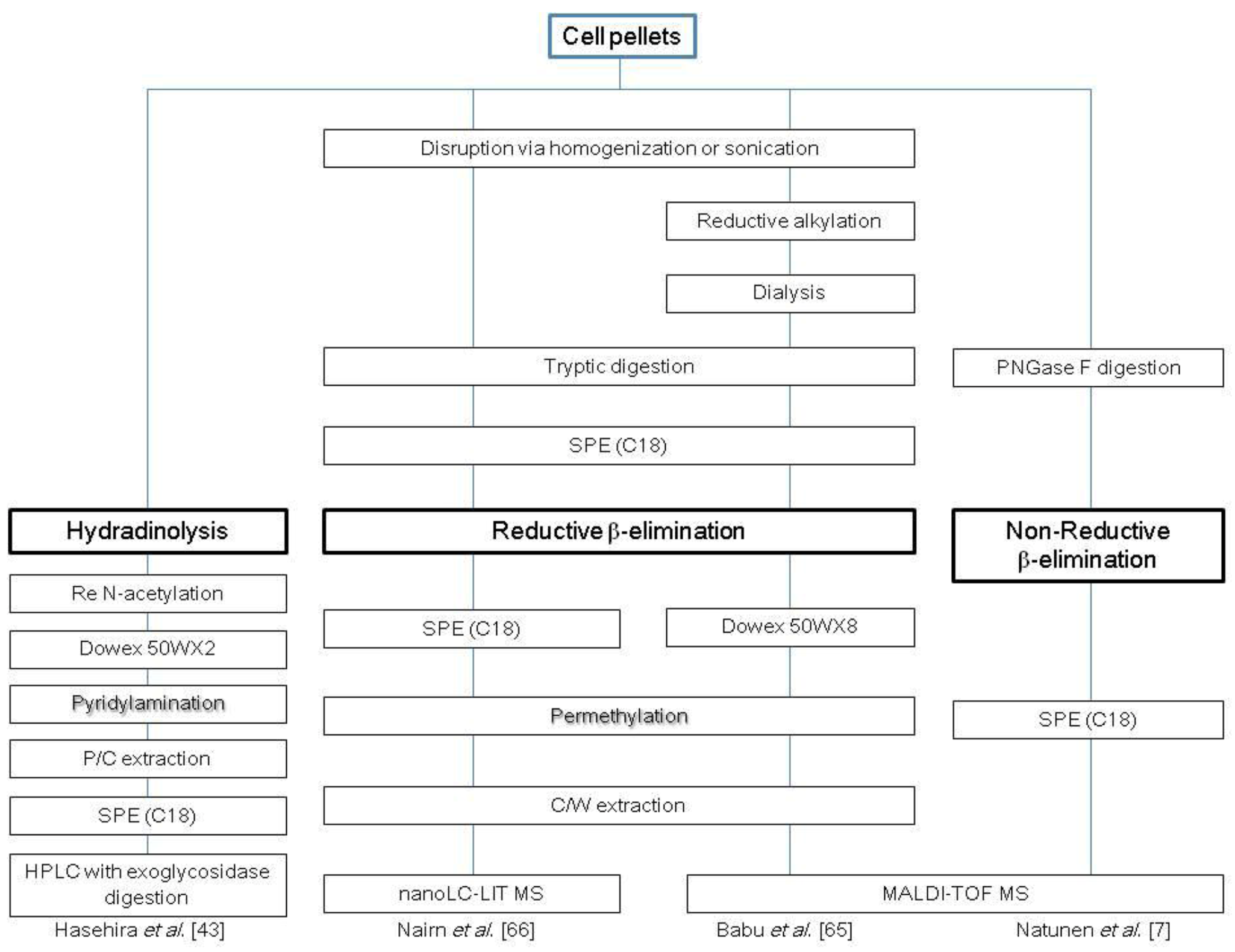
Reductive β-elimination, reported in 1968 by Carlson [64], is still the most reliable technique for liberation of O-glycans and has proven to be a feasible method for cellular O-glycomic analyses. Reductive β-elimination releases O-glycans in their reduced form (alditol form) as a result of immediate in situ reduction by sodium borohydride. Reduction of the innermost sugar to alditol minimizes the side peeling reaction. One limitation of this method is that reduction causes a loss of the reducing end of the carbohydrate, which precludes additional downstream glycomic analysis, such as derivatization suitable for enrichment, chromatographic and mass spectrometric analyses. Using the reductive β-elimination technique combined with MALDI-TOF analysis, Babu et al. reported the structural characterization of neutrophil O-glycomics [65]. Glycoproteins from human neutrophil cells were subjected to reductive carboxymethylation followed by digestion with trypsin. O-glycans were then released from tryptic glycopeptides by reductive β-elimination, and purified glycans by Dowex 50W-X8 were permethylated to enhance the sensitivity of MS detection. The O-glycomic profile consisted of core 1 and core 2 O-glycans with sialyl Lex and Lex as non-reducing terminal epitopes. The same technique was also used to determine the O-glycomic profile of parent and glycosylation mutant CHO cell lines [29]. The O-glycans from both wild-type (parent) and mutant CHO cell lines showed a simple set of core 1 structures comprising T-antigen, sialylated T and di-sialylated T. Nairn et al. reported the O-glycomic alteration of mouse ES cells upon differentiation into embryoid bodies or extraembryonic endodermal cells [66]. In this study, 28 types of O-glycans were identified, including mannosyl-, fucosyl- and glucosyl-type O-glycans. The authors of this study concluded that changes in glycan structures correlated with alterations in transcript abundance of the corresponding biosynthetic enzymes, suggesting that transcriptional regulation contributes significantly to the regulation of glycan expression. Maniatis et al. reported microwave-assisted reductive β-elimination using dimethylamine, which enabled short-time quantitative release of O-glycans from model glycopeptides and glycoproteins [67].
Extensive attempts to establish non-reductive β-elimination methods have been made, with the aim of enabling subsequent derivatization of reducing terminal for high sensitive detection. Huang et al. reported ammonium-based alkali-catalyzed non-reductive β-elimination [68], though it was reported that peeling cannot be completely prevented, and the hydrolysis yield is low compared to the reductive β-elimination method [69]. Natunen et al. used the technique to identify the O-glycomic profile of human ES cells [7]. This study identified (Hex)3(HexNAc)3, which has a Galβ1-3GlcNAc epitope at the non-reducing end and is tentatively assigned to be Galβ1-3GlcNAcβ1-3Galβ1-4GlcNAcβ1-6 (Galβ1-3GlcNAcβ1-3)Galβ1-4Glc, They reported that this structure is the epitope for Tra-1-60 and Tra-1-81 antibodies.
Hydrazinolysis is another chemical de-O-glycosylation method that produces O-glycans in a non-reduced form [70,71,72,73,74]. Hasehira et al. used this method to report the O-glycomic profile of human iPS cells and human dermal fibroblasts [43]. A cell pellet generated from approximately 1 × 107 cells was treated with anhydrous hydrazine at 100 °C for 4 h, and the released glycans were re-N-acetylated. The resulting non-reductive O-glycans were fluorescently tagged with 2-aminopyrydine and then analyzed using HPLC mapping assisted by MALDI-TOF-MS and exoglycosidase digestion. This study identified a total of ten different types of O-glycans and concluded that α-1-2 linked fucosylation and type 1 LAcNAc structures are critical epitopes that are characteristic to iPS cells. Several representative strategies for the analysis of cellular O-glycans are summarized in Scheme 2.
Zauner et al. [75], Wang et al. [76] and Furukawa et al. [77] have introduced a novel one-pot O-glycome analytical method combined with release from glycoproteins and labeling with pyrazolone analogs. This method, namely β-elimination in the presence of pyrazolone analogs (BEP), allows simultaneous labeling of released O-glycans with pyrazolone analogs and, thus, minimizes the undesirable peeling reaction.
Scheme 2.
Representative protocols for cellular O-glycomic analysis. C, chloroform; M, methanol; W, water; P, phenol; SPE, solid-phase extraction.
Scheme 2.
Representative protocols for cellular O-glycomic analysis. C, chloroform; M, methanol; W, water; P, phenol; SPE, solid-phase extraction.
Identification of O-glycosylation sites can be performed by β-elimination and subsequent Michael addition (BEMA). Hedou et al. identified the O-GlcNAc modification sites in purified contractile protein homogenates using an MS-based method that relied on BEMA of DTT, following dephosphorylation to discriminate between phosphorylation and O-glycosylation sites [78]. BEP allows analysis of released O-glycans and formerly O-glycosylated glycopeptides, because both species can be labeled by pyrazolone analogs and are thus amenable to further analysis [77]. It was recently reported that O-GlcNAc regulates pluripotency and reprogramming by acting directly on core components of the pluripotency network [79]. Oct4 was purified by immunoprecipitation of ZHBTc4 F-Oct4 cell lysates with a Flag antibody. O-GlcNAcylated Oct4 was then enriched by WGA pull down. After SDS-PAGE and Colloidal Blue Staining, the Oct4 band was analyzed using nano-LC-ESI-MS/MS. Trinidad et al. identified greater than 1,750 and 16,500 sites of O-GlcNAcylation and phosphorylation, respectively, by using a combination of fragmentation techniques, including ECD, collision-induced dissociation (CID) and higher-energy collisional dissociation (HCD) for GlcNAcylated, phosphor and non-modified peptides, respectively [80]. A technique for extensive O-glycosylation that uses an enzymatically glyco-engineered human cell line (SimpleCell line) expressing homogenous truncated O-glycosylation with GalNAcα (Tn) or NeuAcα2-6GalNAcα (STn) O-glycans was recently reported by Steentoft et al. This technique allows straightforward isolation and sequencing of GalNAc O-glycopeptides from total cell lysates using lectin chromatography and nanoflow liquid chromatography-MS [81]. In total, greater than 100 O-glycoproteins with greater than 350 O-glycan sites were identified.
Recent progress in MS techniques, including ECD and electron-transfer dissociation (ETD) fragmentation, have enabled analysis of both glycan and peptide moieties; these methods have thus provided very powerful measurement tools for O-glycoproteomics [82]. However, extensive direct analysis of cellular O-glycoproteomics, unveiling both glycan and peptide moieties, is still a difficult task, due to the tremendous heterogeneity generated by these moieties.
2.3. Glycosphingolipids
Glycolipids are lipid molecules linked to one or more carbohydrate units. Based on their lipid moiety, glycolipids are classified as either glycosphingolipids (GSLs), containing ceramide, or glycoglycerolipids, containing glycerol. In mammals, glycolipids are predominantly present as GSLs [83]. GSLs are one of the major components of plasma membrane lipid rafts and play important roles in various biological events, such as cell-cell interactions, signal transduction and cellular differentiation [84,85,86,87,88,89,90,91]. The structural classification of GSLs is defined by their glycan structure rather than by their ceramide moieties and includes ganglio-, globo-, isoglobo-, lacto- and neolacto-series. Among these classes, globo-series GSLs contain a treasure trove of undifferentiated cell markers, such as SSEA-3 (Gb5) and SSEA-4 (sialyl Gb5), which were first isolated from teratocarcinoma [5] and form Galβ1-3GalNAcβ1-3Galα1-4Galβ1-4Glcβ1-Cer and NeuAcα2-3Galβ1-3GalNAcβ1-3Galα1-4Galβ1-4Glcβ1-Cer, respectively. Moreover, globo H GSL (fucosyl Gb5), which contains a type IV H-antigen with a Fucα1-2Galβ1-3GalNAcβ1-3Galα1-4Galβ1-4Glcβ1-Cer structure, was also identified as a promising undifferentiated cell marker in studies using breast cancer stem cells [92] and human ES cells [93]. Other cell-specific GSLs, such as cancer-associated gangliosides (e.g., GD2, GD3, GM2, fucosyl GM1 and Neu5GcGM3) have also been reported and are expected to be promising antigens for the development of cancer vaccines [94]. Furthermore, several human cluster of differentiation (CD) markers, such as CD17 (LacCer), CD60a (GD3), CD60b (9-O-acetyl GD3), CD60c (7-O-acetyl GD3) and CD77 (Gb3), are comprised of GSLs [95].
Among various glycoconjugates, glycomic analyses of GSLs have been performed extensively using thin layer chromatography (TLC) since the mid-1960s. Recently, MS analysis has played a principal role in the structural and quantitative analysis of GSLs. Of the two major approaches, which include analysis of the intact form of GSLs and analysis of the glycan moiety upon deglycosylation, the former approach provides valuable information regarding both glycomics and lipidomics simultaneously.
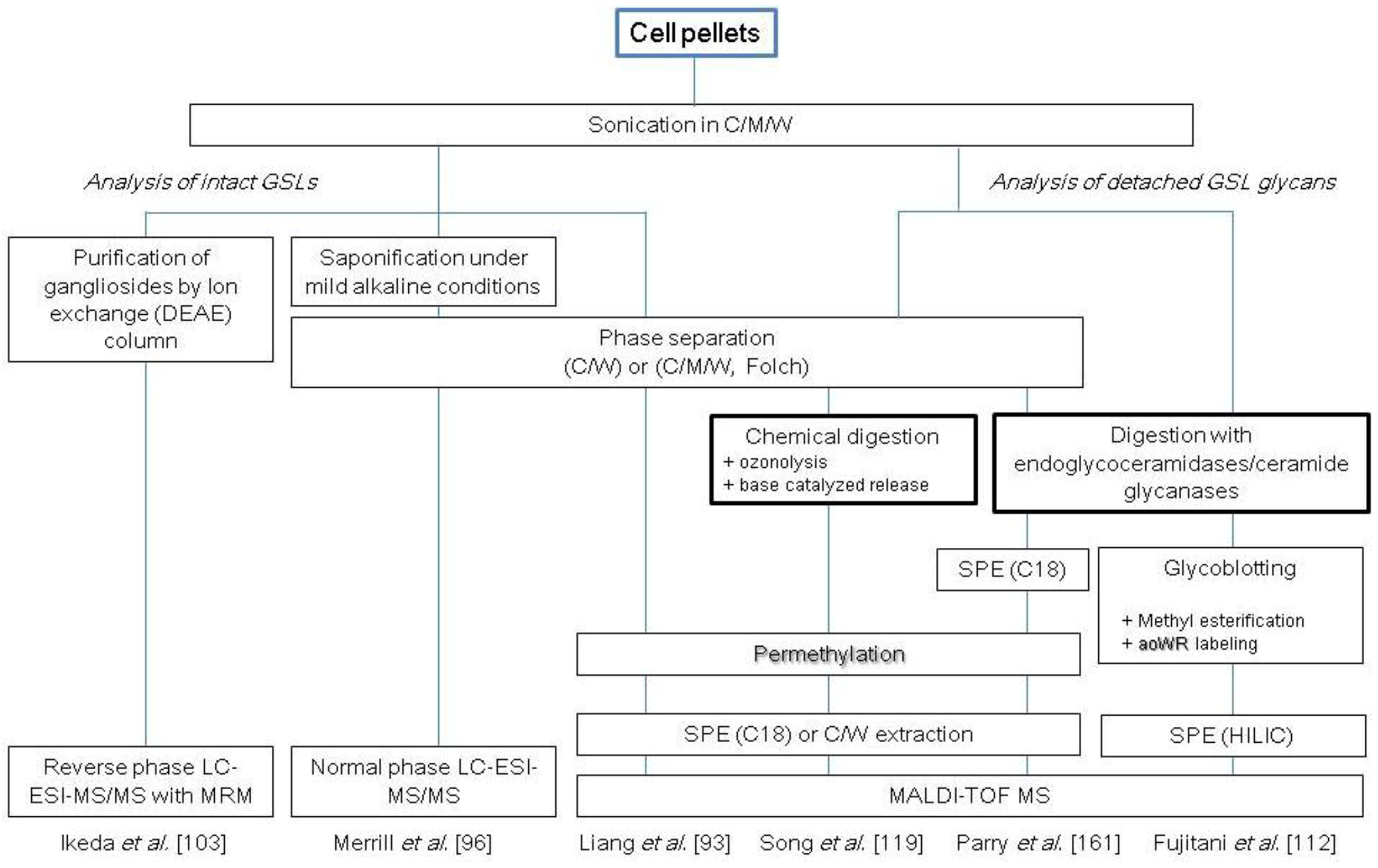
GSLs are generally extracted from cells by homogenization in a solvent consisting of chloroform, methanol and water. Soluble GSLs are recovered from the supernatant by centrifugation. The GSL fraction may be further fractionated into polar and nonpolar GSLs by phase partitioning. Because glycerolipids sometimes interfere with the analysis of GSLs, these compounds are occasionally saponificated under mild alkaline conditions [96]. The extracted and purified GSLs are usually permethylated prior to MALDI-TOF MS analysis [97]. Combination of MS with TLC or LC circumvents the inadequacies of MS analysis alone, such as separation of structural isomers, and increases the quantitative capability of the glycosphingolipidomic technique [98,99,100]. Liang et al. used MALDI-MS(/MS) and gene expression profiling of glycosyltransferases involved in GSL synthesis to identify a drastic structural alteration of cellular GSLs from globo- and lacto-series to ganglio-series during the differentiation of human ES cells to embryoid bodies [93]. MS analyses of GSL-associated glycans that are targeting cancer diagnosis and therapy are also currently in progress. Gupta et al. performed a comparison of GSL-derived glycan structures in cancer stem and non-stem cells and showed that enhanced ceramide glycosylation and the expression level of Gb3 correlate well with the numbers of cancer stem cells in breast cancer cell lines [101]. This result indicates that GSL-glycan profiling has a high potential for determination of the malignancy grading of cancer and for cancer therapy.
Liquid chromatography is useful to discriminate isomers (e.g., GlcCer and GalCer) and to detect low abundant glycan species to reduce ionization suppression. RP chromatography is used for separations based on the length and saturation of acyl chains and normal phase chromatography to separate compounds primarily by their headgroup constituents (for example, distinguish Cer, GlcCer, LacCer, globotriaosylceramide, globotetraosylceramide, sphingomyelin, as well as cholesterol, etc.) [102]. Microfluidic or nanofluidic LC paired with ESI-MS analysis with multiple reaction monitoring (MRM) can be used to quantify GSLs. Ikeda et al. reported a highly effective method using LC/ESI-MS/MS with MRM for quantitative structural analysis of ganglio-series GSLs and sulfatides extracted from mouse brain [103] and mouse cerebellum sections [104].
Another cellular GSL glycomics approach is the analysis of the glycan moiety following release of the head-group glycans from ceramide. By focusing on the glycan moiety alone, it is possible to reduce the complexity of GSLs that arises from the heterogeneity generated by both glycan and ceramide moieties. The oligosaccharides can be detached from GSLs by either enzymatic or chemical digestion. The enzymatic treatment involves the use of endo-type glycosylceramidases (EC 3.2.1.123) that specifically cleave the linkage between glycan and ceramide. These enzymes, called ceramide glycanases (CGases) or endoglycoceramidases (EGCases), have been isolated from some invertebrate species and bacteria [105,106,107,108,109,110,111]. Enzymatic digestion allows analysis of both glycan and ceramides and, therefore, enables simultaneous glycomic and lipidomic analyses. However, one limitation of this approach is the relatively high substrate specificities of endo-type glycosylceramidases. Fujitani et al. reported maximal deglycosylation efficiency by using a mixture of Rhodococcal EGCase I and II [108,109]. In this study, the GSL fraction, which was recovered by traditional homogenization of cells in a solvent consisting of chloroform and methanol, was subjected to Rhodococcal EGCase I and II digestion. The liberated GSL glycans were directly purified by glycoblotting, and their reducing termini were simultaneously labeled with aoWR. The labeled glycans were then subjected to MALDI-TOF MS [112]. The cellular GSL glycomic profiles of 11 different types of cells, including embryonic carcinoma cells, were studied. This study demonstrated that both the relative proportion of GSL glycans and the total amount of expressed GSL glycans are highly cell-specific and, therefore, provide a novel measure for the characterization of cells. Mesenchymal stem cells (MSCs) are an invaluable tool for disease therapy and regenerative medicine studies. Glycomic characterization, including identification of GSL-associated oligosaccharides from bone marrow-derived MSCs, has been performed using a combination of MALDI-MS(/MS), NMR and glycosidase digestions [113]. Glycosphingolipid glycans were detached by Macrobdella decora endoglycoceramidase digestion. The glycome profiles of MSCs and osteoblast cells that differentiate from them displayed distinct differences, indicating that glycosylation analyses can be used to evaluate the differentiation state of MSCs.
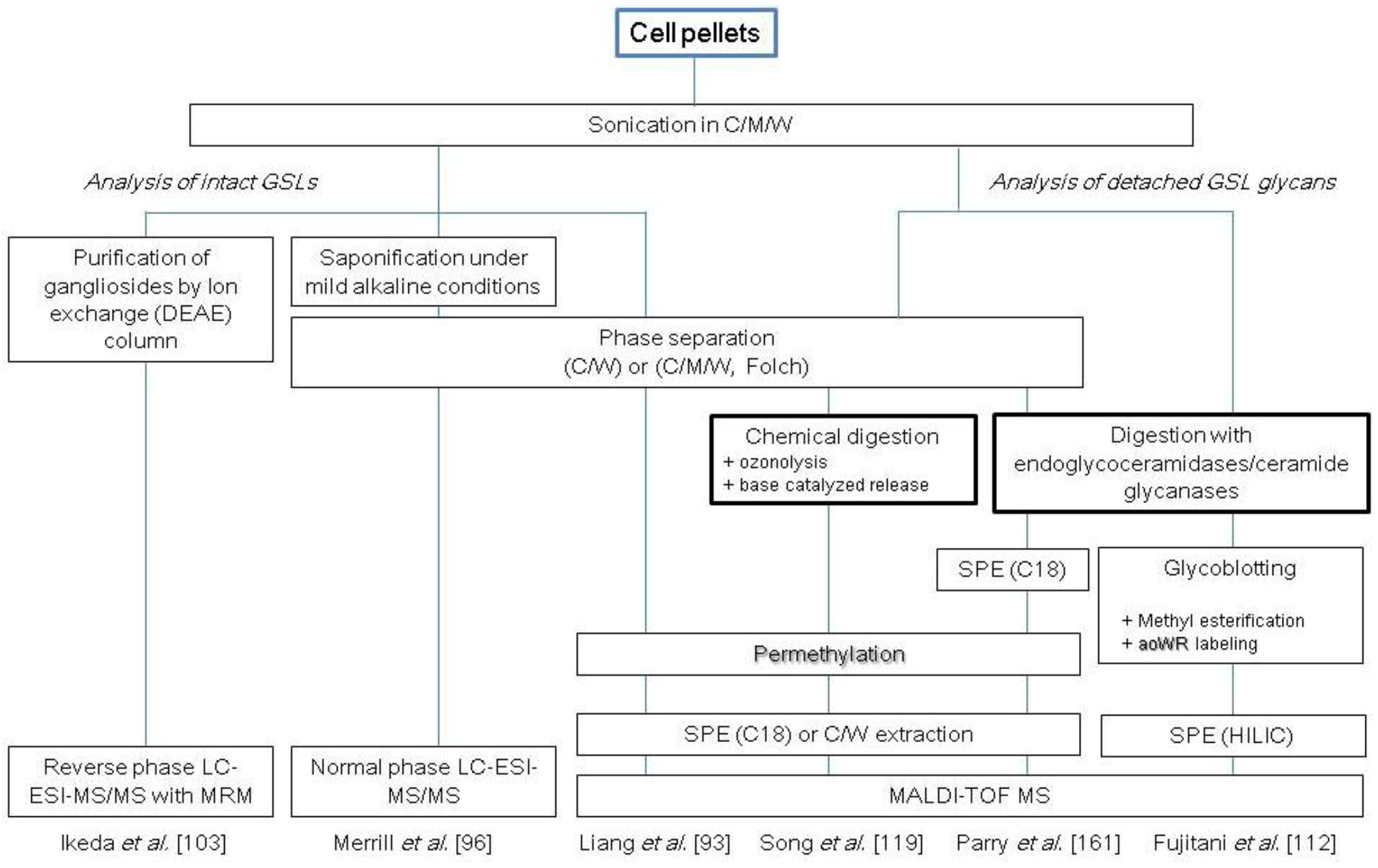
Another deglycosylation method is based on chemical digestion, which is traditionally performed by ozonolysis or osmium-catalyzed periodate oxidation to cleave the olefinic double bond of sphingosine, followed by alkaline treatment to release glycans by a β-elimination-like reaction [114,115,116,117,118]. The major advantage of chemical digestion is the non-specific cleavage of glycan from GSLs, which allows recovery of multiple types of glycan head-groups. To date, few studies have employed chemical digestion for glycomic analyses of cellular GSLs. Song et al. recently reported a novel method to release glycans from GSLs under near neutral pH conditions; this method drastically suppresses the peeling reaction, as demonstrated by analysis of the GSL-derived glycans from human erythrocytes [119]. Several representative strategies for the analysis of cellular GSLs and GSL-linked glycans are summarized in Scheme 3.
Scheme 3.
Representative protocols for the analysis of cellular glycosphingolipids (GSLs) and GSL-linked glycans. C, chloroform; M, methanol; W, water; SPE, solid-phase extraction.
Scheme 3.
Representative protocols for the analysis of cellular glycosphingolipids (GSLs) and GSL-linked glycans. C, chloroform; M, methanol; W, water; SPE, solid-phase extraction.
2.4. Glycosaminoglycans
Proteoglycans (PGs) are a diverse group of glycoconjugates consisting of various core proteins that are post-translationally modified with linear, anionic polysaccharide glycosaminoglycans (GAGs) consisting of repeating disaccharides. PGs are distributed ubiquitously throughout the extracellular matrix and are found on virtually all cell surfaces and basement membranes of different tissues. Through their interaction with proteins, PGs mediate numerous biological processes, including cell-cell and cell-matrix interactions, growth factor sequestration, chemokine and cytokine activation, regulation of stem cell differentiation, tissue morphogenesis during embryonic development, cell migration and cell proliferation [120,121].
Characterization of PGs is challenging due to the tremendous structural diversity of the bioconjugates that arise from multiple isoforms of PG core proteins and variations of their GAG components, although it is noteworthy that analysis of the GAGs is much more difficult than analysis of the core proteins. All GAGs consist of repeating disaccharide structures. The simplest GAG is hyaluronic acid (HA); this non-sulfated free polysaccharide, which is synthesized at and extruded from the plasma membrane, consists of [Galβ1-3GlcNAcβ1-4]n with n > 2,000 in extracellular matrices. Heparin and heparan sulfate (HS) chains are synthesized as [GlcAβ1-4GlcNα1-4]. The GlcN moiety may be 6-O-sulfated, N-sulfated, N-acetylated or sporadically 3-O-sulfated; the GlcA moiety may be epimerized to iduronic acid (IdoA) ; the IdoA may be sulfated at the 2-position. Chondroitin sulfate (CS) consists of repeating units of [GlcAβ1-3GalNAcβ1-4] and may be sulfated at the 4- and/or 6-position of GalNAc. Dermatan sulfate (DS) is a CS variant in which a substantial fraction of GlcA residues are epimerized to IdoA; the IdoA may be sulfated at the 2-position. HS, CS and DS are bound to serine (Ser) residues of proteoglycan core proteins via a [GlcAβ1-3Galβ1,3Galβ1,4Xylβ1-O-Ser] tetrasaccharide linker. Keratan sulfate (KS) is a sulfated polylactosamine chain consisting of repeating units of [Galβ1,4GlcNAcβ1,3]. KS consists of approximately 50 disaccharide residues and is linked to the protein either as an extension to an N-linked glycan (KS I) or through a Ser/threonine (Thr) bound linker structure (KS II) [122].
Ly et al. reported the first direct analysis of purified peptidoglycan (pG) prepared from bikunin, the simplest PG having one CS chain, by MS and CID-MS/MS experiments with both Fourier transform ion cyclotron resonance (FT-ICR) and Fourier transform mass spectrometry (FTMS) instruments [123]. However, structurally intensive analysis of intact PGs has been a virtually impossible task to date, because of the tremendous structural diversity of these molecules. The fine structures of GAGs are assessed by analyzing the disaccharide composition, which typically involves isolation of PGs, recovery of the GAG fraction, enzymatic depolymerization, labeling of disaccharides and then determination of the disaccharide composition.
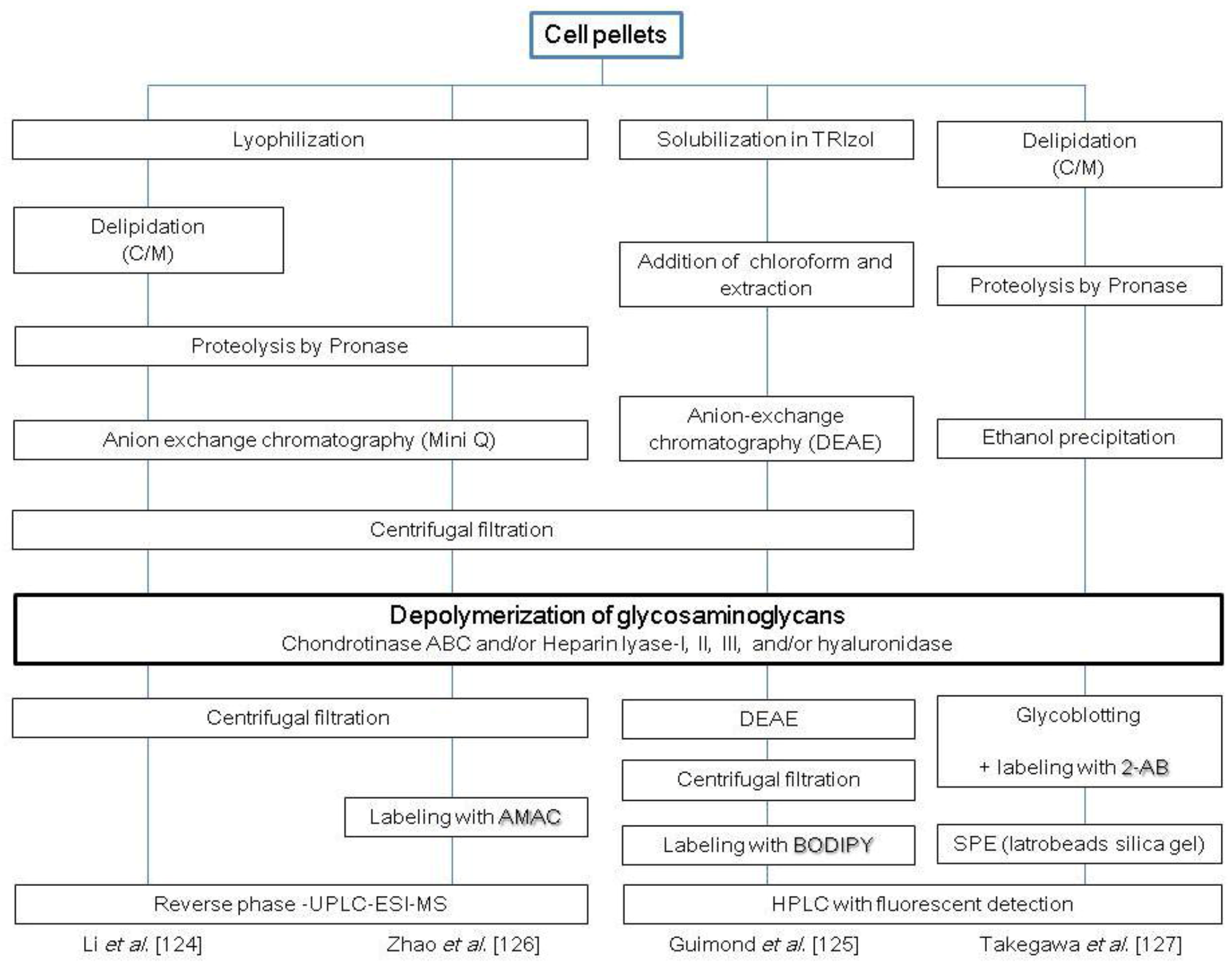
Due to the large hydrodynamic radius and high net negative charge of linear GAG chains, isolation of PGs is a fairly straightforward task. A protocol for the recovery and purification of GAGs established by Linhardt and coworkers consists of homogenization of cells, delipidation, proteolysis of PGs by nonspecific protease (e.g., actinase E) to obtain pGs, extraction of pGs into CHAPS/urea and then recovery of pGs by spin column-based ion chromatography and subsequent membrane-based desalting [124]. Guimond et al. developed a rapid and efficient method for PG extraction in which exclusive partition of PGs into the aqueous phase of TRIzol-chloroform can be achieved in less than one hour [125]. The resulting extract can be applied directly to the anion-exchange medium, which eliminates the requirement for a buffer exchange step to urea. Anion-exchange chromatography is effectively utilized for the purification of GAGs in both cases. Non-sulfated GAGs in biological samples, such as HA and N-acetyl heparosan, are often ignored or underestimated. Zhao et al. employed mini strong anion-exchange spin columns with an optimized concentration of sodium chloride solution to enable high recovery of non-sulfated GAGs [126]. A protocol recently reported by Takegawa et al. consists of homogenization of cells, delipidation, proteolysis by Pronase and then ethanol precipitation. The relatively low purification efficiency, which is possibly caused by eliminating the anion-exchange process, is compensated by the employment of chemoselective glycoblotting for purification of glycans. Since this protocol does not rely on ion exchange chromatography, the loss of non-sulfated GAGs is reduced [127].
Purified pGs can be subjected to PAGE or gel filtration chromatography for fractionation and molecular weight determination of GAGs [124,128]. For disaccharide analysis, GAGs are usually depolymerized by commercially available GAG lyases, which cleave a HexNAc-HexA bond and leave a C4-C5 unsaturated HexA at the new non-reducing terminus. Chondroitinase ABC from Proteus vulgaris is a nonspecific lyase that cleaves all forms of CS. A combination of three heparin lyases (Heparin lyase-I, II and III) from Flavobacterium heparinum is often used for HS-PG glycomic studies [129]. In the case of KS, keratanase (from Pseudomonas) and keratanase II (from Bacillus sp. Ks36) are used; these enzymes act via a hydrolytic mechanism to produce saturated residues. Keratanase is an endo-β-1,4-galactosidase that requires an unsulfated galactosamine (Gal) residue flanked by a 6-O-sulfated GlcNAc residue; keratanase II is an endo-β-1,3-glucosaminidase that requires sulfation at the 6-O position of GlcN and thus cleaves at a majority of 6-O-sulfated GlcNAc residues. HA is typically converted with hyaluronidase SD (from Streptococcus dysgalactiae) to an unsaturated disaccharide. The depolymerized products are usually purified by ultrafiltration [124] or diethylaminoethyl (DEAE) chromatography followed by ultrafiltration [125].
Since a C4-C5 unsaturated double bond is introduced at the non-reducing end of the uronic acid residue upon lyase treatment, direct measurement of absorbance at 232 nm is possible [123,130]. To enable more sensitive detection, the products of GAG depolymerizing enzymes are often labeled with fluorescent tags, such as 2-aminoacridone (AMAC) [131,132], 2-aminobenzamide (2-AB) [133], 2-aminobenzoic acid [134] or boron-dipyrromethene (BODIPY) hydrazide [135], which target newly created reducing ends. The use of isotope-coded tags for MS detection, including [12C6]/[13C6] anilines [136], 2-anthranilic acid-d0 or -d4 [137] and tetraplex stable isotope-coded tags [138], has also been reported. Unlike the analysis of N-, O- and GSL-glycans, where MALDI-TOF MS is often the analytical technique chosen, the separation process is crucial for GAG disaccharide analysis, since many of constituent disaccharides share identical charge and mass. Though several separation techniques, including capillary electrophoresis (CE) [139,140], fluorophore-assisted gel electrophoresis (FACE) [141] and HPLC, have been used for disaccharide compositional analysis, HPLC is the most popular method for cellular GAG disaccharide analysis, due to its general versatility and variety of useful separation modes. The application of ultra-performance liquid chromatography (UPLC) performed at high pressure (up to 108 Pa) with 1.7 µm particles and a proprietary mobile phase pump provides shorter separation times with improved separation [142].
Strong anion-exchange HPLC has conventionally been used to separate disaccharides; however, interfacing this technique with MS is difficult [143]. RP ion-pairing (IP) HPLC, which employs volatile primary, secondary and tertiary amines as post-column additives, provides excellent chromatographic resolution and is compatible with MS detection [144], although the IP reagents seriously contaminate the MS ion source. HILIC separates glycans based on their size and number of acidic groups under solvent conditions appropriate for LC/MS [145]. Takegawa et al. recently reported simultaneous analysis of 2-AB labeled disaccharides from HS/HP, CS/DS and HA by single-step zwitterionic-hydrophilic interaction chromatography (ZIC-HILIC-HPLC) [127]. Volpi reported the analysis of CS/HA disaccharides by employing labeling with 2-aminoacridone (AMAC) and RP chromatography [146]. Yang et al. amended this analytical protocol to enable analysis of 17 AMAC-tagged disaccharides from HS/HP, CS/DS and HA by a single RP-UPLC-MS experiment [126,147]. These studies are the first successful demonstrations of simultaneous analysis of all three families of uronic acid-containing GAGs by a single chromatographic method. Due to their acidity, GAG oligosaccharides are usually analyzed by ESI-MS in the negative ionization mode. The topic of MS analyses of GAGs has been extensively reviewed elsewhere [41,148].
By utilizing the methods described above, GAG disaccharide profiles have been unveiled for many cells, including embryonic-like tetracarcinoma cells [124], mesenchymal cells [149,150,151], murine ESs [152], as well as various other human cells and cell lines from other animal species [127,128,136]. Lawrence et al. reported the HS and CS disaccharides and KS digestion products of various cells, including mutant and wild-type CHO cells and other mammalian cells [136]. GAG chains were extracted from cell pellets by exhaustive digestion with Pronase followed by AEC (DEAE-Sepharose). pGs were digested with GAG degradation enzymes, and the resultant HS disaccharides, CS disaccharides and KS digestion products were labeled with [12C6]/[13C6] anilines, which were then analyzed using RP-IP-ESI-MS. This study demonstrated significant qualitative and quantitative differences in the structures of GAGs among different organisms. Nairn et al. reported the first HS, CS/DS and HA-GAG disaccharide profiles of murine ESs and their differentiated cells by RP-IP-HPLC with post-column fluorescence detection [152]. This group found that the overall GAG content increased and the sulfation patterns changed as ESs differentiated into embryoid bodies and extraembryonic endodermal cells. The same approach was also used to profile human embryonic-like tetracarcinoma cells [124]. Kawabe et al. characterized the epitope of a novel monoclonal antibody (R-10G) specific to human iPS/ES cells by KS glycomic analysis of human iPS cells, which led to the conclusion that the R-10G epitope is a unique KS that lacks oversulfated structures [153]. Several representative strategies for the analysis of cellular GAG disaccharides are summarized in Scheme 3.
Further structural elucidation of PGs/GAGs can be performed by partial enzymatic or chemical depolymerization under controlled conditions [154]. In studies involving PG core protein sequencing or identification, GAGs can be released from their Ser attachment sites using BEMA, as described earlier in this review (refer to “O-glycome” section). The GAG-protein linkage region tetrasaccharide (GlcAβ1-3Galβ1-3Galβ1-4Xylβ1-O-Ser) is the subject of many studies, owing to its proposed role in the initiation of GAG chain biosynthesis. Modifications of the linkage region tetrasaccharide, such as 2-O-phosphorylation of Xyl or 4-O-sulfation and 6-O-sulfonation of Gal residues, have been correlated with changes in the number and type of GAG chains present in PGs [155]. Lawrence et al. reported a novel approach based on the detection of nonreducing ends of the glycosaminoglycans that accumulate in cells, blood and urine of individuals with mucopolysaccharidoses (MPS). After GAG depolymerization, the specific nonreducing ends fragments of the GAGs that are unique in each MPS are quantified [156].
Scheme 4.
Representative protocols for cellular glycosaminoglycans (GAG) disaccharides analysis. C, chloroform; M, methanol; W, water; SPE, solid-phase extraction.
Scheme 4.
Representative protocols for cellular glycosaminoglycans (GAG) disaccharides analysis. C, chloroform; M, methanol; W, water; SPE, solid-phase extraction.
3. Perspective
This review has described the recent advances in elementary glycomic analysis of N- and O-linked glycans derived from glycoproteins, proteoglycans and glycosphingolipids, with a special focus on their application to cellular glycomics. An understanding of the elementary glycome is an important prerequisite for extensive cellular glycomics and precise cellular characterization. To date, reports of extensive cellular glycosylation analyses have been scarce. Wearne et al. employed fluorescently-labeled lectins to classify structural glycosylation motifs on the stem cell surface [157], and the utility of lectin arrays for cell characterization has recently been demonstrated [72,158,159]. Lectin arrays are shown to be capable of profiling unique bacterial cell surface glycomes in part [160]. Glycosylation profiling based on carbohydrate-recognizing molecules, such as lectins and antibodies, provides valuable information about sugar epitopes. However, only a limited number of lectins and glycan-specific antibodies are currently available, and it is often difficult to discriminate between different glycoconjugate species by these methods. In this regard, glycomic analyses based on molecular recognition and structurally intensive studies using sophisticated analytical chemistry (e.g., separation techniques, MS, etc.) are complementary. This review focused on the structurally intensive studies. Analysis of the comprehensive cellular glycome has been addressed in pioneering MS work. Parry et al. reported a mass spectrometric strategy to characterize both glycans from GSLs and glycoproteins using mouse brain as a model case [161]. Heiskanen et al. unveiled N- and O-linked glycans derived from glycoproteins and glycans of GSLs of bone marrow-derived MSCs by MS [113].
No attempts have yet been made to determine all major classes of glycans or the relative concentration of each type of glycan present within a cell. Integration of elementary glycomic analyses to obtain a comprehensive understanding of the glycome requires streamlining the sample preparation protocol as effectively as possible. Development of techniques that enable multiplexing is also required to improve the throughput of glycomic studies, as has been achieved for proteomic and transcriptomic analyses. To compare the relative concentration of each type of glycan derived from various sources of glycoconjugates, absolute quantification, rather than relative quantification, is most important. The number of cells required for the glycomic analyses reviewed in this article is typically in the range of 104 and 108 cells, depending on the specific technique employed and the type of glycoconjugate. Since detection sensitivity is defined by signal-to-noise ratio, it is important to reduce the noise as much as possible, which may be achieved through an optimized sample preparation technique. Increasing the signal may be achieved by employing an optimized labeling strategy and/or highly sensitive analytical technique. Current glycomic analytical protocols do not allow analysis of species with low abundance, which is another issue that needs to be addressed. Another issue to be addressed is the necessity of informatics to link and interpret the data obtained by these different methods.
A panoramic view of the extensive cellular glycome has multiple potential applications. For example, comprehensive identification of pluripotency biomarkers and disease-related biomarkers will be accelerated. In addition, elucidation of the overall picture of cellular glycosylation and its alteration upon differentiation or acquisition of disease would provide a useful foundation for analysis of iPS and differentiated cells during drug discovery studies. An understanding of the entire cellular glycome is also expected to expedite the development of more specific biomarkers that can discriminate between somatic stem cells and cancerous cells, as well as cancer vaccines for which identification of a specific antigenic epitope is most important. Analysis of all major glycans, including those described in this review, as well as other glycans/glycoconjugates, such as GPI anchors, intracellular lipid-linked glycans, free cellular oligosaccharides and sugar nucleotides, will allow deciphering of intra- and inter-glycomic correlations. Overall, the ability to analyze the cellular glycome extensively and completely will open new avenues in systems biology glycomics.
Acknowledgments
The authors thank Ms J. Piao for her assistance in preparing this manuscript.
References
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Karsdal, M.A.; Henriksen, K.; Leeming, D.J.; Mitchell, P.; Duffin, K.; Barascuk, N.; Klickstein, L.; Aggarwal, P.; Nemirovskiy, O.; Byrjalsen, I.; et al. Biochemical markers and the FDA Critical Path: how biomarkers may contribute to the understanding of pathophysiology and provide unique and necessary tools for drug development. Biomarkers 2009, 14, 181–202. [Google Scholar] [CrossRef]
- Bertolini, F.; Mancuso, P.; Shaked, Y.; Kerbel, RS. Molecular and cellular biomarkers for angiogenesis in clinical oncology. Drug Discovery Today 2007, 12, 806–812. [Google Scholar] [CrossRef]
- Kannagi, R.; Cochran, N.A.; Ishigami, F.; Hakomori, S.; Andrews, P.W.; Knowles, B.B.; Solter, D. Stage-specific embryonic antigens (SSEA-3 and -4) are epitopes of a unique globo-series ganglioside isolated from human teratocarcinoma cells. EMBO J. 1983, 2, 2355–2361. [Google Scholar]
- Lee, J.B.; Kim, J.M.; Kim, S.J.; Park, J.H.; Hong, S.H.; Roh, S.I.; Kim, M.K.; Yoon, H.S. Comparative characteristics of three human embryonic stem cell lines. Mol. Cells 2005, 19, 31–38. [Google Scholar]
- Natunen, S.; Satomaa, T.; Pitkänen, V.; Salo, H.; Mikkola, M.; Natunen, J.; Otonkoski, T.; Valmu, L. The binding specificity of the marker antibodies Tra-1-60 and Tra-1-81 reveals a novel pluripotency-associated type 1 lactosamine epitope. Glycobiology 2011, 21, 1125–1130. [Google Scholar]
- Lanctot, P.M.; Gage, F.H.; Varki, A.P. The glycans of stem cells. Curr. Opin. Chem. Biol. 2007, 11, 373–380. [Google Scholar] [CrossRef]
- Angata, T.; Fujinawa, R.; Kurimoto, A.; Nakajima, K.; Kato, M.; Takamatsu, S.; Korekane, H.; Gao, C.X.; Ohtsubo, K.; Kitazume, S.; Taniguchi, N. Integrated approach toward the discovery of glyco-biomarkers of inflammation-related diseases. Ann. N. Y. Acad. Sci. 2012, 1253, 159–169. [Google Scholar]
- Ludwig, J.A.; Weinstein, J.N. Biomarkers in cancer staging; prognosis and treatment selection. Nat. Rev. Cancer 2005, 5, 845–856. [Google Scholar] [CrossRef]
- Rillahan, C.D.; Paulson, J.C. Glycan microarrays for decoding the glycome. Annu. Rev. Biochem. 2011, 80, 797–823. [Google Scholar] [CrossRef]
- Freeze, H.H. Genetic defects in the human glycome. Nat. Rev. Genet. 2006, 7, 537–551. [Google Scholar] [CrossRef]
- Gabius, H.J.; André, S.; Jiménez-Barbero, J.; Romero, A.; Solís, D. From lectin structure to functional glycomics: principles of the sugar code. Trends Biochem. Sci. 2011, 36, 298–313. [Google Scholar] [CrossRef]
- Stanley, P.; Cummings, R.D. Structures Common to Different Glycans. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2009; pp. 175–198. [Google Scholar]
- Furukawa, K.; Takamiya, K.; Okada, M.; Inoue, M.; Fukumoto, S.; Furukawa, K. Novel function of complex carbohydrates elucidated by the mutant mice of glycosyltransferase genes. Biochim. Biophys. Acta 2001, 1525, 1–12. [Google Scholar] [CrossRef]
- Helenius, A.; Aebi, M. Intracellular functions of N-linked glycans. Science 2001, 291, 2364–2369. [Google Scholar] [CrossRef]
- Hebert, D.N.; Molinari, M. Flagging and docking: dual roles for N-glycans in protein quality control and cellular proteostasis. Trends Biochem Sci. 2012, 37, 404–410. [Google Scholar] [CrossRef]
- Wada, Y.; Azadi, P.; Costello, C.E.; Dell, A.; Dwek, R.A.; Geyer, R.; Kakehi, K.; Karisson, N.G.; Kato, K.; Kawasaki, N.; et al. Comparison of the methods for profiling glycoprotein glycans--HUPO Human Disease Glycomics/Proteome Initiative multi-institutional study. Glycobiology 2007, 17, 411–422. [Google Scholar] [CrossRef]
- Naven, T.J.; Harvey, D.J. Effect of structure on the signal strength of oligosaccharides in matrix-assisted laser desorption/ionization mass spectrometry on time-of-flight and magnetic sector instruments. Rapid Commun. Mass Spectrom. 1996, 10, 1361–1366. [Google Scholar] [CrossRef]
- Powell, A.K.; Harvey, D.J. Stabilization of sialic acids in N-linked oligosaccharides and gangliosides for analysis by positive ion matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun. Mass Spectrom. 1996, 10, 1027–1032. [Google Scholar] [CrossRef]
- Miura, Y.; Shinohara, Y.; Furukawa, J.; Nagahori, N.; Nishimura, S. Rapid and simple solid-phase esterification of sialic acid residues for quantitative glycomics by mass spectrometry. Chem. Eur. J. 2007, 13, 4797–4804. [Google Scholar] [CrossRef]
- Dell, A.; Morris, H.R. Glycoprotein structure determination by mass spectrometry. Science 2001, 291, 2351–2356. [Google Scholar] [CrossRef]
- Kita, Y.; Miura, Y.; Furukawa, J.; Nakano, M.; Shinohara, Y.; Ohno, M.; Takimoto, A.; Nishimura, S. Quantitative glycomics of human whole serum glycoproteins based on the standardized protocol for liberating N-glycans. Mol. Cell Proteomics 2007, 6, 1437–1445. [Google Scholar] [CrossRef]
- Krishnamoorthy, L.; Mahal, L.K. Glycomic analysis: an array of technologies. ACS Chem. Biol. 2009, 4, 715–732. [Google Scholar] [CrossRef]
- Ruhaak, L.R.; Zauner, G.; Huhn, C.; Bruggink, C.; Deelder, A.M.; Wuhrer, M. Glycan labeling strategies and their use in identification and quantification. Anal. Bioanal. Chem. 2010, 397, 3457–3481. [Google Scholar] [CrossRef]
- Mechref, Y.; Kang, P.; Novotny, M.V. Solid-phase permethylation for glycomic analysis. Methods Mol. Biol. 2009, 534, 53–64. [Google Scholar] [CrossRef]
- Jeong, H.J.; Kim, Y.G.; Yang, Y.H.; Kim, B.G. High-throughput quantitative analysis of total N-glycans by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal. Chem. 2012, 84, 3453–3460. [Google Scholar] [CrossRef]
- Lemoine, J.; Chirat, F.; Domon, B. Structural analysis of derivatized oligosaccharides using post-source decay matrix-assisted laser desorption/ionization mass spectrometry. J. Mass Spectrom. 1996, 31, 908–912. [Google Scholar] [CrossRef]
- North, S.J.; Huang, H.H.; Sundaram, S.; Jang-Lee, J.; Etienne, A.T.; Trollope, A.; Chalabi, S.; Dell, A.; Stanley, P.; Haslam, S.M. Glycomics profiling of Chinese hamster ovary cell glycosylation mutants reveals N-glycans of a novel size and complexity. J. Biol Chem. 2010, 285, 5759–5775. [Google Scholar]
- Antonopoulos, A.; North, S.J.; Haslam, S.M.; Dell, A. Glycosylation of mouse and human immune cells: insights emerging from N-glycomics analyses. Biochem. Soc. Trans. 2011, 39, 1334–1340. [Google Scholar] [CrossRef]
- Satomaa, T.; Heiskanen, A.; Mikkola, M.; Olsson, C.; Blomqvist, M.; Tiittanen, M.; Jaatinen, T.; Aitio, O.; Olonen, A.; Helin, J.; et al. The N-glycome of human embryonic stem cells. BMC Cell Biol. 2009, 10, 42. [Google Scholar] [CrossRef]
- Furukawa, J.; Shinohara, Y.; Kuramoto, H.; Miura, Y.; Shimaoka, H.; Kurogochi, M.; Nakano, M.; Nishimura, S. Comprehensive approach to structural and functional glycomics based on chemoselective glycoblotting and sequential tag conversion. Anal. Chem. 2008, 80, 1094–1101. [Google Scholar]
- Uematsu, R.; Furukawa, J.; Nakagawa, H.; Shinohara, Y.; Deguchi, K.; Monde, K.; Nishimura, S. High throughput quantitative glycomics and glycoform-focused proteomics of murine dermis and epidermis. Mol. Cell. Proteomics 2005, 4, 1977–1989. [Google Scholar]
- Uematsu, R.; Shinohara, Y.; Nakagawa, H.; Kurogochi, M.; Furukawa, J.; Miura, Y.; Akiyama, M.; Shimizu, H.; Nishimura, S. Glycosylation specific for adhesion molecules in epidermis and its receptor revealed by glycoform-focused reverse genomics. Mol. Cell. Proteomics 2009, 8, 232–244. [Google Scholar]
- Amano, M.; Yamaguchi, M.; Takegawa, Y.; Yamashita, T.; Terashima, M.; Furukawa, J.; Miura, Y.; Shinohara, Y.; Iwasaki, N.; Minami, A.; Nishimura, S. Threshold in stage-specific embryonic glycotypes uncovered by a full portrait of dynamic N-glycan expression during cell differentiation. Mol. Cell Proteomics 2010, 9, 523–537. [Google Scholar] [CrossRef]
- Hase, S.; Hara, S.; Matsushima, Y. Tagging of sugars with a fluorescent compound, 2-aminopyridine. J. Biochem. 1979, 85, 217–220. [Google Scholar]
- Bigge, J.C.; Patel, T.P.; Bruce, J.A.; Goulding, P.N.; Charles, S.M.; Parekh, R.B. Nonselective and efficient fluorescent labeling of glycans using 2-amino benzamide and anthranilic acid. Anal. Biochem. 1995, 230, 229–238. [Google Scholar]
- Wuhrer, M.; de Boer, A.R.; Deelder, A.M. Structural glycomics using hydrophilic interaction chromatography (HILIC) with mass spectrometry. Mass Spectrom Rev. 2009, 28, 192–206. [Google Scholar] [CrossRef]
- Takegawa, Y.; Deguchi, K.; Keira, T.; Ito, H.; Nakagawa, H.; Nishimura, S. Separation of isomeric 2-aminopyridine derivatized N-glycans and N-glycopeptides of human serum immunoglobulin G by using a zwitterionic type of hydrophilic-interaction chromatography. J. Chromatogr. A 2006, 1113, 177–181. [Google Scholar] [CrossRef]
- Packer, N.H.; Lawson, M.A.; Jardine, D.R.; Redmond, J.W. A general approach to desalting oligosaccharides released from glycoproteins. Glycoconj. J. 1998, 15, 737–747. [Google Scholar] [CrossRef]
- Zaia, J. Mass spectrometry and glycomics. OMICS 2010, 14, 401–418. [Google Scholar] [CrossRef]
- An, H.J.; Gip, P.; Kim, J.; Wu, S.; Park, K.W.; McVaugh, C.T.; Schaffer, D.V.; Bertozzi, C.R.; Lebrilla, C.B. Extensive determination of glycan heterogeneity reveals an unusual abundance of high mannose glycans in enriched plasma membranes of human embryonic stem cells. Mol. Cell Proteomics 2012, 11. M111.010660. [Google Scholar] [CrossRef]
- Hasehira, K.; Tateno, H.; Onuma, Y.; Ito, Y.; Asashima, M.; Hirabayashi, J. Structural and quantitative evidence for dynamic glycome shift upon production of human induced pluripotent stem cells. Mol. Cell Proteomics 2012, 11, 1913–1923. [Google Scholar] [CrossRef]
- Zhang, H.; Li, X.J.; Martin, D.B.; Aebersold, R. Entification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotech. 2003, 21, 660–666. [Google Scholar]
- Baycin-Hizal, D.; Tabb, D.L.; Chaerkady, R.; Chen, L.; Lewis, N.E.; Nagarajan, H.; Sarkaria, V.; Kumar, A.; Wolozny, D.; Colao, J.; et al. Proteomic analysis of chinese hamster ovary cells. J. Proteome. Res. 2012, 11, 5265–5276. [Google Scholar]
- Sudhir, P.R.; Chen, C.H.; Pavana Kumari, M.; Wang, M.J.; Tsou, C.C.; Sung, T.Y.; Chen, J.Y.; Chen, C.H. Label-free quantitative proteomics and N-glycoproteomics analysis of KRAS-activated human bronchial epithelial cells. Mol. Cell Proteomics 2012, 11, 901–915. [Google Scholar] [CrossRef]
- Almaraz, R.T.; Tian, Y.; Bhattarcharya, R.; Tan, E.; Chen, S.H.; Dallas, M.R.; Chen, L.; Zhang, Z.; Zhang, H.; Konstantopoulos, K.; et al. Metabolic flux increases glycoprotein sialylation: implications for cell adhesion and cancer metastasis. Mol. Cell Proteomics 2012, 11. M112.017558. [Google Scholar]
- Kurogochi, M.; Matsushista, T.; Amano, M.; Furukawa, J.; Shinohara, Y.; Aoshima, M.; Nishimura, S. Sialic acid-focused quantitative mouse serum glycoproteomics by multiple reaction monitoring assay. Mol. Cell. Proteomics 2010, 9, 2354–2368. [Google Scholar] [CrossRef]
- Zhang, Y.; Yin, H.; Lu, H. Recent progress in quantitative glycoproteomics. Glycoconj. J. 2012, 29, 249–258. [Google Scholar] [CrossRef]
- Li, Y.; Tian, Y.; Rezai, T.; Prakash, A.; Lopez, M.F.; Chan, D.W.; Zhang, H. Simultaneous analysis of glycosylated and sialylated prostate-specific antigen revealing differential distribution of glycosylated prostate-specific antigen isoforms in prostate cancer tissues. Anal. Chem. 2011, 83, 240–245. [Google Scholar]
- Jensen, O.N. Interpreting the protein language using proteomics. Nat. Rev. Mol. Cell. Biol. 2006, 7, 391–403. [Google Scholar] [CrossRef]
- Gram Schjoldager, K.T.; Vester-Christensen, M.B.; Goth, C.K.; Petersen, T.N.; Brunak, S.; Bennett, E.P.; Levery, S.B.; Clausen, H. O-glycosylation modulates proprotein convertase activation of angiopoietin-like protein 3 - possible role of polypeptide GalNAc-transferase-2 in regulation of concentrations of plasma lipids. J. Biol. Chem. 2010, 285, 36293–36303. [Google Scholar]
- Kato, K.; Jeanneau, C.; Tarp, M.A.; Benet-Pages, A.; Lorenz-Depiereux, B.; Bennett, E. P.; Mandel, U.; Strom, T. M.; Clausen, H. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J. Biol. Chem. 2006, 281, 18370–18377. [Google Scholar]
- Gill, D.; Clausen, H.; Bard, F. Location, location, location: new insights into O-GalNAc protein glycosylation. Trends Cell Biol. 2010, 23, 149–158. [Google Scholar]
- Schwientek, T.; Bennett, E.P.; Flores, C.; Thacker, J.; Hollmann, M.; Reis, C.A.; Behrens, J.; Mandel, U.; Keck, B.; Schafer, M.A.; et al. Functional conservation of subfamilies of putative UDP-N-acetylgalactosamine:polypeptide N-acetylgalactosaminyltransferases in Drosophila, Caenorhabditis elegans, and mammals. One subfamily composed of l(2)35Aa is essential in Drosophila. J. Biol. Chem. 2002, 277, 22623–22638. [Google Scholar]
- Storous, G.J.; Dekker, J. Mucin-type glycoproteins. CRC Crit. Rev. Biochem. Mol. Biol. 1992, 27, 57–92. [Google Scholar] [CrossRef]
- Carlstedt, I.; Sheehan, J.K.; Corfield, A.P.; Gallagher, J.T. Mucous glycoproteins: A gel of a problem. Essays Biochem. 1985, 20, 40–76. [Google Scholar]
- Taylor-Papadimitriou, J.; Burchell, J.; Miles, D.W.; Dalziel, M. MUC1 and cancer. Biochim. Biophys. Acta 1999, 1455, 301–313. [Google Scholar]
- Park, J.H.; Nishidate, T.; Kijima, K.; Ohashi, T.; Takegawa, K.; Fujikane, T.; Hirata, K.; Nakamura, Y.; Katagiri, T. Critical roles of mucin 1 glycosylation by transactivated polypeptide N-acetylgalactosaminyltransferase 6 in mammary carcinogenesis. Cancer Res. 2010, 70, 2759–2769. [Google Scholar]
- Pedersen, J.W.; Blixt, O.; Bennett, E.P.; Tarp, M.A.; Dar, I.; Mandel, U.; Poulsen, S.S.; Pedersen, A.E.; Rasmussen, S.; Jess, P.; et al. Seromic profi ling of colorectal cancer patients with novel glycopeptide microarray. Int. J. Cancer 2011, 128, 1860–1871. [Google Scholar]
- Plummer, T.H. Jr.; Elder, J. H.; Alexander, S.; Phelan, A.W.; Tarentino, A.L. Demonstration of peptide: N-glycosidase F activity in endo-beta-N-acetylglucosaminidase F preparations. J. Biol. Chem. 1984, 259, 10700–10704. [Google Scholar]
- Dwek, R.A.; Edge, C.J.; Harvey, D.J.; Wormald, M.R.; Parekh, R.B. Analysis of glycoprotein-associated oligosaccharides. Annu. Rev. Biochem. 1993, 62, 65–100. [Google Scholar] [CrossRef]
- Wada, Y.; Dell, A.; Haslam, S.M.; Tissot, B.; Canis, K.; Azadi, P.; Backstrom, M.; Costello, C.E.; Hansson, G.C.; Hiki, Y.; et al. Comparison of methods for profi ling O-glycosylation: Human Proteome Organisation Human Disease Glycomics/Proteome Initiative multi-institutional study of IgA1. Mol. Cell. Proteomics 2010, 9, 719–727. [Google Scholar] [CrossRef]
- Carlson, D.M. Structures and immunochemical properties of oligosaccharides isolated from pig submaxillary mucins. J. Biol. Chem. 1968, 243, 616–626. [Google Scholar]
- Babu, P.; North, S. J.; Jang-Lee, J.; Chalabi, S.; Mackerness, K.; Stowell, S. R.; Cummings, R. D.; Rankin, S.; Dell, A.; Haslam, S. M. Structural characterization of neutrophil glycans by ultra sensitive mass spectrometric glycomics methodology. Glycoconj. J. 2009, 26, 975–986. [Google Scholar] [CrossRef]
- Nairn, A.V.; Aoki, K.; Dela Rosa, M.; Porterfield, M.; Lim, J.M.; Kulik, M.; Pierce, J.M.; Wells, L.; Dalton, S.; Tiemeyer, M.; et al. Regulation of Glycan Structures in Murine Embryonic Stem Cells: Combined Transcript Profiling of Glycan-Related Genes and Glycan Structural Analysis. J. Biol. Chem. 2012, 287, 37835–37856. [Google Scholar]
- Maniatis, S.; Zhou, H.; Reinhold, V. Rapid de-O-glycosylation concomitant with peptide labeling using microwave radiation and an alkyl amine base. Anal. Chem. 2010, 82, 2421–2425. [Google Scholar] [CrossRef]
- Huang, Y.; Mechref, Y.; Novotny, M.V. Microscale nonreductive release of O-linked glycans for subsequent analysis throught MALDI mass spectrometry and capillary electrophoresis. Anal. Chem. 2001, 73, 6063–6069. [Google Scholar] [CrossRef]
- Yu, G.; Zhang, Y.; Zhang, Z.; Song, L.; Wang, P.; Chai, W. Effect and limitation of excess ammonium on the release of O-glycans in reducing forms from glycoproteins under mild alkaline conditions for glycomic and functional analysis. Anal. Chem. 2010, 82, 9534–9542. [Google Scholar]
- Patel, T.; Bruce, J.; Merry, A.; Bigge, C.; Wormald, M.; Jaques, A.; Parekh, R. Use of hydrazine to release in intact and unreduced form both N- and. O-Linked oligosaccharides from glycoproteins. Biochemistry 1993, 32, 679–693. [Google Scholar]
- Merry, A.H.; Neville, D.C.; Royle, L.; Matthews, B.; Harvey, D.J.; Dwek, R.A.; Rudd, P.M. Recovery of intact 2-aminobenzamide-labeled O-glycans released from glycoproteins by hydrazinolysis. Anal. Biochem. 2002, 304, 91–99. [Google Scholar] [CrossRef]
- Tateno, H.; Toyota, M.; Saito, S.; Onuma, Y.; Ito, Y.; Hiemori, K.; Fukumura, M.; Nakasu, A.; Nakanishi, M.; Ohnuma, K.; et al. Glycome diagnosis of human induced pluripotent stem cells using lectin microarray. J. Biol. Chem. 2011, 286, 20345–20353. [Google Scholar]
- Takasaki, S.; Mizuochi, T.; Kobata, A. Hydrazinolysis of asparagine-linked sugar chains to produce free oligosaccharides. Methods Enzymol. 1982, 83, 263–268. [Google Scholar] [CrossRef]
- Kawashima, H.; Murata, T.; Yamamoto, K.; Tateishi, A.; Irimura, T.; Osawa, T. A simple method for the release of asparagine-linked oligosaccharides from a glycoprotein purified by SDS-polyacrylamide gel electrophoresis. J. Biochem. 1992, 111, 620–622. [Google Scholar]
- Zauner, G.; Koeleman, C.A.; Deelder, A.M.; Wuhrer, M. Mass spectrometric O-glycan analysis after combined O-glycan release by β-elimination and 1-phenyl-3-methyl-5-pyrazolone labeling. Biochim. Biophys. Acta 2011, 1820, 1420–1428. [Google Scholar]
- Wang, C.; Fan, W.; Zhang, P.; Wang, Z.; Huang, L. One-pot nonreductive O-glycan release and labeling with 1-phenyl-3-methyl-5-pyrazolone followed by ESI-MS analysis. Proteomics 2011, 11, 4229–4242. [Google Scholar] [CrossRef]
- Furukawa, J.-i.; Fujitani, N.; Araki, K.; Takegawa, Y.; Kodama, K.; Shinohara, Y. A versatile method for analysis of serine/threonine posttranslational modifications by β-elimination in the presence of pyrazolone analogues. Anal. Chem. 2011, 83, 9060–9067. [Google Scholar]
- Hédou, J.; Bastide, B.; Page, A.; Michalski, J.C.; Morelle, W. Mapping of O-linked beta-N-acetylglucosamine modification sites in key contractile proteins of rat skeletal muscle. Proteomics 2009, 9, 2139–2148. [Google Scholar] [CrossRef]
- Jang, H.; Kim, T.W.; Yoon, S.; Choi, S.Y.; Kang, T.W.; Kim, S.Y.; Kwon, Y.W.; Cho, E.J.; Youn, H.D. O-GlcNAc regulates pluripotency and reprogramming by directly acting on core components of the pluripotency network. Cell Stem Cell 2012, 11, 62–74. [Google Scholar] [CrossRef]
- Trinidad, J.C.; Barkan, D.T.; Gulledge, B.F.; Thalhammer, A.; Sali, A.; Schoepfer, R.; Burlingame, A.L. Global identification and characterization of both O-glcNAcylation and phosphorylation at the murine synapse. Mol. Cell Proteomics 2012, 11, 215–229. [Google Scholar] [CrossRef]
- Steentoft, C.; Vakhrushev, S.Y.; Vester-Christensen, M.B.; Schjoldager, K.T.; Kong, Y.; Bennett, E.P.; Mandel, U.; Wandall, H.; Levery, S.B.; Clausen, H. Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat. Methods 2011, 8, 977–982. [Google Scholar]
- Zauner, G.; Kozak, R.P.; Gardner, R.A.; Fernandes, D.L.; Deelder, A.M.; Wuhrer, M. Protein O-glycosylation analysis. Biol. Chem. 2012, 393, 687–708. [Google Scholar]
- Robinson, L.N.; Artpradit, C.; Raman, R.; Shriver, Z.H.; Ruchirawat, M.; Sasisekharan, R. Harnessing glycomics technologies: integrating structure with function for glycan characterization. Electrophoresis 2012, 33, 797–814. [Google Scholar]
- Hakomori, S. Structure and function of glycosphingolipids and sphingolipids: Recollections and future trends. Biochim. Biophy. Acta 2008, 1780, 325–346. [Google Scholar] [CrossRef]
- Lahiri, S.; Futerman, A.H. The metabolism and function of sphingolipids and glycosphingolipids. Cell Mol. Life Sci. 2007, 64, 2270–2284. [Google Scholar] [CrossRef]
- Hoetzl, S.; Sprong, H.; van Meer, G. van Meer, G. The way we view cellular (glyco)sphingolipids. J. Neurochem. 2007, 103, 3–13. [Google Scholar] [CrossRef]
- Hakomori, S.; Handa, K.; Iwabuchi, K.; Yamamura, S.; Prinetti, A. New insights in glycosphingolipid function: ‘‘glycosignaling domain,’’ a cell surface assembly of glycosphingolipids with signal transducer molecules, involved in cell adhesion coupled with signaling. Glycobiology 1998, 8. xi–xix. [Google Scholar] [CrossRef]
- Sorice, M.; Longo, A.; Garofalo, T.; Mattei, V.; Misasi, R.; Pavan, A. Role of GM3-enriched microdomains in signal transduction regulation in T lymphocytes. Glycoconj. J. 2004, 20, 63–70. [Google Scholar]
- Yu, S.; Withers, D.A.; Hakomori, S. Globoside-dependent adhesion of human embryonal carcinoma cells, based on carbohydrate-carbohydrate interaction, initiates signal transduction and induces enhanced activity of transcription factors AP1 and CREB. Biol. Chem. 1998, 273, 2517–2525. [Google Scholar] [CrossRef]
- Todeschini, A.R.; Hakomori, S.I. Functional role of glycosphingolipids and gangliosides in control of cell adhesion, motility, and growth through glycosynaptic microdomains. Biochim. Biophys. Acta 2008, 1780, 421–433. [Google Scholar] [CrossRef]
- Merrill, A.H. Jr. Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem Rev. 2011, 111, 6387–6422. [Google Scholar] [CrossRef]
- Chang, W.W.; Lee, C.H.; Lee, P.; Lin, J.; Hsu, C.W.; Hung, J.T.; Lin, J.J.; Yu, J.C.; Shao, L.E.; Yu, J.; et al. Expression of Globo H and SSEA3 in breast cancer stem cells and the involvement of fucosyl transferases 1 and 2 in Globo H synthesis. Proc. Natl. Acad. Sci. USA 2008, 105, 11667–11672. [Google Scholar]
- Liang, Y.J.; Kuo, H.H.; Lin, C.H.; Chen, Y.Y.; Yang, B.C.; Cheng, Y.Y.; Yu, A.L.; Khoo, K.H.; Yu, J. Switching of the core structures of glycosphingolipids from globo- and lacto- to ganglio-series upon human embryonic stem cell differentiation. Proc. Natl. Acad. Sci. USA 2010, 107, 22564–22569. [Google Scholar]
- Heimburg-Molinaro, J.; Lum, M.; Vijay, G.; Jain, M.; Almogren, A.; Rittenhouse-Olson, K. Cancer vaccines and carbohydrate epitopes. Vaccine 2011, 29, 8802–8826. [Google Scholar]
- Wuhrer, M. Glycomics using mass spectrometry. Glycoconj. J. 2013, 30, 11–22. [Google Scholar] [CrossRef]
- Merrill, A.H. Jr.; Sullards, M.C.; Allegood, J.C.; Kelly, S.; Wang, E. Sphingolipidomics: High-throughput, structure-specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectrometry. Methods 2005, 36, 207–224. [Google Scholar] [CrossRef]
- Ciucanu, I.; Kerek, F. A simple and rapid method for the permethylation of carbohydrates. Carbohydr. Res. 1984, 131, 209–217. [Google Scholar] [CrossRef]
- Levery, S.B. Glycosphingolipid structural analysis and glycosphingolipidomics. Methods Enzymol. 2005, 405, 300–369. [Google Scholar] [CrossRef]
- Muthing, J.; Distler, U. Advances on the compositional analysis of glycosphingolipids combining thin-layer chromatography with mass spectrometry. Mass. Spectrom. Rev. 2010, 29, 425–479. [Google Scholar]
- Flanqea, C.; Serb, A.; Sisu, E.; Zamfir, A.D. Reprint of: Chip-based nanoelectrospray mass spectrometry of brain gangliosides. Biochim. Biophy. Acta 2011, 1811, 897–917. [Google Scholar] [CrossRef]
- Gupta, V.; Bhinge, K.N.; Hosain, S.B.; Xiong, K.; Gu, X.; Shi, R.; Ho, M.Y.; Khoo, K.H.; Li, S.C.; et al. Ceramide glycosylation by glucosylceramide synthase selectively maintains the properties of breast cancer stem cells. J. Biol. Chem. 2012, 287, 37195–37205. [Google Scholar]
- Sisu, E.; Flangea, C.; Serb, A.; Rizzi, A.; Zamfir, A.D. High-performance separation techniques hyphenated to mass spectrometry for ganglioside analysis. Electrophoresis 2011, 32, 1591–1609. [Google Scholar]
- Ikeda, K.; Shimizu, T.; Taguchi, R. Targeted analysis of ganglioside and sulfatide molecular species by LC/ESI-MS/MS with theoretically expanded multiple reaction monitoring. J. Lipid Res. 2008, 49, 2678–2689. [Google Scholar] [CrossRef]
- Ikeda, K.; Taguchi, R. Highly sensitive localization analysis of gangliosides and sulfatides including structural isomers in mouse cerebellum sections by combination of laser microdissection and hydrophilic interaction liquid chromatography/electrospray ionization mass spectrometry with theoretically expanded multiple reaction monitoring. Rapid Commun. Mass Spectrom. 2010, 24, 2957–2965. [Google Scholar] [CrossRef]
- Li, S.C.; DeGasperi, R.; Muldrey, J.E.; Li, Y.T. A unique glycosphingolipid-splitting enzyme (ceramide-glycanase from leech) cleaves the linkage between the oligosaccharide and the ceramide. Biochem. Biophys. Res. Commun. 1986, 141, 346–352. [Google Scholar] [CrossRef]
- Zhou, B.; Li, S.C.; Laine, R.A.; Huang, R.T.; Li, Y.T. Isolation and characterization of ceramide glycanase from the leech, Macrobdella decora. J. Biol. Chem. 1989, 264, 12272–12277. [Google Scholar]
- Li, Y.T.; Ishikawa, Y.; Li, S.C. Occurrence of ceramide-glycanase in the earthworm; Lumbricus terrestris. Biochem. Biophys. Res. Commun. 1987, 149, 167–172. [Google Scholar] [CrossRef]
- Ito, M.; Yamagata, T. A novel glycosphingolipid-degrading enzyme cleaves the linkage between the oligosaccharide and ceramide of neutral and acidic glycosphingolipids. J. Biol. Chem. 1986, 261, 14278–14282. [Google Scholar]
- Ito, M.; Yamagata, T. Purification; characterization of glycosphingolipid-specific endoglycosidases (endoglycoceramidases) from a mutant strain of Rhodococcus sp. Evidence for three molecular species of endoglycoceramidase with different specificities. J. Biol. Chem. 1989, 264, 9510–9519. [Google Scholar]
- Ashida, H.; Yamamoto, K.; Kumagai, H.; Tochikura, T. Purification; characterization of membrane-bound endoglycoceramidase from Corynebacterium sp. Eur. J. Biochem. 1992, 205, 729–735. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Kobayashi, U.; Hijikata, A.; Sakaguchi, K.; Goda, H.M.; Tamura, T.; Okino, N.; Ito, M. Preparation and characterization of EGCase I, applicable to the comprehensive analysis of GSLs, using a rhodococcal expression system. J. Lipid. Res. 2012, 53, 2242–2251. [Google Scholar] [CrossRef]
- Fujitani, N.; Takegawa, Y.; Ishibashi, Y.; Araki, K.; Furukawa, J.; Mitsutake, S.; Igarashi, Y.; Ito, M.; Shinohara, Y. Qualitative and quantitative cellular glycomics of glycosphingolipids based on rhodococcal endoglycosylceramidase-assisted glycan cleavage; glycoblotting-assisted sample preparation; and matrix-assisted laser desorption ionization tandem time-of-flight mass spectrometry analysis. J. Biol. Chem. 2011, 286, 41669–41679. [Google Scholar]
- Heiskanen, A.; Hirvonen, T.; Salo, H.; Impola, U.; Olonen, A.; Laitinen, A.; Tiitinen, S.; Natunen, S.; Aitio, O.; Miller-Podraza, H.; et al. Glycomics of bone marrow-derived mesenchymal stem cells can be used to evaluate their cellular differentiation stage. Glycoconj. J. 2009, 26, 367–384. [Google Scholar] [CrossRef]
- Hakomori, S. Release of carbohydrates from sphingoglycolipid by osmium-catalyzed periodate oxidation, followed by treatment with mild alkali. J. Lipid Res. 1966, 7, 789–792. [Google Scholar]
- MacDonald, D.L.; Patt, L.M.; Hakomori, S. Notes on improved procedures for the chemical modification and degradation of glycosphingolipids. J. Lipid Res. 1980, 21, 642–645. [Google Scholar]
- Mylvaganam, M.; Meng, L.; Lingwood, C.A. Oxidation of glycosphingolipids under basic conditions: synthesis of glycosyl “serine acids” as opposed to “ceramide acids”. Precursors for neoglycoconjugates with increased ligand binding affinity. Biochemistry 1999, 38, 10885–10897. [Google Scholar] [CrossRef]
- Yowler, B.C.; Stoehr, S.A.; Schengrund, C.L. Oxidation and base-catalyzed elimination of the saccharide portion of GSLs having very different polarities. J. Lipid Res. 2001, 42, 659–662. [Google Scholar]
- Song, X.; Lasanajak, Y.; Xia, B.; Molinaro, J.; Rhea, J.M.; Ju, H.; Zhao, C.; Molinaro, R.J.; Cummings, R.D.; Smith, D.F. Shotgun glycomics: A microarray strategy for functional glycomics. Nat. Methods 2011, 8, 85–90. [Google Scholar]
- Song, X.; Smith, D.F.; Cummings, R.D. Nonenzymatic release of free reducing glycans from glycosphingolipids. Anal. Biochem. 2012, 429, 82–87. [Google Scholar] [CrossRef]
- Smith, R.A.; Meade, K.; Pickford, C.E.; Holley, R.J.; Merry, C.L. Glycosaminoglycans as regulators of stem cell differentiation. Biochem. Soc. Trans. 2011, 39, 383–387. [Google Scholar] [CrossRef]
- Purushothaman, A.; Sugahara, K.; Faissner, A. Chondroitin sulfate "wobble motifs" modulate maintenance and differentiation of neural stem cells and their progeny. J. Biol. Chem. 2012, 287, 2935–2942. [Google Scholar] [CrossRef]
- Quantock, A.J.; Young, R.D.; Akama, T.O. Structural and biochemical aspects of keratan sulphate in the cornea. Cell Mol. Life Sci. 2010, 67, 891–906. [Google Scholar] [CrossRef]
- Ly, M.; Leach, F.E., 3rd.; Laremore, T.N.; Toida, T.; Amster, I.J.; Linhardt, R.J. The proteoglycan bikunin has a defined sequence. Nat. Chem. Biol. 2011, 7, 827–833. [Google Scholar] [CrossRef]
- Li, B.; Liu, H.; Zhang, Z.; Stansfield, H.E.; Dordick, J.S.; Linhardt, R.J. Analysis of glycosaminoglycans in stem cell glycomics. Methods Mol. Biol. 2011, 690, 285–300. [Google Scholar] [CrossRef]
- Guimond, S.E.; Puvirajesinghe, T,M.; Skidmore, M.A.; Kalus, I.; Dierks, T.; Yates, E.A.; Turnbull, J.E. Rapid purification and high sensitivity analysis of heparan sulfate from cells and tissues: toward glycomics profiling. J. Biol. Chem. 2009, 284, 25714–25722. [Google Scholar]
- Zhao, X.; Yang, B.; Datta, P.; Gasmili, L.; Zhang, F.; Linhard, R.J. Cell-based microscale isolation of glycoaminoglycans for glycomics study. J. Carbohydr. Chem. 2012, 31, 420–435. [Google Scholar] [CrossRef]
- Takegawa, Y.; Araki, K.; Fujitani, N.; Furukawa, J.; Sugiyama, H.; Sakai, H.; Shinohara, Y. Simultaneous analysis of heparan sulfate; chondroitin/dermatan sulfates; and hyaluronan disaccharides by glycoblotting-assisted sample preparation followed by single-step zwitter-ionic-hydrophilic interaction chromatography. Anal. Chem. 2011, 83, 9443–9449. [Google Scholar]
- Izumikawa, T.; Uyama, T.; Okuura, Y.; Sugahara, K.; Kitagawa, H. Involvement of chondroitin sulfate synthase-3 (chondroitin synthase-2) in chondroitin polymerization through its interaction with chondroitin synthase-1 or chondroitin-polymerizing factor. Biochem. J. 2007, 403, 545–552. [Google Scholar] [CrossRef]
- Ernst, S.; Langer, R.; Cooney, C.L.; Sasisekharan, R. Enzymatic degradation of glycosaminoglycans. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 387–444. [Google Scholar] [CrossRef]
- Linhardt, R.J.; Rice, K.G.; Kim, Y.S.; Lohse, D.L.; Wang, H.M.; Loganathan, D. Mapping and quantification of the major oligosaccharide components of heparin. Biochem. J. 1988, 254, 781–787. [Google Scholar]
- Kitagawa, H.; Kinoshita, A.; Sugahara, K. Microanalysis of glycosaminoglycan-derived disaccharides labeled with the fluorophore 2-aminoacridone by capillary electrophoresis and high-performance liquid chromatography. Anal. Biochem. 1995, 232, 114–121. [Google Scholar] [CrossRef]
- Militsopoulou, M.; Lamari, F.N.; Hjerpe, A.; Karamanos, N.K. Determination of twelve heparin- and heparan sulfate-derived disaccharides as 2-aminoacridone derivatives by capillary zone electrophoresis using ultraviolet and laser-induced fluorescence detection. Electrophoresis 2002, 23, 1104–1109. [Google Scholar] [CrossRef]
- Kinoshita, A.; Sugahara, K. Microanalysis of glycosaminoglycan-derived oligosaccharides labeled with a fluorophore 2-aminobenzamide by high-performance liquid chromatography: application to disaccharide composition analysis and exosequencing of oligosaccharides. Anal. Biochem. 1999, 269, 367–378. [Google Scholar] [CrossRef]
- Turnbull, J.E.; Hopwood, J.J.; Gallagher, J.T. A strategy for rapid sequencing of heparan sulfate and heparin saccharides. Proc. Natl. Acad. Sci. USA 1999, 96, 2698–2703. [Google Scholar]
- Skidmore, M.A.; Guimond, S.E.; Dumax-Vorzet, A.F.; Yates, E.A.; Turnbull, J.E. Disaccharide compositional analysis of heparan sulfate and heparin polysaccharides using UV or high-sensitivity fluorescence (BODIPY) detection. Nat. Protoc. 2010, 5, 1983–1992. [Google Scholar] [CrossRef]
- Lawrence, R.; Olson, S.K.; Steele, R.E.; Wang, L.; Warrior, R.; Cummings, R.D.; Esko, J.D. Evolutionary differences in glycosaminoglycan fine structure detected by quantitative glycan reductive isotope labeling. J. Biol. Chem. 2008, 283, 33674–33684. [Google Scholar]
- Hitchcock, A.M.; Yates, K.E.; Costello, C.E.; Zaia, J. Comparative glycomics of connective tissue glycosaminoglycans. Proteomics 2008, 8, 1384–1397. [Google Scholar] [CrossRef]
- Bowman, M.J.; Zaia, J. Comparative glycomics using a tetraplex stable-isotope coded tag. Anal. Chem. 2010, 82, 3023–3031. [Google Scholar] [CrossRef]
- Hitchkock, A.M.; Bowman, M.J.; Staples, G.O.; Zaia, J. Improved workup for glycosaminoglycan disaccharide analysis using CE with LIF detection. Electrophoresis 2008, 29, 4538–4548. [Google Scholar] [CrossRef]
- Eldridge, S.L.; Higgins, L.A.; Dickey, B.J.; Larive, C.K. Insights into the capillary electrophoresis separation of heparin disaccharides from nuclear magnetic resonance, pKa, and electrophoretic mobility measurement. Anal. Chem. 2009, 81, 7406–7415. [Google Scholar]
- Plaas, A.H.; West, L.A.; Midura, R.J. Keratan sulfate disaccharide composition determined by FACE analysis of keratanase II and endo-beta-galactosidase digestion products. Glycobiology 2001, 10, 779–790. [Google Scholar] [CrossRef]
- Korir, A.K.; Limtiaco, J.F.; Gutierrez, S.M.; Larive, C.K. Ultraperformance ion-pair liquid chromatography coupled to electrospray time-of-flight mass spectrometry for compositional profiling and quantification of heparin and heparan sulfate. Anal. Chem. 2008, 80, 1297–1306. [Google Scholar]
- Imanari, T.; Toida, T.; Koshiishi, I.; Toyoda, H. High-performance liquid chromatographic analysis of glycosaminoglycan-derived oligosaccharides. J. Chromatogr. A 1996, 720, 275–293. [Google Scholar] [CrossRef]
- Jones, C.J.; Membreno, N.; Larive, C.K. Insights into the mechanism of separation of heparin and heparan sulfate disaccharides by reverse-phase ion-pair chromatography. J. Chromatogr. A 2010, 1217, 479–488. [Google Scholar] [CrossRef]
- Staples, G.O.; Shi, X.; Zaia, J. Extended N-sulfated domains reside at the nonreducing end of heparan sulfate chains. J. Biol. Chem. 2010, 285, 18336–18343. [Google Scholar] [CrossRef]
- Volpi, N. High-performance liquid chromatography and on-line mass spectrometry detection for the analysis of chondroitin sulfates/hyaluronan disaccharides derivatized with 2-aminoacridone. Anal. Biochem. 2010, 397, 12–23. [Google Scholar] [CrossRef]
- Yang, B.; Chang, Y.; Weyers, A.M.; Sterner, E.; Linhardt, R.J. Disaccharide analysis of glycosaminoglycan mixtures by ultra-high-performance liquid chromatography-mass spectrometry. J. Chromatogr. A 2012, 1225, 91–98. [Google Scholar] [CrossRef]
- Laremore, T.N.; Leach, F.E., 3rd.; Solakyildirim, K.; Amster, I.J.; Linhardt, R.J. Glycosaminoglycan characterization by electrospray ionization mass spectrometry including fourier transform mass spectrometry. Methods Enzymol. 2010, 478, 79–108. [Google Scholar] [CrossRef]
- Uygun, B.E.; Stojsih, S.E.; Matthew, H.W. Effects of immobilized glycosaminoglycans on the proliferation and differentiation of mesenchymal stem cells. Tissue Eng. Part A 2009, 15, 3499–3512. [Google Scholar] [CrossRef]
- Kumarasuriyar, A.; Murali, S.; Nurcombe, V.; Cool, S.M. Glycosaminoglycan composition changes with MG-63 osteosarcoma osteogenesis in vitro and induces human mesenchymal stem cell aggregation. J. Cell Physiol. 2009, 218, 501–511. [Google Scholar] [CrossRef]
- Dombrowski, C.; Song, S.J.; Chuan, P.; Lim, X.; Susanto, E.; Sawyer, A.A.; Woodruff, M.A.; Hutmacher, D.W.; Nurcombe, V.; Cool, S.M. Heparan sulfate mediates the proliferation and differentiation of rat mesenchymal stem cells. Stem Cells Dev. 2009, 18, 661–670. [Google Scholar] [CrossRef]
- Nairn, A.V.; Kinoshita-Toyoda, A.; Toyoda, H.; Xie, J.; Harris, K.; Dalton, S.; Kulik, M.; Pierce, J.M.; Toida, T.; Moremen, K.W.; et al. Glycomics of proteoglycan biosynthesis in murine embryonic stem cell differentiation. J. Proteome Res. 2007, 6, 4374–4387. [Google Scholar] [CrossRef]
- Kawabe, K.; Tateyama, D.; Toyoda, H.; Kawasaki, N.; Hashii, N.; Nakao, H.; Matsumoto, S.; Nonaka, M.; Matsumura, H.; Hirose, Y.; et al. Novel Antibody for Human Induced Pluripotent Stem (hiPS) Cells and Embryonic Stem (ES) Cells Recognizes a Type of Keratan Sulfate Lacking Oversulfated Structures. Glycobiology 2012, 23, 322–336. [Google Scholar]
- Toida, T.; Hileman, R.E.; Smith, A.E.; Vlahova, P.I.; Linhardt, R.J. Enzymatic preparation of heparin oligosaccharides containing antithrombin III binding sites. J. Biol. Chem. 1996, 271, 32040–32947. [Google Scholar]
- Ly, M.; Laremore, T.N.; Linhardt, R.J. Proteoglycomics: recent progress and future challenges. OMICS 2010, 14, 389–399. [Google Scholar] [CrossRef]
- Lawrence, R.; Brown, J.R.; Al-Mafraji, K.; Lamanna, W.C,; Beitel, J.R.; Boons, G.J.; Esko, J.D.; Crawford, B.E. Disease-specific non-reducing end carbohydrate biomarkers for mucopolysaccharidoses. Nat. Chem. Biol. 2012, 8, 197–204. [Google Scholar]
- Wearne, K.A.; Winter, H.C.; O'Shea, K.; Goldstein, I.J. Use of lectins for probing differentiated human embryonic stem cells for carbohydrates. Glycobiology 2006, 16, 981–990. [Google Scholar] [CrossRef]
- Toyoda, M.; Yamazaki-Inoue, M.; Itakura, Y.; Kuno, A.; Ogawa, T.; Yamada, M.; Akutsu, H.; Takahashi, Y.; Kanzaki, S.; Narimatsu, H.; et al. Lectin microarray analysis of pluripotent and multipotent stem cells. Genes Cells 2011, 16, 1–11. [Google Scholar]
- Wang, Y.C.; Nakagawa, M.; Garitaonandia, I.; Slavin, I.; Altun, G.; Lacharite, R.M.; Nazor, K.L.; Tran, H.T.; Lynch, C.L.; Leonardo, T.R.; et al. Specific lectin biomarkers for isolation of human pluripotent stem cells identified through array-based glycomic analysis. Cell Res. 2011, 21, 1551–1563. [Google Scholar] [CrossRef]
- Yasuda, E.; Tateno, H.; Hirabayashi, J.; Iino, I.; Sako, T. Lectin microarray reveals binding profiles of Lactobacillus casei strains in a comprehensive analysis of bacterial cell wall polysaccharides. Appl. Eviron Microbiol. 2011, 77, 4539–4546. [Google Scholar] [CrossRef]
- Parry, S.; Ledger, V.; Tissot, B.; Haslam, S.M.; Scott, J.; Morris, H.R. Integrated mass spectrometric strategy for characterizing the glycans from glycosphingolipids and glycoproteins: direct identification of sialyl Lex in mice. Glycobiology 2007, 17, 646–654. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Furukawa, J.-i.; Fujitani, N.; Shinohara, Y. Recent Advances in Cellular Glycomic Analyses. Biomolecules 2013, 3, 198-225. https://doi.org/10.3390/biom3010198
AMA Style
Furukawa J-i, Fujitani N, Shinohara Y. Recent Advances in Cellular Glycomic Analyses. Biomolecules. 2013; 3(1):198-225. https://doi.org/10.3390/biom3010198
Chicago/Turabian StyleFurukawa, Jun-ichi, Naoki Fujitani, and Yasuro Shinohara. 2013. "Recent Advances in Cellular Glycomic Analyses" Biomolecules 3, no. 1: 198-225. https://doi.org/10.3390/biom3010198