Cullin E3 Ligases and Their Rewiring by Viral Factors



Abstract
:1. Introduction
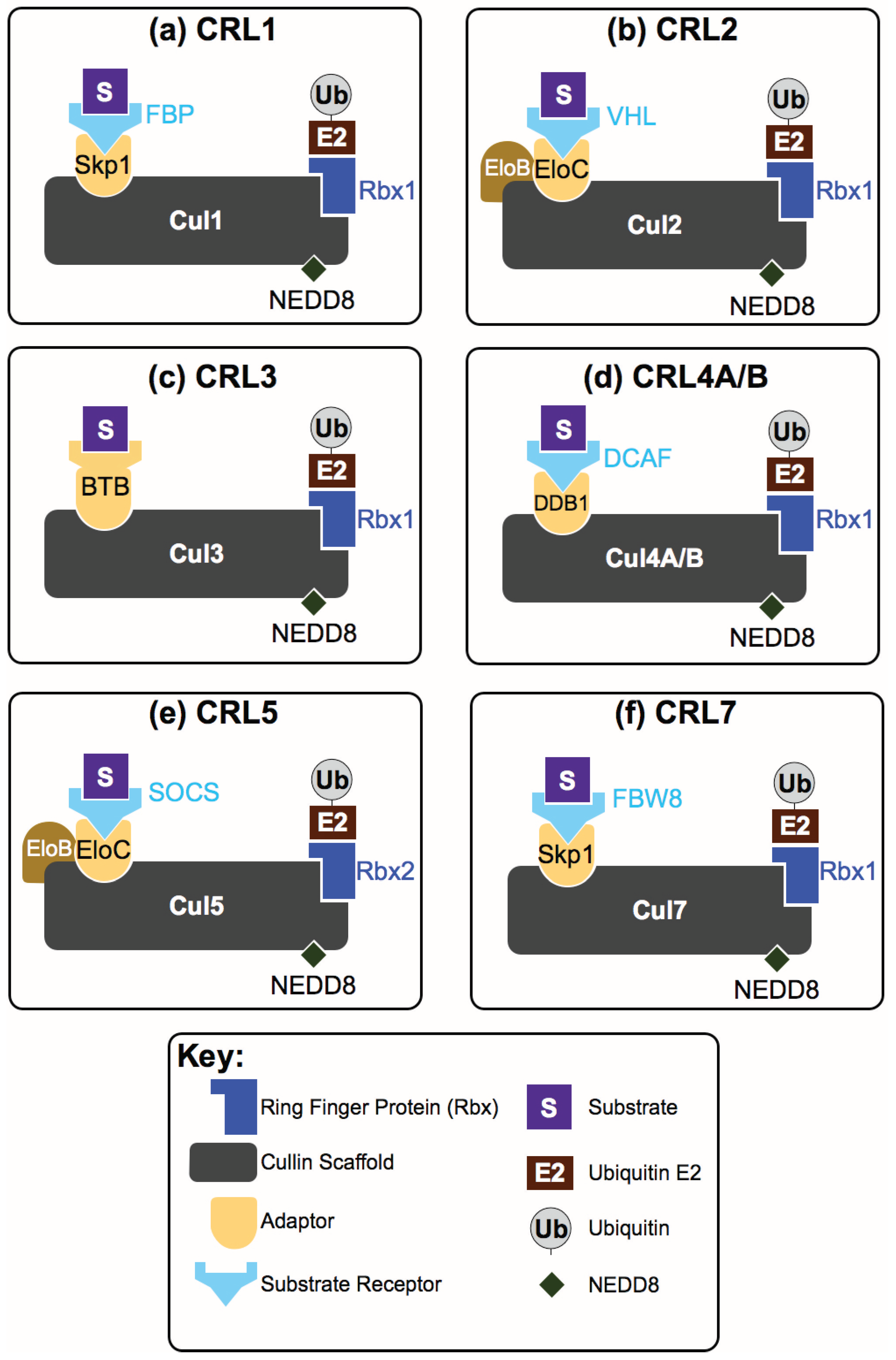

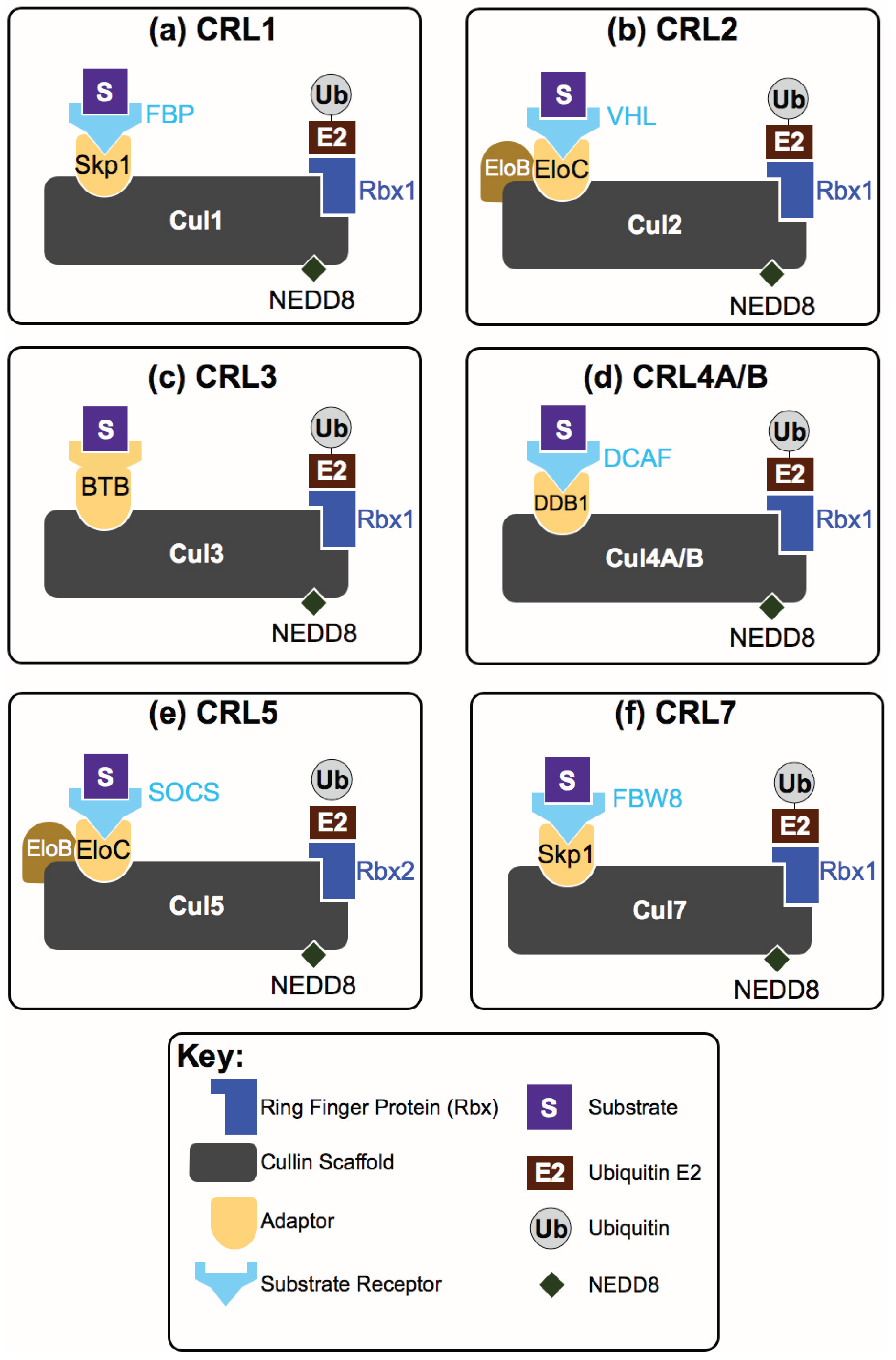{kind=link}
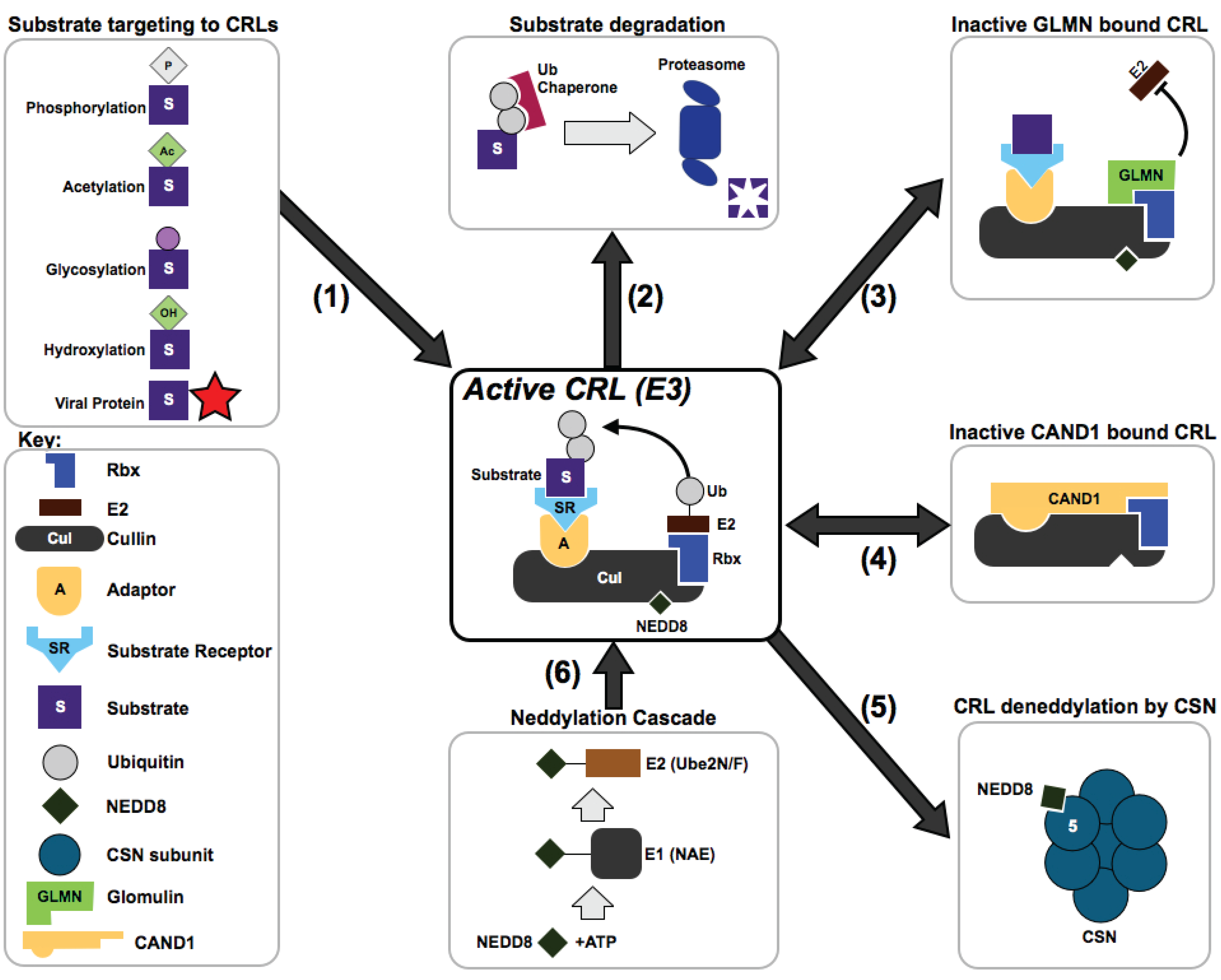
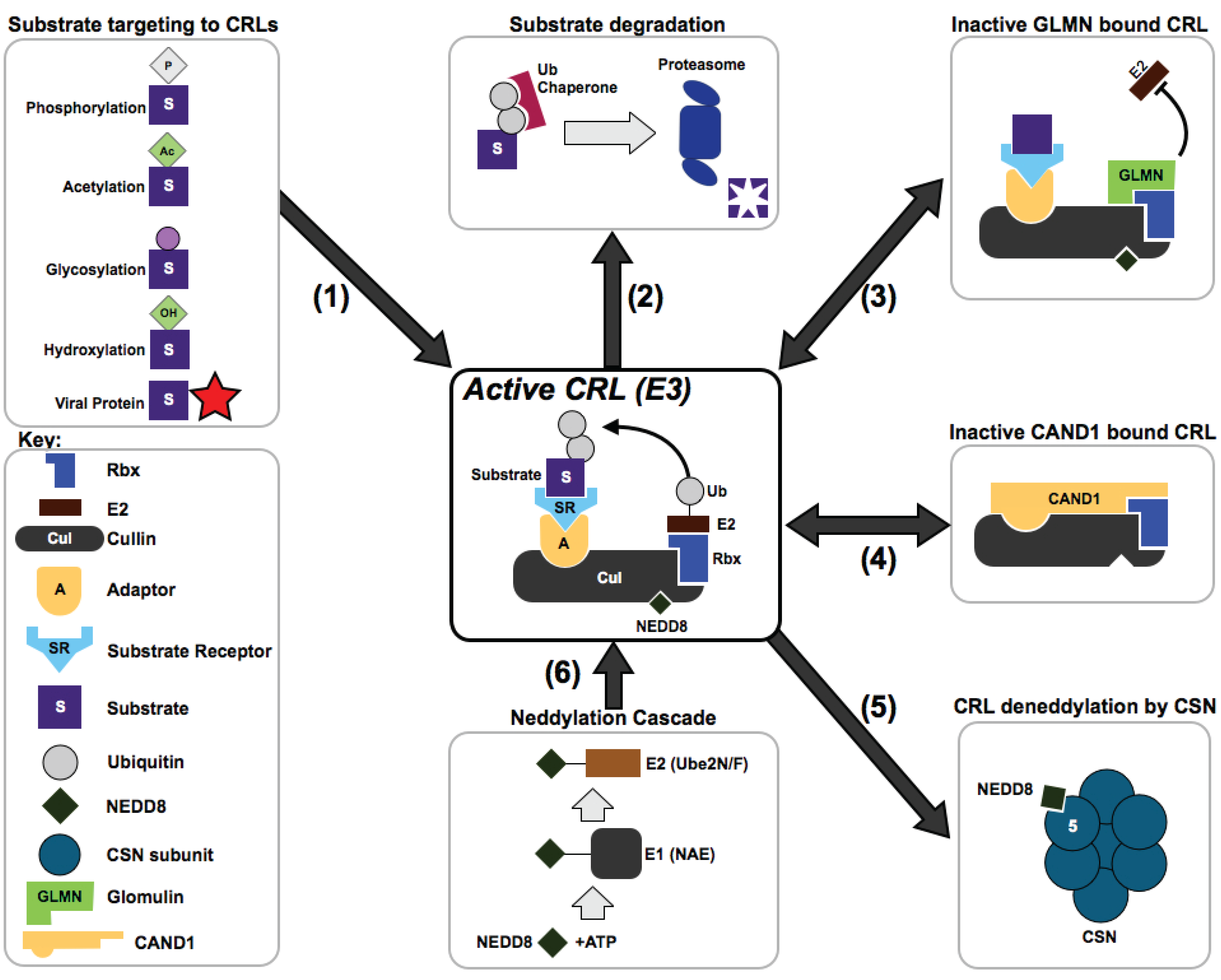{kind=link}
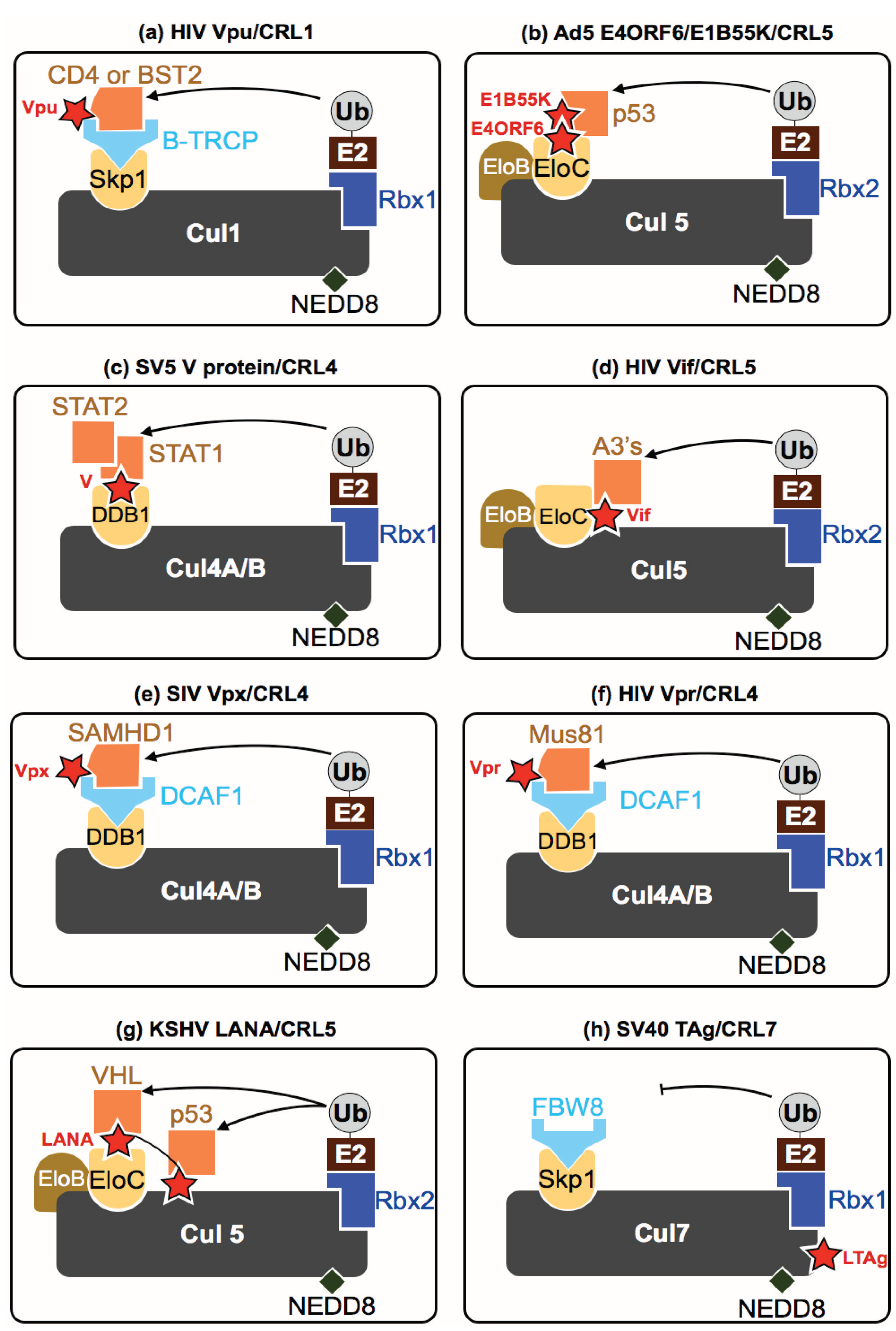{kind=link}
| Virus | Viral Protein | UPS Factor Targeted | Viral Protein Function | Reference |
|---|---|---|---|---|
| Group I | ||||
| Ectromelia | 4 × BTB-kelch type 1 | CRL3 | Form complexes with CRL3, substrates unknown. EVM150 inhibits NFκB independently of CRL3 | [23,24] |
| VACV | Unknown | CRL3 | Unknown | [25] |
| Adenovirus | E4ORF6 & EB155K | CRL2/5 | Serotype specific degradation of p53, TOPBP1, MRE11, BLM, ATRX, integrin α3 & DNA ligase IV | [26,27,28,29,30,31,32] |
| HPV | E7 | CRL2 & proteasome | Degradation of pRb | [33] |
| KSHV | LANA | CRL5 | Acts as a substrate receptor for P53 and VHL degradation | [34,35,36] |
| LANA | CRL1 | Binds to FBW7 inhibiting ICN degradation; Interacts with GSK-3 and inhibits c-Myc degradation by CRL1 | ||
| V-cyclin | CRL1 | Causes phosphorylation of p27 which is then degraded by CRL1 | ||
| Myxoma virus | MT-5 | CRL1 | Drives degradation of p27 regulating cell cycle progression at G0/G1 | [37] |
| AcMNPV | Lef7 | CRL1 | Functions as an F box protein, alters the host DNA damage response through an unknown mechanism | [38] |
| MuHV-4 | ORF73 | CRL5 | Functions as a substrate receptor to degrade p65- inhibiting NFκB signaling | [39] |
| CELO virus | GAM1 | CRL2/5 | Drives SAE1 and SAE degradation | [40] |
| SV40 | LTAg | CRL7 | Inhibits CRL7 degradation of IRS-1 | [41,42] |
| HSV1 | ICPo | Proteasome and E2 | Trans-activates host cellular genes through unknown mechanisms | [43] |
| EBV | BZLF | CRL2 | Functions as a substrate adaptor driving p53 degradation | [44] |
| BPFL1 | CRLs | Acts as a deneddylase inactivating CRLs preventing the degradation of host cell cycle and DNA damage regulators (Cdc25A, CDT1, p21, and p27) | [45] | |
| Group II | ||||
| TYLCSV | C2 | CSN5 | Inhibits CSN activity and deregulates CRL activity | [46] |
| MVM | Unknown | CRL4 | Infection causes CRL4 driven degradation of host p21 | [47] |
| FBNYV | Clink | SKP1 | Contains a functional F box with no known biological role | [48] |
| Group III | ||||
| Rotavirus | NSP1 | CRL1 | Causes degradation of β-TRCP | [49] |
| Group IV | ||||
| HepE | ORF2 | CRL1 | Inhibits IκBa ubiquitination by CRL1 | [50] |
| BNYVV | P25 | CRL1 | Probably interacts with host F box proteins | [51,52,53] |
| Polerovirus | P0 | CRL1 | Targets a yet undiscovered host factor involved in post-transcriptional gene silencing | [54] |
| Group V | ||||
| Paramyxoxy virus family | V proteins | CRL4 | Degrades host STAT proteins— disabling host interferon response | [55,56] |
| RSV | NS1 | CRL2 | Degrades host STAT proteins—disabling host interferon response | [57] |
| Rift Valley Fever | NSs | CRL1 | Binds to FBXO3 driving degradation of TFIIH subunit | [58,59] |
| Group VI | ||||
| HIV-1 | Vif | CRL5 | Acts as a substrate receptor driving degradation of A3 proteins | [60] |
| Vpu | CRL1 | Assembles in a β-TRCP-containing SCF complex driving CD4 and possibly BST2 degradation | [61] | |
| Vpr | CRL4 | Prematurely activates SLX4 complex by driving Mus81 ubiquitination | [62] | |
| HIV-2/SIV | Vpx | CRL4 | Drives SAMHD1 degradation in specific cell types | [63,64,65] |
| Group VII | ||||
| HepB | HBx | CRL4 | Causes genome instability through an unknown mechanism | [64,66] |
2. How Are CRL Complexes Formed?
3. How are CRL Complexes Regulated Within Cells?
4. How Do CRLs Select Their Substrates?
5. How Do Viruses Affect CRLs?
5.1. Group-I (Double Stranded DNA Viruses)
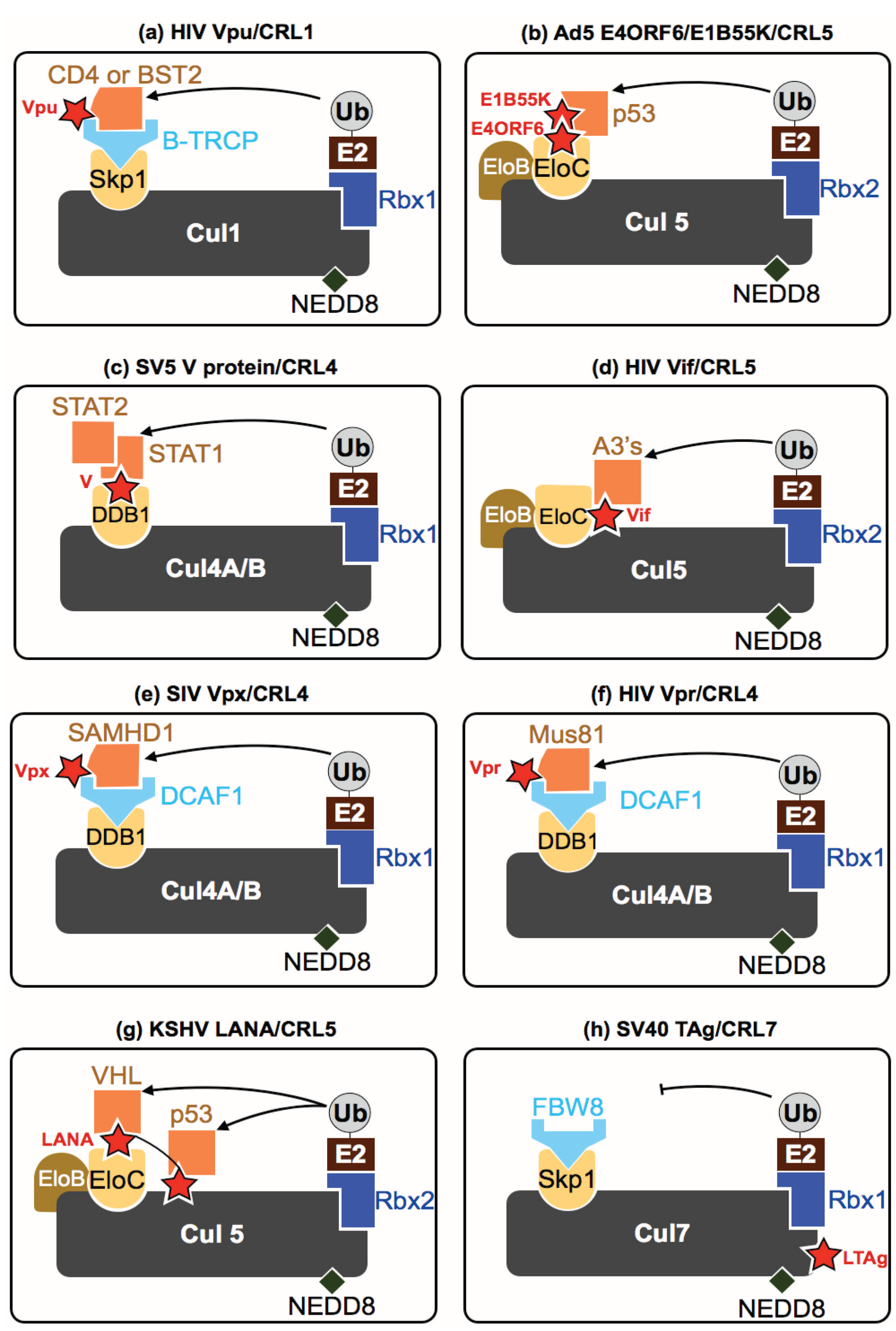
5.2. Group-II (Single Stranded DNA Viruses)
5.3. Group-III (Double Stranded RNA Viruses)
5.4. Group-IV (Positive Sense ssRNA Viruses)
5.5. Group-V (Negative Sense ssRNA Viruses)
5.6. Group-VI (ssRNA-RT Viruses {+ Strand or Sense} RNA with DNA Intermediate in Life-Cycle)
5.7. Group-VII (dsDNA-RT Viruses)
6. Do Viral Proteins Targeting CRLs also Interact with the Proteasome?
7. Could CRL Based Therapeutics Work as Anti-Virals?
8. How to Identify CRL Substrates?
9. What Open Questions Remain Regarding CRLs in Viral Infections?
10. Conclusions
Acknowledgments
Abbreviations
| A3F/G | Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3 F/G |
| AcMNPV | Autographa californica multicapsid nucleo-polyhedrovirus |
| AIDS | Acquired immunodeficiency syndrome |
| ATRX | X-linked α-thalassaemia retardation syndrome protein |
| β-TRCP | Beta-transducin repeat containing E3 ubiquitin protein ligase |
| BLM | Bloom helicase |
| BNYVV | Beet benyvirus necrotic yellow vein virus |
| BST2 | Bone marrow cell stromal antigen 2 |
| CAND1 | Cullin-associated NEDD8-dissociated protein 1 |
| CBF-β | Core-binding factor subunit beta |
| CDT1 | Chromatin licensing and DNA replication factor-1 |
| CDT2 | Chromatin licensing and DNA replication factor-2 |
| CELO | Chicken embryo lethal orphan virus |
| CSN | COP9 signalosome |
| CSA1 | Cockayne syndrome A |
| CRL | Cullin-RING E3 ligase |
| DCAF | DDB1/Cul4A associated factor |
| DCN1 | Defective in cullin neddylation-1 |
| DDA1 | DET1- and DDB1-associated protein 1 |
| DDB1 | DNA damage binding protein-1 |
| DDB2 | DNA damage binding protein-2 |
| EBV | Epstein Barr virus |
| EloB | Elongin B |
| EloC | Elongin C |
| FBNYV | Faba bean necrotic yellows virus |
| FBP | F-box protein |
| GLMN | Glomulin |
| GPS | Global protein stability |
| GSK-3 | Glycogen synthase kinase-3 |
| Hbx | Hepatitis B virus X protein |
| HepB/E | Hepatitis B/E virus |
| HIV-1 | Human immunodeficiency virus-1 |
| HPV | Human papillomavirus virus |
| HSV1 | Herpes simplex virus 1 |
| Human RSV | Human respiratory syncytial virus |
| ICN | Intracellular notch1 |
| IκBa | NF-kappa-B inhibitor alpha-like |
| IPMS | Immuno-precipitation mass spectrometry |
| IRF9 | Interferon regulatory factor 9 |
| KSHV | Kaposi sarcoma-associated herpesvirus |
| LANA | Latency-associated nuclear antigen |
| LTAg | Large T antigen |
| MAPK | Mitogen-activated protein kinase |
| MHC1 | Major histocompatibility complex 1 |
| MPN | Mpr1p and PAD1p N-terminal)/JAMM (JAB1/MPN/Mov34 metalloenzyme) |
| MRN | MRE11A-RAD50-NBS1 complex |
| MuHV-4 | Murid herpesvirus-4 |
| MVM | Minute virus of mice |
| NAE1 | NEDD8 activating enzyme-1 |
| NEDD8 | Neural precursor cell expressed developmentally down-regulated protein 8 |
| Nef | Negative regulatory factor |
| NFκB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NHEJ | Non-homologous end-joining |
| NiV | Nipah virus |
| NS1 | RSV non-structural protein 1 |
| NSP1 | Rotavirus nonstructural protein-1 |
| NSs | Rift valley fever virus non-structural protein |
| PCNA | Proliferating cell nuclear antigen |
| pRB | Phosphorylated retinoblastoma protein |
| PTTG1 | Protooncogene pituitary tumor-transforming gene 1 |
| QUAINT | Quantitative ubiquitination interrogation |
| ROC1/Rbx1 | Regulator of cullins/RING box protein-1 |
| ROC2/Rbx2 | Regulator of cullins/RING box protein-2 |
| SAMHD1 | SAM domain and HD domain-containing protein 1 |
| SCF | Skp1-Cul1-F-box |
| SIV | Simian immunodeficiency virus |
| SKP1 | S-phase kinase-associated protein-1 |
| SOCS | Suppressor of cytokine signaling |
| SMUG1 | Single-strand-selective monofunctional uracil-DNA glycosylase 1 |
| SR | Substrate receptor |
| STAT | Signal transducer and activator of transcription |
| SUMO-1 | Small ubiquitin-like modifier-1 |
| SV40 | Simian Virus 40 |
| SV5 | Simian Virus 5 |
| TFIIB | Transciption factor IIB |
| TOPBP1 | Topoisomerase (DNA) II binding protein |
| TYLCSV | Tomato yellow leaf curl sardinia virus |
| Ube2f | Ubiquitin-conjugating enzyme E2M |
| Ube2n | Ubiquitin-conjugating enzyme E2N |
| UNG2 | Uracil N glycosylase 2 |
| UPS | Ubiquitin-proteasome system |
| VACV | Vaccinia virus |
| VHL | Von Hippel-Lindau tumor suppressor |
| Vif | Viral infectivity factor |
| Vpr | Viral Protein R |
| Vpu | Viral Protein U |
| Vpx | Viral Protein X |
Conflicts of Interest
References
- Ciechanover, A. Intracellular protein degradation: From a vague idea through the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Bioorg. Med. Chem. 2013, 21, 3400–3410. [Google Scholar] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin-proteasome pathway. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A. Ring domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef] [PubMed]
- Spratt, D.E.; Walden, H.; Shaw, G.S. RBR E3 ubiquitin ligases: New structures, new insights, new questions. Biochem. J. 2014, 458, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Lever, A.M. Wrapping up the bad news: HIV assembly and release. Retrovirology 2013. [Google Scholar] [CrossRef]
- Alcaide-Loridan, C.; Jupin, I. Ubiquitin and plant viruses, let’s play together! Plant. Physiol. 2012, 160, 72–82. [Google Scholar]
- Fehr, A.R.; Yu, D. Control the host cell cycle: Viral regulation of the anaphase-promoting complex. J. Virol. 2013, 87, 8818–8825. [Google Scholar] [CrossRef] [PubMed]
- Rajsbaum, R.; Garcia-Sastre, A. Viral evasion mechanisms of early antiviral responses involving regulation of ubiquitin pathways. Trends Microbiol. 2013, 21, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.G.; Wong, J.; Marchant, D.; Luo, H. The ubiquitin-proteasome system in positive-strand RNA virus infection. Rev. Med. Virol. 2013, 23, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, J.; Pagano, J.S. Targeting of host-cell ubiquitin pathways by viruses. Essays Biochem. 2005, 41, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, J.; Pagano, J.S. Role of the ubiquitin system and tumor viruses in AIDS-related cancer. BMC Biochem. 2007. [Google Scholar] [CrossRef]
- Wise-Draper, T.M.; Wells, S.I. Papillomavirus E6 and E7 proteins and their cellular targets. Front. Biosci. 2008, 13, 1003–1017. [Google Scholar] [CrossRef] [PubMed]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef] [PubMed]
- Lydeard, J.R.; Schulman, B.A.; Harper, J.W. Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep. 2013, 14, 1050–1061. [Google Scholar] [CrossRef] [PubMed]
- Lyapina, S.; Cope, G.; Shevchenko, A.; Serino, G.; Tsuge, T.; Zhou, C.; Wolf, D.A.; Wei, N.; Shevchenko, A.; Deshaies, R.J. Promotion of NEDD-cul1 conjugate cleavage by COP9 signalosome. Science 2001, 292, 1382–1385. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.W.; McQuary, P.R.; Wee, S.; Hofmann, K.; Wolf, D.A. F-box-directed CRl complex assembly and regulation by the CSN and CAND1. Mol. Cell 2009, 35, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Gummlich, L.; Rabien, A.; Jung, K.; Dubiel, W. Deregulation of the COP9 signalosome-Cullin-RING ubiquitin-ligase pathway: Mechanisms and roles in urological cancers. Int. J. Biochem. Cell Biol. 2013, 45, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H.; Bieniasz, P.D. HIV restriction factors and mechanisms of evasion. Cold Spring Harbor Perspect. Med. 2012, 2, a006940. [Google Scholar] [CrossRef]
- Kirchhoff, F. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell. Host Microbe 2010, 8, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.R.; Collins, K.L. HIV-1 accessory proteins adapt cellular adaptors to facilitate immune evasion. PLoS Pathog. 2014, 10, e1003851. [Google Scholar] [CrossRef] [PubMed]
- Irigoyen, M.L.; Iniesto, E.; Scrima, A.; Bohm, K.; Matsumoto, S.; Lingaraju, G.M.; Faty, M.; Yasuda, T.; Cavadini, S.; Wakasugi, M.; Hanaoka, F.; et al. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell 2011, 147, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Enchev, R.I.; Scott, D.C.; da Fonseca, P.C.; Schreiber, A.; Monda, J.K.; Schulman, B.A.; Peter, M.; Morris, E.P. Structural basis for a reciprocal regulation between SCF and CSN. Cell Rep. 2012, 2, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Wilton, B.A.; Campbell, S.; van Buuren, N.; Garneau, R.; Furukawa, M.; Xiong, Y.; Barry, M. Ectromelia virus BTB/kelch proteins, EVM150 and EVM167, interact with Cullin-3-based ubiquitin ligases. Virology 2008, 374, 82–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Burles, K.; Couturier, B.; Randall, C.M.; Shisler, J.; Barry, M. Ectromelia virus encodes a BTB/kelch protein, EVM150, that inhibits NFkappab signaling. J. Virol. 2014, 88, 4853–4865. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Snijder, B.; Sacher, R.; Burkard, C.; Bleck, C.K.; Stahlberg, H.; Pelkmans, L.; Helenius, A. RNAi screening reveals proteasome- and Cullin3-dependent stages in vaccinia virus infection. Cell Rep. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Gilson, T.; Cheng, C.Y.; Hur, W.S.; Blanchette, P.; Branton, P.E. Analysis of the Cullin binding sites of the E4orf6 proteins of human adenovirus E3 ubiquitin ligases. J. Virol. 2014, 88, 3885–3897. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.Y.; Wimmer, P.; Schreiner, S.; Ketner, G.; Dobner, T.; Branton, P.E.; Blanchette, P. Role of E1b55k in E4orf6/E1b55k E3 ligase complexes formed by different human adenovirus serotypes. J. Virol. 2013, 87, 6232–6245. [Google Scholar] [PubMed]
- Querido, E.; Blanchette, P.; Yan, Q.; Kamura, T.; Morrison, M.; Boivin, D.; Kaelin, W.G.; Conaway, R.C.; Conaway, J.W.; Branton, P.E. Degradation of p53 by adenovirus E4orf6 and E1b55k proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 2001, 15, 3104–3117. [Google Scholar] [CrossRef] [PubMed]
- Querido, E.; Morrison, M.R.; Chu-Pham-Dang, H.; Thirlwell, S.W.; Boivin, D.; Branton, P.E. Identification of three functions of the adenovirus E4orf6 protein that mediate p53 degradation by the E4orf6-E1b55k complex. J. Virol. 2001, 75, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Patel, R.N.; Forrester, N.A.; Theil, K.; Groitl, P.; Stewart, G.S.; Taylor, A.M.; Morgan, I.M.; Dobner, T.; Grand, R.J.; et al. Adenovirus 12 E4orf6 inhibits atr activation by promoting topbp1 degradation. Proc. Natl. Acad. Sci. USA 2010, 107, 12251–12256. [Google Scholar] [CrossRef] [PubMed]
- Orazio, N.I.; Karlseder, J.; Weitzman, M.D. The adenovirus E1b55k/E4orf6 complex induces degradation of the bloom helicase during infection. J. Virol. 2011, 85, 1887–1892. [Google Scholar] [CrossRef] [PubMed]
- Dallaire, F.; Blanchette, P.; Groitl, P.; Dobner, T.; Branton, P.E. Identification of integrin alpha3 as a new substrate of the adenovirus E4orf6/E1b 55-kilodalton E3 ubiquitin ligase complex. J. Virol. 2009, 83, 5329–5338. [Google Scholar] [CrossRef] [PubMed]
- Huh, K.; Zhou, X.; Hayakawa, H.; Cho, J.Y.; Libermann, T.A.; Jin, J.; Harper, J.W.; Munger, K. Human papillomavirus type 16 E7 oncoprotein associates with the Cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J. Virol. 2007, 81, 9737–9747. [Google Scholar] [CrossRef] [PubMed]
- Ashizawa, A.; Higashi, C.; Masuda, K.; Ohga, R.; Taira, T.; Fujimuro, M. The ubiquitin system and kaposi’s sarcoma-associated herpesvirus. Front. Microbiol. 2012. [Google Scholar] [CrossRef]
- Ellis, M.; Peng Chew, Y.; Fallis, L.; Freddersdorf, S.; Boshoff, C.; Weiss, RA.; Lu, X.; Mittnacht, S. Degradation of p27(Kip) Cdk inhibitor triggered by kaposi’s sarcoma virus Cyclin-Cdk6 complex. EMBO J. 1999, 18, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Martin, H.; Shamay, M.; Woodard, C.; Tang, Q.Q.; Hayward, S.D. Kaposi’s sarcoma-associated herpesvirus lana protein downregulates nuclear glycogen synthase kinase 3 activity and consequently blocks differentiation. J. Virol. 2007, 81, 4722–4731. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.B.; Wang, G.; Barrett, J.W.; Nazarian, S.H.; Colwill, K.; Moran, M.; McFadden, G. Myxoma virus M-t5 protects infected cells from the stress of cell cycle arrest through its interaction with host cell Cullin-1. J. Virol. 2005, 79, 10750–10763. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.K.; Byers, N.M.; Friesen, P.D. Baculovirus F-box protein Lef-7 modifies the host DNA damage response to enhance virus multiplication. J. Virol. 2013, 87, 12592–12599. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.; Filipe, J.; Seldon, M.P.; Fonseca, L.; Anrather, J.; Soares, M.P.; Simas, J.P. Termination of NF-kappaB activity through a gammaherpesvirus protein that assembles an EC5S ubiquitin-ligase. EMBO J. 2009, 28, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Boggio, R.; Passafaro, A.; Chiocca, S. Targeting SUMO E1 to ubiquitin ligases: A viral strategy to counteract sumoylation. J. Biol. Chem. 2007, 282, 15376–15382. [Google Scholar]
- Hartmann, T.; Xu, X.; Kronast, M.; Muehlich, S.; Meyer, K.; Zimmermann, W.; Hurwitz, J.; Pan, Z.Q.; Engelhardt, S.; Sarikas, A. Inhibition of Cullin-RING E3 ubiquitin ligase 7 by simian virus 40 large T antigen. Proc. Natl. Acad. Sci. USA 2014, 111, 3371–3376. [Google Scholar] [PubMed]
- Kasper, J.S.; Kuwabara, H.; Arai, T.; Ali, S.H.; DeCaprio, J.A. Simian virus 40 large T antigen’s association with the Cul7 SCF complex contributes to cellular transformation. J. Virol. 2005, 79, 11685–11692. [Google Scholar] [CrossRef] [PubMed]
- Van Sant, C.; Hagglund, R.; Lopez, P.; Roizman, B. The infected cell protein 0 of herpes simplex virus 1 dynamically interacts with proteasomes, binds and activates the Cdc34 E2 ubiquitin-conjugating enzyme, and possesses in vitro E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA 2001, 98, 8815–8820. [Google Scholar]
- Sato, Y.; Kamura, T.; Shirata, N.; Murata, T.; Kudoh, A.; Iwahori, S.; Nakayama, S.; Isomura, H.; Nishiyama, Y.; Tsurumi, T. Degradation of phosphorylated p53 by viral protein-Ecs E3 ligase complex. PLoS Pathog. 2009. [Google Scholar] [CrossRef]
- Gastaldello, S.; Hildebrand, S.; Faridani, O.; Callegari, S.; Palmkvist, M.; di Guglielmo, C.; Masucci, M.G. A deneddylase encoded by epstein-barr virus promotes viral DNA replication by regulating the activity of Cullin-RING ligases. Nat. Cell. Biol. 2010, 12, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Duran, R.; Bejarano, E.R. Geminivirus C2 protein might be the key player for geminiviral co-option of Scf-mediated ubiquitination. Plant. Signal. Behav. 2011, 6, 999–1001. [Google Scholar]
- Adeyemi, R.O.; Fuller, M.S.; Pintel, D.J. Efficient parvovirus replication requires CRL4cdt2-targeted depletion of p21 to prevent its inhibitory interaction with PCNA. PLoS Pathog. 2014, 10, e1004055. [Google Scholar] [PubMed]
- Aronson, M.N.; Meyer, A.; Györgyey, J.; Katul, L.; Vetten, H.J.; Gronenborn, B.; Timchenko, T. Clink, a nanovirus-encoded protein, binds both pRB and SKP1. J. Virol. 2000, 74, 2967–2972. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.W.; Ettayebi, K.; Hardy, M.E. Rotavirus NSP1 inhibits NFkappaB activation by inducing proteasome-dependent degradation of beta-TrCP: A novel mechanism of IFN antagonism. PLoS Pathog. 2009, 5, e1000280. [Google Scholar] [CrossRef] [PubMed]
- Surjit, M.; Varshney, B.; Lal, S.K. The ORF2 glycoprotein of hepatitis E virus inhibits cellular NF-kappaB activity by blocking ubiquitination mediated proteasomal degradation of Ikappabalpha in human hepatoma cells. BMC Biochem. 2012. [Google Scholar] [CrossRef]
- Thiel, H.; Varrelmann, M. Identification of Beet necrotic yellow vein virus P25 pathogenicity factor-interacting sugar beet proteins that represent putative virus targets or components of plant resistance. Mol. Plant. Microbe Interact. 2009, 22, 999–1010. [Google Scholar] [PubMed]
- Thiel, H.; Hleibieh, K.; Gilmer, D.; Varrelmann, M. The P25 pathogenicity factor of Beet necrotic yellow vein virus targets the sugar Beet 26S proteasome involved in the induction of a hypersensitive resistance response via interaction with an F-box protein. Mol. Plant. Microbe Interact. 2012, 25, 1058–1072. [Google Scholar] [PubMed]
- Chiu, M.H.; Chen, I.H.; Baulcombe, D.C.; Tsai, C.H. The silencing suppressor P25 of potato virus X interacts with argonaute1 and mediates its degradation through the proteasome pathway. Mol. Plant. Pathol. 2010, 11, 641–649. [Google Scholar]
- Pazhouhandeh, M.D.M.; Marrocco, K.; Lechner, E.; Berry, B.; Brault, V.; Hemmer, O.; Kretsch, T.; Richards, K.E.; Genschik, P.; Ziegler-Graff, V. F-box-like domain in the polerovirus protein p0 is required for silencing suppressor function. Proc. Natl. Acad. Sci. USA 2006, 103, 1994–1999. [Google Scholar] [CrossRef] [PubMed]
- Precious, B.; Childs, K.; Fitzpatrick-Swallow, V.; Goodbourn, S.; Randall, R.E. Simian virus 5 V protein acts as an adaptor, linking Ddb1 to Stat2, to facilitate the ubiquitination of Stat1. J. Virol. 2005, 79, 13434–13441. [Google Scholar] [CrossRef] [PubMed]
- Ulane, C.M.; Horvath, C.M. Paramyxoviruses SV5 and HPIV2 assemble stat protein ubiquitin ligase complexes from cellular components. Virology 2002, 304, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.; Lynch, O.T.; Suessmuth, Y.; Qian, P.; Boyd, C.R.; Burrows, J.F.; Buick, R.; Stevenson, N.J.; Touzelet, O.; Gadina, M.; et al. Respiratory syncytial virus NS1 protein degrades Stat2 by using the elongin-Cullin E3 ligase. J. Virol. 2007, 81, 3428–3436. [Google Scholar] [CrossRef]
- Le May, N.; Dubaele, S.; Proietti De Santis, L.; Billecocq, A.; Bouloy, M.; Egly, J.M. TFIIH transcription factor, a target for the Rift valley hemorrhagic fever virus. Cell 2004, 116, 541–550. [Google Scholar] [CrossRef]
- Kainulainen, M.; Habjan, M.; Hubel, P.; Busch, L.; Lau, S.; Colinge, J.; Superti-Furga, G.; Pichlmair, A.; Weber, F. Virulence factor NSS of Rift valley fever virus recruits the F-box protein Fbxo3 to degrade subunit p62 of general transcription factor TFIIH. J. Virol. 2014, 88, 3464–3473. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Kim, D.Y.; Hultquist, J.F.; Shindo, K.; LaRue, R.S.; Kwon, E.; Li, M.; Anderson, B.D.; Yen, L.; Stanley, D.; et al. VIF hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 2012, 481, 371–375. [Google Scholar]
- Zhang, S.; Zhang, B.; Xu, X.; Wang, L.; Feng, X.; Wang, Q.; Huang, H.; Wu, J.; Li, P.; Wang, J. HIV-1 viral protein R downregulates Ebp1 and stabilizes p53 in glioblastoma U87MG cells. Clin. Transl. Oncol. 2014, 16, 293–300. [Google Scholar] [CrossRef]
- Leupin, O.; Bontron, S.; Schaeffer, C.; Strubin, M. Hepatitis B virus X protein stimulates viral genome replication via a DDB1-dependent pathway distinct from that leading to cell death. J. Virol. 2005, 79, 4238–4245. [Google Scholar] [PubMed]
- Kim, B.; Nguyen, L.A.; Daddacha, W.; Hollenbaugh, J.A. Tight interplay among SAMHD1 protein level, cellular Dntp levels, and HIV-1 proviral DNA synthesis kinetics in human primary monocyte-derived macrophages. J. Biol. Chem. 2012, 287, 21570–21574. [Google Scholar] [PubMed]
- Sitterlin, D.; Bergametti, F.; Transy, C. UVDDB p127-binding modulates activities and intracellular distribution of hepatitis B virus X protein. Oncogene 2000, 19, 4417–4426. [Google Scholar] [PubMed]
- Sitterlin, D.; Bergametti, F.; Tiollais, P.; Tennant, B.C.; Transy, C. Correct binding of viral X protein to Uvddb-p127 cellular protein is critical for efficient infection by hepatitis B viruses. Oncogene 2000, 19, 4427–4431. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Elledge, S.J.; Butel, J.S. Hepatitis B virus X protein interacts with a probable cellular DNA repair protein. J. Virol. 1995, 69, 1107–1114. [Google Scholar] [PubMed]
- Zimmerman, E.S.; Schulman, B.A.; Zheng, N. Structural assembly of Cullin-RING ubiquitin ligase complexes. Curr. Opin. Struct. Biol. 2010, 20, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Sarikas, A.; Hartmann, T.; Pan, Z.Q. The cullin protein family. Genome Biol. 2011. [Google Scholar] [CrossRef]
- Skaar, J.R.; Pagan, J.K.; Pagano, M. Mechanisms and function of substrate recruitment by F-box proteins. Nat. Rev. Mol. Cell Biol. 2013, 14, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Skaar, J.R.; D’Angiolella, V.; Pagan, J.K.; Pagano, M. Snapshot: F box proteins II. Cell 2009. [Google Scholar] [CrossRef]
- Xu, L.; Wei, Y.; Reboul, J.; Vaglio, P.; Shin, T.H.; Vidal, M.; Elledge, S.J.; Harper, J.W. BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature 2003, 425, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Irigoyen, M.L.; Iniesto, E.; Rodriguez, L.; Puga, M.I.; Yanagawa, Y.; Pick, E.; Strickland, E.; Paz-Ares, J.; Wei, N.; de Jaeger, G.; et al. Targeted degradation of abscisic acid receptors is mediated by the ubiquitin ligase substrate adaptor DDA1 in arabidopsis. Plant. Cell 2014, 26, 712–728. [Google Scholar] [CrossRef] [PubMed]
- Pick, E.; Lau, O.S.; Tsuge, T.; Menon, S.; Tong, Y.; Dohmae, N.; Plafker, S.M.; Deng, X.W.; Wei, N. Mammalian DET1 regulates Cul4A activity and forms stable complexes with E2 ubiquitin-conjugating enzymes. Mol. Cell. Biol. 2007, 27, 4708–4719. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.C.; Matak-Vinkovic, D.; van Molle, I.; Ciulli, A. Multimeric complexes among ankyrin-repeat and SOCS-box protein 9 (ASB9), elonginBC, and Cullin 5: Insights into the structure and assembly of ECS-type Cullin-RING E3 ubiquitin ligases. Biochemistry 2013, 52, 5236–5246. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Novince, Z.; Concel, J.; Byeon, C.H.; Makhov, A.M.; Byeon, I.J.; Zhang, P.; Gronenborn, A.M. The Cullin-RING E3 ubiquitin ligase CRL4-DCAF1 complex dimerizes via a short helical region in DCAF1. Biochemistry 2011, 50, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Choo, Y.Y.; Hagen, T. Mechanism of Cullin3 E3 ubiquitin ligase dimerization. PLoS One 2012, 7, e41350. [Google Scholar] [CrossRef] [PubMed]
- Duda, D.M.; Scott, D.C.; Calabrese, M.F.; Zimmerman, E.S.; Zheng, N.; Schulman, B.A. Structural regulation of Cullin-RING ubiquitin ligase complexes. Curr. Opin. Struct. Biol. 2011, 21, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.C.; Monda, J.K.; Grace, C.R.; Duda, D.M.; Kriwacki, R.W.; Kurz, T.; Schulman, B.A. A dual E3 mechanism for Rub1 ligation to Cdc53. Mol. Cell 2010, 39, 784–796. [Google Scholar] [CrossRef] [PubMed]
- Kohroki, J.; Nishiyama, T.; Nakamura, T.; Masuho, Y. ASB proteins interact with Cullin5 and RBX2 to form E3 ubiquitin ligase complexes. FEBS Lett. 2005, 579, 6796–6802. [Google Scholar] [CrossRef] [PubMed]
- Hotton, S.K.; Callis, J. Regulation of Cullin RING ligases. Ann. Rev. Plant Biol. 2008, 59, 467–489. [Google Scholar] [CrossRef]
- Wee, S.; Geyer, R.K.; Toda, T.; Wolf, D.A. Csn facilitates Cullin-RING ubiquitin ligase function by counteracting autocatalytic adapter instability. Nat. Cell Biol. 2005, 7, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Schmaler, T.; Dubiel, W. Control of deneddylation by the COP9 signalosome. Sub Cell. Biochem. 2010, 54, 57–68. [Google Scholar]
- Ambroggio, X.I.; Rees, D.C.; Deshaies, R.J. Jamm: A metalloprotease-like zinc site in the proteasome and signalosome. PLoS Biol. 2004, 2, e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cope, G.A.; Suh, G.S.; Aravind, L.; Schwarz, S.E.; Zipursky, S.L.; Koonin, E.V.; Deshaies, R.J. Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of NEDD8 from Cul1. Science 2002, 298, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Wei, N.; Serino, G.; Deng, X.W. The COP9 signalosome: More than a protease. Trends Biochem. Sci. 2008, 33, 592–600. [Google Scholar] [PubMed]
- Peth, A.; Berndt, C.; Henke, W.; Dubiel, W. Downregulation of COP9 signalosome subunits differentially affects the Csn complex and target protein stability. BMC Biochem. 2007, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Cope, G.A.; Deshaies, R.J. Targeted silencing of Jab1/Csn5 in human cells downregulates SCF activity through reduction of F-box protein levels. BMC Biochem. 2006, 7, 1. [Google Scholar] [CrossRef]
- Denti, S.; Fernandez-Sanchez, M.E.; Rogge, L.; Bianchi, E. The COP9 signalosome regulates Skp2 levels and proliferation of human cells. J. Biol. Chem. 2006, 281, 32188–32196. [Google Scholar] [PubMed]
- Zhou, Z.; Wang, Y.; Cai, G.; He, Q. Neurospora COP9 signalosome integrity plays major roles for hyphal growth, conidial development, and circadian function. PLoS Genet. 2012, 8, e1002712. [Google Scholar] [CrossRef] [PubMed]
- Pierce, N.W.; Lee, J.E.; Liu, X.; Sweredoski, M.J.; Graham, R.L.; Larimore, E.A.; Rome, M.; Zheng, N.; Clurman, B.E.; Hess, S.; et al. Cand1 promotes assembly of new SCF complexes through dynamic exchange of F box proteins. Cell 2013, 153, 206–215. [Google Scholar]
- Tron, A.E.; Arai, T.; Duda, D.M.; Kuwabara, H.; Olszewski, J.L.; Fujiwara, Y.; Bahamon, B.N.; Signoretti, S.; Schulman, B.A.; DeCaprio, J.A. The glomuvenous malformation protein glomulin binds Rbx1 and regulates Cullin RING ligase-mediated turnover of Fbw7. Mol. Cell 2012, 46, 67–78. [Google Scholar] [CrossRef]
- Guo, Y.; Dong, L.; Qiu, X.; Wang, Y.; Zhang, B.; Liu, H.; Yu, Y.; Zang, Y.; Yang, M.; Huang, Z. Structural basis for hijacking Cbf-beta and Cul5 E3 ligase complex by HIV-1 Vif. Nature 2014, 505, 229–233. [Google Scholar] [CrossRef]
- Schwefel, D.; Groom, H.C.; Boucherit, V.C.; Christodoulou, E.; Walker, P.A.; Stoye, J.P.; Bishop, K.N.; Taylor, I.A. Structural basis of lentiviral subversion of a cellular protein degradation pathway. Nature 2014, 505, 234–238. [Google Scholar] [PubMed]
- Liu, J.; Nussinov, R. The mechanism of ubiquitination in the Cullin-RING E3 ligase machinery: Conformational control of substrate orientation. PLoS Comput. Biol. 2009, 5, e1000527. [Google Scholar]
- Yoshida, Y.; Chiba, T.; Tokunaga, F.; Kawasaki, H.; Iwai, K.; Suzuki, T.; Ito, Y.; Matsuoka, K.; Yoshida, M.; Tanaka, K.; et al. E3 ubiquitin ligase that recognizes sugar chains. Nature 2002, 418, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Calderon-Villalobos, L.I.; Sharon, M.; Zheng, C.; Robinson, C.V.; Estelle, M.; Zheng, N. Mechanism of auxin perception by the Tir1 ubiquitin ligase. Nature 2007, 446, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Cimermancic, P.; Gulbahce, N.; Johnson, J.R.; McGovern, K.E.; Clarke, S.C.; Shales, M.; Mercenne, G.; Pache, L.; Li, K.; et al. Global landscape of HIV-human protein complexes. Nature 2012, 481, 365–370. [Google Scholar]
- Baltimore, D. Expression of animal virus genomes. Bacteriol. Rev. 1971, 35, 235–241. [Google Scholar] [PubMed]
- Baker, A.; Rohleder, K.J.; Hanakahi, L.A.; Ketner, G. Adenovirus E4 34k and E1b 55k oncoproteins target host DNA ligase IV for proteasomal degradation. J. Virol. 2007, 81, 7034–7040. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.A.; Blackford, A.N.; Niedzwiedz, W. ATR activation and replication fork restart are defective in FANCM-deficient cells. EMBO J. 2010, 29, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, S.; Bürck, C.; Glass, M.; Groitl, P.; Wimmer, P.; Kinkley, S.; Mund, A.; Everett, R.D.; Dobner, T. Control of human adenovirus type 5 gene expression by cellular daxx/atrx chromatin-associated complexes. Nucleic Acid Res. 2013, 41, 3532–3550. [Google Scholar] [PubMed]
- Berezutskaya, E.; Bagchi, S. The human papillomavirus E7 oncoprotein functionally interacts with the S4 subunit of the 26S proteasome. J. Biol. Chem. 1997, 272, 30135–30140. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, S.L.; Stremlau, M.; He, X.; Basile, J.R.; Munger, K. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J. Virol. 2001, 75, 7583–7591. [Google Scholar] [CrossRef] [PubMed]
- Winter, K.; von Kietzell, K.; Heilbronn, R.; Pozzuto, T.; Fechner, H.; Weger, S. Roles of E4orf6 and va I RNA in adenovirus-mediated stimulation of human parvovirus b19 DNA replication and structural gene expression. J. Virol. 2012, 86, 5099–5109. [Google Scholar] [PubMed]
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.T.; Zheng, N. Molecular architecture and assembly of the Ddb1-Cul4A ubiquitin ligase machinery. Nature 2006, 443, 590–593. [Google Scholar]
- Precious, B.; Young, D.F.; Andrejeva, L.; Goodbourn, S.; Randall, R.E. In vitro and in vivo specificity of ubiquitination and degradation of Stat1 and Stat2 by the V proteins of the paramyxoviruses simian virus 5 and human parainfluenza virus type 2. J. Gen. Virol. 2005, 86, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Precious, B.L.; Carlos, T.S.; Goodbourn, S.; Randall, R.E. Catalytic turnover of Stat1 allows PIV5 to dismantle the interferon-induced anti-viral state of cells. Virology 2007, 368, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Albin, J.S.; Anderson, J.S.; Johnson, J.R.; Harjes, E.; Matsuo, H.; Krogan, N.J.; Harris, R.S. Dispersed sites of HIV VIF-dependent polyubiquitination in the DNA deaminase APOBEC3F. J. Mol. Biol. 2013, 425, 1172–1182. [Google Scholar] [CrossRef] [PubMed]
- Magadan, J.G.; Perez-Victoria, F.J.; Sougrat, R.; Ye, Y.; Strebel, K.; Bonifacino, J.S. Multilayered mechanism of CD4 downregulation by HIV-1 Vpu involving distinct ER retention and ERAD targeting steps. PLoS Pathog. 2010, 6, e1000869. [Google Scholar] [CrossRef] [PubMed]
- Dube, M.; Bego, M.G.; Paquay, C.; Cohen, E.A. Modulation of HIV-1-host interaction: Role of the Vpu accessory protein. Retrovirology 2010, 7, 114. [Google Scholar] [CrossRef] [PubMed]
- Arias, E.E.; Walter, J.C. PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nat. Cell Biol. 2006, 8, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Groisman, R.; Polanowska, J.; Kuraoka, I.; Sawada, J.; Saijo, M.; Drapkin, R.; Kisselev, A.F.; Tanaka, K.; Nakatani, Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 2003, 113, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Schrofelbauer, B.; Hakata, Y.; Landau, N.R. HIV-1 Vpr function is mediated by interaction with the damage-specific DNA-binding protein DDB1. Proc. Natl. Acad. Sci. USA 2007, 104, 4130–4135. [Google Scholar] [CrossRef] [PubMed]
- Benko, Z.; Liang, D.; Agbottah, E.; Hou, J.; Taricani, L.; Young, P.G.; Bukrinsky, M.; Zhao, R.Y. Antagonistic interaction of HIV-1 VPR with Hsf-mediated cellular heat shock response and hsp16 in fission yeast (Schizosaccharomyces pombe). Retrovirology 2007. [Google Scholar] [CrossRef]
- Cherrier, T.; Suzanne, S.; Redel, L.; Calao, M.; Marban, C.; Samah, B.; Mukerjee, R.; Schwartz, C.; Gras, G.; Sawaya, B.E.; et al. P21(Waf1) gene promoter is epigenetically silenced by Ctip2 and SUV39H1. Oncogene 2009, 28, 3380–3389. [Google Scholar] [PubMed]
- Maudet, C.; Sourisce, A.; Dragin, L.; Lahouassa, H.; Rain, J.C.; Bouaziz, S.; Ramirez, B.C.; Margottin-Goguet, F. HIV-1 Vpr induces the degradation of ZIP and sZIP, adaptors of the NuRD chromatin remodeling complex, by hijacking DCAF1/VprBP. PLoS One 2013, 8, e77320. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Casey Klockow, L.; Nekorchuk, M.; Sharifi, H.J.; de Noronha, C.M. The HIV1 protein Vpr acts to enhance constitutive Dcaf1-dependent UNG2 turnover. PLoS One 2012, 7, e30939. [Google Scholar] [CrossRef] [PubMed]
- Schrofelbauer, B.; Yu, Q.; Zeitlin, S.G.; Landau, N.R. Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and smug Uracil-DNA glycosylases. J. Virol. 2005, 79, 10978–10987. [Google Scholar] [CrossRef] [PubMed]
- Fregoso, O.I.; Ahn, J.; Wang, C.; Mehrens, J.; Skowronski, J.; Emerman, M. Evolutionary toggling of Vpx/Vpr specificity results in divergent recognition of the restriction factor SAMHD1. PLoS Pathog. 2013, 9, e1003496. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Bregnard, C.; Hue, P.; Basbous, J.; Yatim, A.; Larroque, M.; Kirchhoff, F.; Constantinou, A.; Sobhian, B.; Benkirane, M. Premature activation of the Slx4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell 2014, 156, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Rahm, N.; Sobhian, B.; Chable-Bessia, C.; Munch, J.; Snoeck, J.; Sauter, D.; Switzer, W.M.; Heneine, W.; Kirchhoff, F.; et al. Evolutionary and functional analyses of the interaction between the myeloid restriction factor SAMHD1 and the lentiviral Vpx protein. Cell Host Microbe 2012, 11, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, J.; Choi, J.; Oh, C.; Kim, S.; Seo, M.; Kim, S.Y.; Seo, D.; Kim, J.; White, T.E.; Brandariz-Nunez, A.; et al. The ribonuclease activity of samhd1 is required for HIV-1 restriction. Nat. Med. 2014, 20, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Turrini, F.; di Pietro, A.; Vicenzi, E. Lentiviral effector pathways of TRIM proteins. DNA Cell Biol. 2014, 33, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Bontron, S.; Lin-Marq, N.; Strubin, M. Hepatitis B virus X protein associated with UV-Ddb1 induces cell death in the nucleus and is functionally antagonized by UV-Ddb2. J. Biol. Chem. 2002, 277, 38847–38854. [Google Scholar] [CrossRef] [PubMed]
- Martin-Lluesma, S.; Schaeffer, C.; Robert, E.I.; van Breugel, P.C.; Leupin, O.; Hantz, O.; Strubin, M. Hepatitis B virus X protein affects S phase progression leading to chromosome segregation defects by binding to damaged DNA binding protein 1. Hepatology 2008, 48, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Kew, M.C. Hepatitis B virus X protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2011, 26, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.W.; Bae, I.H.; Park, Y.H.; Chung, J.H.; Lim, K.M.; Kang, K.S. Expression levels of pituitary tumor transforming 1 and glutathione-S-transferase theta 3 are associated with the individual susceptibility to D-galactosamine-induced hepatotoxicity. Toxicol. Appl. Pharmacol. 2010, 242, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Torii, N.; Furusaka, A.; Malayaman, N.; Hu, Z.; Liang, T.J. Structural and functional characterization of interaction between hepatitis B virus X protein and the proteasome complex. J. Biol. Chem. 2000, 275, 15157–15165. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Ma, D.; Dong, J.; Jin, J.; Li, D.; Deng, C.; Wang, T. HC-pro protein of potato virus Y can interact with three arabidopsis 20S proteasome subunits in planta. J. Virol. 2007, 81, 12881–12888. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, C.; Zhang, Q.; Wang, T.; Li, J.; Guan, W.; Yu, J.; Liang, M.; Li, D. Interactions of Sars coronavirus nucleocapsid protein with the host cell proteasome subunit p42. Virol. J. 2010. [Google Scholar] [CrossRef]
- Li, G.; Elder, R.T.; Dubrovsky, L.; Liang, D.; Pushkarsky, T.; Chiu, K.; Fan, T.; Sire, J.; Bukrinsky, M.; Zhao, R.Y. HIV-1 replication through Hhr23A-mediated interaction of Vpr with 26S proteasome. PLoS One 2010, 5, e11371. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Byeon, I.J.; DeLucia, M.; Koharudin, L.M.; Ahn, J.; Gronenborn, A.M. Binding of HIV-1 Vpr protein to the human homolog of the yeast DNA repair protein Rad23 (Hhr23A) requires its xeroderma pigmentosum complementation group C binding (Xpcb) domain as well as the ubiquitin-associated 2 (Uba2) domain. J. Biol. Chem. 2014, 289, 2577–2588. [Google Scholar] [PubMed]
- Dou, Q.P.; Goldfarb, R.H. Bortezomib (millennium pharmaceuticals). IDrugs 2002, 5, 828–834. [Google Scholar] [PubMed]
- Gustin, J.K.; Moses, A.V.; Fruh, K.; Douglas, J.L. Viral takeover of the host ubiquitin system. Front. Microbiol. 2011. [Google Scholar] [CrossRef]
- Raaben, M.; Grinwis, G.C.; Rottier, P.J.; de Haan, C.A. The proteasome inhibitor velcade enhances rather than reduces disease in mouse hepatitis coronavirus-infected mice. J. Virol. 2010, 84, 7880–7885. [Google Scholar] [CrossRef] [PubMed]
- Lupfer, C.; Patton, K.M.; Pastey, M.K. Treatment of human respiratory syncytial virus infected Balb/C mice with the proteasome inhibitor bortezomib (velcade, PS-341) results in increased inflammation and mortality. Toxicology 2010, 268, 25–30. [Google Scholar] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Anderica-Romero, A.C.; Gonzalez-Herrera, I.G.; Santamaria, A.; Pedraza-Chaverri, J. Cullin 3 as a novel target in diverse pathologies. Redox Biol. 2013, 1, 366–372. [Google Scholar] [PubMed]
- Zhao, Y.; Sun, Y. Cullin-RING ligases as attractive anti-cancer targets. Curr. Pharm. Design 2013, 19, 3215–3225. [Google Scholar] [CrossRef]
- Sharma, P.; Nag, A. Cul4A ubiquitin ligase: A promising drug target for cancer and other human diseases. Open Biol. 2014, 4, 130217. [Google Scholar] [PubMed]
- Nekorchuk, M.D.; Sharifi, H.J.; Furuya, A.K.; Jellinger, R.; de Noronha, C.M. HIV relies on neddylation for ubiquitin ligase-mediated functions. Retrovirology 2013, 10, 138. [Google Scholar] [CrossRef] [PubMed]
- Kramer, V.G.; Schader, S.M.; Oliveira, M.; Colby-Germinario, S.P.; Donahue, D.A.; Singhroy, D.N.; Tressler, R.; Sloan, R.D.; Wainberg, M.A. Maraviroc and other HIV-1 entry inhibitors exhibit a class-specific redistribution effect that results in increased extracellular viral load. Antimicrob. Agents Chemother. 2012, 56, 4154–4160. [Google Scholar] [CrossRef] [PubMed]
- Nathans, R.; Cao, H.; Sharova, N.; Ali, A.; Sharkey, M.; Stranska, R.; Stevenson, M.; Rana, T.M. Small-molecule inhibition of HIV-1 Vif. Nat. Biotechnol. 2008, 26, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Cen, S.; Peng, Z.G.; Li, X.Y.; Li, Z.R.; Ma, J.; Wang, Y.M.; Fan, B.; You, X.F.; Wang, Y.P.; Liu, F.; et al. Small molecular compounds inhibit HIV-1 replication through specifically stabilizing APOBEC3G. J. Biol. Chem. 2010, 285, 16546–16552. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Tan, M.K. Understanding Cullin-RING E3 biology through proteomics-based substrate identification. Mol. Cell. Proteomics 2012, 11, 1541–1550. [Google Scholar] [CrossRef]
- Emanuele, M.J.; Elia, A.E.; Xu, Q.; Thoma, C.R.; Izhar, L.; Leng, Y.; Guo, A.; Chen, Y.N.; Rush, J.; Hsu, P.W.; et al. Global identification of modular Cullin-RING ligase substrates. Cell 2011, 147, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Mark, K.G.; Simonetta, M.; Maiolica, A.; Seller, C.A.; Toczyski, D.P. Ubiquitin ligase trapping identifies an Scf(Saf1) pathway targeting unprocessed vacuolar/lysosomal proteins. Mol. Cell 2014, 53, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Salahudeen, A.A.; Thompson, J.W.; Ruiz, J.C.; Ma, H.W.; Kinch, L.N.; Li, Q.; Grishin, N.V.; Bruick, R.K. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 2009, 326, 722–726. [Google Scholar] [PubMed]
- Vashisht, A.A.; Zumbrennen, K.B.; Huang, X.; Powers, D.N.; Durazo, A.; Sun, D.; Bhaskaran, N.; Persson, A.; Uhlen, M.; Sangfelt, O.; et al. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 2009, 326, 718–721. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Khadka, S.; Vangeloff, A.D.; Zhang, C.; Siddavatam, P.; Heaton, N.S.; Wang, L.; Sengupta, R.; Sahasrabudhe, S.; Randall, G.; Gribskov, M.; et al. A physical interaction network of dengue virus and human proteins. Mol. Cell. Proteomics 2011. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahon, C.; Krogan, N.J.; Craik, C.S.; Pick, E. Cullin E3 Ligases and Their Rewiring by Viral Factors. Biomolecules 2014, 4, 897-930. https://doi.org/10.3390/biom4040897
Mahon C, Krogan NJ, Craik CS, Pick E. Cullin E3 Ligases and Their Rewiring by Viral Factors. Biomolecules. 2014; 4(4):897-930. https://doi.org/10.3390/biom4040897
Chicago/Turabian StyleMahon, Cathal, Nevan J. Krogan, Charles S. Craik, and Elah Pick. 2014. "Cullin E3 Ligases and Their Rewiring by Viral Factors" Biomolecules 4, no. 4: 897-930. https://doi.org/10.3390/biom4040897
APA StyleMahon, C., Krogan, N. J., Craik, C. S., & Pick, E. (2014). Cullin E3 Ligases and Their Rewiring by Viral Factors. Biomolecules, 4(4), 897-930. https://doi.org/10.3390/biom4040897