
Redox-Regulated Pathway of Tyrosine Phosphorylation Underlies NF-κB Induction by an Atypical Pathway Independent of the 26S Proteasome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
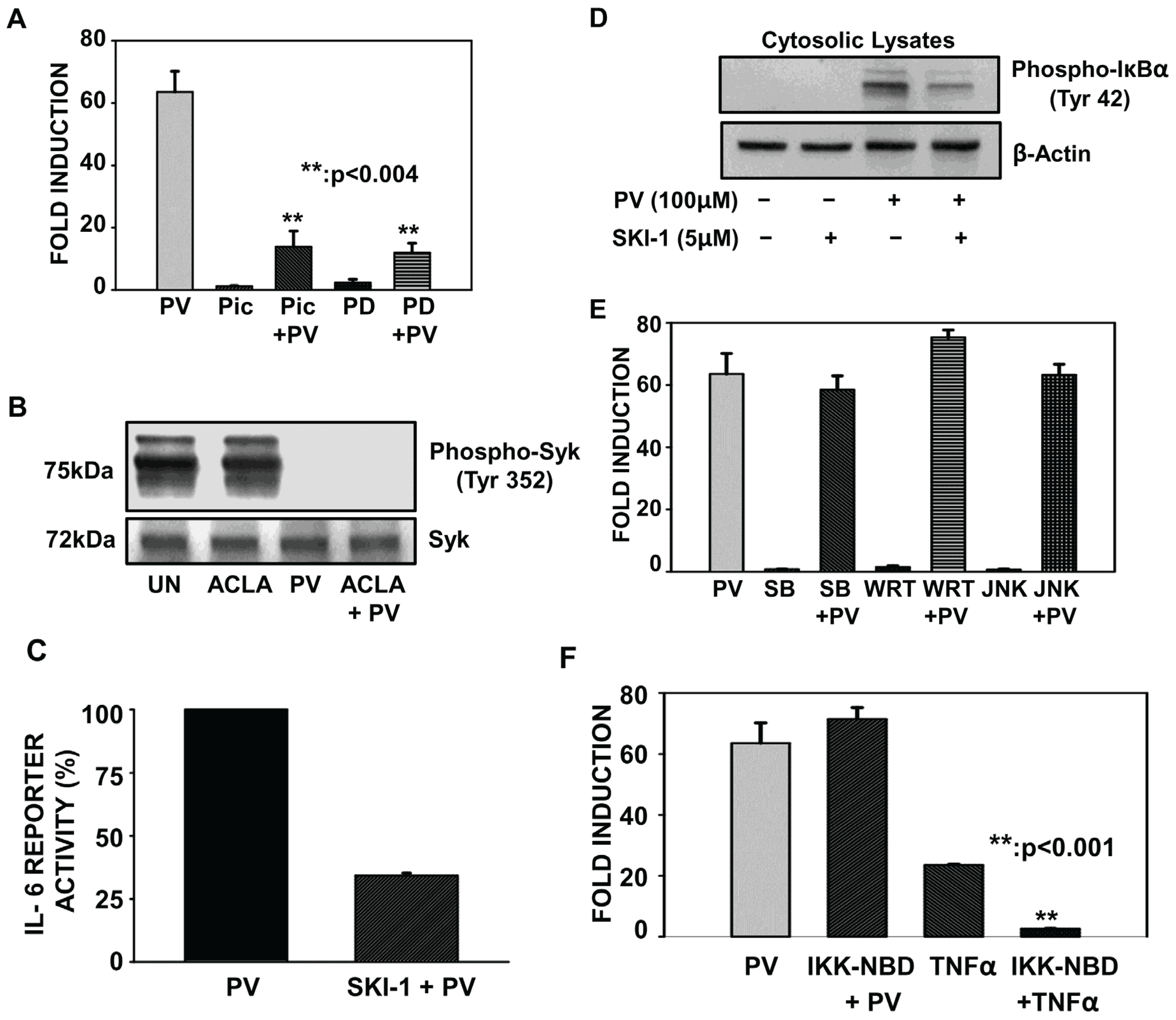
2.1. Pervanadate Stimulation Induces Tyrosine Phosphorylation of IκBα But Not Its Proteolytic Degradation

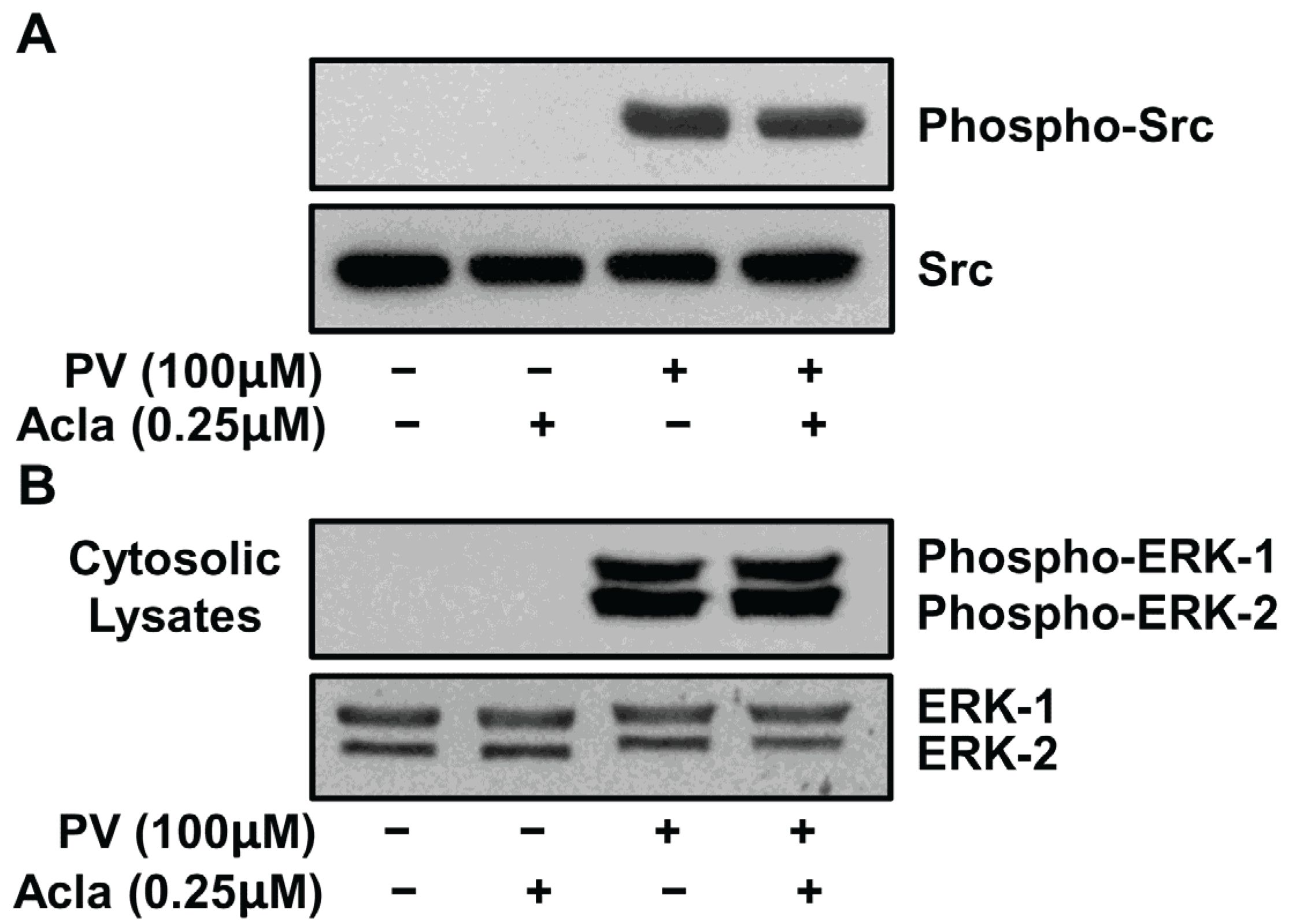
2.2. Pretreatment with Inhibitors of Src and MEK but not p38, PI3K, JNK and IKK Complex Interfere with PV-Induced NF-κB Activity

2.3. Proteasome Inhibition Does Not Alter PV-Mediated Activation of Src or MEK Kinases
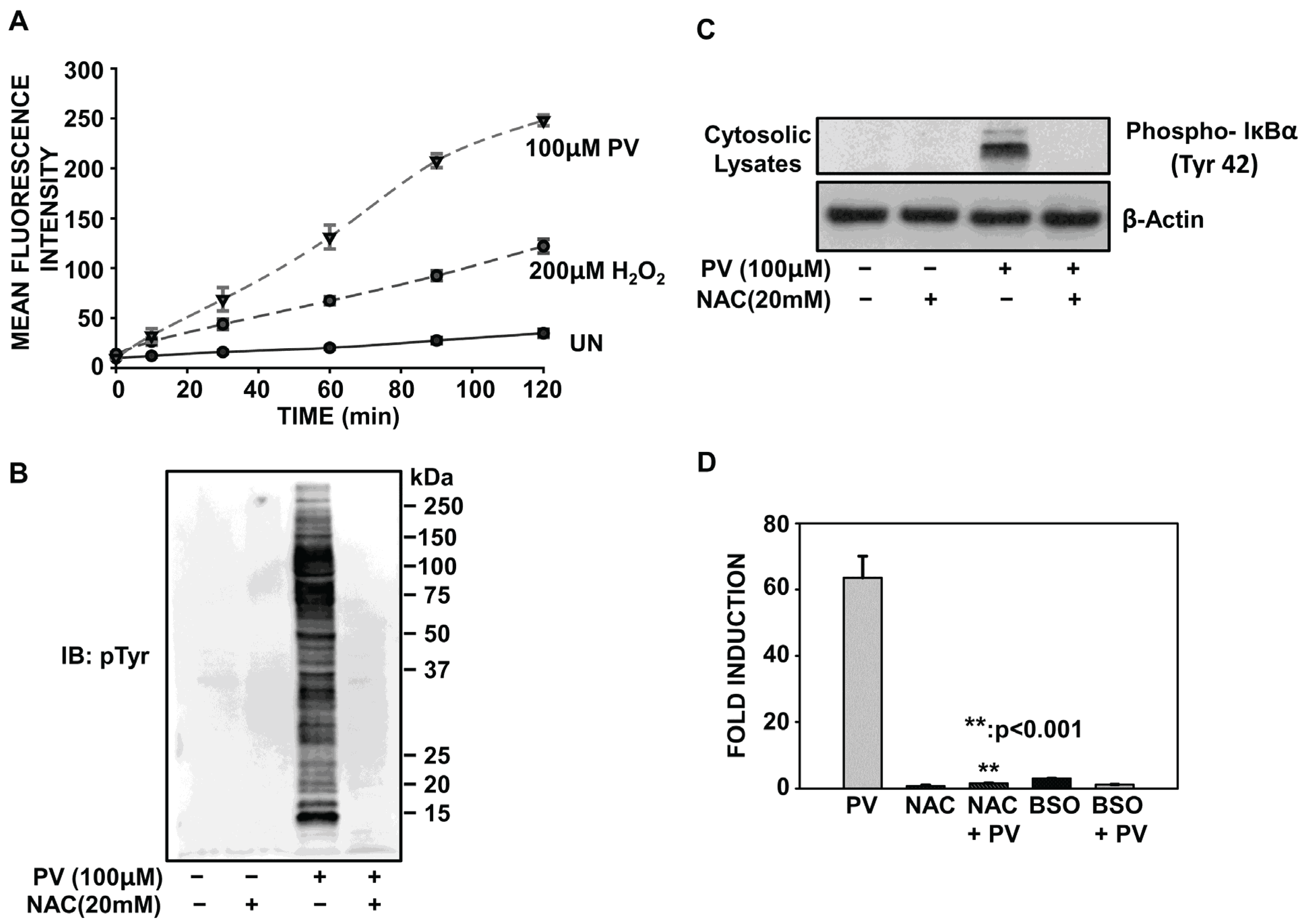
2.4. PV-Mediated Activation of NF-κB Involves Oxidative Stress


2.5. Proteasome Inhibition Potentiates Oxidative Stress Associated with PV-Stimulation

3. Discussion
4. Experimental Section
4.1. Antibodies & Reagents
4.2. Cell Culture
4.3. Preparation of Cytosolic and Nuclear Extracts and Western Blotting
4.4. Measurement of Intracellular Oxidation Using H2DCF-DA
4.5. Measurement of Intracellular GSH
4.6. Luciferase Reporter Assay
4.7. Statistical Analyses
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pahl, H.L. Activators and Target Genes of Rel/NF-κB Transcription Factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Karin, M. Is NF-κB the Sensor of Oxidative Stress? FASEB J. 1999, 13, 1137–1143. [Google Scholar] [PubMed]
- Janssens, S.; Tschopp, J. Signals From Within: The DNA-Damage-Induced NF-κB Response. Cell Death Differ. 2006, 13, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Pahl, H.L.; Baeuerle, P.A. A novel signal transduction pathway from the endoplasmic reticulum to the nucleus is mediated by transcription factor NF-κB. EMBO J. 1995, 14, 2580–2588. [Google Scholar] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared Principles in NF-κB Signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Canty, T.G., Jr.; Boyle, E.M., Jr.; Farr, A.; Morgan, E.N.; Verrier, E.D.; Pohlman, T.H. Oxidative stress induces NF-κB nuclear translocation without degradation of IκBα. Circulation 1999, 100, II361–II364. [Google Scholar] [CrossRef] [PubMed]
- Imbert, V.; Rupec, R.A.; Livolsi, A.; Pahl, H.L.; Traenckner, E.B.; Mueller-Dieckmann, C.; Farahifar, D.; Rossi, B.; Auberger, P.; Baeuerle, P.A.; et al. Tyrosine phosphorylation of IκBα activates NF-κB without proteolytic degradation of IκBα. Cell 1996, 86, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Beraud, C.; Henzel, W.J.; Baeuerle, P.A. Involvement of regulatory and catalytic subunits of phosphoinositide 3-kinase in NF-κB activation. Proc. Natl. Acad. Sci. USA 1999, 96, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Heffetz, D.; Bushkin, I.; Dror, R.; Zick, Y. The insulinomimetic agents H2O2 and vanadate stimulate protein tyrosine phosphorylation in intact cells. J. Biol. Chem. 1990, 265, 2896–2902. [Google Scholar] [PubMed]
- Yaname, H.; Fukunaga, T.; Nigorikawa, K.; Okamura, N.; Ishibashi, S. Pervanadate activates NADPH oxidase via protein kinase C-independent phosphorylation of p47-phox. Arch. Biochem. Biophys. 1999, 361, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P.; Buricchi, F. Protein tyrosine phosphorylation and reversible oxidation: Two cross-talking posttranslation modifications. Antioxid. Redox Signal. 2007, 9, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Li, Q.; Ross, D.; Engelhardt, J.F. Tyrosine Phosphorylation of IκBα Activates NF κB through a redox-regulated and c-Src-dependent mechanism following hypoxia/reoxygenation. J. Biol. Chem. 2003, 278, 2072–2080. [Google Scholar] [CrossRef] [PubMed]
- Bui, N.T.; Livolsi, A.; Peyron, J.F.; Prehn, J.H. Activation of nuclear factor κB and Bcl-x Survival gene expression by nerve growth factor requires tyrosine phosphorylation of IκBα. J. Cell Biol. 2001, 152, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, D.; Gutierrez, H.; Gavalda, N.; O’Keeffe, G.; Hay, R.; Davies, A.M. Nuclear Factor-κB activation via tyrosine phosphorylation of inhibitor κB-α is crucial for ciliary neurotrophic factor-promoted neurite growth from developing neurons. J. Neurosci. 2007, 27, 9664–9669. [Google Scholar] [CrossRef] [PubMed]
- Mahabeleshwar, G.H.; Kundu, G.C. Tyrosine kinase P56lck regulates cell motility and nuclear factor κB-mediated secretion of urokinase type plasminogen activator through tyrosine phosphorylation of IκBα following hypoxia/reoxygenation. J. Biol. Chem. 2003, 278, 52598–52612. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, R.; Fisher, B.J.; Jones, D.G.; Fowler, A.A., III. Atypical Mechanism of NF-κB activation during reoxygenation stress in microvascular endothelium: A role for tyrosine kinases. Free Radical Biol. Med. 2002, 33, 962–973. [Google Scholar] [CrossRef]
- Sethi, G.; Ahn, K.S.; Chaturvedi, M.M.; Aggarwal, B.B. Epidermal growth factor (EGF) activates nuclear factor-κB through IκBα kinase-independent but EGF receptor-kinase dependent tyrosine 42 phosphorylation of IκBα. Oncogene 2007, 26, 7324–7332. [Google Scholar] [CrossRef] [PubMed]
- Markovina, S.; Callander, N.S.; O’Connor, S.L.; Kim, J.; Werndli, J.E.; Raschko, M.; Leith, C.P.; Kahl, B.S.; Kim, K.; Miyamoto, S. Bortezomib-resistant nuclear factor-κB activity in multiple myeloma cells. Mol. Cancer Res. 2008, 6, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.T.; Young, K.H.; Kahl, B.S.; Markovina, S.; Miyamoto, S. Prevalence of bortezomib-resistant constitutive NF-κB activity in mantle cell lymphoma. Mol. Cancer 2008, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Cullen, S.J.; Ponnappan, S.; Ponnappan, U. Proteasome inhibition up-regulates inflammatory gene transcription induced by an atypical pathway of NF-κB activation. Biochem. Pharmacol. 2010, 79, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Livolsi, A.; Busuttil, V.; Imbert, V.; Abraham, R.T.; Peyron, J.F. Tyrosine phosphorylation-dependent activation of NF-κB. Requirement for p56 LCK and ZAP-70 protein tyrosine kinases. Eur. J. Biochem. 2001, 268, 1508–1515. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Tan, Z.; Diltz, C.D.; You, M.; Fischer, E.H. Activation of mitogen-activated protein (MAP) kinase pathway by pervanadate, a potent inhibitor of tyrosine phosphatases. J. Biol. Chem. 1996, 271, 22251–22255. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, N.J.; Ikeyama, S. Age-related decline in cellular response to oxidative stress: Links to growth factor signaling pathways with common defects. Biochem. Pharmacol. 2002, 64, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Storz, P.; Toker, A. Protein kinase D mediates a stress-induced NF-κB activation and survival pathway. EMBO J. 2003, 22, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Cullen, S.; Ponnappan, S.; Ponnappan, U.; Department of Microbiology and Immunology, UAMS, Little Rock, AR, USA. Unpublished data. 2009.
- Krejsa, C.M.; Nadler, S.G.; Esselstyn, J.M.; Kavanagh, T.J.; Ledbetter, J.A.; Schieven, G.L. Role of oxidative stress in the action of vanadium phosphotyrosine phosphatase inhibitors. Redox independent activation of NF-κB. J. Biol. Chem. 1997, 272, 11541–11549. [Google Scholar] [CrossRef] [PubMed]
- Hakak, Y.; Martin, G.S. Ubiquitin-dependent degradation of active Src. Curr. Biol. 1999, 9, 1039–1042. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.F.; Shoji, I.; Cooper, E.M.; Kumar, S.; Oda, H.; Howley, P.M. Ubiquitin-mediated degradation of active Src tyrosine kinase. Proc. Natl. Acad. Sci. USA 1999, 96, 13738–13743. [Google Scholar] [CrossRef] [PubMed]
- Crevecoeur, J.; Merville, M.P.; Piette, J.; Gloire, G. Geldanamycin inhibits tyrosine phosphorylation-dependent NF-κB activation. Biochem. Pharmacol. 2008, 75, 2183–2191. [Google Scholar] [CrossRef] [PubMed]
- Kawai, H.; Nie, L.; Yuan, Z.M. Inactivation of NF-κB-dependent cell survival, a novel mechanism for the proapoptotic function of C-Abl. Mol. Cell. Biol. 2002, 22, 6079–6088. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Livolsi, A.; Imbert, V.; Peyron, J.F.; Siddiqui, A. Hepatitis C virus NS5A and subgenomic replicon activate NF-κB via tyrosine phosphorylation of IκBα and its degradation by calpain protease. J. Biol. Chem. 2003, 278, 40778–40787. [Google Scholar] [CrossRef] [PubMed]
- Desterro, J.M.; Rodriguez, M.S.; Hay, R.T. SUMO-1 Modification of IκBα inhibits NF-κB activation. Mol. Cell 1998, 2, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Darnay, B.G.; Aggarwal, B.B. Site-specific tyrosine phosphorylation of IκBα negatively regulates its inducible phosphorylation and degradation. J. Biol. Chem. 1996, 271, 31049–31054. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Reinacker, K.; Dimayuga, E.; Nukala, V.; Drake, J.; Butterfield, D.A.; Dunn, J.C.; Martin, S.; Bruce-Keller, A.J.; Keller, J.N. Role of the proteasome in protein oxidation and neural viability following low-level oxidative stress. FEBS Lett. 2003, 546, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.X.; Zhang, H.Y.; Meng, X.; Guan, Y.; Wang, H.Q. Role of oxidative stress and intracellular glutathione in the sensitivity to apoptosis induced by proteasome inhibitor in thyroid cancer cells. BMC Cancer 2009, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.F.; Zhou, J.; Zhang, X.; Bian, Q.; Sparrow, J.; Taylor, A.; Pereira, P.; Shang, F. Oxidative inactivation of the proteasome in retinal pigment epithelial cells. A potential link between oxidative stress and up-regulation of Interleukin-8. J. Biol. Chem. 2008, 283, 20745–20753. [Google Scholar] [CrossRef] [PubMed]
- Papa, L.; Rockwell, P. Persistent mitochondrial dysfunction and oxidative stress hinder neuronal cell recovery from reversible proteasome inhibition. Apoptosis 2008, 13, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.G.; Dragicevic, N.B.; Deng, J.H.; Bai, Y.; Dimayuga, E.; Ding, Q.; Chen, Q.; Bruce-Keller, A.J.; Keller, J.N. Proteasome inhibition alters neural mitochondrial homeostasis and mitochondria turnover. J. Biol. Chem. 2004, 279, 20699–20707. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, A.; Petrov, L.; Georgieva, A.; Kessiova, M.; Tzvetanova, E.; Kirkova, M.; Kukan, M. Effect of MG132 on proteasome activity and prooxidant/antioxidant status of rat liver subjected to ischemia/reperfusion injury. Hepatol. Res. 2008, 38, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Ponnappan, S.; Ponnappan, U. Redox regulation of the proteasome in T lymphocytes during aging. Free Radical Biol. Med. 2007, 42, 541–551. [Google Scholar] [CrossRef]
- Wood, K.V. Firefly luciferase: A new tool for molecular biologists. Promega Notes 1990, 28, 1–3. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cullen, S.; Ponnappan, S.; Ponnappan, U. Redox-Regulated Pathway of Tyrosine Phosphorylation Underlies NF-κB Induction by an Atypical Pathway Independent of the 26S Proteasome. Biomolecules 2015, 5, 95-112. https://doi.org/10.3390/biom5010095
Cullen S, Ponnappan S, Ponnappan U. Redox-Regulated Pathway of Tyrosine Phosphorylation Underlies NF-κB Induction by an Atypical Pathway Independent of the 26S Proteasome. Biomolecules. 2015; 5(1):95-112. https://doi.org/10.3390/biom5010095
Chicago/Turabian StyleCullen, Sarah, Subramaniam Ponnappan, and Usha Ponnappan. 2015. "Redox-Regulated Pathway of Tyrosine Phosphorylation Underlies NF-κB Induction by an Atypical Pathway Independent of the 26S Proteasome" Biomolecules 5, no. 1: 95-112. https://doi.org/10.3390/biom5010095
APA StyleCullen, S., Ponnappan, S., & Ponnappan, U. (2015). Redox-Regulated Pathway of Tyrosine Phosphorylation Underlies NF-κB Induction by an Atypical Pathway Independent of the 26S Proteasome. Biomolecules, 5(1), 95-112. https://doi.org/10.3390/biom5010095