
Impact of Oxidative Stress on Exercising Skeletal Muscle



Department of Cell Biology, University of Salzburg, A-5020 Salzburg, Austria
*
Author to whom correspondence should be addressed.
Biomolecules 2015, 5(2), 356-377; https://doi.org/10.3390/biom5020356
Submission received: 13 January 2015
/
Revised: 24 March 2015
/
Accepted: 30 March 2015
/
Published: 10 April 2015
(This article belongs to the Special Issue Oxidative Stress and Oxygen Radicals)
{kind=link}
{kind=link}
Abstract
:It is well established that muscle contractions during exercise lead to elevated levels of reactive oxygen species (ROS) in skeletal muscle. These highly reactive molecules have many deleterious effects, such as a reduction of force generation and increased muscle atrophy. Since the discovery of exercise-induced oxidative stress several decades ago, evidence has accumulated that ROS produced during exercise also have positive effects by influencing cellular processes that lead to increased expression of antioxidants. These molecules are particularly elevated in regularly exercising muscle to prevent the negative effects of ROS by neutralizing the free radicals. In addition, ROS also seem to be involved in the exercise-induced adaptation of the muscle phenotype. This review provides an overview of the evidences to date on the effects of ROS in exercising muscle. These aspects include the sources of ROS, their positive and negative cellular effects, the role of antioxidants, and the present evidence on ROS-dependent adaptations of muscle cells in response to physical exercise.
1. Introduction
Skeletal muscle is a highly specialized tissue with excellent plasticity in response to external stimuli such as exercise and training. The repetitive muscle contractions conducted during endurance training lead to a variety of phenotypic and physiological responses. These responses include activation of mitochondrial biogenesis, fiber type transformation and angiogenesis. Together, they increase the muscle’s capacity of aerobic metabolism and its resistance to fatigue. High muscle activity also involves a strong increase in reactive oxygen species (ROS) production. These unstable molecules and ions contain oxygen and are extremely reactive due to an unpaired electron. Among these oxygen intermediates are the free radicals superoxide, peroxide and the hydroxyl radicals and other highly reactive oxidants, such as singlet oxygen and hypochlorous acid. They promote oxidation reactions with other molecules, such as proteins, lipids and DNA and can thus be highly detrimental. However, recent research has demonstrated that ROS also have a beneficial role in promoting the adaptive responses of muscle to training.
More than three decades ago it was established that muscle activity leads to an increase in ROS production and concentration of free radicals [1,2]. Since then numerous investigations in rodents and humans have confirmed these early observations. Thus, it is generally accepted that single bouts of aerobic or anaerobic exercise, as well as chronic exercise promote the generation of ROS (summarized in the reviews [3,4]; and more recently e.g., [5,6,7]).
The great interest in this topic also stems from data that show that ROS levels are increased in subjects with aging-related sarcopenia, cardiac reperfusion injuries or muscular diseases, i.e., muscle dystrophies. Thus, it was assumed that exercise-induced ROS are potentially detrimental to muscle function and lead to muscle fatigue and muscle atrophy. Hence, many investigations focused on ways to prevent ROS production and accumulation and subsequent oxidative damage during and following physical exercise.
2. Sources of ROS in Muscle
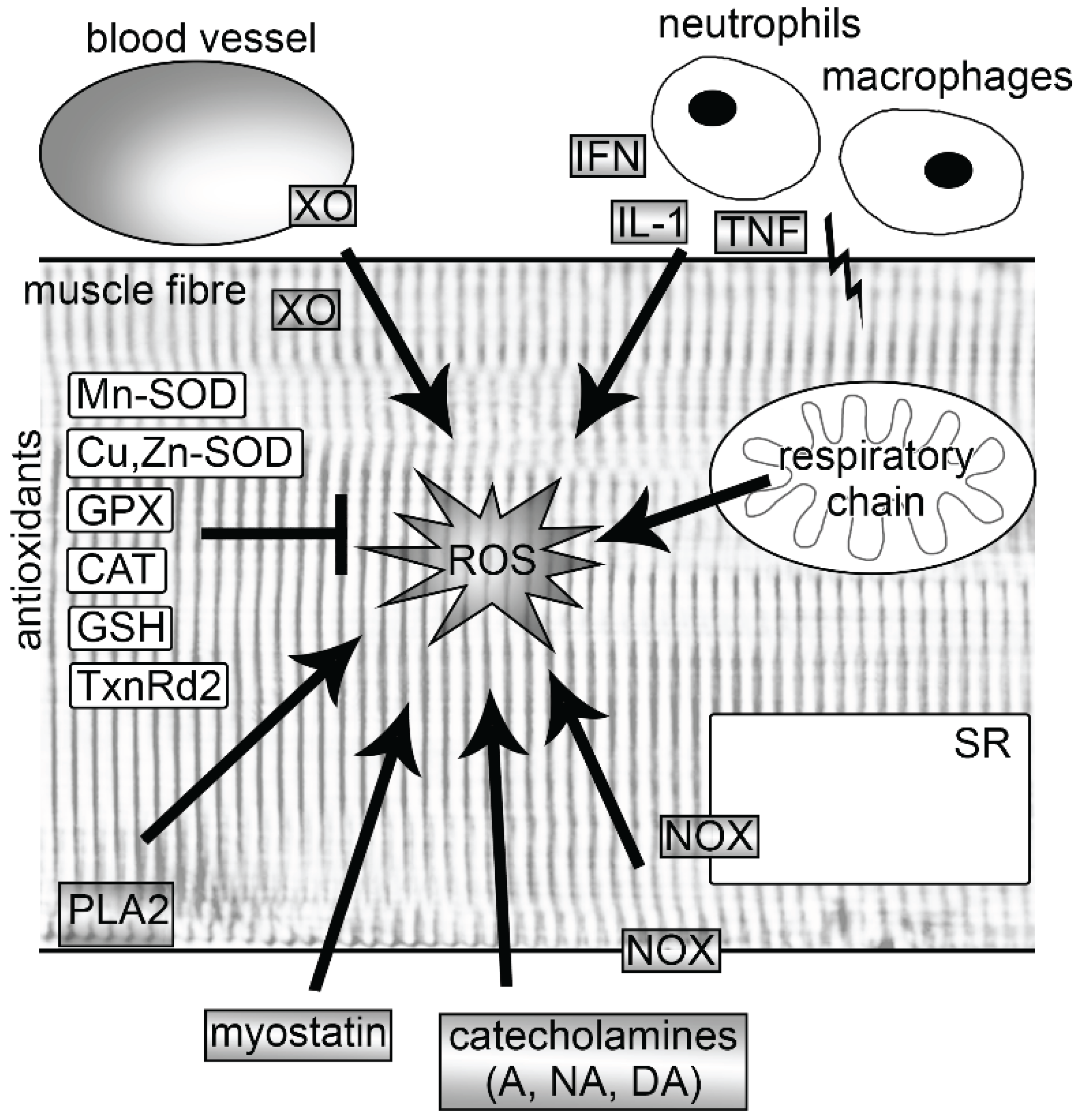
It has consistently been shown that muscle activity leads to a strong increase in ROS production [8]. However, there is a large debate about the sources and the extent of ROS that these sources produce. Several potential producers of ROS have been identified in muscle cells which are likely to be activated by different stimuli. Among these are mitochondria, nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs), phospholipase A2 (PLA2), xanthine oxidase (XO) and lipoxygenases (Figure 1). Some of these are discussed in more detail below. In addition to these intracellular sources, ROS has been shown to be produced from non-muscle sources. Strenuous exercise can elicit muscle injuries, which then lead to the activation of the neutrophils and macrophages via interferon-γ (IFN-γ), interleukin-1 (IL-1) and tumor necrosis factor (TNF) (for more detailed information see reviews [9,10]). These immune cells excessively produce ROS (oxidative burst), which is a central component of neutrophil defense mechanism. In addition, the exercise-induced increase of catecholamines (adrenaline, noradrenaline, dopamine) also play a role in the generation of ROS [11], as well as ROS derived from endothelium [12] (Figure 1).
2.1. Mitochondria
For a long period of time, mitochondria were regarded as the main producer of cellular ROS with an estimated superoxide production rate of approximately 1%–4% of total mitochondrial O2 consumption (see reviews [8,13,14]). More recent data demonstrate that the production of ROS in mitochondria is by an order of magnitude smaller than originally expected and is approximately 0.15% [15]. Mitochondria are thought to produce ROS by a leak of single electrons in the respiratory chain in the mitochondrial inner membrane of the contracting muscle cells. Ten different sites of superoxide/H2O2 generation have been found as yet in mammalian mitochondria [16,17]. Superoxide production mainly occurs from complexes I (NADH dehydrogenase) and III (coenzyme Q and cytochrome C oxidoreductase) of the electron transport chain [18,19]. New findings also identify complex II (succinate dehydrogenase) as a major source of superoxide production [16]. Using isolated mitochondria, the contribution of each site to total H2O2 production has recently been quantified and shown to strongly depend on the substrate being oxidized [20]. At rest, H2O2 was predominantly produced from the quinol site in complex I (site IQ) and flavin site in complex II (site IIF), followed by sites IF and IIIQo. Under conditions that mimic mild and intense aerobic exercise, total production is much less and the low capacity site IF dominates [20].
Figure 1.
Sources of reactive oxygen species (ROS) and endogenous antioxidants in skeletal muscle fibers. Following exercise, ROS are produced endogenously by mitochondria, NOXs, PLA2 and XO. In addition, exercise increases ROS production also in activated neutrophils and macrophages, endothelia of blood vessels and by catecholamines. Regular exercise leads to an increase of endogenous antioxidants, which are able to neutralize free radicals. A, adrenaline; CAT, catalase; Cu, Zn-SOD, copper-zinc superoxide dismutase; DA, dopamine; GPX, glutathione peroxidase; GSH, glutathione; IFN, interferon γ; IL-1, interleukin-1; Mn-SOD, manganese superoxide dismutase; NA, noradrenaline; NOX, nicotinamide adenine dinucleotide phosphate oxidase; PLA2, phospholipase A2; SR, sarcoplasmic reticulum; TNF, tumor necrosis factor; TxnRd2, thioredoxin reductase 2; XO, xanthine oxidase.
Figure 1.
Sources of reactive oxygen species (ROS) and endogenous antioxidants in skeletal muscle fibers. Following exercise, ROS are produced endogenously by mitochondria, NOXs, PLA2 and XO. In addition, exercise increases ROS production also in activated neutrophils and macrophages, endothelia of blood vessels and by catecholamines. Regular exercise leads to an increase of endogenous antioxidants, which are able to neutralize free radicals. A, adrenaline; CAT, catalase; Cu, Zn-SOD, copper-zinc superoxide dismutase; DA, dopamine; GPX, glutathione peroxidase; GSH, glutathione; IFN, interferon γ; IL-1, interleukin-1; Mn-SOD, manganese superoxide dismutase; NA, noradrenaline; NOX, nicotinamide adenine dinucleotide phosphate oxidase; PLA2, phospholipase A2; SR, sarcoplasmic reticulum; TNF, tumor necrosis factor; TxnRd2, thioredoxin reductase 2; XO, xanthine oxidase.

2.2. NADPH Oxidases
NADPH oxidases (NOXs) are flavoprotein enzymes that are activated by calcium, free fatty acids, protein-protein interactions and posttranslational modifications and use NADPH as electron donors [21,22]. They are transmembrane proteins in the transverse tubules and the sarcoplasmic reticulum and transport electrons across biological membranes to reduce oxygen to superoxide or H2O2 [21,22]. It was shown that NOX family members contribute to cytosolic superoxide production in skeletal muscle both at rest and during contractile activity to a larger extent than mitochondria [23,24,25]. ROS generated by NOXs activates ryanodine receptors (RyR), which leads to an intracellular Ca2+ release [26,27,28]. More recently, it was found that insulin induces ROS generation through NOX activation and that this ROS increase is required for the intracellular Ca2+ rise mediated by inositol triphosphate (IP3) receptors [29].
2.3. Xanthine Oxidase
Xanthine oxidase (XO) is a cytosolic molybdoflavoenzyme that is recognized as a key enzyme in purine catabolism in which it catalyzes the hydroxylation of hypoxanthine to xanthine and of xanthine to uric acid [30]. In muscle, XO is present in the cytosol but also in the associated endothelial cells [8]. Upon contraction, XO activity is significantly increased and leads to increased lipid peroxidation, protein oxidation, muscle damage and edema [31]. During intense exercise in which large amounts of ATP are consumed, hypoxanthine and xanthine levels are rising and serve as substrates for XO to generate ROS [32]. Interestingly, ROS generated by XO appears to be involved in the regulation of exercise-induced mitochondrial biogenesis via peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) [33].
2.4. Myostatin
Recently, it was demonstrated that myostatin, a blocker of muscle differentiation, is capable of signaling ROS production via canonical Smad3, nuclear factor (NF)-κB and TNF-α in muscle cells [34]. In the absence of Smad3, myostatin induces ROS production through the activation of p38 and ERK mitogen-activated protein kinase (MAPK) pathways mediated via TNF- α, IL-6, NOX and XO [35].
2.5. Phospholipase A2
Enzymes of the phospholipase A2 (PLA2) family also contribute to intra- and extracellular ROS increase during muscle contraction. They cleave arachidonic acid from phospholipids in the plasma membrane, sarcoplasmic reticulum or mitochondrial membranes. Arachidonic acid is an important lipid-signaling molecule and is a substrate for lipoxygenases for the production of ROS [36]. In addition, the cytosolic PLA2 enzyme has been demonstrated to increase ROS by stimulating NOXs [37]. Human muscle is known to contain approximately 15 different PLA2 isoforms that are either Ca2+-sensitive or Ca2+-insensitive [38]. The Ca2+-independent and dependent enzymes are supposed to produce ROS under resting and activity conditions, respectively [39].
3. Effects of ROS on Force Generation and Muscle Atrophy
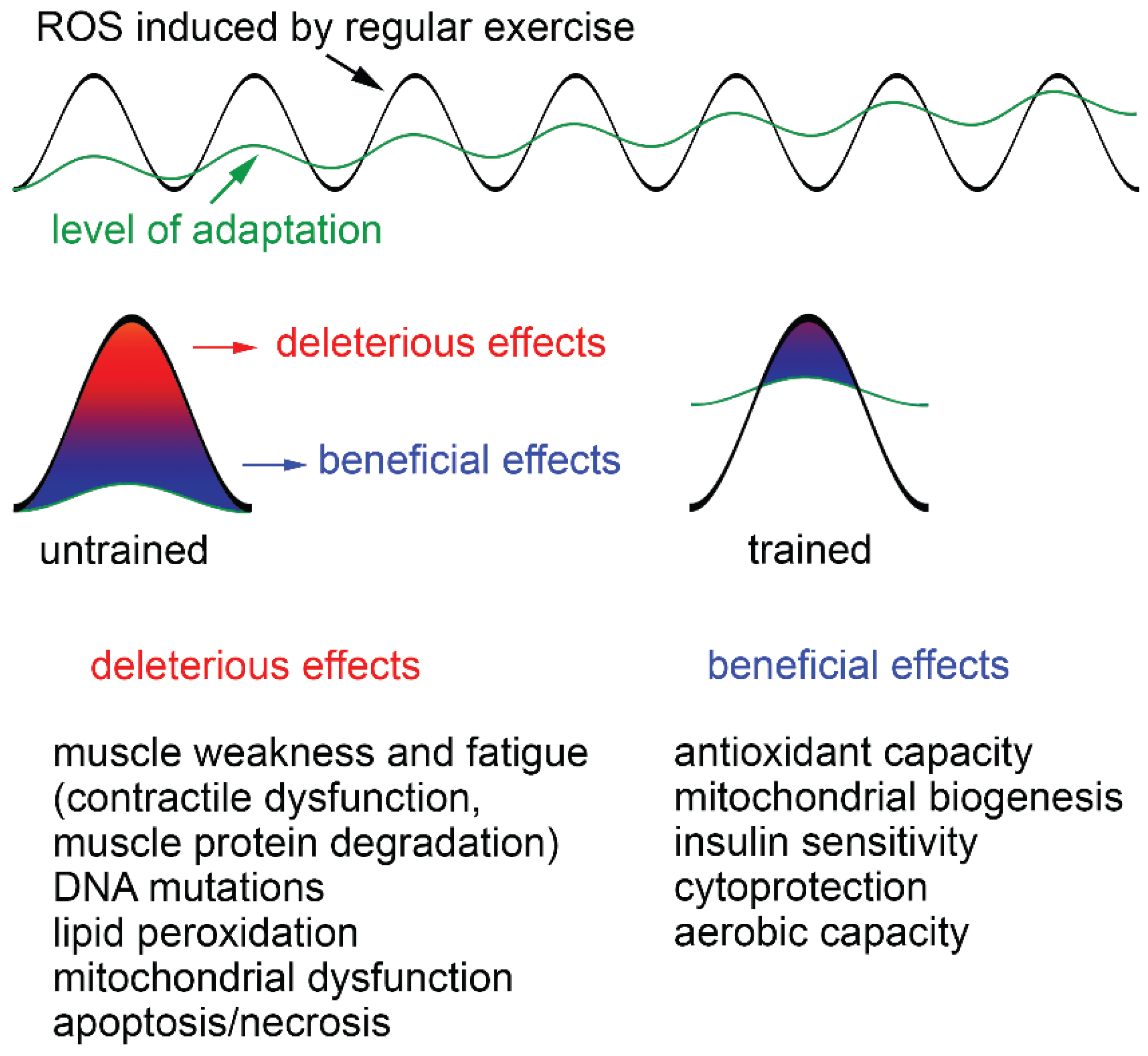
In unfatigued muscle, intracellular ROS appear to be essential for normal force generation. Low-level ROS supplementation even increases force production [40]. A stronger increase of ROS due to intense exercise leads to a variety of adaptations of the muscle cells. Dependent upon the ROS concentration, duration of ROS exposure and training status of the individual, ROS can have beneficial and detrimental effects (Figure 2). Thus, a single bout of exhaustive exercise has been shown to cause oxidative damage in untrained persons while in trained subjects, no such effects are observed due to an increased resistance of such persons to oxidative stress [41]. Strong increases in ROS after strenuous exercise, aging and/or disease (e.g., chronic heart failure, COPD, cancer) can cause contractile dysfunction and muscle atrophy, which both promote muscle weakness and fatigue [3,42].
Figure 2.
Deleterious and beneficial effects of exercise-induced ROS increase. Exercise produces ROS and whether they are beneficial or detrimental to health is dependent upon the ROS concentration, duration of ROS exposure and training status of the individual. A single bout of exhaustive exercise leads to strong increases of ROS, which cannot be buffered by endogenous antioxidants, particularly in untrained individuals. This results in severe oxidative damage, including muscle weakness and fatigue, DNA mutations, lipid peroxidation, mitochondrial dysfunction and apoptosis/necrosis. Trained persons have a higher level of adaptation and less health risks. ROS produced during regular exercise continuously increase the level of adaptation by improving antioxidant capacity, mitochondrial biogenesis, insulin sensitivity, cytoprotection and aerobic capacity of skeletal muscle.
Figure 2.
Deleterious and beneficial effects of exercise-induced ROS increase. Exercise produces ROS and whether they are beneficial or detrimental to health is dependent upon the ROS concentration, duration of ROS exposure and training status of the individual. A single bout of exhaustive exercise leads to strong increases of ROS, which cannot be buffered by endogenous antioxidants, particularly in untrained individuals. This results in severe oxidative damage, including muscle weakness and fatigue, DNA mutations, lipid peroxidation, mitochondrial dysfunction and apoptosis/necrosis. Trained persons have a higher level of adaptation and less health risks. ROS produced during regular exercise continuously increase the level of adaptation by improving antioxidant capacity, mitochondrial biogenesis, insulin sensitivity, cytoprotection and aerobic capacity of skeletal muscle.

3.1. Contractile Dysfunction
Contractile dysfunction may result from oxidative modifications of a variety of proteins in diverse intracellular components [43,44]. However, our understanding of the processes involved is still limited and many data are equivocal. In the sarcoplasmic reticulum, the ryanodine receptor (RyR), which is the Ca2+ release channel, was shown to be oxidized by ROS and it was hypothesized to contribute to muscle fatigue [26,27,28]. However, other work demonstrated that this oxidation resulted only in increased Ca2+-induced Ca2+ release, whereas the Ca2+ release triggered by action potentials was not affected [45,46]. From this it was inferred that ROS-mediated effects on Ca2+ release in the sarcoplasmic reticulum are unlikely to contribute to muscle fatigue. This goes in line with recent findings that demonstrate that the mitochondrial antioxidant SS-31 restored the decrease in sarcoplasmic reticulum Ca2+ release while force recovery was not improved [47].
Similar uncertainty surrounds the effect of ROS on the force generating myofilaments. Initial data have shown that brief exposure to low concentrations of H2O2 increased force by 27%, while a longer exposure results in force decline [48]. By contrast, a short exposure of skinned fibers to 10 mM H2O2 had no effect on maximum force [45], while longer exposure to 50 mM H2O2 inhibited contractility [49]. Although the exact mechanisms are unknown, it is generally assumed that changes in force generation are the result of changes in the myofibrillar Ca2+ sensitivity [8,44,47]. In this regard, it was most recently suggested that troponin I, which is involved in sensing the intracellular Ca2+ levels, is a target of ROS. Oxidized cysteine residues of troponin I can react with the antioxidant glutathione, which helps protect the molecule from oxidative stress and make the contractile apparatus much more sensitive to Ca2+ [50]. However, it must be mentioned that ROS may also lead to changes in the contractile proteins. In this regard, it was demonstrated that H2O2 is able to modify the S1 fragment in the myosin head, which then leads to a restriction of the myosin-actin dynamics in the presence of ATP [49].
3.2. Muscle Atrophy
Besides their effects on the contractile kinetics, ROS are also able to modulate various signaling pathways, such as calcium, protein tyrosine kinases and phosphatases, serine/threonine kinases, and phospholipases [51]. This then leads to changes in gene expression, cell function, metabolism or cell damage. Chronic oxidative stress is associated with an increase in protein loss and muscle atrophy. High ROS levels cause a sustained activation of NF-κB and of FoxO which then activate two muscle-specific E3 ubiquitin ligases, atrogin-1 or muscle atrophy F-box (MAFbx) and muscle RING (Really Interesting New Gene)-finger protein 1 (MuRF-1) [52]. MAFbx and MuRF-1 then degrade various proteins, such as titin, nebulin, troponin, myosin-binding protein C, myosin light chains 1 and 2 and myosin heavy chain [53,54]. Recently, it was demonstrated that excessive oxidative stress also enhances the transcription factor C/EBP homology protein (CHOP). This transcription factor also enhances expression of MuRF1, which again results in increased protein degradation [35].
4. Antioxidants in Muscle
4.1. Enzymatic and Nonenzymatic Antioxidants
Muscle activity increases ROS but simultaneously also the body’s antioxidant defense system. These molecules are able to neutralize free radicals by accepting the unpaired electron and thereby inhibit the oxidation of other molecules. Depending on the oxygen consumption rate, cells constitutively express different levels of antioxidant enzymes, including mitochondrial antioxidant manganese superoxide dismutase (Mn-SOD, SOD2), cytosolic copper-zinc superoxide dismutase (Cu, Zn-SOD, SOD1), glutathione peroxidase (GPX) and catalase (CAT), and the nonenzymatic antioxidant glutathione (GSH) [55] (Figure 1).
GSH is the most abundant nonprotein thiol in cells with intracellular concentrations of 1–15 mM [56]. It plays a major role in the detoxification of electrophilic xenobiotics, such as chemical carcinogens, environmental pollutants, and the inactivation of endogenous α,β-unsaturated aldehydes, quinones, epoxides, and hydroperoxides, which are formed as secondary metabolites during oxidative stress via members of the glutathione transferase family [57]. It also protects from oxidative stress by reducing hydrogen peroxide and organic peroxides levels via a reaction catalyzed by GSH peroxidase thus keeping the intracellular environment in the reduced state [55,58]. In addition, GSH is a substrate for dehydroascorbate reductase enabling the recycling of ascorbic acid, and it is a scavenger of hydroxyl radicals and singlet oxygen [59].
The aforementioned enzymatic reactions lead to the oxidation of GSH to glutathione disulfide (GSSG). This molecule can inactivate a number of enzymes by reacting with protein thiols leading to the formation of mixed disulfides (e.g., [60]). To avoid damage to intracellular constituents, GSSG is efficiently reduced to GSH by glutathione reductase (GR) utilizing NADPH. This action of GR is also very important during and after exercise in which a substantial amount of GSH is oxidized due to the elevated ROS levels to keep the GSH/GSSG ratio constant thereby maintaining homeostasis. Furthermore, exercising skeletal muscle appears to increase GSH import from plasma [61,62], and liver can synthesize GSH de novo and supply it [58]. But also increased muscle glutathione synthetase activities have been observed after treadmill training [63]. These exercise responses are tissue- and fiber-specific [64].
The antioxidant enzymes SOD, CAT and GPX are the primary defense against ROS generated during exercise and increase in response to exercise [65,66]. Recent work identified thioredoxin reductase-2 (TxnRd2) as another key player to decrease the exercise-induced content of mitochondrial H2O2 in skeletal muscle [67]. The same authors have shown that TxnRd2 is also able to control mitochondrial H2O2 levels after a high-fat, high-sucrose diet in the heart but not in skeletal muscle. Antioxidant enzyme levels vary considerably with respect to muscle fiber types, i.e., type I muscle fibers possess higher activity of all antioxidant enzymes than the type IIA and type IIB fibers [68].
4.2. Adaptive Responses to Exercise
In general, it was found that there is an exercise-induced increase in antioxidant protein levels and antioxidant activity. Thus, endurance training in rats leads to an increase in Mn-SOD, GPX and CAT, while the data on Cu, Zn-SOD are somewhat less clear [69,70,71,72,73]. Note that from the above studies, it is likely that upregulation of these antioxidants is muscle- and/or fiber type-specific. Many studies have shown that even an acute bout of exercise increases SOD activity in skeletal muscle ([74,75,76,77]; for review see [55]), and it has further been shown that Cu, Zn-SOD and Mn-SOD contents are increased. While the Cu, Zn-SOD enzyme activity gradually returns to resting levels within three days, Mn-SOD activity and protein content continues to increase in the post-exercise period [78]. GPX activity after acute exercise on the other hand appears to depend on the muscle type, i.e., GPX activity was increased a day after an acute bout of treadmill running to exhaustion in rat soleus but not tibialis muscle [78], and CAT activity appears not to be altered by acute exercise [65].
ROS generated by acute exercise can lead to increased lipid peroxidation as measured by the formation of malondialdehyde [79]. Interestingly, this effect was only found in liver and fast skeletal muscle in the sedentary group, whereas the endurance-trained group did not show increases in lipid peroxidation after exercise. Lipid peroxidation generates a vast number of oxidative lipid breakdown products for which more or less specific tests are available (for review see [59]. Some investigators determined plasma isoprostane levels in athletes performing either a 50 km ultramarathon [80] or exercise for 2.5 h on a treadmill [81]. Peak levels of isoprostanes were found directly post-exercise, followed by a return to baseline within one day or one hour, respectively. Other investigators found increased levels of pentane in the breath after exercise [1], increased levels of lipid hydroperoxides [82], conjugated dienes [83] and oxidative DNA damage as measured by 8-hydroxydeoxyguanosine [84] or single cell gel electrophoresis [85]. The latter investigation gave results similar to those of Alessio and Goldfarb [79], namely a significant reduction of oxidative DNA damage in the trained compared to the untrained group indicating that exercise training causes an adaptive response to elevated oxidative stress by increased antioxidant enzyme activity. Powers et al. [86] studied the influence of training and observed significantly increased SOD activity in the soleus following exercise up to 60 min/d. Conversely, training induced significant increases in GPX activity in slow gastrocnemius only, and the magnitude of the GPX increase was directly related to exercise duration but relatively independent of intensity. However, CAT activity was not increased in any muscle with training. In addition, Radák et al. [87] observed decreased DNA damage and increased DNA repair levels as well as resistance against oxidative stress of proteins in aged rat skeletal muscle upon training. The obviously paradoxical situation that increased exercise-induced oxidative stress causes beneficial effects is interpreted in terms of hormesis—beneficial effects of potentially harmful agents—which apart from providing an adaptation to the damaging agent provide also systemic beneficial effects, including improved physiological function, decreased incidence of disease and a higher quality of life [88]. However, the beneficial effects appear to depend upon the duration of the exercise. While a single bout of exercise is suggested to lead to a limited adaptive response, regular exercise appears to gradually increase the level of adaptation by the repeated activation of antioxidant genes and proteins [32]. These authors hypothesize that it is the increased ROS level that is the important stimulus for the muscle cells to adapt to chronic exercise. The improved capabilities to decrease ROS may then provide a better protection from ROS during subsequent trainings but also attenuate the aging process and promote health with increased functional capacities [32]. Therefore, exercise is very similar to the adaptive ischemic preconditioning response [89]. Restoration of perfusion to ischemic organs results in increased ROS levels that can lead to tissue damage, myocardial infarction and stroke. However, such deleterious effects can be avoided by short intermittent bouts of reperfusion in which the transiently elevated ROS levels are important mediators of a cardioprotective response [89,90].
Important mediators of the adaptive responses are the adenosine monophosphate-activated protein kinase (AMPK), the transcription factors NF-κB, together with p38 MAPK, and members of the FoxO transcription factor family [91,92,93]. At low ROS levels, they promote adaptation by increasing gene expression of antioxidant enzymes, such as Mn-SOD and Cu, Zn-SOD, CAT and GPX1 [94]. High antioxidant capacities then diminish the deleterious effects of subsequent increases in ROS [95]. In addition, products of radical reactions are also suggested to be the mediator of this adaptation. Of special interest in this context are lipid peroxidation products, in particular 4-hydroxynonenal (HNE), which has been shown to both induce DNA damage [96] but also to be involved in the regulation of cell proliferation and growth as well as necrotic or apoptotic cell death by its marked ability to modulate several major pathways of cell signaling and, consequently, gene expression (for review see [97]). With respect to antioxidant gene expression it has been shown to be one of the most effective activators of nuclear factor erythroid-derived 2-like 2 (Nrf2) [98] which on stimulation dissociates from its cytoplasmic inhibitor Keap1, translocates to the nucleus and transactivates antioxidant-responsive elements (ARE)-dependent genes [99]. In addition, HNE has been demonstrated to cause mitochondrial uncoupling and thus protection from ROS specifically via the induction of the uncoupling proteins UCP1, UCP2 and UCP3 and the adenine nucleotide translocase [100].
4.3. Exogenous Antioxidants and Exercise
Apart from the endogenous antioxidants, which are obviously regulated by exercise, exogenous antioxidants such as vitamin C, E, and carotenoids are taken up with the food or are used as dietary supplements. The question therefore arises whether such supplements can be considered beneficial during exercise. To address this question, Ristow et al. [73] investigated the effects of a diet supplemented with vitamin C and E on exercise-induced insulin sensitivity as measured by glucose infusion rates during a hyperinsulinemic, euglycemic clamp in previously untrained and pre-trained healthy young men. Interestingly, exercise was found to increase parameters of insulin sensitivity (including adiponectin) only in the absence of antioxidants in both previously untrained and pretrained individuals. This was paralleled by increased expression of ROS-sensitive transcriptional regulators of insulin sensitivity and ROS defense capacity, peroxisome proliferator-activated receptor γ (PPARγ) and PPARγ coactivators PGC-1α and PGC-1β only in the absence of antioxidants. Molecular mediators of endogenous ROS defense (Mn-SOD, Cu, Zn-SOD and GPX) were also induced by exercise, and this effect was again blocked by antioxidant supplementation. The authors concluded that exercise-induced oxidative stress ameliorates insulin resistance and causes an adaptive response promoting endogenous antioxidant defense capacity and that supplementation with antioxidants may preclude these health-promoting effects of exercise in humans. It was demonstrated that exercise causes an activation of mitogen-activated protein kinases (MAPKs: p38, ERK 1 and ERK 2), which in turn activates nuclear factor κB (NF-κB) in rat gastrocnemius muscle and consequently the expression of important enzymes associated with defense against ROS (SOD) and adaptation to exercise—endothelial nitric oxide synthase (eNOS) and inducible nitric oxide synthase (iNOS) [101,102,103]. The expression of these enzymes can be inhibited by allopurinol, an inhibitor of XO indicating also that the prevention of ROS formation causes an inhibition of an adaptive response. The authors therefore conclude that in all likelihood, antioxidant supplements should not be recommended before training as they interfere with muscle cell adaptation. Thus, physical exercise is considered a double-edged sword: when practiced strenuously it causes oxidative stress and cell damage; in this case application of antioxidants may be helpful. But when practiced in moderation, it increases the expression of antioxidant enzymes and thus should be considered an antioxidant [101,103]. Supportive evidence for this assumption comes from studies on physical overtraining. Margonis et al. [104] examined the responses of oxidative stress biomarkers to a resistance training protocol of progressively increased and decreased volume/intensity in male test persons and observed significantly increased levels of urinary isoprostanes (7-fold), serum levels of thiobarbituric acid reactive substances (TBARS), protein carbonyls, CAT, GPX, and GSSG and significantly decreased levels of GSH, the GSH/GSSG ratio, and total antioxidant capacity in blood serum of over-trained individuals. Similarly, Palazzetti et al. [105] investigated the effects of overloaded training (OT) with athletes exercising for a duathlon before and after a four week OT and found that at rest conditions, OT induced an increased plasma GPX activity and a decreased plasma total antioxidant status, while OT resulted in higher exercise-induced variations of blood GSH/GSSG ratios, TBARS levels and decreased total antioxidant status in exercise conditions indicating that OT could compromise the antioxidant defense mechanisms. By comparing the oxidative stress response in control athletes and athletes with overtraining syndrome Tanskanen et al. [106] were further able to show that exercise to exhaustion led to an increase in oxygen radical absorbance (antioxidant) capacity and malondialdehyde in the controls but not in the over-trained athletes. Instead, over-trained athletes showed negative correlations between oxygen radical absorbance capacity at rest and protein carbonyls after exhaustive exercise indicating that increased oxidative stress may play a role in the pathophysiology of overtraining syndrome. Although these observations are not yet conclusive they indicate that adaptation to exercise is limited and that its protective effect can be exceeded leading to oxidative stress that cannot be dealt with by the endogenous antioxidant system. Whether it is helpful to apply exogenous antioxidants under such conditions as suggested still has to be elucidated.
5. Training-Induced Muscular Adaptation, PGC-1α and ROS
In addition to the above-described effects of exercise on contents and activities of antioxidant enzymes, regularly performed exercise in the form of endurance training leads to well described adaptations of the cardiovascular and muscular system. Important responses at the intramyocellular level include increases in size and number of mitochondria as well as such in the activities of oxidative enzymes [107,108,109]. In support of the increased oxidation of fatty acids, the content of intramyocellular lipid is also elevated [110]. Endurance exercise is also known to improve insulin sensitivity and muscular glucose uptake [108,111]. Recent research has demonstrated that ROS also have a beneficial role in promoting these adaptive responses of muscle to training.
5.1. Role of PGC-1α in Exercise
In rodents and humans, it has been demonstrated that peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) is a key regulator of the exercise-induced changes of muscle fibers towards a slow phenotype, as well as in the protection from muscle atrophy [108,112,113]. Several studies have shown that PGC-1α is upregulated after high-intensity training [114,115,116,117,118]. Activation of PGC-1α is likely to occur by phosphorylation of the PGC-1α protein by p38 MAPK together with NF-κB [119], both of which are known to be activated by ROS [91,92]. PGC-1α has been demonstrated to regulate lipid and carbohydrate metabolism, and to improve the oxidative capacity of the muscle fibers by increasing the amount and activity of mitochondria through upregulation of nuclear respiratory factors (NRF-1, 2) and mitochondrial transcription factor A (TFAM) [120,121]. Furthermore, PGC-1α regulates genes involved in the determination of muscle fiber type. Overexpression of PGC-1α increases the proportion of oxidative type I fibers [122] while PGC-1α knock-out (KO) mice exhibit a shift from oxidative type I and IIA toward glycolytic type IID/X and IIB fibers [123]. This regulatory diversity of PGC-1α is enabled by its broad binding capacity to transcription factors in various signaling pathways. PGC-1α has multiple binding sites for the interactions with diverse coactivators. A domain between amino acids 200 and 400 interacts with the nuclear receptors PPARγ and NRF-1 [124], which are considered as master regulators of mitochondrial biogenesis [125]. PGC-1α binds to and activates the transcriptional function of NRF-1 on the promoter for TFAM, a direct regulator of mitochondrial DNA replication and transcription [120]. Another domain that predominantly binds to nuclear hormone receptors such as ERR-α, PPARs, RXR, glucocorticoid receptor, HNF4, and probably others, is an LXXLL sequence in the N-terminal region of PGC-1α [124]. This sequence is necessary for the coactivation of the nuclear receptor liver x receptor α (LXRα) [126]. The transcription complex of LXRα and PGC-1α then activates fatty acid synthase (FAS), a multifunctional enzyme that catalyzes all reactions required for the de novo biosynthesis of lipid [127]. The binding site of the nuclear receptor estrogen-related receptor α (ERR-α) is also in the LXXLL region of PGC-1α [124]. The transcription complex formed by ERR-α and PGC-1α induces the expression of vascular endothelial growth factor (VEGF), a potent stimulator of angiogenesis [128,129]. Between amino acids 400 to 500 of the PGC-1α protein is the binding site for myocyte enhancer factor 2 (MEF2). This transcription factor is a key regulator of slow muscle identity [130]. MEF2 proteins are activated through the calcium-regulated calcineurin signaling pathway [130,131]. When overexpressed, MEF2C promotes the formation of slow fibers, thus enhancing running endurance in mice [132]. Genetic deletion of Mef2c has been shown to block activity-dependent (exercise-induced) fast-to-slow fiber type transition [132]. This is in line with the proposed role of PGC-1α in such transitions. Muscle-specific overexpression of PGC-1α has been shown to evoke a transition of glycolytic type II in oxidative type I fibers [122]. This shift is initiated by the formation of a PGC-1α/MEF2 transcription complex, which then activates the expression of slow muscle genes [133]. Handschin et al. [123] have shown that PGC-1α deficient mice display a significant shift from slow oxidative type I and fast oxidative IIA toward fast glycolytic type IIX and IIB fibers, resulting in a reduced endurance capacity.
5.2. PGC-1α Regulates ROS Defense
It has been shown that oxidative stress increases the expression of PGC-1α [134]. Similarly, depleting the endogenous antioxidant glutathione augments exercise-mediated induction of PGC-1α expression [135]. Upregulation of PGC-1α possibly involves the transcription factor Cre-binding protein (CREB) [136]. PGC-1α then induces an increase of ROS-detoxifying enzymes, including GPX1 and Mn-SOD [136]. Only recently, new light has been shed on the molecular mechanisms involved in antioxidant activation. Therefore, it is highly likely that PGC-1α binds to ERR-α and activates the NAD+-dependent histone deacetylase silent information regulator 3 (SIRT3) in the mitochondrial matrix [137]. SIRT3 is also known to regulate ROS production by directly binding and deacetylating mitochondrial complex I and II [138,139]. Previous studies have also shown that SIRT3 is able to deacetylate the mitochondrial enzyme Mn-SOD, thereby promoting its antioxidative activity [140,141,142]. Thus, it appears that PGC-1α is a powerful suppressor of ROS production mainly by upregulation of antioxidant expression. Correspondingly, it was demonstrated that PGC-1α knock-out (KO) mice have reduced expression levels of Mn-SOD, Cu, Zn-SOD and GPX1 and are thus more sensitive to oxidative stressors [136]. By contrast, overexpression of PGC-1α enhances antioxidant defense by upregulation of Mn-SOD expression and a higher catalase activity [143]. Further, PGC-1α increases the expression of uncoupling proteins 2 and 3 (UCP2, UCP3) and thereby concomitantly reduces mitochondrial ROS production [144].
6. Conclusions
There is rapidly growing evidence that ROS have both positive and negative effects in contracting skeletal muscle cells. The deleterious effects such as a reduction of force generation and increased muscle atrophy appear to occur particularly after non-regular strenuous exercise, while regular training has positive effects by influencing cellular processes that lead to increased expression of antioxidants. These molecules then provide a better protection from ROS during subsequent trainings. However, a diet supplemented with exogenous antioxidants such as vitamins appears to prevent health-promoting effects of physical exercise in humans. The exercise-induced production of ROS may also be an important signal to activate PGC-1α, a key player in the adaption of muscle cells to exercise.
Author Contributions
Both authors contributed interactively to explore the background literature and to the writing procedure. Peter Steinbacher took responsibility for preparing the final version of the manuscript.
Abbreviations
| AMPK | adenosine monophosphate-activated protein kinase |
| ARE | antioxidant-responsive element |
| CAT | catalase |
| CHOP | C/EBP homology protein |
| CREB | Cre-binding protein |
| Cu, Zn-SOD | copper-zinc superoxide dismutase |
| eNOS | endothelial nitric oxide synthase |
| ERR-α | estrogen-related receptor α |
| FAS | fatty acid synthase |
| GPX | glutathione peroxidase |
| GR | glutathione reductase |
| GSH | glutathione |
| GSSG | glutathione disulfide |
| HNE | 4-hydroxynonenal |
| IFN-γ | interferon-γ |
| IL-1 | interleukin-1 |
| iNOS | inducible nitric oxide synthase |
| LXRα | nuclear receptor liver x receptor α |
| MAFbx | muscle atrophy F-box |
| MAPK | mitogen-activated protein kinase |
| MEF2 | myocyte enhancer factor 2 |
| Mn-SOD | manganese superoxide dismutase |
| MuRF-1 | muscle RING-finger protein 1 |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NF-κB | nuclear factor-κB |
| NOX | nicotinamide adenine dinucleotide phosphate oxidase |
| NRF | nuclear respiratory factor |
| Nrf2 | nuclear factor erythroid-derived 2-like 2 |
| OT | overloaded training |
| PGC-1α | peroxisome proliferator-activated receptor-γ coactivator-1α |
| PLA2 | phospholipase A2 |
| PPARγ | peroxisome proliferator-activated receptor γ |
| ROS | reactive oxygen species |
| RyR | ryanodine receptor |
| SIRT3 | silent information regulator 3 |
| TBARS | thiobarbituric acid reactive substance |
| TFAM | mitochondrial transcription factor A |
| TNF | tumor necrosis factor |
| TxnRd2, | thioredoxin reductase-2 |
| UCP | uncoupling protein |
| VEGF | vascular endothelial growth factor |
| XO | xanthine oxidase |
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dillard, C.J.; Litov, R.E.; Savin, W.M.; Dumelin, E.E.; Tappel, A.L. Effects of exercise, vitamin E, and ozone on pulmonary function and lipid peroxidation. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1978, 45, 927–932. [Google Scholar] [PubMed]
- Davies, K.J.; Quintanilha, A.T.; Brooks, G.A.; Packer, L. Free radicals and tissue damage produced by exercise. Biochem. Biophys. Res. Commun. 1982, 107, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Vollaard, N.B.; Shearman, J.P.; Cooper, C.E. Exercise-induced oxidative stress: Myths, realities and physiological relevance. Sports Med. 2005, 35, 1045–1062. [Google Scholar] [CrossRef] [PubMed]
- Fisher-Wellman, K.; Bloomer, R.J. Acute exercise and oxidative stress: A 30 year history. Dyn. Med. 2009. [Google Scholar] [CrossRef]
- Sahlin, K.; Shabalina, I.G.; Mattsson, C.M.; Bakkman, L.; Fernström, M.; Rozhdestvenskaya, Z.; Enqvist, J.K.; Nedergaard, J.; Ekblom, B.; Tonkonogi, M. Ultraendurance exercise increases the production of reactive oxygen species in isolated mitochondria from human skeletal muscle. J. Appl. Physiol. 2010, 108, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Hey-Mogensen, M.; Højlund, K.; Vind, B.F.; Wang, L.; Dela, F.; Beck-Nielsen, H.; Fernström, M.; Sahlin, K. Effect of physical training on mitochondrial respiration and reactive oxygen species release in skeletal muscle in patients with obesity and type 2 diabetes. Diabetologia 2010, 53, 1976–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Lertwattanarak, R.; Lefort, N.; Molina-Carrion, M.; Joya-Galeana, J.; Bowen, B.P.; Garduno-Garcia Jde, J.; Abdul-Ghani, M.; Richardson, A.; DeFronzo, R.A.; et al. Reduction in reactive oxygen species production by mitochondria from elderly subjects with normal and impaired glucose tolerance. Diabetes 2011, 60, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef] [PubMed]
- Peake, J.; Suzuki, K. Neutrophil activation, antioxidant supplements and exercise-induced oxidative stress. Exerc. Immunol. Rev. 2004, 10, 129–141. [Google Scholar] [PubMed]
- Moylan, J.S.; Reid, M.B. Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve 2007, 35, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Gomes, E.C.; Silva, A.N.; de Oliveira, M.R. Oxidants, antioxidants, and the beneficial roles of exercise-induced production of reactive species. Oxid. Med. Cell Longev. 2012, 2012, 756132. [Google Scholar] [PubMed]
- Duarte, J.A.; Appell, H.J.; Carvalho, F.; Bastos, M.L.; Soares, J.M. Endothelium-derived oxidative stress may contribute to exercise-induced muscle damage. Int. J. Sports Med. 1993, 14, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed]
- Perevoshchikova, I.V.; Quinlan, C.L.; Orr, A.L.; Gerencser, A.A.; Brand, M.D. Sites of superoxide and hydrogen peroxide production during fatty acid oxidation in rat skeletal muscle mitochondria. Free Radic. Biol. Med. 2013, 61, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Hey-Mogensen, M.; Goncalves, R.L.; Orr, A.L.; Brand, M.D. Production of superoxide/H2O2 by dihydroorotate dehydrogenase in rat skeletal muscle mitochondria. Free Radic. Biol. Med. 2014, 72, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Barja, G. Mitochondrial oxygen radical generation and leak: Sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J. Bioenerg. Biomembr. 1999, 31, 347–366. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Liu, Y.; van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, R.L.; Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Brand, M.D. Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J. Biol. Chem. 2015, 290, 209–227. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Webb, J.A.; Gnall, L.L.; Cutler, K.; Abramson, J.J. Skeletal muscle sarcoplasmic reticulum contains a NADH-dependent oxidase that generates superoxide. Am. J. Physiol. Cell Physiol. 2003, 285, C215–C221. [Google Scholar] [CrossRef] [PubMed]
- Shkryl, V.M.; Martins, A.S.; Ullrich, N.D.; Nowycky, M.C.; Niggli, E.; Shirokova, N. Reciprocal amplification of ROS and Ca2+ signals in stressed mdx dystrophic skeletal muscle fibers. Pflugers Arch. 2009, 458, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Sakellariou, G.K.; Vasilaki, A.; Palomero, J.; Kayani, A.; Zibrik, L.; McArdle, A.; Jackson, M.J. Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid. Redox Signal. 2013, 18, 603–621. [Google Scholar] [CrossRef] [PubMed]
- Cherednichenko, G.; Zima, A.V.; Feng, W.; Schaefer, S.; Blatter, L.A.; Pessah, I.N. NADH oxidase activity of rat cardiac sarcoplasmic reticulum regulates calcium-induced calcium release. Circ. Res. 2004, 94, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, A.; Leiva, A.; Peña, M.; Müller, M.; Debandi, A.; Hidalgo, C.; Carrasco, M.A.; Jaimovich, E. Myotube depolarization generates reactive oxygen species through NAD(P)H oxidase; ROS-elicited Ca2+ stimulates ERK, CREB, early genes. J. Cell Physiol. 2006, 209, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, C.; Sánchez, G.; Barrientos, G.; Aracena-Parks, P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S-glutathionylation. J. Biol. Chem. 2006, 281, 26473–26482. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, A.; García, A.; Härtel, S.; Hidalgo, C.; Jaimovich, E. NADPH oxidase and hydrogen peroxide mediate insulin-induced calcium increase in skeletal muscle cells. J. Biol. Chem. 2009, 284, 2568–2575. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R. Structure and function of xanthine oxidoreductase: Where are we now? Free Radic. Biol. Med. 2002, 33, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Judge, A.R.; Dodd, S.L. Xanthine oxidase and activated neutrophils cause oxidative damage to skeletal muscle after contractile claudication. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H252–H256. [Google Scholar] [CrossRef] [PubMed]
- Radák, Z.; Zhao, Z.; Koltai, E.; Ohno, H.; Atalay, M. Oxygen consumption and usage during physical exercise: The balance between oxidative stress and ROS-dependent adaptive signaling. Antioxid. Redox Signal. 2013, 18, 1208–1246. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Chung, E.; Diffee, G.; Ji, L.L. Exercise training attenuates aging-associated mitochondrial dysfunction in rat skeletal muscle: Role of PGC-1α. Exp. Gerontol. 2013, 48, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Sriram, S.; Subramanian, S.; Sathiakumar, D.; Venkatesh, R.; Salerno, M.S.; McFarlane, C.D.; Kambadur, R.; Sharma, M. Modulation of reactive oxygen species in skeletal muscle by myostatin is mediated through NF-κB. Aging Cell 2011, 10, 931–948. [Google Scholar] [CrossRef] [PubMed]
- Sriram, S.; Subramanian, S.; Juvvuna, P.K.; Ge, X.; Lokireddy, S.; McFarlane, C.D.; Wahli, W.; Kambadur, R.; Sharma, M. Myostatin augments muscle-specific ring finger protein-1 expression through an NF-κB independent mechanism in SMAD3 null muscle. Mol. Endocrinol. 2014, 28, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Christofi, F.L.; Wright, V.P.; Bao, S.; Clanton, T.L. Lipoxygenase-dependent superoxide release in skeletal muscle. J. Appl. Physiol. 2004, 97, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Bey, E.A.; Wientjes, F.B.; Cathcart, M.K. Cytosolic phospholipase A2 (cPLA2) regulation of human monocyte NADPH oxidase activity. cPLA2 affects translocation but not phosphorylation of p67(phox) and p47(phox). J. Biol. Chem. 2002, 277, 25385–25392. [Google Scholar] [PubMed]
- Ježek, J.; Jaburek, M.; Zelenka, J.; Ježek, P. Mitochondrial phospholipase A2 activated by reactive oxygen species in heart mitochondria induces mild uncoupling. Physiol. Res. 2010, 59, 737–747. [Google Scholar] [PubMed]
- Gong, M.C.; Arbogast, S.; Guo, Z.; Mathenia, J.; Su, W.; Reid, M.B. Calcium-independent phospholipase A2 modulates cytosolic oxidant activity and contractile function in murine skeletal muscle cells. J. Appl. Physiol. 2006, 100, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Reid, M.B. Redox modulation of contractile function in respiratory and limb skeletal muscle. Respir. Physiol. Neurobiol. 2006, 151, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Radák, Z.; Taylor, A.W.; Ohno, H.; Goto, S. Adaptation to exercise-induced oxidative stress: From muscle to brain. Exerc. Immunol. Rev. 2001, 7, 90–107. [Google Scholar] [PubMed]
- Reid, M.B. Free radicals and muscle fatigue: Of ROS, canaries, and the IOC. Free Radic. Biol. Med. 2008, 44, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Lamb, G.D.; Westerblad, H. Skeletal muscle fatigue: Cellular mechanisms. Physiol. Rev. 2008, 88, 287–332. [Google Scholar] [CrossRef] [PubMed]
- Lamb, G.D.; Westerblad, H. Acute effects of reactive oxygen and nitrogen species on the contractile function of skeletal muscle. J. Physiol. 2011, 589, 2119–2127. [Google Scholar] [CrossRef] [PubMed]
- Lamb, G.D.; Posterino, G.S. Effects of oxidation and reduction on contractile function in skeletal muscle fibres of the rat. J. Physiol. 2003, 546, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Posterino, G.S.; Cellini, M.A.; Lamb, G.D. Effects of oxidation and cytosolic redox conditions on excitation-contraction coupling in rat skeletal muscle. J. Physiol. 2003, 547, 807–823. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.J.; Bruton, J.D.; Lanner, J.T.; Westerblad, H. Antioxidant treatments do not improve force recovery after fatiguing stimulation of mouse skeletal muscle fibres. J. Physiol. 2015, 593, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Andrade, F.H.; Reid, M.B.; Allen, D.G.; Westerblad, H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J. Physiol. 1998, 509, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Prochniewicz, E.; Spakowicz, D.; Thomas, D.D. Changes in actin structural transitions associated with oxidative inhibition of muscle contraction. Biochemistry 2008, 47, 11811–11817. [Google Scholar] [CrossRef] [PubMed]
- Mollica, J.P.; Dutka, T.L.; Merry, T.L.; Lamboley, C.R.; McConell, G.K.; McKenna, M.J.; Murphy, R.M.; Lamb, G.D. S-glutathionylation of troponin I (fast) increases contractile apparatus Ca2+ sensitivity in fast-twitch muscle fibres of rats and humans. J. Physiol. 2012, 590, 1443–1463. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J.; Papa, S.; Bolaños, J.; Bruckdorfer, R.; Carlsen, H.; Elliott, R.M.; Flier, J.; Griffiths, H.R.; Heales, S.; Holst, B.; et al. Antioxidants, reactive oxygen and nitrogen species, gene induction and mitochondrial function. Mol. Aspects Med. 2002, 23, 209–285. [Google Scholar] [CrossRef] [PubMed]
- Gumucio, J.P.; Mendias, C.L. Atrogin-1, MuRF-1, and sarcopenia. Endocrine 2013, 43, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Witt, S.H.; Granzier, H.; Witt, C.C.; Labeit, S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: Towards understanding MURF-dependent muscle ubiquitination. J. Mol. Biol. 2005, 350, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Brault, J.J.; Gygi, S.P.; Glass, D.J.; Valenzuela, D.M.; Gartner, C.; Latres, E.; Goldberg, A.L. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J. Cell Biol. 2009, 185, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L. Antioxidants and oxidative stress in exercise. Proc. Soc. Exp. Biol. Med. 1999, 222, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.V.; Jones, D.P.; Guenther, T.M.; Lash, L.H.; Lauterburg, B.H. Compartmentation of glutathione: Implications for the study of toxicity and disease. Toxicol. Appl. Pharmacol. 1996, 140, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Ann. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J. Free Radicals in Biology and Medicine; Oxford University Press: New York, NY, USA, 2007. [Google Scholar]
- Bellomo, G.; Mirabelli, F.; DiMonte, D.; Richelmi, P.; Thor, H.; Orrenius, C.; Orrenius, S. Formation and reduction of glutathione-protein mixed disulfides during oxidative stress. A study with isolated hepatocytes and menadione (2-methyl-1,4-naphthoquinone). Biochem. Pharmacol. 1987, 36, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L.; Fu, R.; Mitchell, E.W. Glutathione and antioxidant enzymes in skeletal muscle: Effects of fiber type and exercise intensity. J. Appl. Physiol. 1992, 73, 1854–1859. [Google Scholar] [PubMed]
- Lew, H.; Pyke, S.; Quintanilha, A. Changes in the glutathione status of plasma, liver and muscle following exhaustive exercise in rats. FEBS Lett. 1985, 185, 262–266. [Google Scholar] [PubMed]
- Marin, E.; Kretzschmar, M.; Arokoski, J.; Hänninen, O.; Klinger, W. Enzymes of glutathione synthesis in dog skeletal muscles and their response to training. Acta Physiol. Scand. 1993, 147, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Leeuwenburgh, C.; Hollander, J.; Leichtweis, S.; Griffiths, M.; Gore, M.; Ji, L.L. Adaptations of glutathione antioxidant system to endurance training are tissue and muscle fiber specific. Am. J. Physiol. 1997, 272, R363–R369. [Google Scholar] [PubMed]
- Ji, L.L. Exercise and oxidative stress: Role of the cellular antioxidant systems. Exerc. Sport Sci. Rev. 1995, 23, 135–166. [Google Scholar] [PubMed]
- Sen, C.K. Oxidants and antioxidants in exercise. J. Appl. Physiol. 1995, 79, 675–686. [Google Scholar] [PubMed]
- Fisher-Wellman, K.H.; Mattox, T.A.; Thayne, K.; Katunga, L.A.; la Favor, J.D.; Neufer, P.D.; Hickner, R.C.; Wingard, C.J.; Anderson, E.J. Novel role for thioredoxin reductase-2 in mitochondrial redox adaptations to obesogenic diet and exercise in heart and skeletal muscle. J. Physiol. 2013, 591, 3471–3486. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L. Modulation of skeletal muscle antioxidant defense by exercise: Role of redox signaling. Free Radic. Biol. Med. 2008, 44, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Criswell, D.; Lawler, J.; Martin, D.; Ji, L.L.; Herb, R.A.; Dudley, G. Regional training-induced alterations in diaphragmatic oxidative and antioxidant enzymes. Respir. Physiol. 1994, 95, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Gore, M.; Fiebig, R.; Hollander, J.; Leeuwenburgh, C.; Ohno, H.; Ji, L.L. Endurance training alters antioxidant enzyme gene expression in rat skeletal muscle. Can. J. Physiol. Pharmacol. 1998, 76, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Hollander, J.; Fiebig, R.; Gore, M.; Bejma, J.; Ookawara, T.; Ohno, H.; Ji, L.L. Superoxide dismutase gene expression in skeletal muscle: Fiber-specific adaptation to endurance training. Am. J. Physiol. 1999, 277, R856–R862. [Google Scholar] [PubMed]
- Lambertucci, R.H.; Levada-Pires, A.C.; Rossoni, L.V.; Curi, R.; Pithon-Curi, T.C. Effects of aerobic exercise training on antioxidant enzyme activities and mRNA levels in soleus muscle from young and aged rats. Mech. Ageing Dev. 2007, 128, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K.; Oberbach, A.; Klöting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Blüher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L.; Fu, R. Responses of glutathione system and antioxidant enzymes to exhaustive exercise and hydroperoxide. J. Appl. Physiol. 1992, 72, 549–554. [Google Scholar] [PubMed]
- Lawler, J.M.; Powers, S.K.; Visser, T.; van Dijk, H.; Kordus, M.J.; Ji, L.L. Acute exercise and skeletal muscle antioxidant and metabolic enzymes: Effects of fiber type and age. Am. J. Physiol. 1993, 265, R1344–R1350. [Google Scholar] [PubMed]
- Hollander, J.; Fiebig, R.; Gore, M.; Ookawara, T.; Ohno, H.; Ji, L.L. Superoxide dismutase gene expression is activated by a single bout of exercise in rat skeletal muscle. Pflugers Arch. 2001, 442, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, Y.; Watanabe, S.; Kizaki, T.; Sakurai, T.; Takemasa, T.; Haga, S.; Ookawara, T.; Suzuki, K.; Ohno, H. Acute exercise increases expression of extracellular superoxide dismutase in skeletal muscle and the aorta. Redox. Rep. 2008, 13, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Radák, Z.; Asano, K.; Inoue, M.; Kizaki, T.; Oh-Ishi, S.; Suzuki, K.; Taniguchi, N.; Ohno, H. Superoxide dismutase derivative reduces oxidative damage in skeletal muscle of rats during exhaustive exercise. J. Appl. Physiol. 1995, 79, 129–135. [Google Scholar] [PubMed]
- Alessio, H.M.; Goldfarb, A.H. Lipid peroxidation and scavenger enzymes during exercise: Adaptive response to training. J. Appl. Physiol. 1988, 64, 1333–1336. [Google Scholar] [PubMed]
- Mastaloudis, A.; Leonard, S.W.; Traber, M.G. Oxidative stress in athletes during extreme endurance exercise. Free Radic. Biol. Med. 2001, 31, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Steensberg, A.; Morrow, J.; Toft, A.D.; Bruunsgaard, H.; Pedersen, B.K. Prolonged exercise, lymphocyte apoptosis and F2-isoprostanes. Eur. J. Appl. Physiol. 2002, 87, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Ashton, T.; Rowlands, C.C.; Jones, E.; Young, I.S.; Jackson, S.K.; Davies, B.; Peters, J.R. Electron spin resonance spectroscopic detection of oxygen-centred radicals in human serum following exhaustive exercise. Eur. J. Appl. Physiol. Occup. Physiol. 1998, 77, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Vasankari, T.; Kujala, U.; Heinonen, O.; Kapanen, J.; Ahotupa, M. Measurement of serum lipid peroxidation during exercise using three different methods: Diene conjugation, thiobarbituric acid reactive material and fluorescent chromolipids. Clin. Chim. Acta 1995, 234, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Umegaki, K.; Daohua, P.; Sugisawa, A.; Kimura, M.; Higuchi, M. Influence of one bout of vigorous exercise on ascorbic acid in plasma and oxidative damage to DNA in blood cells and muscle in untrained rats. J. Nutr. Biochem. 2000, 11, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Niess, A.M.; Hartmann, A.; Grünert-Fuchs, M.; Poch, B.; Speit, G. DNA damage after exhaustive treadmill running in trained and untrained men. Int. J. Sports Med. 1996, 17, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Criswell, D.; Lawler, J.; Ji, L.L.; Martin, D.; Herb, R.A.; Dudley, G. Influence of exercise and fiber type on antioxidant enzyme activity in rat skeletal muscle. Am. J. Physiol. 1994, 266, R375–R380. [Google Scholar] [PubMed]
- Radák, Z.; Naito, H.; Kaneko, T.; Tahara, S.; Nakamoto, H.; Takahashi, R.; Cardozo-Pelaez, F.; Goto, S. Exercise training decreases DNA damage and increases DNA repair and resistance against oxidative stress of proteins in aged rat skeletal muscle. Pflugers Arch. 2002, 445, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Radák, Z.; Chung, H.Y.; Goto, S. Systemic adaptation to oxidative challenge induced by regular exercise. Free Radic. Biol. Med. 2008, 44, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Alleman, R.J.; Katunga, L.A.; Nelson, M.A.M.; Brown, D.A.; Anderson, E.J. The “Goldilocks Zone” from a redox perspective—Adaptive vs. deleterious responses to oxidative stress in striated muscle. Front. Physiol. 2014. [Google Scholar] [CrossRef]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs. preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Dodd, S.L.; Gagnon, B.J.; Senf, S.M.; Hain, B.A.; Judge, A.R. Ros-mediated activation of NF-kappaB and Foxo during muscle disuse. Muscle Nerve 2010, 41, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Derbre, F.; Ferrando, B.; Gomez-Cabrera, M.C.; Sanchis-Gomar, F.; Martinez-Bello, V.E.; Olaso-Gonzalez, G.; Diaz, A.; Gratas-Delamarche, A.; Cerda, M.; Viña, J. Inhibition of xanthine oxidase by allopurinol prevents skeletal muscle atrophy: Role of p38 MAPKinase and E3 ubiquitin ligases. PLoS ONE 2012, 7, e46668. [Google Scholar] [CrossRef] [PubMed]
- Kröller-Schön, S.; Jansen, T.; Hauptmann, F.; Schüler, A.; Heeren, T.; Hausding, M.; Oelze, M.; Viollet, B.; Keaney, J.F., Jr.; Wenzel, P.; et al. α1AMP-activated protein kinase mediates vascular protective effects of exercise. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- McArdle, F.; Spiers, S.; Aldemir, H.; Vasilaki, A.; Beaver, A.; Iwanejko, L.; McArdle, A.; Jackson, M.J. Preconditioning of skeletal muscle against contraction-induced damage: The role of adaptations to oxidants in mice. J. Physiol. 2004, 561, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Eckl, P.M.; Ortner, A.; Esterbauer, H. Genotoxic properties of 4-hydroxyalkenals and analogous aldehydes. Mutat. Res. 1993, 290, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Schaur, R.J.; Siems, W.G.; Leonarduzzi, G. 4-hydroxynonenal: A membrane lipid oxidation product of medicinal interest. Med. Res. Rev. 2008, 28, 569–631. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Ruiz, E.; Leake, D.S.; Unoki, H.; Yamamoto, M.; Mann, G.E. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: Activation by oxidatively modified LDL and 4-hydroxynonenal. Circ. Res. 2004, 94, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Levonen, A.L.; Landar, A.; Ramachandra, A.; Cease, E.K.; Dickinson, D.A.; Zanoni, G.; Morrow, J.D.; Darley-Usmar, V.M. Cellular mechanisms of redox cell signalling: Role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem. J. 2004, 378, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Echtay, K.S.; Esteves, T.C.; Pakay, J.L.; Jekabsons, M.B.; Lambert, A.J.; Portero-Otín, M.; Pamplona, R.; Vidal-Puig, A.J.; Wang, S.; Roebuck, S.J.; et al. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J. 2003, 22, 4103–4110. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.C.; Borrás, C.; Pallardó, F.V.; Sastre, J.; Ji, L.L.; Viña, J. Decreasing xanthine oxidase-mediated oxidative stress prevents useful cellular adaptations to exercise in rats. J. Physiol. 2005, 567, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.C.; Domenech, E.; Viña, J. Moderate exercise is an antioxidant: Upregulation of antioxidant genes by training. Free Radic. Biol. Med. 2008, 44, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L.; Gomez-Cabrera, M.C.; Steinhafel, N.; Viña, J. Acute exercise activates nuclear factor (NF)-kappaB signaling pathway in rat skeletal muscle. FASEB J. 2004, 18, 1499–1506. [Google Scholar] [CrossRef]
- Margonis, K.; Fatouros, I.G.; Jamurtas, A.Z.; Nikolaidis, M.G.; Douroudos, I.; Chatzinikolaou, A.; Mitrakou, A.; Mastorakos, G.; Papassotiriou, I.; Taxildaris, K.; et al. Oxidative stress biomarkers responses to physical overtraining: Implications for diagnosis. Free Radic. Biol. Med. 2007, 43, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Palazzetti, S.; Richard, M.J.; Favier, A.; Margaritis, I. Overloaded training increases exercise-induced oxidative stress and damage. Can. J. Appl. Physiol. 2003, 28, 588–604. [Google Scholar] [CrossRef] [PubMed]
- Tanskanen, M.; Atalay, M.; Uusitalo, A. Altered oxidative stress in overtrained athletes. J. Sports Sci. 2010, 28, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Irrcher, I.; Adhihetty, P.J.; Sheehan, T.; Joseph, A.M.; Hood, D.A. PPARγ coactivator-1α expression during thyroid hormone- and contractile activity-induced mitochondrial adaptations. Am. J. Physiol. Cell Physiol. 2003, 284, C1669–C1677. [Google Scholar] [CrossRef] [PubMed]
- Holloszy, J.O. Regulation by exercise of skeletal muscle content of mitochondria and GLUT4. J. Physiol. Pharmacol. 2008, 59, 5–18. [Google Scholar] [PubMed]
- Yan, Z.; Okutsu, M.; Akhtar, Y.N.; Lira, V.A. Regulation of exercise-induced fiber type transformation, mitochondrial biogenesis, and angiogenesis in skeletal muscle. J. Appl. Physiol. 2011, 110, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, L.J.C.; Goodpaster, B.H. Increased intramuscular lipid storage in the insulin-resistant and endurance-trained state. Pflugers Arch. 2006, 451, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Hawley, J.A.; Lessard, S.J. Exercise training-induced improvements in insulin action. Acta Physiol. 2008, 192, 127–135. [Google Scholar] [CrossRef]
- Olesen, J.; Kiilerich, K.; Pilegaard, H. PGC-1α-mediated adaptations in skeletal muscle. Pflugers Arch. 2010, 460, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Ji, L.L. Role of PGC-1α in muscle function and aging. J. Sport Health Sci. 2013, 2, 81–86. [Google Scholar] [CrossRef]
- Pilegaard, H.; Osada, T.; Andersen, L.T.; Helge, J.W.; Saltin, B.; Neufer, P.D. Substrate availability and transcriptional regulation of metabolic genes in human skeletal muscle during recovery from exercise. Metabolism 2005, 54, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Krämer, D.K.; Ahlsen, M.; Norrbom, J.; Jansson, E.; Hjeltnes, N.; Gustafsson, T.; Krook, A. Human skeletal muscle fibre type variations correlate with PPARα, PPARδ and PGC-1α mRNA. Acta Physiol. 2006, 188, 207–216. [Google Scholar]
- Mathai, A.S.; Bonen, A.; Benton, C.R.; Robinson, D.L.; Graham, T.E. Rapid exercise-induced changes in PGC-1α mRNA and protein in human skeletal muscle. J. Appl. Physiol. 2008, 105, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Little, J.P.; Safdar, A.; Bishop, D.; Tarnopolsky, M.A.; Gibala, M.J. An acute bout of high-intensity interval training increases the nuclear abundance of PGC-1α and activates mitochondrial biogenesis in human skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1303–R1310. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N.; Bartlett, J.D.; Kayani, A.; Murray, S.W.; Louhelainen, J.; Donovan, T.; Waldron, S.; Gregson, W.; Burniston, J.G.; Morton, J.P.; et al. PGC-1α transcriptional response and mitochondrial adaptation to acute exercise is maintained in skeletal muscle of sedentary elderly males. Biogerontology 2012, 13, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.C.; Han, D.H.; Garcia-Roves, P.M.; Geiger, P.C.; Jones, T.E.; Holloszy, J. Exercise-induced mitochondrial biogenesis begins before the increase in muscle PGC-1α expression. J. Biol. Chem. 2007, 282, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Koves, T.R.; Li, P.; An, J.; Akimoto, T.; Slentz, D.; Ilkayeva, O.; Dohm, G.L.; Yan, Z.; Newgard, C.B.; Muoio, D.M. Peroxisome proliferator-activated receptor-γ co-activator 1α-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J. Biol. Chem. 2005, 280, 33588–33598. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Chin, S.; Li, P.; Liu, F.; Maratos-Flier, E.; Lebrasseur, N.K.; Yan, Z.; Spiegelman, B.M. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1α muscle-specific knock-out animals. J. Biol. Chem. 2007, 282, 30014–30021. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 2012, 23, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Oberkofler, H.; Schraml, E.; Krempler, F.; Patsch, W. Potentiation of liver X receptor transcriptional activity by peroxisome-proliferator-activated receptor γ co-activator 1α. Biochem. J. 2003, 371, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Summermatter, S.; Baum, O.; Santos, G.; Hoppeler, H.; Handschin, C. Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) promotes skeletal muscle lipid refueling in vivo by activating de novo lipogenesis and the pentose phosphate pathway. J. Biol. Chem. 2010, 285, 32793–32800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arany, Z.; Foo, S.Y.; Ma, Y.; Ruas, J.L.; Bommi-Reddy, A.; Girnun, G.; Cooper, M.; Laznik, D.; Chinsomboon, J.; Rangwala, S.M.; et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1α. Nature 2008, 451, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- Chinsomboon, J.; Ruas, J.; Gupta, R.K.; Thom, R.; Shoag, J.; Rowe, G.C.; Sawada, N.; Raghuram, S.; Arany, Z. The transcriptional coactivator PGC-1α mediates exercise-induced angiogenesis in skeletal muscle. Proc. Natl. Acad. Sci. USA 2009, 106, 21401–21406. [Google Scholar] [CrossRef] [PubMed]
- Chin, E.R.; Olson, E.N.; Richardson, J.A.; Yang, Q.; Humphries, C.; Shelton, J.M.; Wu, H.; Zhu, W.; Bassel-Duby, R.; Williams, R.S. A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Genes Dev. 1998, 12, 2499–2509. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Naya, F.J.; McKinsey, T.A.; Mercer, B.; Shelton, J.M.; Chin, E.R.; Simard, A.R.; Michel, R.N.; Bassel-Duby, R.; Olson, E.N.; et al. MEF2 responds to multiple calcium-regulated signals in the control of skeletal muscle fiber type. EMBO J. 2000, 19, 1963–1973. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Wu, H.; Arnold, M.A.; Shelton, J.M.; Backs, J.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J. Clin. Investig. 2007, 117, 2459–2467. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Rhee, J.; Lin, J.; Tarr, P.T.; Spiegelman, B.M. An autoregulatory loop controls peroxisome proliferator-activated receptor γ coactivator 1α expression in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 7111–7116. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; O’Moore, K.M.; Dickman, J.R.; Ji, L.L. Exercise activation of muscle peroxisome proliferator-activated receptor-gamma coactivator-1α signaling is redox sensitive. Free Radic. Biol. Med. 2009, 47, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Strobel, N.A.; Matsumoto, A.; Peake, J.M.; Marsh, S.A.; Peternelj, T.T.; Briskey, D.; Fassett, R.G.; Coombes, J.S.; Wadley, G.D. Altering the redox state of skeletal muscle by glutathione depletion increases the exercise-activation of PGC-1α. Physiol. Rep. 2014, 2, e12224. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a new target of PGC-1α, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS ONE 2010, 5, e11707. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef] [PubMed]
- Finley, L.W.; Haas, W.; Desquiret-Dumas, V.; Wallace, D.C.; Procaccio, V.; Gygi, S.P.; Haigis, M.C. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS ONE 2011, 6, e23295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [PubMed]
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.; Guan, K.L.; Zhao, S.; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011, 12, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Wenz, T.; Rossi, S.G.; Rotundo, R.L.; Spiegelman, B.M.; Moraes, C.T. Increased muscle PGC-1α expression protects from sarcopenia and metabolic disease during aging. Proc. Natl. Acad. Sci. USA 2009, 106, 20405–20410. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Lin, J.; Krauss, S.; Tarr, P.T.; Yang, R.; Newgard, C.B.; Spiegelman, B.M. Bioenergetic analysis of peroxisome proliferator-activated receptor γ coactivators 1α and 1β (PGC-1α and PGC-1β) in muscle cells. J. Biol. Chem. 2003, 278, 26597–26603. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Steinbacher, P.; Eckl, P. Impact of Oxidative Stress on Exercising Skeletal Muscle. Biomolecules 2015, 5, 356-377. https://doi.org/10.3390/biom5020356
AMA Style
Steinbacher P, Eckl P. Impact of Oxidative Stress on Exercising Skeletal Muscle. Biomolecules. 2015; 5(2):356-377. https://doi.org/10.3390/biom5020356
Chicago/Turabian StyleSteinbacher, Peter, and Peter Eckl. 2015. "Impact of Oxidative Stress on Exercising Skeletal Muscle" Biomolecules 5, no. 2: 356-377. https://doi.org/10.3390/biom5020356