
The Interplay between Alpha-Synuclein Clearance and Spreading

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Parkinson’s Disease
1.2. Alpha-Synuclein
2. Protein Degradation Systems
2.1. α-Syn and CMA—A Symbiotic Relation with a “Do not Disturb” Sign

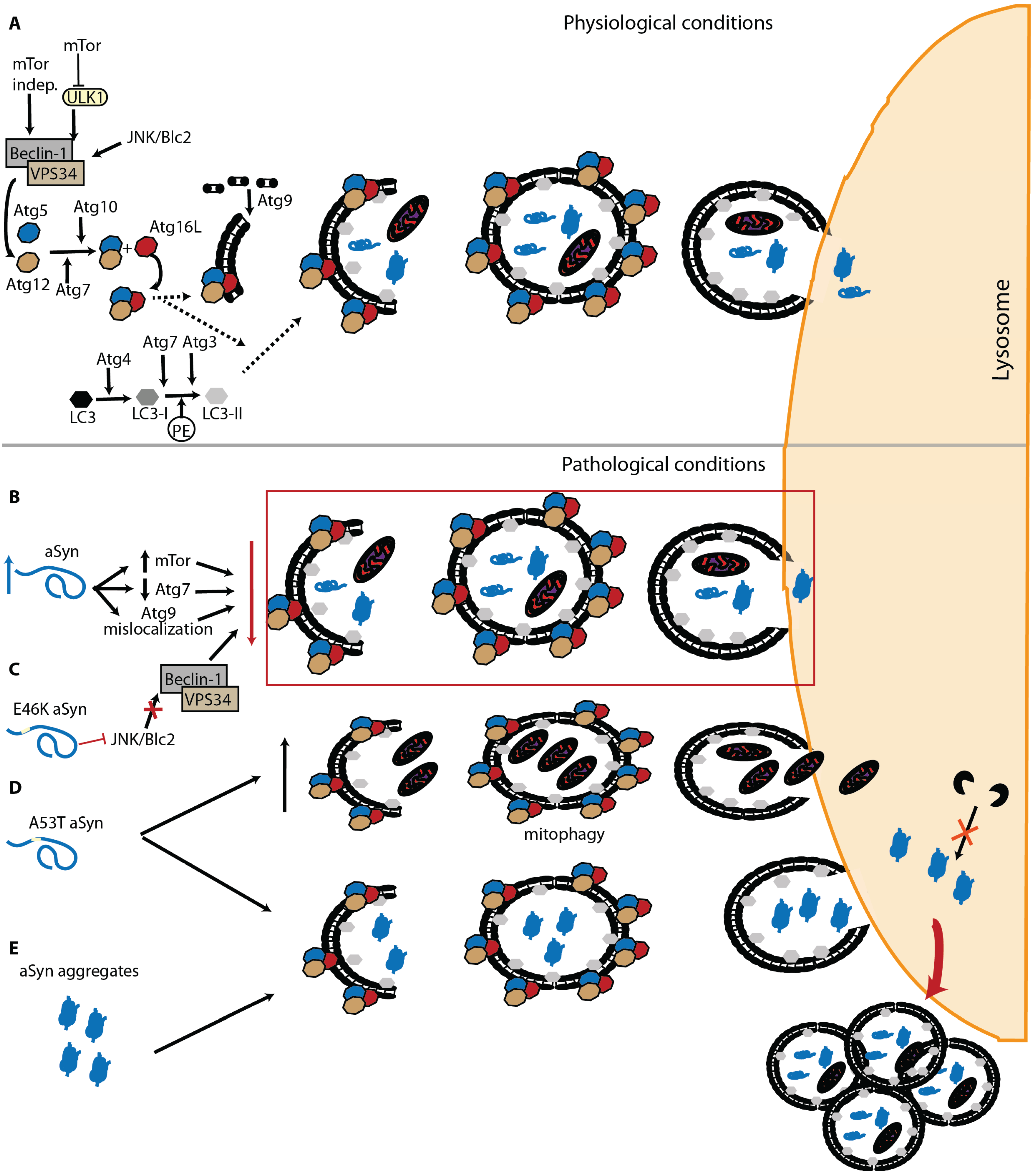
2.2. Macroautophagy of α-Syn—The Last Resource to Avoid Protein Aggregation

3. Prion-Like Spreading of α-Syn Pathology
3.1. Insight from Pathological Studies
3.2. The Proof of Concept: in Vivo α-Syn Propagation Experiments
3.3. Mechanisms of α-Syn Cell-to-Cell Propagation
3.4. What Species of α-Syn are Transmitted and Spread the Pathology?
4. Interplay between α-Syn Clearance Mechanisms and Spreading
4.1. Lysosomal Activity and the Generation of Spreading-Competent α-Syn Species

4.2. Macrosecretion and Lysosome-Mediated Exocytosis

4.3. Extracellular α-Syn Species
4.4. The Putative Role of Glial Cells in the Spreading of α-Syn Pathology
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Marti, M.J.; Tolosa, E.; Campdelacreu, J. Clinical overview of the synucleinopathies. Mov. Disord. 2003, 18, S21–S27. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, K.R.; Naidu, Y. Early Parkinson’s disease and non-motor issues. J. Neurol. 2008, 255, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Harding, A.J.; Stimson, E.; Henderson, J.M.; Halliday, G.M. Clinical correlates of selective pathology in the amygdala of patients with Parkinson’s disease. Brain 2002, 125, 2431–2445. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Kikuchi, A.; Hirayama, K.; Nishio, Y.; Hosokai, Y.; Kanno, S.; Hasegawa, T.; Sugeno, N.; Konno, M.; Suzuki, K.; et al. Severe olfactory dysfunction is a prodromal symptom of dementia associated with Parkinson’s disease: A 3 year longitudinal study. Brain 2012, 135, 161–169. [Google Scholar] [CrossRef]
- Moessnang, C.; Frank, G.; Bogdahn, U.; Winkler, J.; Greenlee, M.W.; Klucken, J. Altered activation patterns within the olfactory network in Parkinson’s disease. Cereb. Cortex 2011, 21, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; You, H.; Liu, J.F.; Ni, D.F.; Zhang, Z.X.; Guan, J. Association of olfactory bulb volume and olfactory sulcus depth with olfactory function in patients with Parkinson disease. Am. J. Neuroradiol. 2011, 32, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Ansari, K.A.; Johnson, A. Olfactory function in patients with Parkinson’s disease. J. Chronic Dis. 1975, 28, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Simuni, T.; Sethi, K. Nonmotor manifestations of Parkinson’s disease. Ann. Neurol. 2008, 64, S65–S80. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Stacy, M. Non-motor symptoms in Parkinson’s disease. J. Neurol. 2009, 256, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, D.A.; Lees, A.J.; Schrag, A. What are the most important nonmotor symptoms in patients with Parkinson’s disease and are we missing them? Mov. Disord. 2010, 25, 2493–2500. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Goetz, C.G.; Kordower, J.H.; Stoessl, A.J.; Sossi, V.; Brin, M.F.; Shannon, K.M.; Nauert, G.M.; Perl, D.P.; Godbold, J.; et al. A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann. Neurol. 2003, 54, 403–414. [Google Scholar] [CrossRef]
- Lindvall, O.; Sawle, G.; Widner, H.; Rothwell, J.C.; Bjorklund, A.; Brooks, D.; Brundin, P.; Frackowiak, R.; Marsden, C.D.; Odin, P.; et al. Evidence for long-term survival and function of dopaminergic grafts in progressive Parkinson’s disease. Ann. Neurol. 1994, 35, 172–180. [Google Scholar] [CrossRef]
- Kordower, J.H.; Freeman, T.B.; Snow, B.J.; Vingerhoets, F.J.; Mufson, E.J.; Sanberg, P.R.; Hauser, R.A.; Smith, D.A.; Nauert, G.M.; Perl, D.P.; et al. Neuropathological evidence of graft survival and striatal reinnervation after the transplantation of fetal mesencephalic tissue in a patient with Parkinson’s disease. N. Engl. J. Med. 1995, 332, 1118–1124. [Google Scholar] [CrossRef]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Bjorklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Wales, P.; Pinho, R.; Lazaro, D.F.; Outeiro, T.F. Limelight on alpha-synuclein: Pathological and mechanistic implications in neurodegeneration. J. Parkinsons Dis. 2013, 3, 415–459. [Google Scholar] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of alpha-synuclein causes parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honore, A.; Rozas, N.; Pieri, L.; Madiona, K.; Durr, A.; Melki, R.; et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef] [Green Version]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Poyhonen, M.; Paetau, A. A novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1–2180.e5. [Google Scholar] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef]
- Simon-Sanchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef]
- Edwards, T.L.; Scott, W.K.; Almonte, C.; Burt, A.; Powell, E.H.; Beecham, G.W.; Wang, L.; Zuchner, S.; Konidari, I.; Wang, G.; et al. Genome-wide association study confirms snps in SNCA and the mapt region as common risk factors for Parkinson disease. Ann. Hum. Genet. 2010, 74, 97–109. [Google Scholar] [CrossRef]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef] [Green Version]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [PubMed]
- Surewicz, W.K.; Epand, R.M.; Pownall, H.J.; Hui, S.W. Human apolipoprotein A–I forms thermally stable complexes with anionic but not with zwitterionic phospholipids. J. Biol. Chem. 1986, 261, 16191–16197. [Google Scholar] [PubMed]
- Vamvaca, K.; Volles, M.J.; Lansbury, P.T., Jr. The first N-terminal amino acids of alpha-synuclein are essential for alpha-helical structure formation in vitro and membrane binding in yeast. J. Mol. Biol. 2009, 389, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Ahlstrom, L.S.; Leftin, A.; Kamp, F.; Haass, C.; Brown, M.F.; Beyer, K. The N-terminus of the intrinsically disordered protein alpha-synuclein triggers membrane binding and helix folding. Biophys. J. 2010, 99, 2116–2124. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Murray, I.V.; Trojanowski, J.Q.; Lee, V.M. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef] [PubMed]
- Izawa, Y.; Tateno, H.; Kameda, H.; Hirakawa, K.; Hato, K.; Yagi, H.; Hongo, K.; Mizobata, T.; Kawata, Y. Role of C-terminal negative charges and tyrosine residues in fibril formation of alpha-synuclein. Brain Behav. 2012, 2, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.S.; Vorum, H.; Lindersson, E.; Jensen, P.H. Ca2+ binding to alpha-synuclein regulates ligand binding and oligomerization. J. Biol. Chem. 2001, 276, 22680–22684. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.; Pountney, D.L.; Jensen, P.H.; Gai, W.P.; Voelcker, N.H. Calcium(II) selectively induces alpha-synuclein annular oligomers via interaction with the C-terminal domain. Protein Sci. 2004, 13, 3245–3252. [Google Scholar] [CrossRef] [PubMed]
- Souza, J.M.; Giasson, B.I.; Lee, V.M.; Ischiropoulos, H. Chaperone-like activity of synucleins. FEBS Lett. 2000, 474, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. Alpha-synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Perovic, I.; Chittuluru, J.; Kaganovich, A.; Nguyen, L.T.; Liao, J.; Auclair, J.R.; Johnson, D.; Landeru, A.; Simorellis, A.K.; et al. A soluble alpha-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. USA 2011, 108, 17797–17802. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, U.; Newman, A.J.; Luth, E.S.; Bartels, T.; Selkoe, D. In vivo cross-linking reveals principally oligomeric forms of alpha-synuclein and beta-synuclein in neurons and non-neural cells. J. Biol. Chem. 2013, 288, 6371–6385. [Google Scholar] [CrossRef] [PubMed]
- Fauvet, B.; Mbefo, M.K.; Fares, M.B.; Desobry, C.; Michael, S.; Ardah, M.T.; Tsika, E.; Coune, P.; Prudent, M.; Lion, N.; et al. Alpha-synuclein in central nervous system and from erythrocytes, mammalian cells, and escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 2012, 287, 15345–15364. [Google Scholar] [CrossRef] [PubMed]
- Outeiro, T.F.; Putcha, P.; Tetzlaff, J.E.; Spoelgen, R.; Koker, M.; Carvalho, F.; Hyman, B.T.; McLean, P.J. Formation of toxic oligomeric alpha-synuclein species in living cells. PLOS ONE 2008, 3, e1867. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed]
- Karpinar, D.P.; Balija, M.B.; Kugler, S.; Opazo, F.; Rezaei-Ghaleh, N.; Wender, N.; Kim, H.Y.; Taschenberger, G.; Falkenburger, B.H.; Heise, H.; et al. Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson’s disease models. MBO J. 2009, 28, 3256–3268. [Google Scholar]
- El-Agnaf, O.M.; Jakes, R.; Curran, M.D.; Middleton, D.; Ingenito, R.; Bianchi, E.; Pessi, A.; Neill, D.; Wallace, A. Aggregates from mutant and wild-type alpha-synuclein proteins and NAC peptide induce apoptotic cell death in human neuroblastoma cells by formation of beta-sheet and amyloid-like filaments. FEBS Lett. 1998, 440, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Tanik, S.A.; Schultheiss, C.E.; Volpicelli-Daley, L.A.; Brunden, K.R.; Lee, V.M. Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 2013, 288, 15194–15210. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.K.; Razzaq, A.; Ghetti, B.; Lilley, K.S.; Spillantini, M.G. Ubiquitination of alpha-synuclein in lewy bodies is a pathological event not associated with impairment of proteasome function. J. Biol. Chem. 2003, 278, 44405–44411. [Google Scholar] [CrossRef] [PubMed]
- Rott, R.; Szargel, R.; Haskin, J.; Shani, V.; Shainskaya, A.; Manov, I.; Liani, E.; Avraham, E.; Engelender, S. Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 2008, 283, 3316–3328. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Klucken, J.; Patterson, C.; Hyman, B.T.; McLean, P.J. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J. Biol. Chem. 2005, 280, 23727–23734. [Google Scholar] [CrossRef] [PubMed]
- Krumova, P.; Meulmeester, E.; Garrido, M.; Tirard, M.; Hsiao, H.H.; Bossis, G.; Urlaub, H.; Zweckstetter, M.; Kugler, S.; Melchior, F.; et al. Sumoylation inhibits alpha-synuclein aggregation and toxicity. J. Cell Biol. 2011, 194, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Jang, W.H.; Quezado, M.M.; Oh, Y.; Chung, K.C.; Junn, E.; Mouradian, M.M. Proteasome inhibition induces alpha-synuclein sumoylation and aggregate formation. J. Neurol. Sci. 2011, 307, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Shahpasandzadeh, H.; Popova, B.; Kleinknecht, A.; Fraser, P.E.; Outeiro, T.F.; Braus, G.H. Interplay between sumoylation and phosphorylation for protection against alpha-synuclein inclusions. J. Biol. Chem. 2014, 289, 31224–31240. [Google Scholar] [CrossRef] [PubMed]
- Dikiy, I.; Eliezer, D. N-terminal acetylation stabilizes N-terminal helicity in lipid- and micelle-bound alpha-synuclein and increases its affinity for physiological membranes. J. Biol. Chem. 2014, 289, 3652–3665. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, A.S.; Ying, J.; Bax, A. Impact of N-terminal acetylation of alpha-synuclein on its random coil and lipid binding properties. Biochemistry 2012, 51, 5004–5013. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Kim, N.C.; Luth, E.S.; Selkoe, D.J. N-alpha-acetylation of alpha-synuclein increases its helical folding propensity, gm1 binding specificity and resistance to aggregation. PLOS ONE 2014, 9, e103727. [Google Scholar] [CrossRef] [PubMed]
- Okochi, M.; Walter, J.; Koyama, A.; Nakajo, S.; Baba, M.; Iwatsubo, T.; Meijer, L.; Kahle, P.J.; Haass, C. Constitutive phosphorylation of the Parkinson’s disease associated alpha-synuclein. J. Biol. Chem. 2000, 275, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. Alpha-synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Pronin, A.N.; Morris, A.J.; Surguchov, A.; Benovic, J.L. Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J. Biol. Chem. 2000, 275, 26515–26522. [Google Scholar] [CrossRef] [PubMed]
- Arawaka, S.; Wada, M.; Goto, S.; Karube, H.; Sakamoto, M.; Ren, C.H.; Koyama, S.; Nagasawa, H.; Kimura, H.; Kawanami, T.; et al. The role of G-protein-coupled receptor kinase 5 in pathogenesis of sporadic Parkinson’s disease. J. Neurosci. 2006, 26, 9227–9238. [Google Scholar] [CrossRef] [PubMed]
- Inglis, K.J.; Chereau, D.; Brigham, E.F.; Chiou, S.S.; Schobel, S.; Frigon, N.L.; Yu, M.; Caccavello, R.J.; Nelson, S.; Motter, R.; et al. Polo-like kinase 2 (PLK2) phosphorylates alpha-synuclein at serine 129 in central nervous system. J. Biol. Chem. 2009, 284, 2598–2602. [Google Scholar] [CrossRef] [PubMed]
- Mbefo, M.K.; Paleologou, K.E.; Boucharaba, A.; Oueslati, A.; Schell, H.; Fournier, M.; Olschewski, D.; Yin, G.; Zweckstetter, M.; Masliah, E.; et al. Phosphorylation of synucleins by members of the polo-like kinase family. J. Biol. Chem. 2010, 285, 2807–2822. [Google Scholar] [CrossRef] [PubMed]
- Ishii, A.; Nonaka, T.; Taniguchi, S.; Saito, T.; Arai, T.; Mann, D.; Iwatsubo, T.; Hisanaga, S.; Goedert, M.; Hasegawa, M. Casein kinase 2 is the major enzyme in brain that phosphorylates ser129 of human alpha-synuclein: Implication for alpha-synucleinopathies. FEBS Lett. 2007, 581, 4711–4717. [Google Scholar] [CrossRef] [PubMed]
- Basso, E.; Antas, P.; Marijanovic, Z.; Goncalves, S.; Tenreiro, S.; Outeiro, T.F. PLK2 modulates alpha-synuclein aggregation in yeast and mammalian cells. Mol. Neurobiol. 2013, 48, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Ryu, M.Y.; Kim, D.W.; Arima, K.; Mouradian, M.M.; Kim, S.U.; Lee, G. Localization of CKII beta subunits in lewy bodies of Parkinson’s disease. J. Neurol. Sci. 2008, 266, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Feany, M.B. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, S.; Reimao-Pinto, M.M.; Antas, P.; Rino, J.; Wawrzycka, D.; Macedo, D.; Rosado-Ramos, R.; Amen, T.; Waiss, M.; Magalhaes, F.; et al. Phosphorylation modulates clearance of alpha-synuclein inclusions in a yeast model of Parkinson’s disease. PLOS Genet. 2014, 10, e1004302. [Google Scholar] [CrossRef] [PubMed]
- Paleologou, K.E.; Oueslati, A.; Shakked, G.; Rospigliosi, C.C.; Kim, H.Y.; Lamberto, G.R.; Fernandez, C.O.; Schmid, A.; Chegini, F.; Gai, W.P.; et al. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J. Neurosci. 2010, 30, 3184–3198. [Google Scholar] [CrossRef] [PubMed]
- Oueslati, A.; Fournier, M.; Lashuel, H.A. Role of post-translational modifications in modulating the structure, function and toxicity of alpha-synuclein: Implications for Parkinson’s disease pathogenesis and therapies. Prog. Brain Res. 2010, 183, 115–145. [Google Scholar] [PubMed]
- Sato, H.; Kato, T.; Arawaka, S. The role of Ser129 phosphorylation of alpha-synuclein in neurodegeneration of Parkinson’s disease: A review of in vivo models. Rev. Neurosci. 2013, 24, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, S.; Eckermann, K.; Outeiro, T.F. Protein phosphorylation in neurodegeneration: Friend or foe? Front. Mol. Neurosci. 2014. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qiang, M.; Wei, Y.; He, R. A novel molecular mechanism for nitrated α-synuclein-induced cell death. J. Mol. Cell Biol. 2011, 3, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Xu, X.; Xiang, Z.; Zhou, J.; Zhang, Z.; Hu, C.; He, C. Nitrated alpha-synuclein induces the loss of dopaminergic neurons in the substantia nigra of rats. PLOS ONE 2010, 5, e9956. [Google Scholar] [CrossRef] [PubMed]
- Hodara, R.; Norris, E.H.; Giasson, B.I.; Mishizen-Eberz, A.J.; Lynch, D.R.; Lee, V.M.; Ischiropoulos, H. Functional consequences of alpha-synuclein tyrosine nitration: Diminished binding to lipid vesicles and increased fibril formation. J. Biol. Chem. 2004, 279, 47746–47753. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; West, N.; Colla, E.; Pletnikova, O.; Troncoso, J.C.; Marsh, L.; Dawson, T.M.; Jakala, P.; Hartmann, T.; Price, D.L.; et al. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 2162–2167. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in lewy bodies of sporadic Parkinson’s disease and dementia with lewy bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar] [PubMed]
- Murray, I.V.; Giasson, B.I.; Quinn, S.M.; Koppaka, V.; Axelsen, P.H.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry 2003, 42, 8530–8540. [Google Scholar] [CrossRef] [PubMed]
- Ulusoy, A.; Febbraro, F.; Jensen, P.H.; Kirik, D.; Romero-Ramos, M. Co-expression of C-terminal truncated alpha-synuclein enhances full-length alpha-synuclein-induced pathology. Eur. J. Neurosci. 2010, 32, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Mishizen-Eberz, A.J.; Guttmann, R.P.; Giasson, B.I.; Day, G.A., 3rd; Hodara, R.; Ischiropoulos, H.; Lee, V.M.; Trojanowski, J.Q.; Lynch, D.R. Distinct cleavage patterns of normal and pathologic forms of alpha-synuclein by calpain I in vitro. J. Neurochem. 2003, 86, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Mishizen-Eberz, A.J.; Norris, E.H.; Giasson, B.I.; Hodara, R.; Ischiropoulos, H.; Lee, V.M.; Trojanowski, J.Q.; Lynch, D.R. Cleavage of alpha-synuclein by calpain: Potential role in degradation of fibrillized and nitrated species of alpha-synuclein. Biochemistry 2005, 44, 7818–7829. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Maruyama, M.; Akagi, T.; Hashikawa, T.; Kanazawa, I.; Tsuji, S.; Nukina, N. Alpha-synuclein degradation by serine protease neurosin: Implication for pathogenesis of synucleinopathies. Hum. Mol. Genet. 2003, 12, 2625–2635. [Google Scholar] [CrossRef] [PubMed]
- Kasai, T.; Tokuda, T.; Yamaguchi, N.; Watanabe, Y.; Kametani, F.; Nakagawa, M.; Mizuno, T. Cleavage of normal and pathological forms of alpha-synuclein by neurosin in vitro. Neurosci. Lett. 2008, 436, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.; Stefanis, L.; Fredenburg, R.; Lansbury, P. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.W.; Corboy, M.J.; DeMartino, G.N.; Thomas, P.J. Endoproteolytic activity of the proteasome. Science 2003, 299, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Yang, Y.P.; Mao, C.J.; Liu, L.; Zheng, H.F.; Hu, L.F.; Liu, C.F. Crosstalk between the proteasome system and autophagy in the clearance of alpha-synuclein. Acta Pharmacol. Sin. 2013, 34, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Khoshaghideh, F.; Patel, S.; Lee, S.J. Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J. Neurosci. 2004, 24, 1888–1896. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Klionsky, D.J. Protein turnover via autophagy: Implications for metabolism. Annu. Rev. Nutr. 2007, 27, 19–40. [Google Scholar] [CrossRef] [PubMed]
- Dice, J.F. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem. Sci. 1990, 15, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.L.; Terlecky, S.R.; Plant, C.P.; Dice, J.F. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 1989, 246, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L.; Cuervo, A.M.; Taylor, M.R.; Nishino, I.; Blum, J.S.; Dice, J.F.; Sandoval, I.V.; Lippincott-Schwartz, J.; August, J.T.; Saftig, P. Unifying nomenclature for the isoforms of the lysosomal membrane protein LAMP-2. Traffic 2005, 6, 1058–1061. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F. A receptor for the selective uptake and degradation of proteins by lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Agarraberes, F.A.; Terlecky, S.R.; Dice, J.F. An intralysosomal Hsp70 is required for a selective pathway of lysosomal protein degradation. J. Cell Biol. 1997, 137, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [PubMed]
- Mak, S.K.; McCormack, A.L.; Manning-Bog, A.B.; Cuervo, A.M.; di Monte, D.A. Lysosomal degradation of alpha-synuclein in vivo. J. Biol. Chem. 2010, 285, 13621–13629. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Talloczy, Z. Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest. 2008, 118, 777–788. [Google Scholar] [PubMed]
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Park, D.; Stefanis, L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLOS ONE 2009, 4, e5515. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Brekk, O.R.; Landeck, N.; Pitychoutis, P.M.; Papasilekas, T.; Papadopoulou-Daifoti, Z.; Kirik, D.; Stefanis, L. Boosting chaperone-mediated autophagy in vivo mitigates alpha-synuclein-induced neurodegeneration. Brain 2013, 136, 2130–2146. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F. Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L.; Schmidt, C.K.; Neu, S.; Willenborg, M.; Fuertes, G.; Salvador, N.; Tanaka, Y.; Lullmann-Rauch, R.; Hartmann, D.; Heeren, J.; et al. Disturbed cholesterol traffic but normal proteolytic function in LAMP-1/LAMP-2 double-deficient fibroblasts. Mol. Biol. Cell 2004, 15, 3132–3145. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E.; Hannon, G.; Abeliovich, A. A microRNA feedback circuit in midbrain dopamine neurons. Science 2007, 317, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yang, H.; Zhu, D.; Huang, H.; Liu, G.; Lun, P. Targeted suppression of chaperone-mediated autophagy by miR-320A promotes alpha-synuclein aggregation. Int. J. Mol. Sci. 2014, 15, 15845–15857. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Rodriguez-Oroz, M.C.; Obeso, J.A.; Cooper, J.M. Influence of microRNA deregulation on chaperone-mediated autophagy and alpha-synuclein pathology in Parkinson’s disease. Cell Death Dis. 2013, 4, e545. [Google Scholar] [CrossRef] [PubMed]
- Bjersing, J.L.; Bokarewa, M.I.; Mannerkorpi, K. Profile of circulating microRNAs in fibromyalgia and their relation to symptom severity: An exploratory study. Rheumatol. Int. 2015, 35, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Kubiczkova-Besse, L.; Sedlarikova, L.; Kryukov, F.; Nekvindova, J.; Radova, L.; Almasi, M.; Pelcova, J.; Minarik, J.; Pika, T.; Pikalova, Z.; et al. Combination of serum microRNA-320A and microRNA-320B as a marker for waldenstrom macroglobulinemia. Am. J. Hematol. 2015, 90, E51–E52. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Zhong, Y.; Chen, Y.; Du, J.; Ju, X.; Zhao, C.; Zhang, G.; Zhang, L.; Liu, K.; Yang, N.; et al. Hsa-miR-1246, Hsa-miR-320a and Hsa-miR-196B-5P inhibitors can reduce the cytotoxicity of ebola virus glycoprotein in vitro. Sci. China Life Sci. 2014, 57, 959–972. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Suzuki, K.; Ohsumi, Y. Yeast autophagosomes: De novo formation of a membrane structure. Trends Cell Biol. 2002, 12, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Kraft, C.; Martens, S. Mechanisms and regulation of autophagosome formation. Curr. Opin. Cell Biol. 2012, 24, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Tucker, K.A.; Stromhaug, P.E.; Klionsky, D.J. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev. Cell 2004, 6, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Shintani, T.; Nair, U.; Klionsky, D.J. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy 2005, 1, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Orsi, A.; Razi, M.; Dooley, H.C.; Robinson, D.; Weston, A.E.; Collinson, L.M.; Tooze, S.A. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 2012, 23, 1860–1873. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.; Chan, E.Y.; Hu, X.W.; Kochl, R.; Crawshaw, S.G.; High, S.; Hailey, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006, 119, 3888–3900. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, M.E.; Cutler, N.S.; Lorenz, M.C.; di Como, C.J.; Heitman, J. The TOR signaling cascade regulates gene expression in response to nutrients. Genes Dev. 1999, 13, 3271–3279. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Overmeyer, J.H.; Maltese, W.A. Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J. Cell Sci. 2006, 119, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Thumm, M.; Egner, R.; Koch, B.; Schlumpberger, M.; Straub, M.; Veenhuis, M.; Wolf, D.H. Isolation of autophagocytosis mutants of saccharomyces cerevisiae. FEBS Lett. 1994, 349, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Cregg, J.M.; Dunn, W.A., Jr.; Emr, S.D.; Sakai, Y.; Sandoval, I.V.; Sibirny, A.; Subramani, S.; Thumm, M.; Veenhuis, M.; et al. A unified nomenclature for yeast autophagy-related genes. Dev. Cell 2003, 5, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Tanida-Miyake, E.; Ueno, T.; Kominami, E. The human homolog of saccharomyces cerevisiae Apg7p is a protein-activating enzyme for multiple substrates including human Apg12p, GATE-16, GABARAP, and MAP-LC3. J. Biol. Chem. 2001, 276, 1701–1706. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Noda, T.; Ohsumi, Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J. 1999, 18, 3888–3896. [Google Scholar] [CrossRef] [PubMed]
- Romanov, J.; Walczak, M.; Ibiricu, I.; Schuchner, S.; Ogris, E.; Kraft, C.; Martens, S. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012, 31, 4304–4317. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Sou, Y.S.; Ezaki, J.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. HsAtg4b/Hsapg4b/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J. Biol. Chem. 2004, 279, 36268–36276. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Tanida-Miyake, E.; Komatsu, M.; Ueno, T.; Kominami, E. Human Apg3p/Aut1p homologue is an authentic E2 enzyme for multiple substrates, GATE-16, GABARAP, and MAP-LC3, and facilitates the conjugation of Hapg12p to Hapg5p. J. Biol. Chem. 2002, 277, 13739–13744. [Google Scholar] [CrossRef] [PubMed]
- Otomo, C.; Metlagel, Z.; Takaesu, G.; Otomo, T. Structure of the human Atg12~Atg5 conjugate required for LC3 lipidation in autophagy. Nat. Struct. Mol. Biol. 2013, 20, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16l complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Li, L.; Chudaev, M.V.; Chen, J.; Perez, F.A.; Palmiter, R.D.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 2007, 178, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Perander, M.; Outzen, H.; Kristiansen, K.; Overvatn, A.; Michaelsen, E.; Bjorkoy, G.; Johansen, T. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J. Biol. Chem. 2003, 278, 34568–34581. [Google Scholar] [CrossRef] [PubMed]
- Noda, N.N.; Kumeta, H.; Nakatogawa, H.; Satoo, K.; Adachi, W.; Ishii, J.; Fujioka, Y.; Ohsumi, Y.; Inagaki, F. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells Devoted Mol. Cell. Mech. 2008, 13, 1211–1218. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. P62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; Lamark, T.; Sou, Y.S.; Bjorkoy, G.; Nunn, J.L.; Bruun, J.A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.M.; Wong, E.S.; Kirkpatrick, D.S.; Pletnikova, O.; Ko, H.S.; Tay, S.P.; Ho, M.W.; Troncoso, J.; Gygi, S.P.; Lee, M.K.; et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 2008, 17, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Seibenhener, M.L.; Babu, J.R.; Geetha, T.; Wong, H.C.; Krishna, N.R.; Wooten, M.W. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell. Biol. 2004, 24, 8055–8068. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Qian, S.B. Chaperone-mediated hierarchical control in targeting misfolded proteins to aggresomes. Mol. Biol. Cell 2011, 22, 3277–3288. [Google Scholar] [CrossRef] [PubMed]
- Tetzlaff, J.E.; Putcha, P.; Outeiro, T.F.; Ivanov, A.; Berezovska, O.; Hyman, B.T.; McLean, P.J. CHIP targets toxic alpha-synuclein oligomers for degradation. J. Biol. Chem. 2008, 283, 17962–17968. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Kalia, S.K.; Chau, H.; Lozano, A.M.; Hyman, B.T.; McLean, P.J. Ubiquitinylation of alpha-synuclein by carboxyl terminus Hsp70-interacting protein (CHIP) is regulated by Bcl-2-associated athanogene 5 (BAG5). PLOS ONE 2011, 6, e14695. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Shi, J.J.; Yang, Y.P.; Li, J.; Zhang, Y.L.; Chen, J.; Hu, L.F.; Liu, C.F. HDAC6 regulates aggresome-autophagy degradation pathway of alpha-synuclein in response to MPP+-induced stress. J. Neurochem. 2011, 117, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Spencer, B.; Potkar, R.; Trejo, M.; Rockenstein, E.; Patrick, C.; Gindi, R.; Adame, A.; Wyss-Coray, T.; Masliah, E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 2009, 29, 13578–13588. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.K.; Kim, H.T.; Hourez, R.; Jung, J.W.; Kim, K.P.; Goldberg, A.L. Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 17004–17009. [Google Scholar] [CrossRef] [PubMed]
- Paxinou, E.; Chen, Q.; Weisse, M.; Giasson, B.I.; Norris, E.H.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M.; Ischiropoulos, H. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J. Neurosci. 2001, 21, 8053–8061. [Google Scholar] [PubMed]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Salvador, N.; Aguado, C.; Horst, M.; Knecht, E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J. Biol. Chem. 2000, 275, 27447–27456. [Google Scholar] [PubMed]
- Sevlever, D.; Jiang, P.; Yen, S.H. Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 2008, 47, 9678–9687. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, D.; Dodson, M.; Ouyang, X.; Boyer-Guittaut, M.; Liang, Q.; Ballestas, M.E.; Fineberg, N.; Zhang, J. Over-expression of an inactive mutant cathepsin D increases endogenous alpha-synuclein and cathepsin B activity in SH-SY5Y cells. J. Neurochem. 2014, 128, 950–961. [Google Scholar] [CrossRef] [PubMed]
- Cullen, V.; Lindfors, M.; Ng, J.; Paetau, A.; Swinton, E.; Kolodziej, P.; Boston, H.; Saftig, P.; Woulfe, J.; Feany, M.B.; et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol. Brain 2009. [Google Scholar] [CrossRef]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. Alpha-synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Crews, L.; Spencer, B.; Desplats, P.; Patrick, C.; Paulino, A.; Rockenstein, E.; Hansen, L.; Adame, A.; Galasko, D.; Masliah, E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLOS ONE 2010, 5, e9313. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.Q.; Yuan, Y.H.; Gao, Y.N.; Huang, J.Y.; Ma, K.L.; Gao, Y.; Zhang, W.Q.; Guo, X.F.; Chen, N.H. Overexpression of human E46K mutant alpha-synuclein impairs macroautophagy via inactivation of JNK1-Bcl-2 pathway. Mol. Neurobiol. 2014, 50, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Choubey, V.; Safiulina, D.; Vaarmann, A.; Cagalinec, M.; Wareski, P.; Kuum, M.; Zharkovsky, A.; Kaasik, A. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem. 2011, 286, 10814–10824. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L.; Larsen, K.; Rideout, H. Expression of A53T mutant but not wild-type α-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J. Neurosci. 2001, 21, 9549–9560. [Google Scholar] [PubMed]
- Braak, H.; Rüb, U.; del Tredici, K. Cognitive decline correlates with neuropathological stage in Parkinson’s disease. J. Neurol. Sci. 2006, 248, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Rüb, U.; Gai, W.P.; del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s disease: The dual hit theory revisited. Ann. NY Acad. Sci. 2009, 1170, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Olanow, C.W.; Freeman, T.B. Transplanted dopaminergic neurons develop PD pathologic changes: A second case report. Mov. Disord. 2008, 23, 2303–2306. [Google Scholar] [CrossRef] [PubMed]
- Brundin, P.; Li, J.-Y.; Holton, J.L.; Lindvall, O.; Revesz, T. Research in motion: The enigma of Parkinson’s disease pathology spread. Nat. Rev. Neurosci. 2008, 9, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Englund, E.; Widner, H.; Rehncrona, S.; Björklund, A.; Lindvall, O.; Brundin, P. Characterization of Lewy body pathology in 12- and 16-year-old intrastriatal mesencephalic grafts surviving in a patient with Parkinson’s disease. Mov. Disord. 2010, 25, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Kordower, J.H. Lewy body pathology in fetal grafts. Ann. NY Acad. Sci. 2010, 1184, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Kurowska, Z.; Englund, E.; Widner, H.; Lindvall, O.; Li, J.Y.; Brundin, P. Signs of degeneration in 12–22-year old grafts of mesencephalic dopamine neurons in patients with Parkinson’s disease. J. Parkinsons Dis. 2011, 1, 83–92. [Google Scholar] [PubMed]
- Hansen, C.; Angot, E.; Bergström, A. Α-synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Invest. 2011, 121, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Angot, E.; Steiner, J.A.; Tomé, C.M.; Ekström, P.; Mattsson, B.; Björklund, A.; Brundin, P. Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLOS ONE 2012, 7, e39465. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Dodiya, H.B.; Kordower, A.M.; Terpstra, B.; Paumier, K.; Madhavan, L.; Sortwell, C.; Steece-Collier, K.; Collier, T.J. Transfer of host-derived alpha synuclein to grafted dopaminergic neurons in rat. Neurobiol. Dis. 2011, 43, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Borghi, R.; Marchese, R.; Negro, A.; Marinelli, L.; Forloni, G.; Zaccheo, D.; Abbruzzese, G.; Tabaton, M. Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neurosci. Lett. 2000, 287, 65–67. [Google Scholar] [CrossRef] [PubMed]
- El-Agnaf, O.M.A.; Salem, S.A.; Paleologou, K.E.; Cooper, L.J.; Fullwood, N.J.; Gibson, M.J.; Curran, M.D.; Court, J.A.; Mann, D.M.A.; Ikeda, S.; et al. Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003, 17, 1945–1947. [Google Scholar] [PubMed]
- Paleologou, K.E.; Kragh, C.L.; Mann, D.M.A.; Salem, S.A.; Al-Shami, R.; Allsop, D.; Hassan, A.H.; Jensen, P.H.; El-Agnaf, O.M.A. Detection of elevated levels of soluble alpha-synuclein oligomers in post-mortem brain extracts from patients with dementia with lewy bodies. Brain 2009, 132, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Parnetti, L.; Chiasserini, D.; Persichetti, E.; Eusebi, P.; Varghese, S.; Qureshi, M.M.; Dardis, A.; Deganuto, M.; de Carlo, C.; Castrioto, A.; et al. Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov. Disord. 2014, 29, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.Y. Intracerebral inoculation of pathological-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Mougenot, A.L.; Nicot, S.; Bencsik, A.; Morignat, E.; Verchère, J.; Lakhdar, L.; Legastelois, S.; Baron, T. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol. Aging 2012, 33, 2225–2228. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.-Y. Pathological α-synuclein transmission initiates parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Oikawa, T.; Arai, T.; Akiyama, H.; Mann, D.M.A.; Hasegawa, M. Prion-like spreading of pathological α-synuclein in brain. Brain 2013, 136, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Recasens, A.; Dehay, B.; Bové, J.; Carballo-Carbajal, I.; Dovero, S.; Pérez-Villalba, A.; Fernagut, P.O.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2014, 75, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Covell, D.J.; Daniels, J.P.; Iba, M.; Stieber, A.; Zhang, B.; Riddle, D.M.; Kwong, L.K.; Xu, Y.; Trojanowski, J.Q.; et al. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 2013, 154, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Sacino, A.N.; Brooks, M.; Thomas, M.A.; McKinney, A.B.; McGarvey, N.H.; Rutherford, N.J.; Ceballos-Diaz, C.; Robertson, J.; Golde, T.E.; Giasson, B.I. Amyloidogenic α-synuclein seeds do not invariably induce rapid, widespread pathology in mice. Acta Neuropathol. 2014, 127, 645–665. [Google Scholar] [CrossRef] [PubMed]
- Recasens, A.; Dehay, B. Alpha-synuclein spreading in Parkinson disease. Front. Neuroanat. 2014. [Google Scholar] [CrossRef]
- Costanzo, M.; Zurzolo, C. The cell biology of prion-like spread of protein aggregates: Mechanisms and implication in neurodegeneration. Biochem. J. 2013, 452, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Petzold, M.; Magdalinou, N. Cerebrospinal fluid α-synuclein levels in Parkinson’s disease—Changed or unchanged? Eur. J. Neurol. 2014, 21, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Patel, S.; Lee, S.-J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Simons, M. Exosomes: Vesicular carriers for intercellular communication in neurodegenerative disorders. Cell Tissue Res. 2013, 352, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Liu, C.; Cook, T.J.; Bullock, K.M.; Zhao, Y.; Ginghina, C.; Li, Y.; Aro, P.; Dator, R.; He, C.; et al. Plasma exosomal α-synuclein is likely CNS-derived and increased in Parkinson’s disease. Acta neuropathol. 2014, 128, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Freundt, E.C.; Maynard, N.; Clancy, E.K.; Roy, S.; Bousset, L.; Sourigues, Y.; Covert, M.; Melki, R.; Kirkegaard, K.; Brahic, M. Neuron-to-neuron transmission of a-synuclein fibrils through axonal transport. Ann. Neurol. 2012, 72, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Kubo, M.; Shimozawa, A.; Akiyama, H.; Hasegawa, M. Pathological alpha-synuclein propagates through neural networks. Acta Neuropathol. Commun. 2014. [Google Scholar] [CrossRef]
- Jang, A.; Lee, H.J.; Suk, J.E.; Jung, J.W.; Kim, K.P.; Lee, S.J. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J. Neurochem. 2010, 113, 1263–1274. [Google Scholar] [PubMed]
- Lee, H.-J.; Baek, S.M.; Ho, D.-H.; Suk, J.-E.; Cho, E.-D.; Lee, S.-J. Dopamine promotes formation and secretion of non-fibrillar alpha-synuclein oligomers. Exp. Mol. Med. 2011, 43, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Obitsu, S.; Teshima, R. Α-synuclein aggregation and transmission are enhanced by leucine-rich repeat kinase 2 in human neuroblastoma SH-SY5Y cells. Biol. Pharm. Bull. 2011, 34, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Fares, M.B.; Ait-Bouziad, N.; Dikiy, I.; Mbefo, M.K.; Jovičić, A.; Kiely, A.; Holton, J.L.; Lee, S.J.; Gitler, A.D.; Eliezer, D.; et al. The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet. 2014, 23, 4491–4509. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, O.; Fauvet, B.; Oueslati, A.; Dikiy, I.; Mahul-Mellier, A.L.; Ruggeri, F.S.; Mbefo, M.K.; Vercruysse, F.; Dietler, G.; Lee, S.J.; et al. The H50Q mutation enhances alpha-synuclein aggregation, secretion, and toxicity. J. Biol. Chem. 2014, 289, 21856–21876. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Clarke, A.R. A general model of prion strains and their pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Mondal, M.; Mohite, G.M.; Singh, P.K.; Ranjan, P.; Anoop, A.; Ghosh, S.; Jha, N.N.; Kumar, A.; Maji, S.K. The parkinson's disease-associated H50Q mutation accelerates α-synuclein aggregation in vitro. Biochemistry 2013, 52, 6925–6927. [Google Scholar] [CrossRef] [PubMed]
- Heise, H.; Celej, M.S.; Becker, S.; Riedel, D.; Pelah, A.; Kumar, A.; Jovin, T.M.; Baldus, M. Solid-state nmr reveals structural differences between fibrils of wild-type and disease-related A53T mutant alpha-synuclein. J. Mol. Biol. 2008, 380, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Ikeda, T.; Takasaki, J.; Yamada, M. Familial Parkinson disease mutations influence alpha-synuclein assembly. Neurobiol. Dis. 2011, 43, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Bharathi; Indi, S.S.; Rao, K.S.J. Copper- and iron-induced differential fibril formation in alpha-synuclein: Tem study. Neurosci. Lett. 2007, 424, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Binolfi, A.; Rasia, R.M.; Bertoncini, C.W.; Ceolin, M.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernández, C.O. Interaction of α-synuclein with divalent metal ions reveals key differences: A link between structure, binding specificity and fibrillation enhancement. J. Am. Chem. Soc. 2006, 128, 9893–9901. [Google Scholar] [CrossRef] [PubMed]
- Comellas, G.; Lemkau, L.R.; Zhou, D.H.; George, J.M.; Rienstra, C.M. Structural intermediates during α-synuclein fibrillogenesis on phospholipid vesicles. J. Am. Chem. Soc. 2012, 134, 5090–5099. [Google Scholar] [CrossRef] [PubMed]
- Hellstrand, E.; Nowacka, A.; Topgaard, D.; Linse, S.; Sparr, E. Membrane lipid co-aggregation with α-synuclein fibrils. PLOS ONE 2013, 8, e77235. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, W.; Antony, T.; Cherny, D.; Heim, G.; Jovin, T.M.; Subramaniam, V. Dependence of alpha-synuclein aggregate morphology on solution conditions. J. Mol. Biol. 2002, 322, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Haasen, D.; Karow, A.R.; Moussaud, S.; Habeck, M.; Giese, A.; Kretzschmar, H.; Hengerer, B.; Kostka, M. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 2007, 27, 9220–9232. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Krebs, S.K.; Wolff, M.; Birk, G.; Hengerer, B. Seeding induced by alpha-synuclein oligomers provides evidence for spreading of alpha-synuclein pathology. J. Neurochem. 2009, 111, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Böckmann, A.; Meier, B.H.; et al. Structural and functional characterization of two alpha-synuclein strains. Nat. Commun. 2013. [Google Scholar] [CrossRef]
- Cuanalo-Contreras, K.; Mukherjee, A.; Soto, C. Role of protein misfolding and proteostasis deficiency in protein misfolding diseases and aging. Int. J. Cell Biol. 2013. [Google Scholar] [CrossRef]
- Lee, H.J.; Cho, E.D.; Lee, K.W.; Kim, J.H.; Cho, S.G.; Lee, S.J. Autophagic failure promotes the exocytosis and intercellular transfer of alpha-synuclein. Exp. Mol. Med. 2013. [Google Scholar] [CrossRef]
- Piper, R.C.; Katzmann, D.J. Biogenesis and function of multivesicular bodies. Annu. Rev. Cell Dev. Biol. 2007, 23, 519–547. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.M.; Chan, B.K.; Park, J.S.; Hill, K.J.; Aitken, J.B.; Cottle, L.; Farghaian, H.; Cole, A.R.; Lay, P.A.; Sue, C.M.; et al. Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes alpha-synuclein externalization via exosomes. Hum. Mol. Genet. 2014, 23, 2816–2833. [Google Scholar] [CrossRef] [PubMed]
- Tsunemi, T.; Hamada, K.; Krainc, D. ATP13A2/PARK9 regulates secretion of exosomes and alpha-synuclein. J. Neurosci. 2014, 34, 15281–15287. [Google Scholar] [CrossRef] [PubMed]
- Tsunemi, T.; Krainc, D. Zn2+ dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum. Mol. Genet. 2014, 23, 2791–2801. [Google Scholar] [CrossRef] [PubMed]
- Leyk, J.; Goldbaum, O.; Noack, M.; Richter-Landsberg, C. Inhibition of HDAC6 modifies Tau inclusion body formation and impairs autophagic clearance. J. Mol. Neurosci. MN 2015, 55, 1031–1046. [Google Scholar] [CrossRef]
- Fader, C.M.; Sanchez, D.; Furlan, M.; Colombo, M.I. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in K562 cells. Traffic 2008, 9, 230–250. [Google Scholar] [CrossRef] [PubMed]
- Poehler, A.M.; Xiang, W.; Spitzer, P.; May, V.; Meixner, H.; Rockenstein, E.; Chutna, O.; Outeiro, T.; Winkler, J.; Masliah, E.; et al. Autophagy modulates SNCA/α-synuclein release, thereby generating a hostile microenvironment. Autophagy 2014, 10, 2171–2192. [Google Scholar] [CrossRef] [PubMed]
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Ginns, E.I.; Mak, S.K.; Ko, N.; Karlgren, J.; Akbarian, S.; Chou, V.P.; Guo, Y.; Lim, A.; Samuelsson, S.; LaMarca, M.L.; et al. Neuroinflammation and alpha-synuclein accumulation in response to glucocerebrosidase deficiency are accompanied by synaptic dysfunction. Mol. Genet. Metab. 2014, 111, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.J.; Yang, N.Y.; Song, M.; Lee, C.S.; Lee, J.S.; Jung, B.C.; Lee, H.J.; Kim, S.; Masliah, E.; Sardi, S.P.; et al. Glucocerebrosidase depletion enhances cell-to-cell transmission of alpha-synuclein. Nat. Commun. 2014, 5, 4755. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Mantle, D.; Falkous, G.; Ishiura, S.; Perry, R.H.; Perry, E.K. Comparison of cathepsin protease activities in brain tissue from normal cases and cases with Alzheimer’s disease, Lewy body dementia, Parkinson’s disease and Huntington’s disease. J. Neurol. Sci. 1995, 131, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, K.D.; Persichetti, E.; Chiasserini, D.; Eusebi, P.; Beccari, T.; Calabresi, P.; Berendse, H.W.; Parnetti, L.; van de Berg, W.D. Changes in endolysosomal enzyme activities in cerebrospinal fluid of patients with Parkinson’s disease. Mov. Disord. 2013, 28, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Yacoubian, T.A.; Wilson, S.; Xie, Z.L.; Speake, L.D.; Parks, R.; Crabtree, D.; et al. Lysosomal enzyme cathepsin d protects against alpha-synuclein aggregation and toxicity. Mol. Brain 2008, 1, 17. [Google Scholar] [CrossRef] [PubMed]
- Tsujimura, A.; Taguchi, K.; Watanabe, Y.; Tatebe, H.; Tokuda, T.; Mizuno, T.; Tanaka, M. Lysosomal enzyme cathepsin B enhances the aggregate forming activity of exogenous α-synuclein fibrils. Neurobiol. Dis. 2014, 73, 244–253. [Google Scholar] [CrossRef]
- Freeman, D.; Cedillos, R.; Choyke, S.; Lukic, Z.; McGuire, K.; Marvin, S.; Burrage, A.M.; Sudholt, S.; Rana, A.; O’Connor, C.; et al. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLOS ONE 2013, 8, e62143. [Google Scholar] [CrossRef] [PubMed]
- Pratt, M.R.; Sekedat, M.D.; Chiang, K.P.; Muir, T.W. Direct measurement of cathepsin B activity in the cytosol of apoptotic cells by an activity-based probe. Chem. Biol. 2009, 16, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Dufty, B.M.; Warner, L.R.; Hou, S.T.; Jiang, S.X.; Gomez-Isla, T.; Leenhouts, K.M.; Oxford, J.T.; Feany, M.B.; Masliah, E.; Rohn, T.T. Calpain-cleavage of alpha-synuclein: Connecting proteolytic processing to disease-linked aggregation. Am. J. Pathol. 2007, 170, 1725–1738. [Google Scholar] [CrossRef] [PubMed]
- Diepenbroek, M.; Casadei, N.; Esmer, H.; Saido, T.C.; Takano, J.; Kahle, P.J.; Nixon, R.A.; Rao, M.V.; Melki, R.; Pieri, L.; et al. Overexpression of the calpain-specific inhibitor calpastatin reduces human alpha-synuclein processing, aggregation and synaptic impairment in [A30P]αSyn transgenic mice. Hum. Mol. Genet. 2014, 23, 3975–3989. [Google Scholar] [CrossRef] [PubMed]
- Crocker, S.J.; Smith, P.D.; Jackson-Lewis, V.; Lamba, W.R.; Hayley, S.P.; Grimm, E.; Callaghan, S.M.; Slack, R.S.; Melloni, E.; Przedborski, S.; et al. Inhibition of calpains prevents neuronal and behavioral deficits in an mptp mouse model of Parkinson’s disease. J. Neurosci. 2003, 23, 4081–4091. [Google Scholar] [PubMed]
- Games, D.; Valera, E.; Spencer, B.; Rockenstein, E.; Mante, M.; Adame, A.; Patrick, C.; Ubhi, K.; Nuber, S.; Sacayon, P.; et al. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J. Neurosci. 2014, 34, 9441–9454. [Google Scholar] [CrossRef] [PubMed]
- Levesque, S.; Wilson, B.; Gregoria, V.; Thorpe, L.B.; Dallas, S.; Polikov, V.S.; Hong, J.S.; Block, M.L. Reactive microgliosis: Extracellular micro-calpain and microglia-mediated dopaminergic neurotoxicity. Brain 2010, 133, 808–821. [Google Scholar] [CrossRef] [PubMed]
- Rcom-H’cheo-Gauthier, A.; Goodwin, J.; Pountney, D.L. Interactions between calcium and α-synuclein in neurodegeneration. Biomolecules 2014, 4, 795–811. [Google Scholar] [CrossRef] [PubMed]
- Reznichenko, L.; Cheng, Q.; Nizar, K.; Gratiy, S.L.; Saisan, P.A.; Rockenstein, E.M.; Gonzalez, T.; Patrick, C.; Spencer, B.; Desplats, P.; et al. In vivo alterations in calcium buffering capacity in transgenic mouse model of synucleinopathy. J. Neurosci. 2012, 32, 9992–9998. [Google Scholar] [CrossRef] [PubMed]
- Melachroinou, K.; Xilouri, M.; Emmanouilidou, E.; Masgrau, R.; Papazafiri, P.; Stefanis, L.; Vekrellis, K. Deregulation of calcium homeostasis mediates secreted alpha-synuclein-induced neurotoxicity. Neurobiol. Aging 2013, 34, 2853–2865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashima, T. Reconsider Alzheimer’s disease by the “calpain-cathepsin hypothesis”—A perspective review. Prog. Neurobiol. 2013, 105, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Syntichaki, P.; Tavernarakis, N. The biochemistry of neuronal necrosis: Rogue biology? Nat. Rev. Neurosci. 2003, 4, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Bruns, C.; McCaffery, J.M.; Curwin, A.J.; Duran, J.M.; Malhotra, V. Biogenesis of a novel compartment for autophagosome-mediated unconventional protein secretion. J. Cell Biol. 2011, 195, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [PubMed]
- Koprich, J.B.; Reske-Nielsen, C.; Mithal, P.; Isacson, O. Neuroinflammation mediated by IL-1β increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson’s disease. J. Neuroinflammation 2008. [Google Scholar] [CrossRef]
- Nilsson, P.; Loganathan, K.; Sekiguchi, M.; Matsuba, Y.; Hui, K.; Tsubuki, S.; Tanaka, M.; Iwata, N.; Saito, T.; Saido, T.C. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013, 5, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.L.; Fraldi, A.; Bouche, V.; Annunziata, F.; Mansueto, G.; Spampanato, C.; Puri, C.; Pignata, A.; Martina, J.A.; Sardiello, M.; et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell 2011, 21, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.W. Regulated secretion of conventional lysosomes. Trends Cell Biol. 2000, 10, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.; Caler, E.V.; Andrews, N.W. Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell 2001, 106, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.W. Membrane repair and immunological danger. EMBO Rep. 2005, 6, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Bjorklund, A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef] [PubMed]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ninan, I.; Antonova, I.; Battaglia, F.; Trinchese, F.; Narasanna, A.; Kolodilov, N.; Dauer, W.; Hawkins, R.D.; Arancio, O. Alpha-synuclein produces a long-lasting increase in neurotransmitter release. EMBO J. 2004, 23, 4506–4516. [Google Scholar] [CrossRef] [PubMed]
- Tatebe, H.; Watanabe, Y.; Kasai, T.; Mizuno, T.; Nakagawa, M.; Tanaka, M.; Tokuda, T. Extracellular neurosin degrades alpha-synuclein in cultured cells. Neurosci. Res. 2010, 67, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Choi, Y.R.; Park, J.Y.; Lee, J.H.; Kim, D.K.; Lee, S.J.; Paik, S.R.; Jou, I.; Park, S.M. Proteolytic cleavage of extracellular alpha-synuclein by plasmin: Implications for Parkinson disease. J. Biol. Chem. 2012, 287, 24862–24872. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.Y.; Park, S.M.; Lee, C.H.; Um, J.W.; Lee, H.J.; Kim, J.; Oh, Y.J.; Lee, S.T.; Paik, S.R.; Chung, K.C. Proteolytic cleavage of extracellular secreted α-synuclein via matrix metalloproteinases. J. Biol. Chem. 2005, 280, 25216–25224. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Kim, K.S. Proteolytic clearance of extracellular alpha-synuclein as a new therapeutic approach against Parkinson disease. Prion 2013, 7, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, S.J. Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun. 2008, 372, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Suk, J.E.; Patrick, C.; Bae, E.J.; Cho, J.H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.J. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef] [PubMed]
- Deleidi, M.; Gasser, T. The role of inflammation in sporadic and familial Parkinson’s disease. Cell. Mol. Life Sci. 2013, 70, 4259–4273. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Esparcia, P.; Llorens, F.; Carmona, M.; Ferrer, I. Complex deregulation and expression of cytokines and mediators of the immune response in Parkinson’s disease brain is region dependent. Brain Pathol. 2014, 24, 584–598. [Google Scholar] [CrossRef] [PubMed]
- Damier, P.; Hirsch, E.C.; Zhang, P.; Agid, Y.; Javoy-Agid, F. Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience 1993, 52, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Mythri, R.B.; Venkateshappa, C.; Harish, G.; Mahadevan, A.; Muthane, U.B.; Yasha, T.C.; Srinivas Bharath, M.M.; Shankar, S.K. Evaluation of markers of oxidative stress, antioxidant function and astrocytic proliferation in the striatum and frontal cortex of Parkinson’s disease brains. Neurochem. Res. 2011, 36, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of major histocompatibility complex class ii-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.M.; Stevens, C.H. Glia: Initiators and progressors of pathology in Parkinson’s disease. Mov. Disord. 2011, 26, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Stefanova, N. The role of glia in alpha-synucleinopathies. Mol. Neurobiol. 2013, 47, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; de Silva, H.A.; Kittel, A.; Saitoh, T. The precursor protein of non-Aβ component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Hayashi, S.; Yoshimoto, M.; Kudo, H.; Takahashi, H. NACP/α-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 2000, 99, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Hishikawa, N.; Hashizume, Y.; Yoshida, M.; Sobue, G. Widespread occurrence of argyrophilic glial inclusions in Parkinson’s disease. Neuropathol. Appl. Neurobiol. 2001, 27, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Sastre, M.; del Tredici, K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.F.; Rey, N.L.; Bousset, L.; Melki, R.; Brundin, P.; Angot, E. Alpha-synuclein transfers from neurons to oligodendrocytes. Glia 2014, 62, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Radford, R.; Rcom-H’cheo-Gauthier, A.; Wong, M.B.; Eaton, E.D.; Quilty, M.; Blizzard, C.; Norazit, A.; Meedeniya, A.; Vickers, J.C.; Gai, W.P.; et al. The degree of astrocyte activation in multiple system atrophy is inversely proportional to the distance to alpha-synuclein inclusions. Mol. Cell. Neurosci. 2015, 65, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Croisier, E.; Moran, L.B.; Dexter, D.T.; Pearce, R.K.; Graeber, M.B. Microglial inflammation in the parkinsonian substantia nigra: Relationship to alpha-synuclein deposition. J. Neuroinflammation 2005. [Google Scholar] [CrossRef] [Green Version]
- Klegeris, A.; Pelech, S.; Giasson, B.I.; Maguire, J.; Zhang, H.; McGeer, E.G.; McGeer, P.L. Alpha-synuclein activates stress signaling protein kinases in Thp-1 cells and microglia. Neurobiol. Aging 2008, 29, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.J.; Lee, H.J.; Rockenstein, E.; Ho, D.H.; Park, E.B.; Yang, N.Y.; Desplats, P.; Masliah, E.; Lee, S.J. Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J. Neurosci. 2012, 32, 13454–13469. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, N.; Fellner, L.; Reindl, M.; Masliah, E.; Poewe, W.; Wenning, G.K. Toll-like receptor 4 promotes alpha-synuclein clearance and survival of nigral dopaminergic neurons. Am. J. Pathol. 2011, 179, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013. [Google Scholar] [CrossRef]
- Park, J.Y.; Paik, S.R.; Jou, I.; Park, S.M. Microglial phagocytosis is enhanced by monomeric alpha-synuclein, not aggregated alpha-synuclein: Implications for Parkinson’s disease. Glia 2008, 56, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopes da Fonseca, T.; Villar-Piqué, A.; Outeiro, T.F. The Interplay between Alpha-Synuclein Clearance and Spreading. Biomolecules 2015, 5, 435-471. https://doi.org/10.3390/biom5020435
Lopes da Fonseca T, Villar-Piqué A, Outeiro TF. The Interplay between Alpha-Synuclein Clearance and Spreading. Biomolecules. 2015; 5(2):435-471. https://doi.org/10.3390/biom5020435
Chicago/Turabian StyleLopes da Fonseca, Tomás, Anna Villar-Piqué, and Tiago Fleming Outeiro. 2015. "The Interplay between Alpha-Synuclein Clearance and Spreading" Biomolecules 5, no. 2: 435-471. https://doi.org/10.3390/biom5020435