
4-Hydroxy-nonenal—A Bioactive Lipid Peroxidation Product †



Abstract
:| Table of Contents | |
| Preface………………………………………………………………………………………………………………………………… | 2251 |
| 1. Lipid Peroxidation as a Free Radical Amplification Process………………………………………………………………… | 2252 |
| 2. Structure, Properties and Generation of HNE………………………………………………………………………………… | 2255 |
| 3. Major Reaction Mechanisms…………………………………………………………………………………………………… | 2257 |
| 3.1. Reactions of the C=C Double Bond…………………………………………………………………………………………… | 2257 |
| 3.1.1. Michael Additions………………………………………………………………………………………………………… | 2257 |
| 3.1.2. Reduction………………………………………………………………………………………………………………… | 2258 |
| 3.1.3. Epoxidation……………………………………………………………………………………………………………… | 2259 |
| 3.2. Reactions of the Carbonyl Group…………………………………………………………………………………………… | 2259 |
| 3.2.1. Acetal and Thio-Acetal Formation……………………………………………………………………………………… | 2259 |
| 3.2.2. Schiff-Base Formation…………………………………………………………………………………………………… | 2259 |
| 3.2.3. Oxidation………………………………………………………………………………………………………………… | 2259 |
| 3.2.4. Reduction………………………………………………………………………………………………………………… | 2260 |
| 3.3. Reactions of the Hydroxy Group……………………………………………………………………………………………… | 2262 |
| 4. Biophysical Effects……………………………………………………………………………………………………………… | 2262 |
| 5. Biochemical Targets of HNE…………………………………………………………………………………………………… | 2262 |
| 5.1. Reactions with Peptides and Proteins………………………………………………………………………………………… | 2263 |
| 5.1.1. Substrates………………………………………………………………………………………………………………… | 2265 |
| 5.1.1.1. Glutathione……………………………………………………………………………………………………………… | 2265 |
| 5.1.1.2. Carnosine……………………………………………………………………………………………………………… | 2267 |
| 5.1.1.3. Thioredoxin…………………………………………………………………………………………………………… | 2267 |
| 5.1.1.4. Cytochrome c…………………………………………………………………………………………………………… | 2268 |
| 5.1.2. Enzymes………………………………………………………………………………………………………………… | 2268 |
| 5.1.2.1. Oxidoreductases………………………………………………………………………………………………………… | 2269 |
| 5.1.2.1.1. Lactate Dehydrogenase……………………………………………………………………………………………… | 2269 |
| 5.1.2.1.2. Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH)………………………………………………………… | 2269 |
| 5.1.2.2. Transferases…………………………………………………………………………………………………………… | 2270 |
| 5.1.2.2.1. Glutathione-S-Transferase (GST)…………………………………………………………………………………… | 2270 |
| 5.1.2.2.2. Liver Kinase B1 (LKB1)……………………………………………………………………………………………… | 2270 |
| 5.1.2.2.3. 5'-AMP-Activated Protein Kinase (AMPK)………………………………………………………………………… | 2271 |
| 5.1.2.2.4. ZAK Kinase (Sterile Alpha Motif and Leucine Zipper Containing Kinase AZK)………………………………… | 2271 |
| 5.1.2.2.5. Serine/Threonine-Protein Kinase AKT2 (Proteinkinase B2)……………………………………………………… | 2271 |
| 5.1.2.3. Hydrolases……………………………………………………………………………………………………………… | 2271 |
| 5.1.2.3.1. ATP Synthase………………………………………………………………………………………………………… | 2271 |
| 5.1.2.3.2. Phosphatase and Tensin Homolog Deleted on Chromosome 10 (PTEN)…………………………………………… | 2272 |
| 5.1.2.3.3. Sirtuin 3 (SIRT3)…………………………………………………………………………………………………… | 2272 |
| 5.1.2.3.4. Cathepsins…………………………………………………………………………………………………………… | 2273 |
| 5.1.2.3.5. Neprilysin (NEP)…………………………………………………………………………………………………… | 2273 |
| 5.1.2.4. Lyases…………………………………………………………………………………………………………………… | 2273 |
| 5.1.2.4.1. Mitochondrial Aconitase (ACO2)…………………………………………………………………………………… | 2273 |
| 5.1.2.4.2. α-Enolase……………………………………………………………………………………………………………… | 2273 |
| 5.1.2.5. Isomerases……………………………………………………………………………………………………………… | 2274 |
| 5.1.2.5.1. Protein Disulfide Isomerase (PDI)…………………………………………………………………………………… | 2274 |
| 5.1.2.5.2. Peptidyl-Prolyl Cis/Trans-Isomerase A1 (Pin1)……………………………………………………………………… | 2274 |
| 5.1.2.6. Ligases: Glutamine Synthetase………………………………………………………………………………………… | 2274 |
| 5.1.3. Carriers…………………………………………………………………………………………………………………… | 2274 |
| 5.1.3.1. Albumin………………………………………………………………………………………………………………… | 2274 |
| 5.1.3.2. Hemoglobin and Myoglobin…………………………………………………………………………………………… | 2275 |
| 5.1.3.3. Liver and Adipocyte Fatty Acid-Binding Protein (FABP)…………………………………………………………… | 2275 |
| 5.1.3.4. Apolipoprotein B-100 (ApoB)………………………………………………………………………………………… | 2275 |
| 5.1.3.5. β-Lactoglobulin………………………………………………………………………………………………………… | 2276 |
| 5.1.4. Transporters and Channels……………………………………………………………………………………………… | 2276 |
| 5.1.4.1. Glutamate Transport Protein…………………………………………………………………………………………… | 2276 |
| 5.1.4.2. α-Synuclein (α-Syn)…………………………………………………………………………………………………… | 2276 |
| 5.1.4.3. Sarco/Endoplasmic Reticulum Ca2+-ATPase (SERCA1a)…………………………………………………………… | 2277 |
| 5.1.4.4. Transient Receptor Potential Vanilloid 1 (TRPV1)…………………………………………………………………… | 2277 |
| 5.1.4.5. Dopamine Transporter………………………………………………………………………………………………… | 2278 |
| 5.1.5. Receptors………………………………………………………………………………………………………………… | 2278 |
| 5.1.5.1. Platelet-Derived Growth Factor Receptor-β (PDGFR-β)……………………………………………………………… | 2278 |
| 5.1.5.2. Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1 (LOX-1)……………………………………………… | 2278 |
| 5.1.5.3. Toll-Like Receptor 4 (TLR4)…………………………………………………………………………………………… | 2278 |
| 5.1.6. Cytoskeletal Proteins……………………………………………………………………………………………………… | 2279 |
| 5.1.6.1. Tau Proteins…………………………………………………………………………………………………………… | 2279 |
| 5.1.6.2. Ankyrin………………………………………………………………………………………………………………… | 2279 |
| 5.1.6.3. Spectrins………………………………………………………………………………………………………………… | 2280 |
| 5.1.7. Chaperones: Heat Shock Proteins 70 and 90…………………………………………………………………………… | 2280 |
| 5.1.8. Uncoupling Proteins 2 and 3 (UCP2 and UCP3)………………………………………………………………………… | 2282 |
| 5.1.9. Growth Factors: Platelet-Derived Growth Factor (PDGF)……………………………………………………………… | 2283 |
| 5.1.10. Peptide Hormones……………………………………………………………………………………………………… | 2283 |
| 5.1.10.1. Insulin………………………………………………………………………………………………………………… | 2283 |
| 5.1.10.2. Angiotensin II………………………………………………………………………………………………………… | 2283 |
| 5.1.11. Extracellular Matrix Proteins: Collagen………………………………………………………………………………… | 2283 |
| 5.1.12. Histones: Histone-H2A………………………………………………………………………………………………… | 2284 |
| 5.2. Reactions with Lipids………………………………………………………………………………………………………… | 2284 |
| 5.3. Reactions with Cofactors and Vitamins……………………………………………………………………………………… | 2284 |
| 5.3.1. Vitamin C (Ascorbic Acid)……………………………………………………………………………………………… | 2284 |
| 5.3.2. Pyridoxamine…………………………………………………………………………………………………………… | 2285 |
| 5.3.3. Lipoic Acid……………………………………………………………………………………………………………… | 2285 |
| 5.4. Reactions with Nucleic Acids………………………………………………………………………………………………… | 2285 |
| 6. Formation of HNE in Mammalian Cells and Tissues………………………………………………………………………… | 2287 |
| 6.1. HNE Formation in Cellular and Organ Systems……………………………………………………………………………… | 2287 |
| 6.2. HNE in the Whole Healthy Organism………………………………………………………………………………………… | 2289 |
| 6.3. Influence of Nutrition………………………………………………………………………………………………………… | 2290 |
| 7. Metabolism of HNE……………………………………………………………………………………………………………… | 2291 |
| 7.1. HNE Metabolism in Mammalian Cells and Organs………………………………………………………………………… | 2293 |
| 7.2. HNE Metabolism in Subcellular Organelles………………………………………………………………………………… | 2294 |
| 7.3. HNE Metabolism in Whole Animals and Interorgan Relationships………………………………………………………… | 2295 |
| 7.4. Primary HNE Intermediates—Enzymatic Reactions and Quantitative Results……………………………………………… | 2295 |
| 7.5. Secondary HNE Intermediates—Enzymatic Reactions and Quantitative Results…………………………………………… | 2301 |
| 7.6. HNE Metabolism as a Component of the Antioxidative Defense System…………………………………………………… | 2306 |
| 7.7. HNE Intermediates as Potential Biomarkers of LPO………………………………………………………………………… | 2307 |
| 7.8. Further Medical Applications of HNE Metabolism………………………………………………………………………… | 2307 |
| 8. Conclusions……………………………………………………………………………………………………………………… | 2309 |
| Conflicts of Interest………………………………………………………………………………………………………………… | 2309 |
| Abbreviations……………………………………………………………………………………………………………………… | 2309 |
| References…………………………………………………………………………………………………………………………… | 2313 |
| Tables and Figures | |
| Table 1. HNE concentrations in cells, tissues and organs…………………………………………………………………………… | 2289 |
| Table 2. Maximal velocity of total HNE degradation in cells, subcellular organelles, and perfused organs………………………………………………………………………………………………………………………………… | 2292 |
| Table 3. Primary HNE metabolites in different cells and tissues after the addition of 100 μM HNE to the biological system………………………………………………………………………………………………………………………………… | 2301 |
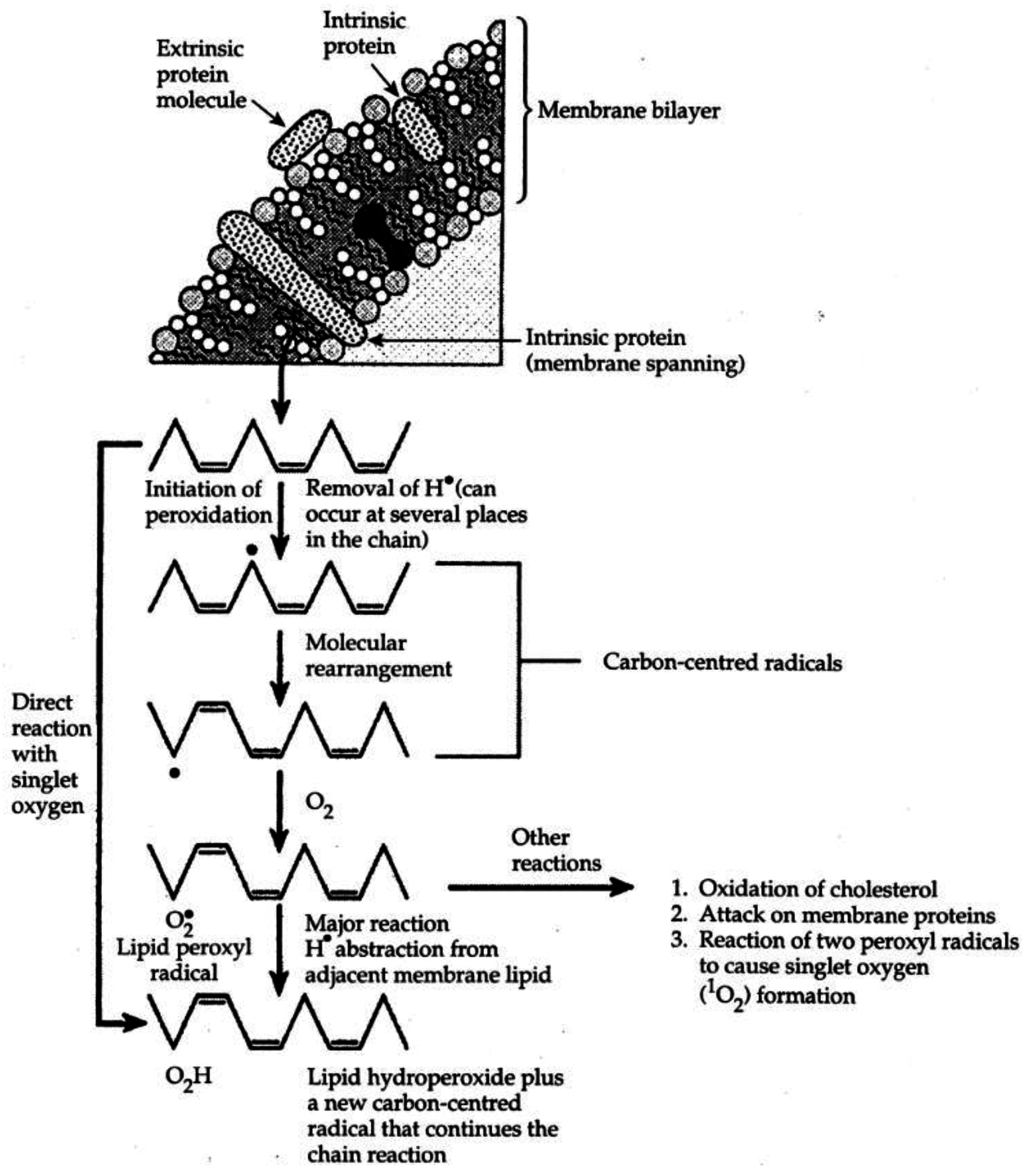
| Figure 1. Idealized representation of the initiation and propagation reactions of lipid peroxidation | 2253 |
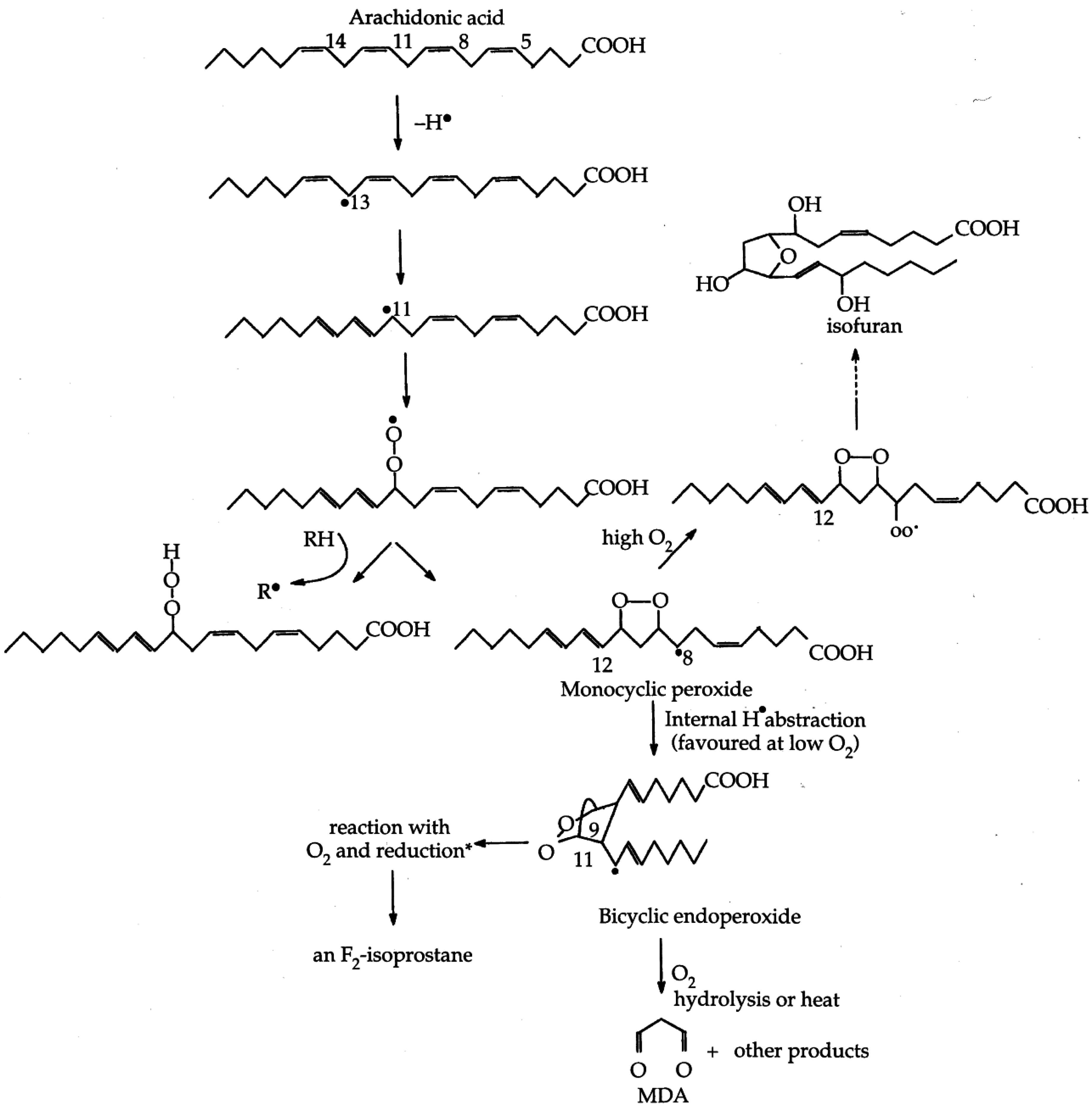
| Figure 2. Formation of lipid hydroperoxides and cyclic peroxides from arachidonic acid.………………………………………… | 2254 |
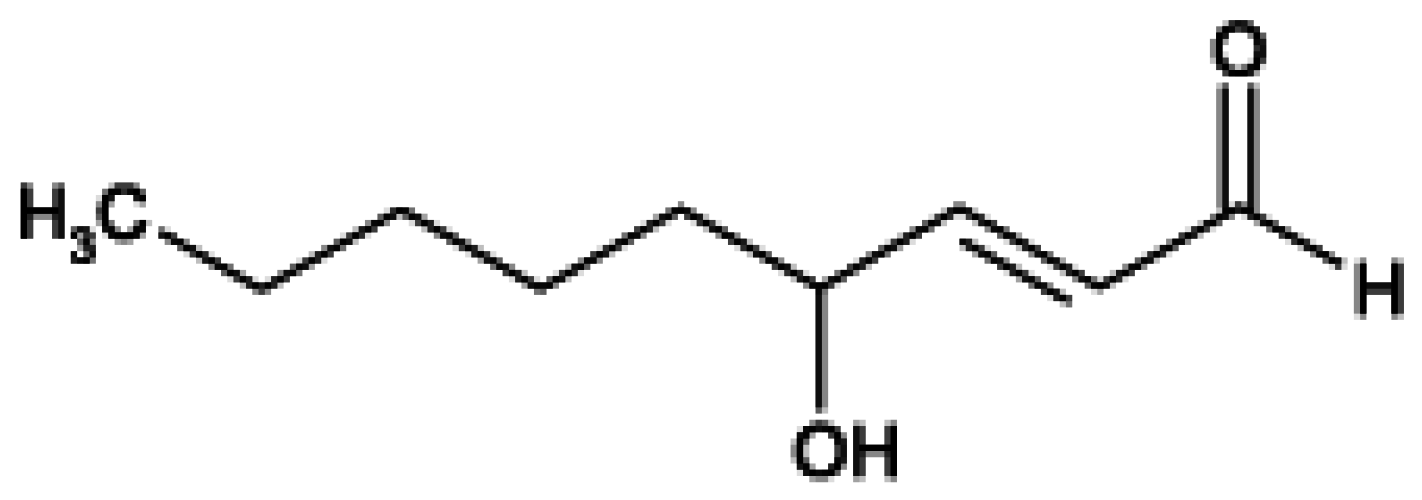
| Figure 3. Chemical structure of 4-hydroxy-2-trans-nonenal (HNE)………………………………………………………………… | 2255 |
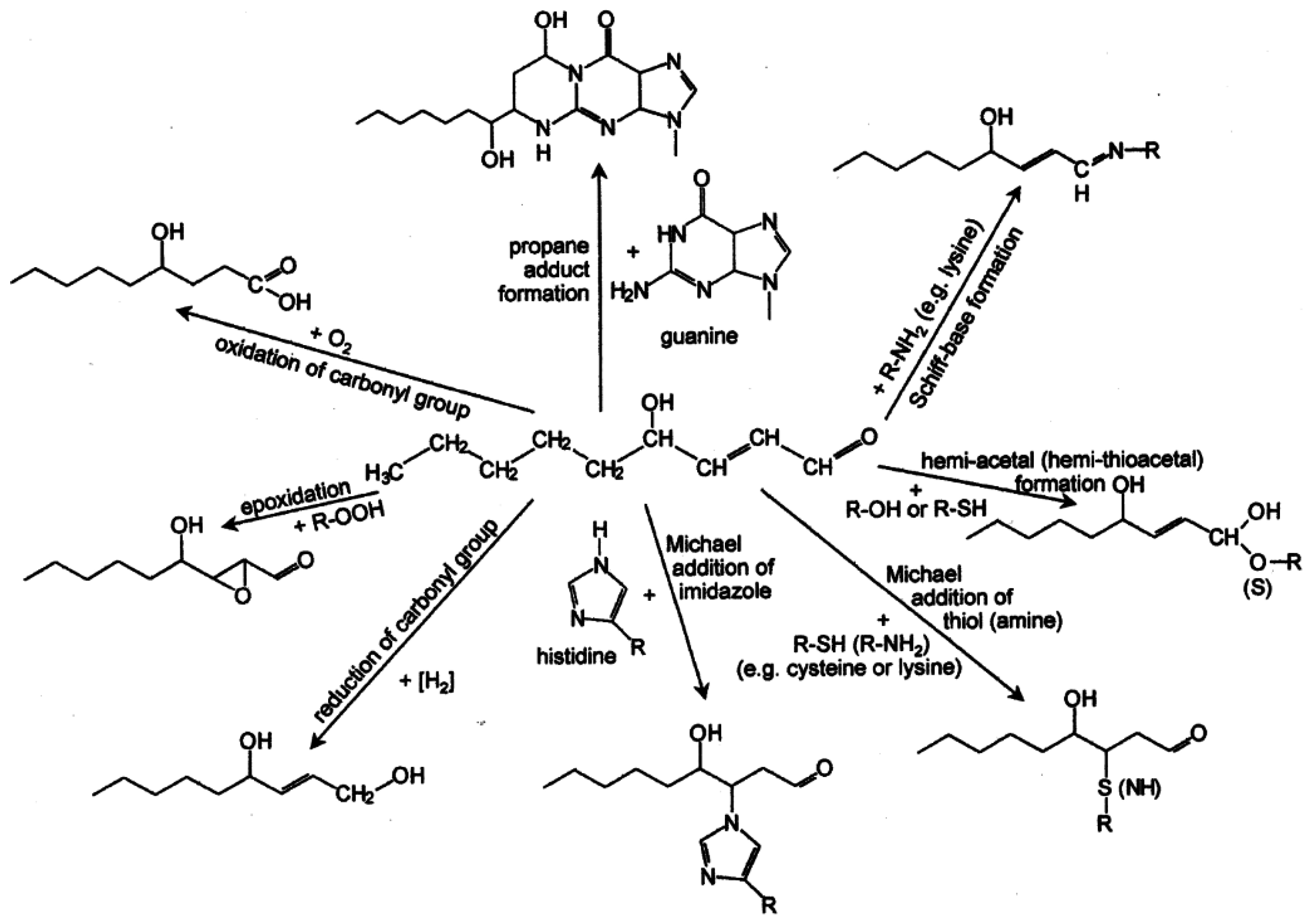
| Figure 4. Overview of the reactions of 4-hydroxy-nonenal with different biomolecules…………………………………………… | 2257 |
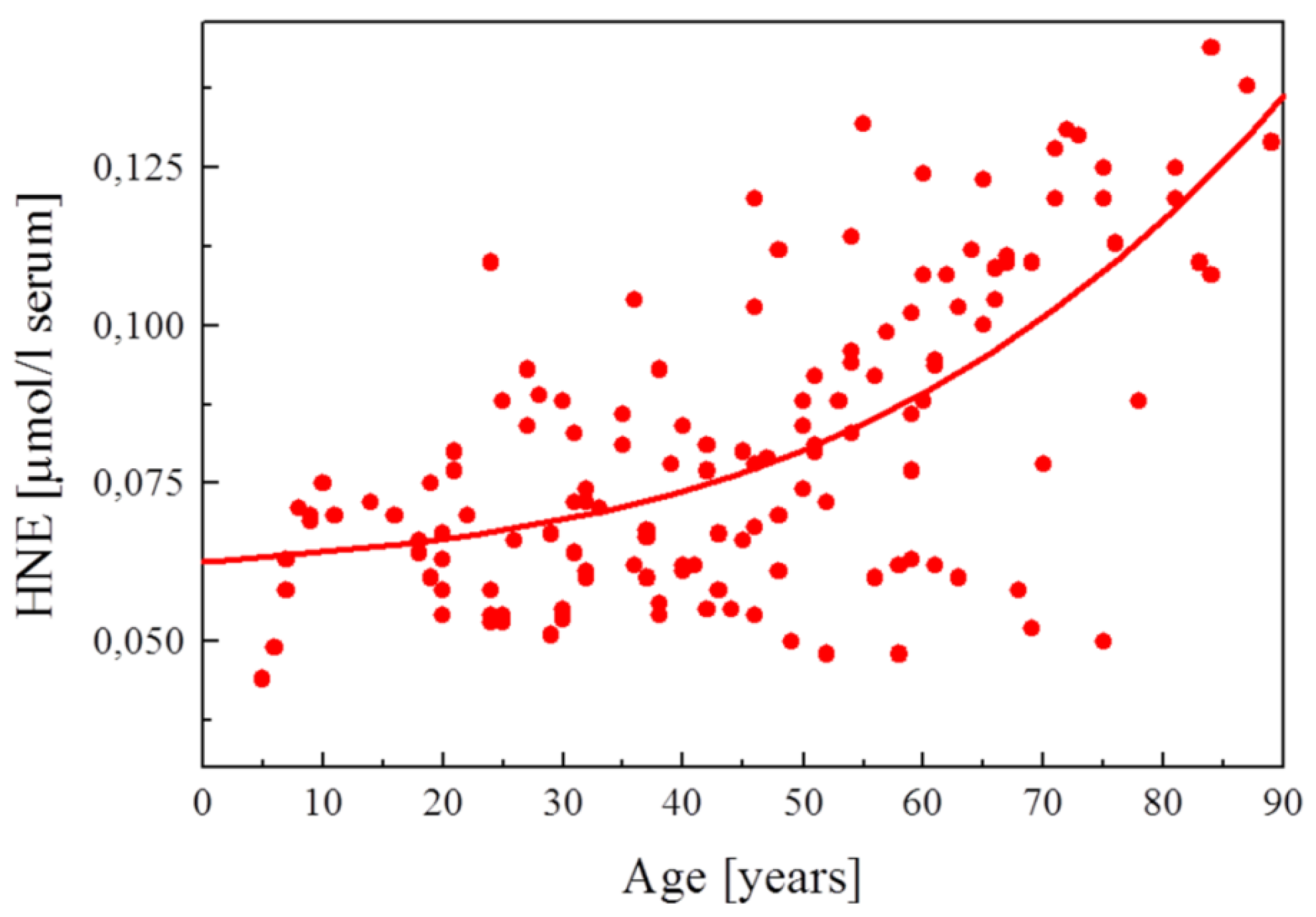
| Figure 5. HNE plasma concentration in dependence on age of the blood donor (5 to 90 years)…………………………………… | 2288 |
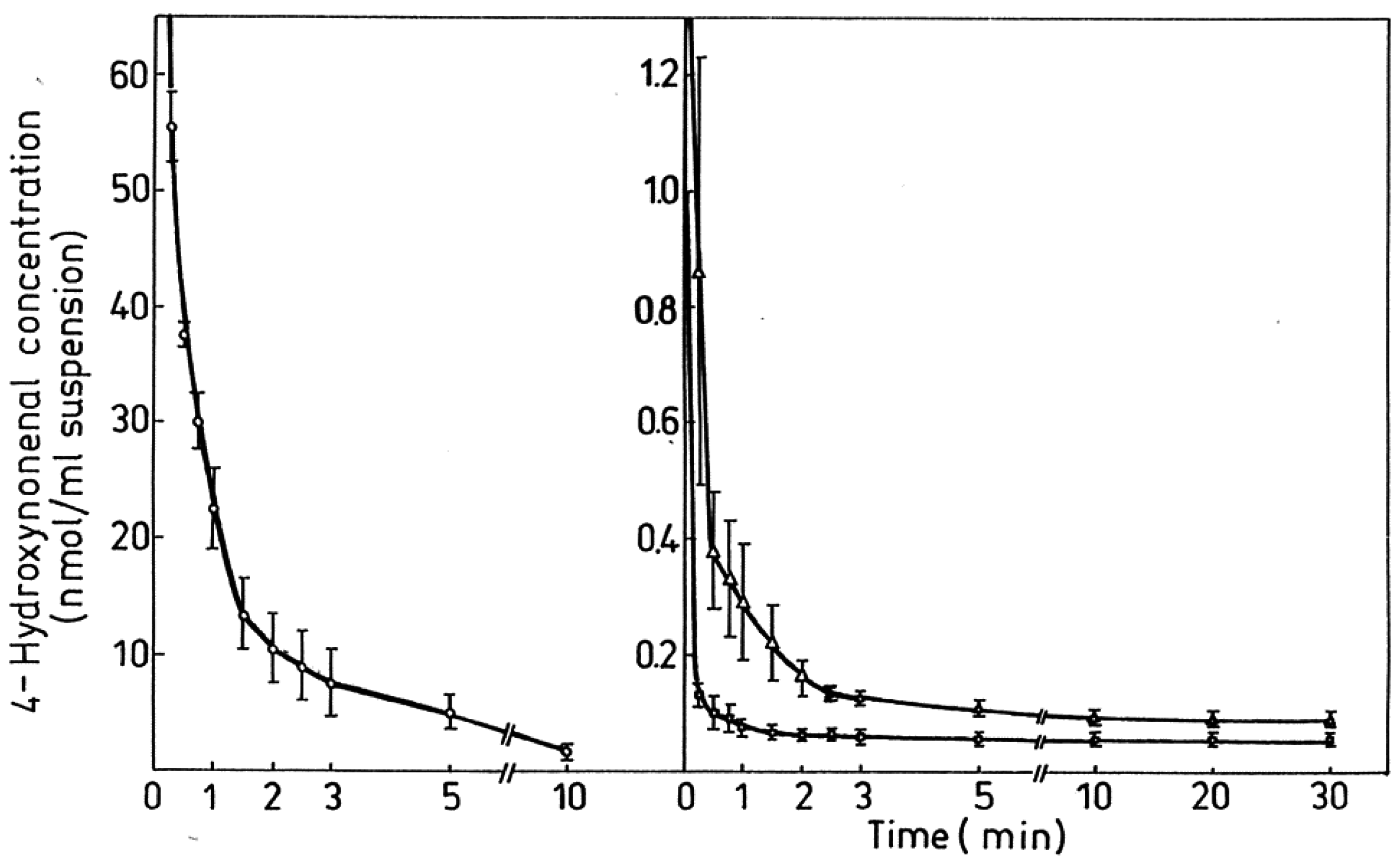
| Figure 6. Degradation/metabolism of 4-HNE in rat hepatocytes…………………………………………………………………… | 2292 |
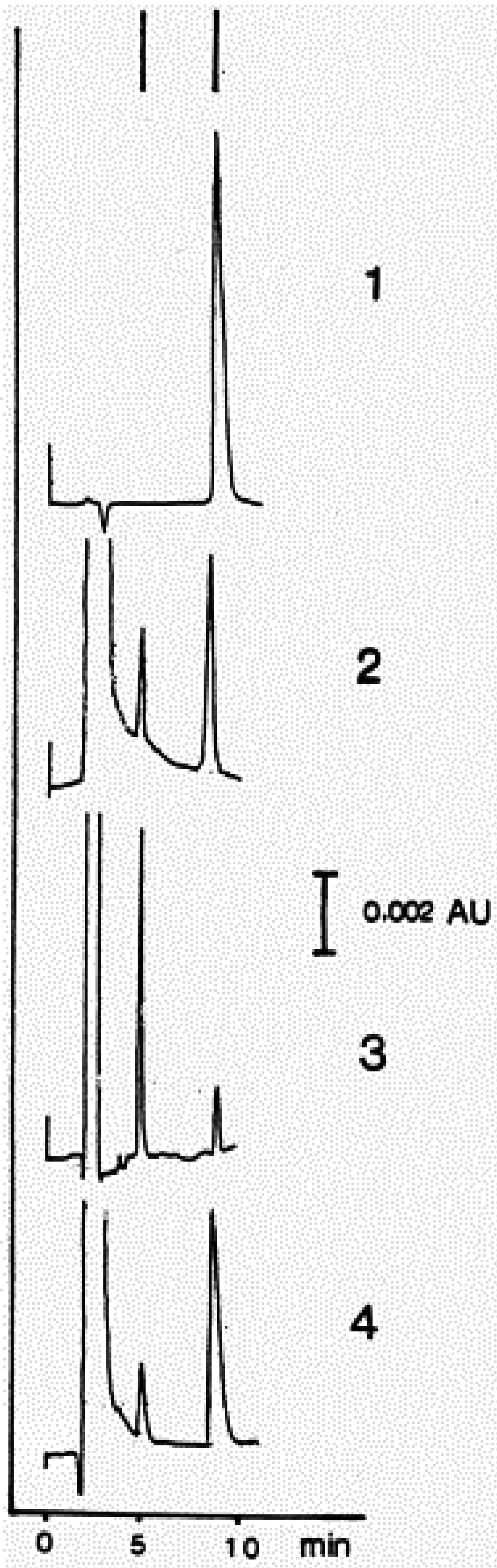
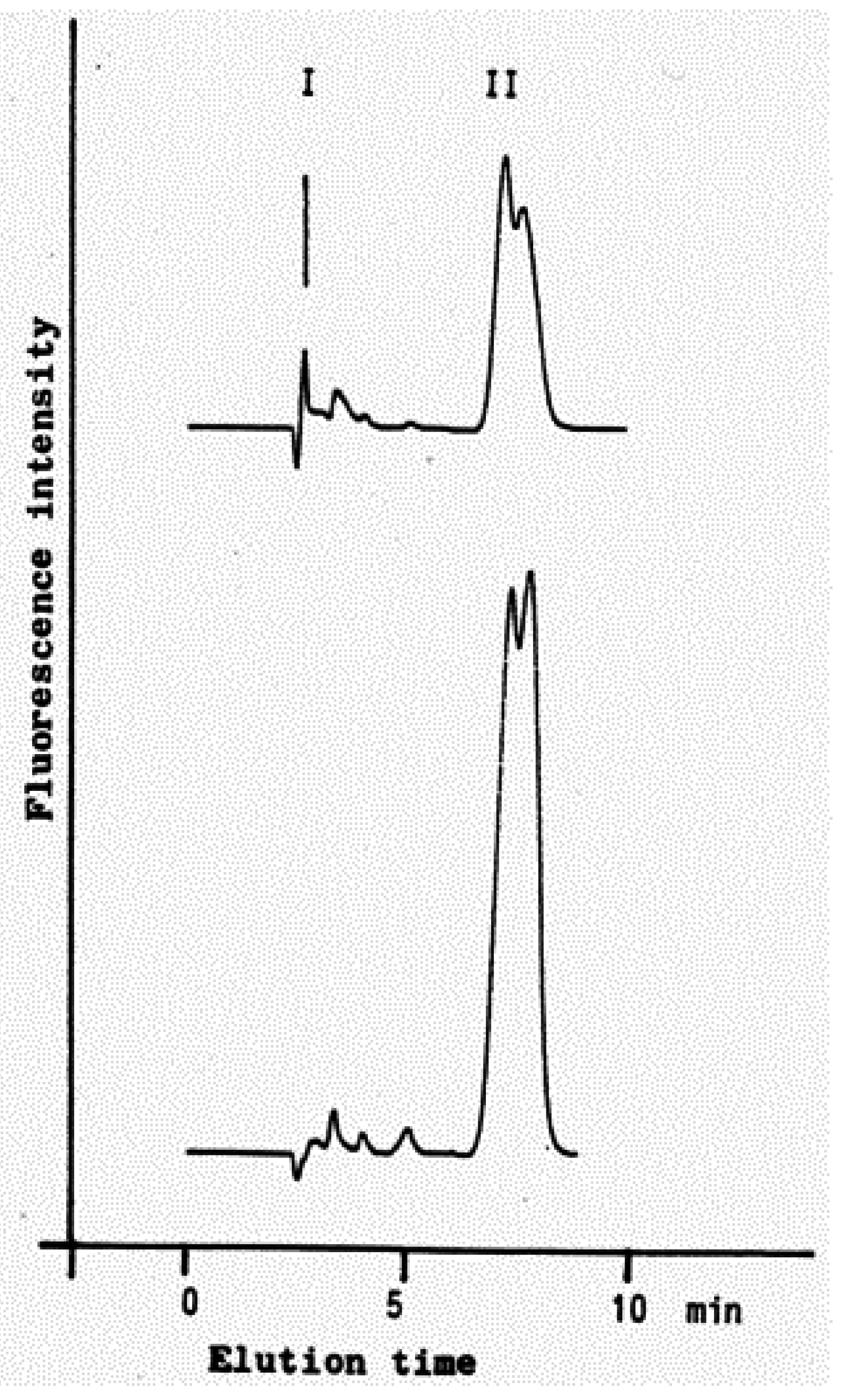
| Figure 7. Identification of HNE and 4-hydroxynonenoic acid (HNA) by isocratic HPLC separation……………………………… | 2297 |
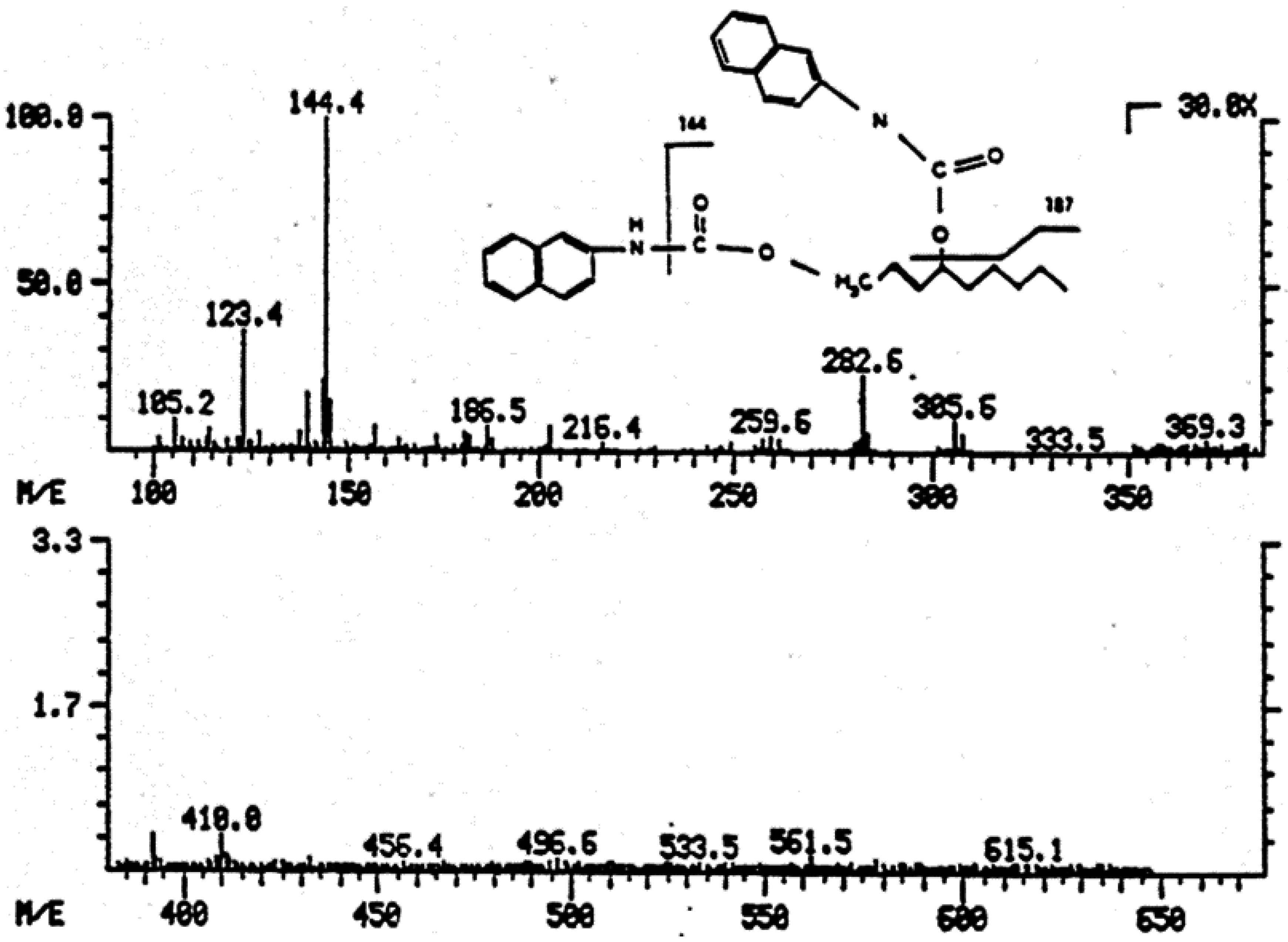
| Figure 8. Mass spectrum of dihydroxynonene urethane (HPLC plus MS) with fluorimetric detection…………………………… | 2298 |
| Figure 9. HNE metabolites.…………………………………………………………………………………………………………… | 2299 |
| Figure 10. HPLC chromatogram of the isoindol derivative of the HNE-GSH conjugate (reaction product in presence of o-phthalaldehyde)……………………………………………………………………………………………………………………… | 2306 |
Preface
1. Lipid Peroxidation as a Free Radical Amplification Process


2. Structure, Properties and Generation of HNE

3. Major Reaction Mechanisms

3.1. Reactions of the C=C Double Bond
3.1.1. Michael Additions
3.1.2. Reduction
3.1.3. Epoxidation
3.2. Reactions of the Carbonyl Group
3.2.1. Acetal and Thio-Acetal Formation
3.2.2. Schiff-Base Formation
3.2.3. Oxidation
3.2.4. Reduction
3.3. Reactions of the Hydroxy Group
4. Biophysical Effects
5. Biochemical Targets of HNE
5.1. Reactions with Peptides and Proteins
5.1.1. Substrates
5.1.1.1. Glutathione
5.1.1.2. Carnosine
5.1.1.3. Thioredoxin
5.1.1.4. Cytochrome c
5.1.2. Enzymes
5.1.2.1. Oxidoreductases
5.1.2.1.1. Lactate Dehydrogenase
5.1.2.1.2. Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH)
5.1.2.2. Transferases
5.1.2.2.1. Glutathione-S-Transferase (GST)
5.1.2.2.2. Liver Kinase B1 (LKB1)
5.1.2.2.3. 5'-AMP-Activated Protein Kinase (AMPK)
5.1.2.2.4. ZAK Kinase (Sterile Alpha Motif and Leucine Zipper Containing Kinase AZK)
5.1.2.2.5. Serine/Threonine-Protein Kinase AKT2 (Proteinkinase B2)
5.1.2.3. Hydrolases
5.1.2.3.1. ATP Synthase
5.1.2.3.2. Phosphatase and Tensin Homolog Deleted on Chromosome 10 (PTEN)
5.1.2.3.3. Sirtuin 3 (SIRT3)
5.1.2.3.4. Cathepsins
5.1.2.3.5. Neprilysin (NEP)
5.1.2.4. Lyases
5.1.2.4.1. Mitochondrial Aconitase (ACO2)
5.1.2.4.2. α-Enolase
5.1.2.5. Isomerases
5.1.2.5.1. Protein Disulfide Isomerase (PDI)
5.1.2.5.2. Peptidyl-Prolyl Cis/Trans-Isomerase A1 (Pin1)
5.1.2.6. Ligases: Glutamine Synthetase
5.1.3. Carriers
5.1.3.1. Albumin
5.1.3.2. Hemoglobin and Myoglobin
5.1.3.3. Liver and Adipocyte Fatty Acid-Binding Protein (FABP)
5.1.3.4. Apolipoprotein B-100 (ApoB)
5.1.3.5. β-Lactoglobulin
5.1.4. Transporters and Channels
5.1.4.1. Glutamate Transport Protein
5.1.4.2. α-Synuclein (α-Syn)
5.1.4.3. Sarco/Endoplasmic Reticulum Ca2+-ATPase (SERCA1a)
5.1.4.4. Transient Receptor Potential Vanilloid 1 (TRPV1)
5.1.4.5. Dopamine Transporter
5.1.5. Receptors
5.1.5.1. Platelet-Derived Growth Factor Receptor-β (PDGFR-β)
5.1.5.2. Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1 (LOX-1)
5.1.5.3. Toll-Like Receptor 4 (TLR4)
5.1.6. Cytoskeletal Proteins
5.1.6.1. Tau Proteins
5.1.6.2. Ankyrin
5.1.6.3. Spectrins
5.1.7. Chaperones: Heat Shock Proteins 70 and 90
5.1.8. Uncoupling Proteins 2 and 3 (UCP2 and UCP3)
5.1.9. Growth Factors: Platelet-Derived Growth Factor (PDGF)
5.1.10. Peptide Hormones
5.1.10.1. Insulin
5.1.10.2. Angiotensin II
5.1.11. Extracellular Matrix Proteins: Collagen
5.1.12. Histones: Histone-H2A
5.2. Reactions with Lipids
5.3. Reactions with Cofactors and Vitamins
5.3.1. Vitamin C (Ascorbic Acid)
5.3.2. Pyridoxamine
5.3.3. Lipoic Acid
5.4. Reactions with Nucleic Acids
6. Formation of HNE in Mammalian Cells and Tissues
6.1. HNE Formation in Cellular and Organ Systems


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells, Tissue | HNE-Concentration | Comment | Reference |
|---|---|---|---|
| Kidney tubular cells (human) | 102 pmol/106 cells | Praeanoxic control | [262] |
| 307 pmol/106 cells | 30 min reoxygenation after 60 min anoxia | ||
| Hepatocytes (rat) | 13 ± 5 pmol/106 cells | Freshly prepared | [267] |
| 27 ± 18 pmol/106 cells | CCl4-treated for 1 h | ||
| 40 ± 14 pmol/106 cells | ADP-Fe3+-treated for 1 h | ||
| 0.43 pmol/5.2 × 106 cells | Freshly prepared | [271] | |
| 1.42 pmol/5.2 × 106 cells | 15 min reoxygenation after 60 min anoxia | ||
| Monocytes (human) | 0.5 pmol/106 cells (1 μM) * | Basic value | [272] |
| 4 pmol/106 cells (8 μM) * | Hemozoin fed at 2 h | ||
| 23 pmol/106 cells (46 μM) * | Hemozoin fed at 5 h | ||
| 7.9 pmol/106 cells (16 μM) * | Hemozoin fed at 12 h | ||
| Small intestine (rat) | 0.68 nmol/g w.w. | perfusion, normoxia | [266] |
| 3.02 nmol/g w.w. | reperfusion after 60 min ischemia | ||
| Blood plasma (human) | 0.074 ± 0.028 μM | 194 healthy woman and men | [273] |
| 0.069 ± 0.015 μM | 18 to 29 years aged | ||
| 0.070 ± 0.014 μM | 30 to 39 years aged | ||
| 0.072 ± 0.020 μM | 40 to 49 years aged | ||
| 0.083 ± 0.020 μM | 50 to 59 years aged | ||
| 0.096 ± 0.022 μM | 60 to 69 years aged | ||
| 0.107 ± 0.027 μM | 70 to 84 years aged | ||
| 106.3 ± 65.8 ng/mL | O-pentafluorobenzyl oxime | [274] | |
| Umbilical cord plasma (human) | 0.3 μM | Full-term healthy neonates | [275] |
| 0.5 μM | Term neonates with acidosis | ||
| 0.6 µM | Term neonates with asphyxia | ||
| 0.1 µM | Preterm neonates (healthy) | ||
| 0.4 µM | Preterm neonates (asphyxia) |
6.2. HNE in the Whole Healthy Organism
6.3. Influence of Nutrition
7. Metabolism of HNE

| Biological System | Maximal Rate of HNE Catabolism | Reference |
|---|---|---|
| Kidney cortex mitochondria, rat | 112.2 nmol/mg protein/min | [292] |
| Hepatocytes, rat | 28.4 nmol/mg w.w./min | [293] |
| Ascites tumor cells | 9 nmol/mg w.w./min | [264] |
| Thymocytes, mouse | 27.7 nmol/mg w.w./min | [294] |
| Synovial fibroblasts, rabbit | 27.3 nmol/106 cells/min | [295] |
| Perfused kidney, rat | 160–190 nmol/g w.w./min | [296] |
| Perfused intestine, rat | 22 nmol/g w.w./min | [266] |
| Perfused heart, rat | 50 nmol/g w.w./min | [264] |
7.1. HNE Metabolism in Mammalian Cells and Organs
7.2. HNE Metabolism in Subcellular Organelles
7.3. HNE Metabolism in Whole Animals and Interorgan Relationships
7.4. Primary HNE Intermediates—Enzymatic Reactions and Quantitative Results



| Biological System | DHN | GSH-HNE | HNA | HNE-P | Reference |
|---|---|---|---|---|---|
| Kidney mitochondria (rat) | 9.3 ± 1.8 | 9.8 ± 1.2 | 5.9 ± 0.7 | 8.4 ± 1.4 | [292] |
| Hepatocytes (rat) | 8.1 ± 2.1 | 27.5 ± 2.5 | 25.3 ± 5.6 | 2.0 ± 0.6 | [293] |
| Enterocytes (rat) | 5.4 ± 0.6 | 11.0 ± 0.5 | 4.2 ± 0.6 | 1.3 ± 0.2 | [297] |
| Thymocytes (mouse) | 6.9 | 10.2 | 6.8 | 2.8 | [294] |
| Synovial fibroblasts (rabbit) | 0.3 ± 0.1 | 29.1 ± 1.9 | 1.0 ± 0.3 | 4.3 ± 0.5 | [295] |
| Kidney venous effluent (rat) | 24 ± 5 | 14 ± 4 | 5 ± 1 | n.d. | [296] |
| Ehrlich ascites cells, proliferating | 2.0 | 10.5 | 3.0 | 7.2 | [264] |
| Ehrlich ascites cells, resting | 1.5. | 8.0 | 3.8 | 1.5 |
7.5. Secondary HNE Intermediates—Enzymatic Reactions and Quantitative Results
Determination of HNE Metabolic Products and Pathways

7.6. HNE Metabolism as a Component of the Antioxidative Defense System
7.7. HNE Intermediates as Potential Biomarkers of LPO
7.8. Further Medical Applications of HNE Metabolism
8. Conclusions
Conflicts of Interest
Abbreviations
| Aβ | amyloid beta peptide |
| ACC | acetyl CoA carboxylase |
| AcLDL | acetylated low-density lipoprotein |
| ACO2 | mitochondrial aconitase |
| Acr-dG | adduct of deoxyguanosine and acrolein |
| AD | Alzheimer disease |
| ADH | alcohol dehydrogenase |
| ADP-iron | adenosine diphosphate-iron |
| AICAR | 5-aminoimidazole-4-carboxyamide ribonucleoside |
| AIDS | acquired immuno-deficiency syndrome |
| AIE | approximate intracellular concentration (extinction) |
| AKR | aldo-keto reductase |
| ALD | alcoholic liver disease |
| ALDH | aldehyde dehydrogenase |
| Al-PEs | aldehyde-modified PEs |
| ALS | amyotrophic lateral sclerosis |
| AMD | age-related macular degeneration |
| AMPK | AMP-activated protein kinase |
| ANT | adenine nucleotide translocase |
| ApoB | apolipoprotein B-100 |
| AR | aldose reductase |
| ARE | antioxidant response element |
| BAG3 | Bcl-2- associated athanogene 3 |
| BER | base excision repair |
| BSA | bovine serum albumin |
| BSO | l-buthionine-S,R-sulfoximine |
| CAR | carnosine |
| CCl4 | carbon tetrachloride |
| CERAD | consortium to establish a registry for Alzheimer disease |
| CH2Cl2 | dichloromethane |
| CHO | Chinese-hamster ovary |
| CHOP | C/EBP homologous protein |
| Col II | collagen type II |
| COX-2 | cyclooxygenase-2 |
| CYP | cytochrome P450 |
| ɛ-dA | 1,N(6)-etheno-2'-deoxyadenosine |
| DHN | dihydroxynonene, non-2-ene-1,4-diol |
| DHN-MA | dihydroxynonene-mercapturic acid |
| DNA | desoxy-ribonucleic acid |
| DNA-PK | DNA-dependent protein kinase |
| DOX | doxorubicin |
| D3T | 3H-1,2-dithiole-3-thione, inducing detoxification enzymes |
| eEF-2 | Eukaryotic Elongation factor 2 |
| EGFP | enhanced green fluorescent protein |
| EGFR | epidermal growth factor receptor |
| EHN | 2,3-epoxy-4-hydroxy-nonanal |
| ERK1 | extracellular signal-regulated kinase 1 |
| ESI-MS-MS | electrospray ionization triple quadrupole mass spectrometry |
| FABP | fatty acid-binding protein |
| FAK | α1β1-integrin-focal adhesion kinase |
| FID | flame ionization detection |
| FITC | fluorescein isothiocyanate |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| GC | gas chromatography |
| GC/MS | gas chromatography/mass spectrometry |
| GERD | gastroesophageal reflux disease |
| GRP78 | glucose-regulated protein 78 |
| GRX | glutaredoxin |
| GSH | glutathione |
| GSH-DHN | glutathione-dihydroxynonene conjugate |
| GSK3β | glycogen synthase kinase-3-β |
| GSSG | glutathione in the oxidized form, glutathione disulfide |
| GST | glutathione-S-transferase |
| HA-1 | Chinese hamster fibroblast control cells |
| HAEC | human aortic endothelial cell |
| Hb | hemoglobin |
| HCC | hepatocellular carcinoma |
| HCV | hepatitis C virus |
| HMEC-1 | immortalized human microvascular endothelial cells |
| HHE | 4-hydroxy-hexenal |
| [3H]HNE | tritium labeled hydroxynonenal |
| HNA | hydroxynonenoic acid |
| HNA-GSH | hydroxynonenoic acid-glutathione conjugate |
| HNE | 4-hydroxy-nonenal |
| HNE-dG | 1,N-2-propane adduct of guanine and HNE |
| HNE-H2A | HNE-modified histone-H2A |
| H2O2 | hydrogen peroxide |
| HPLC | high performance liquid chromatography |
| HPNE | 4-hydroperoxy-2-nonenal |
| HO-1 | heme oxygenase-1 |
| HSA | human serum albumin |
| HU | HU hydroxyurea |
| HUVEC | human umbilical vein endothelial cell |
| ICAM-1 | intercellular adhesion molecule-1 |
| IsoLGs | isolevuglandins |
| iTRAQ | isobaric Tags for Relative and Absolute Quantitation |
| LA | linolenic acid |
| LAD | late Alzheimer disease |
| LADH | lipoamide dehydrogenase |
| LCA | retinal dystrophy Leber congenital amaurosis |
| LC-MS/MS | liquid chromatography-tandem mass spectrometry |
| LDH | lactate dehydrogenase |
| LDL | low density lipoprotein |
| LKB1 | liver kinase B1 |
| LOX1 | lectin-like oxidized low-density lipoprotein receptor-1 |
| LPO | lipid peroxidation |
| MAL-6 | 2,2,6,6-tetramethyl-4-maleimidopiperidin-1-oxyl |
| MALDI-TOF | matrix-assisted laser desorption/ionization-time of flight |
| MAPK | mitogen-activated protein kinase |
| MBP-1 | c-myc binding protein-1 |
| MCI | mild cognitive impairment |
| MDA | malondialdehyde |
| MDM2 | mouse double minute 2 |
| MH1C1 | rat hepatoma cells |
| MMP-13 | matrix metalloproteinase 13 |
| MRP-1 | multidrug resistance protein-1 |
| NAD+ | nicotinamid dinucleotide, oxidized form |
| NADH | nicotinamid dinucleotide, reduced form |
| NADP | nicotinamid dinucleotide phosphate |
| NAFLD | non-alcoholic fatty liver disease |
| NEP | neprilysin |
| NER | nucleotide excision repair |
| Nrf2 | NF-E2-related factor 2 |
| OA | osteoarthritic |
| OC5 and OC14 | Chinese hamster fibroblast H2O2-resistant cell lines |
| ONE | 4-oxo-nonenal |
| oxLDL | oxidized low-density lipoprotein |
| ox-PAPC | oxidized PAPC |
| PAF | 1-O-alkyl-2-acetyl-sn-glycero-3-phospho-choline |
| PAPC | 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phospho-choline |
| PCAD | preclinical Alzheimer disease |
| PDGF | platelet-derived growth factor |
| PDI | protein disulfide isomerase |
| PE | phosphatidyl-ethanolamine |
| PGE | prostaglandin E |
| Pin1 | peptidyl-prolyl cis/trans-isomerase A1 |
| PMF | peptide mass fingerprinting |
| POS | photoreceptor outer segment |
| PG(E2) | prostaglandin E2 |
| PtdIns(3,4,5)P-3 | phosphatidylinositol-3,4,5-trisphosphate |
| PTEN | phosphatase and tensin homolog deleted on chromosome 10 |
| RAS | rennin/angiotensin system |
| RBC | erythrocytes |
| RDH | retinol dehydrogenase |
| RLIP76 | Ral-binding GTPase activating protein |
| ROC | receiver operator curves |
| ROOH | lipid hydroperoxide |
| ROS | reactive oxygen species |
| RPE | retinal pigment epithelial |
| SAM | sterile-alpha motif |
| SCFAs | short chain fatty acids |
| SERCA1a | serco/endoplasmic reticulum Ca2+-ATPase |
| SIRT3 | sirtuin 3 |
| SLE | systemic lupus erythematosus |
| SMC | smooth muscle cell |
| sXBP1 | spliced X-box-binding protein-1 |
| α-Syn | α-synuclein |
| TBA-RS | thiobarbituric acid-reactive substances |
| TBI | traumatic brain injury |
| TLC | thin layer chromatography |
| TLR | toll-like receptor |
| TRPV1 | transient receptor potential vanilloid 1 |
| TRX | thioredoxin |
| TrxR | thioredoxin reductase |
| UCP | uncoupling protein |
| UPR | unfolded protein response |
| VSL#3 | a medical food delivering high amount of beneficial live bacteria |
| YAP1 | Yeast Activator Protein 1 |
| ZAK kinase | sterile alpha motif and leucine zipper containing kinase (AZK) |
References
- Schauenstein, E.; Esterbauer, H. Ueber die Reaktion von 9,12 Linolsaeureester in Wasser 6. Mitteilung: Chromatographische Untersuchungen ueber die Zusammensetzung des Praeparates LHPO und die Isolierung der einzelnen tumor-wirksamen Hydroperoxid-Komponenten. Chem. Mon. 1963, 94, 11. (In Germany) [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J. Free Radicals in Biology and Medicine, 4th ed.; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Catala, A. Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem. Phys. Lipids 2009, 157, 1–11. [Google Scholar] [CrossRef]
- Poli, G.; Schaur, R.J.; Siems, W.G.; Leonarduzzi, G. 4-Hydroxynonenal: A membrane lipid oxidation product of medicinal interest. Med. Res. Rev. 2008, 28, 569–631. [Google Scholar] [PubMed]
- Schaur, J.R. Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Mol. Asp. Med. 2003, 24, 149–159. [Google Scholar] [CrossRef]
- Dubinina, E.E.; Dadali, V.A. Role of 4-hydroxy-trans-2-nonenal in cell functions. Biochem. Moscow 2010, 75, 1069–1087. [Google Scholar]
- Fritz, K.S.; Petersen, D.R. An overview of the chemistry and biology of reactive aldehydes. Free Rad. Biol. Med. 2013, 59, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Zarkovic, N.; Cipak, A.; Jaganjac, M.; Borovic, S.; Zarkovic, K. Pathophysiological relevance of aldehydic protein modifications. J. Proteomics 2013, 92, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Coccia, R.; Butterfield, D.A. 4-hydroxy-2-nonenal, a reactive product of lipid peroxidation, and neurodegenerative diseases: A toxic combination illuminated by redox proteomics studies. Antioxid. Redox Signal. 2012, 17, 1590–1609. [Google Scholar] [CrossRef] [PubMed]
- Riahi, Y.; Cohen, G.; Shamni, O.; Sasson, S. Signaling and cytotoxic functions of 4-hydroxyalkenals. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E879–E886. [Google Scholar] [CrossRef]
- Forman, H.J. Reactive oxygen species and α,β-unsaturated aldehydes as second messengers in signal transduction. Oxid. Nitrosative Stress Dis. 2010, 1203, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Higdon, A.; Diers, A.R.; Oh, J.Y.; Landar, A.; Darley-Usmar, V.M. Cell signalling by reactive lipid species: New concepts and molecular mechanisms. Biochem. J. 2012, 442, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, S.; Sharma, A.; Patrick, B.; Sharma, R.; Awasthi, Y.C. Role of 4-hydroxynonenal and its metabolites in signaling. Redox Rep. 2007, 12, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Munoz, M.F.; Argueelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014. [Google Scholar] [CrossRef] [PubMed]
- Spickett, C.M. The lipid peroxidation product 4-hydroxy-2-nonenal: Advances in chemistry and analysis. Redox Biol. 2013, 1, 145–152. [Google Scholar] [CrossRef] [PubMed]
- LoPachin, R.M.; Gavin, T.; Petersen, D.R.; Barber, D.S. Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: Nucleophilic targets and adduct formation. Chem. Res. Toxicol. 2009, 22, 1499–1508. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp. Gerontol. 2009, 44, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Guichardant, M.; Lagarde, M. Analysis of biomarkers from lipid peroxidation: A comparative study. Eur. J. Lipid Sci. Technol. 2009, 111, 75–82. [Google Scholar] [CrossRef]
- Tappel, A.L. Measurement and protection from in vivo lipid peroxidation. In Free Radicals in Biology; Pryor, W.A., Ed.; Academic Press: New York, NY, USA, 1980; pp. 1–47. [Google Scholar]
- Aikens, J.; Dix, T.A. Perhydroxyl radical (HOO.) initiated lipid-peroxidation—The role of fatty-acid hydroperoxides. J. Biol. Chem. 1991, 266, 15091–15098. [Google Scholar] [PubMed]
- Tsunada, S.; Iwakiri, R.; Noda, T.; Fujimoto, K.; Fuseler, J.; Rhoads, C.A.; Aw, T.Y. Chronic exposure to subtoxic levels of peroxidized lipids suppresses mucosal cell turnover in rat small intestine and reversal by glutathione. Dig. Dis. Sci. 2003, 48, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Bochkov, V.N.; Kadl, A.; Huber, J.; Gruber, F.; Binder, B.R.; Leitinger, N. Protective role of phospholipid oxidation products in endotoxin-induced tissue damage. Nature 2002, 419, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Morrow, J.D.; Porter, N.A. Identification of a novel class of endoperoxides from arachidonate autoxidation. J. Biol. Chem. 2004, 279, 3766–3776. [Google Scholar] [PubMed]
- Poli, G.; Schaur, R.J. 4-Hydroxynonenal in the pathomechanisms of oxidative stress. Iubmb Life 2000, 50, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Pillon, N.J.; Soulere, L.; Vella, R.E.; Croze, M.; Care, B.R.; Soula, H.A.; Doutheau, A.; Lagarde, M.; Soulage, C.O. Quantitative structure-activity relationship for 4-hydroxy-2-alkenal induced cytotoxicity in L6 muscle cells. Chem. Biol. Interact. 2010, 188, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Vazdar, M.; Jurkiewicz, P.; Hof, M.; Jungwirth, P.; Cwiklik, L. Behavior of 4-hydroxynonenal in phospholipid membranes. J. Phys. Chem. B 2012, 116, 6411–6415. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, M.J.; Zolnerciks, J.K.; Balogh, L.M.; Greene, R.J.; Kavanagh, T.J.; Atkins, W.M. Stereoselective Effects of 4-hydroxynonenal in cultured mouse hepatocytes. Chem. Res. Toxicol. 2010, 23, 1601–1607. [Google Scholar] [CrossRef]
- Hiratsuka, A.; Saito, H.; Watabe, T. (S)-preferential cytotoxicity of 4-hydroxy-2(E)-nonenal enantiomers in rat Clone 9 cells. Toxicology 2001, 164, 199. [Google Scholar]
- Gueraud, F.; Crouzet, F.; Alary, J.; Rao, D.; Debrauwer, L.; Laurent, F.; Cravedi, J.P. Enantioselective metabolism of (R)- and (S)-4-hydroxy-2-nonenal in rat. Biofactors 2005, 24, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Sadhukhan, S.; Han, Y.; Jin, Z.; Tochtrop, G.P.; Zhang, G.-F. Glutathionylated 4-hydroxy-2-(E)-alkenal enantiomers in rat organs and their contributions toward the disposal of 4-hydroxy-2-(E)-nonenal in rat liver. Free Rad. Biol. Med. 2014, 70, 78–85. [Google Scholar] [CrossRef]
- Wakita, C.; Maeshima, T.; Yamazaki, A.; Shibata, T.; Ito, S.; Akagawa, M.; Ojika, M.; Yodoi, J.; Uchida, K. Stereochemical configuration of 4-hydroxy-2-nonenal-cysteine adducts and their stereoselective formation in a redox-regulated protein. J. Biol. Chem. 2009, 284, 28810–28822. [Google Scholar] [CrossRef] [PubMed]
- Komisarski, M.; Kaczmarska, Z.; Kusmierek, J.T. Practical highly enantioselective synthesis of (R)- and (S)-(E)-4-hydroxynon-2-enal. Acta Biochim. Polonica 2009, 56, 189–193. [Google Scholar]
- Yadav, U.C.S.; Ramana, K.V. Regulation of NF-kappaB-induced inflammatory signaling by lipid peroxidation-derived aldehydes. Oxid. Med. Cell. Longevity 2013. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Niemczyk, M.; Zimniak, L.; Zimniak, P. Fat accumulation in Caenorhabditis elegans triggered by the electrophilic lipid peroxidation product 4-Hydroxynonenal (4-HNE). Aging 2009, 1, 68–80. [Google Scholar]
- Singh, S.P.; Niemczyk, M.; Saini, D.; Awasthi, Y.C.; Zimniak, L.; Zimniak, P. Role of the electrophilic lipid peroxidation product 4-hydroxynonenal in the development and maintenance of obesity in mice. Biochemistry 2008, 47, 3900–3911. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Brash, A.R. Monomeric and dimeric routes to formation of 4-hydroxynonenal during lipid peroxidation. Abstr. Papers Am. Chem. Soc. 2013, 246. Abstract 41-AGFD. [Google Scholar]
- Long, E.K.; Picklo, M.J., Sr. Trans-4-hydroxy-2-hexenal, a product of n-3 fatty acid peroxidation: Make some room HNE. Free Rad. Biol. Med. 2010, 49, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Allen, T.D.; Yang, Y.; Moore, D.R.; Huycke, M.M. Cyclooxygenase-2 generates the endogenous mutagen trans-4-hydroxy-2-nonenal in Enterococcus faecalis-infected macrophages. Cancer Prev. Res. 2013, 6, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.C.; Kim, H.-W.; Kim, K.B.; Kwack, S.J.; Ahn, I.Y.; Bae, J.Y.; Lim, S.K.; Lee, B.M. Hepatotoxicity and nephrotoxicity produced by 4-hydroxy-2-nonenal (4-HNE) following 4-week oral administration to sprague-dawley rats. J. Toxicol. Environ. Health Part A 2011, 74, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sano, M.; Shinmura, K.; Tamaki, K.; Katsumata, Y.; Matsuhashi, T.; Morizane, S.; Ito, H.; Hishiki, T.; Endo, J.; et al. 4-Hydroxy-2-nonenal protects against cardiac ischemia-reperfusion injury via the Nrf2-dependent pathway. J. Mol. Cell. Cardiol. 2010, 49, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Mannervik, B. Five decades with glutathione and the GSTome. J. Biol. Chem. 2012, 287, 6072–6083. [Google Scholar] [CrossRef] [PubMed]
- Balogh, L.M.; Atkins, W.M. Interactions of glutathione transferases with 4-hydroxynonenal. Drug Metab. Rev. 2011, 43, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Rudd, L.P.; Kabler, S.L.; Morrow, C.S.; Townsend, A.J. Enhanced glutathione depletion, protein adduct formation, and cytotoxicity following exposure to 4-hydroxy-2-nonenal (HNE) in cells expressing human multidrug resistance protein-1 (MRP1) together with human glutathione S-transferase-M1 (GSTM1). Chem. Biol. Interact. 2011, 194, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Shireman, L.M.; Kripps, K.A.; Balogh, L.M.; Conner, K.P.; Whittington, D.; Atkins, W.M. Glutathione transferase A4-4 resists adduction by 4-hydroxynonenal. Arch. Biochem. Biophys. 2010, 504, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.Y.; Decker, E.A. Ability of carnosine and other skeletal muscle components to quench unsaturated aldehydic lipid oxidation products. J. Agric. Food Chem. 1999, 47, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Shearn, C.T.; Smathers, R.L.; Backos, D.S.; Reigan, P.; Orlicky, D.J.; Petersen, D.R. Increased carbonylation of the lipid phosphatase PTEN contributes to Akt2 activation in a murine model of early alcohol-induced steatosis. Free Rad. Biol. Med. 2013, 65, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Zheng, Z.-P.; Cheng, K.-W.; Wu, J.-J.; Zhang, S.; Tang, Y.S.; Sze, K.-H.; Chen, J.; Chen, F.; Wang, M. Natural polyphenols as direct trapping agents of lipid peroxidation-derived acrolein and 4-hydroxy-trans-2-nonenal. Chem. Res. Toxicol. 2009, 22, 1721–1727. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.F.; Sowell, J.D.; Frei, B. Ascorbic Acid Conjugates. US Patent US20090104705A1.
- Uchida, K. Immunochemical detection of lipid peroxidation-specific epitopes. In Biomarkers for Antioxidative Defense and Oxidative Damage; Aldini, G., Yeum, K.J., Niki, E., Russell, R.M., Eds.; Blackwell Publishing: Hoboken, NJ, USA, 2010; pp. 157–171. [Google Scholar]
- Wakita, C.; Honda, K.; Shibata, T.; Akagawa, M.; Uchida, K. A method for detection of 4-hydroxy-2-nonenal adducts in proteins. Free Radic. Biol. Med. 2011, 51, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Shi, Z.; Duncan, D.T.; Prodduturi, N.; Marnett, L.J.; Liebler, D.C. Relating protein adduction to gene expression changes: A systems approach. Mol. Biosyst. 2011, 7, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Dick, R.A.; Kwak, M.K.; Sutter, T.R.; Kensler, T.W. Antioxidative function and substrate specificity of NAD(P)H-dependent alkenal/one oxidoreductase—A new role for leukotriene B-4 12-hydroxydehydrogenase/15-oxoprostaglandin 13-reductase. J. Biol. Chem. 2001, 276, 40803–40810. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.J.C.; Chung, F.L. Epoxidation of trans-4-hydroxy-2-nonenal by fatty acid hydroperoxides and hydrogen peroxide. Chem. Res. Toxicol. 1996, 9, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.A.; Castellani, R.J.; Moreira, P.I.; Nunomura, A.; Lui, Q.; Harris, P.L.R.; Sayre, L.M.; Szweda, P.A.; Szweda, L.I.; Zhu, X.; et al. Neurofilaments are the major neuronal target of hydroxynonenal-mediated protein cross-links. Free Radic. Res. 2013, 47, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.N.; Gilmer, L.K.; Miller, D.M.; Cebak, J.E.; Wang, J.A.; Hall, E.D. Phenelzine mitochondrial functional preservation and neuroprotection after traumatic brain injury related to scavenging of the lipid peroxidation-derived aldehyde 4-hydroxy-2-nonenal. J. Cerebral Blood Flow Metab. 2013, 33, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Fournet, G.; Martin, G.; Quash, G. α,β-Acetylenic amino thiolester inhibitors of aldehyde dehydrogenases 1&3: Suppressors of apoptogenic aldehyde oxidation and activators of apoptosis. Curr. Med. Chem. 2013, 20, 527–533. [Google Scholar] [PubMed]
- Guo, J.-M.; Liu, A.-J.; Zang, P.; Dong, W.-Z.; Ying, L.; Wang, W.; Xu, P.; Song, X.-R.; Cai, J.; Zhang, S.-Q.; et al. ALDH2 protects against stroke by clearing 4-HNE. Cell Res. 2013, 23, 915–930. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Guo, R.; Yu, L.; Zhang, Y.; Ren, J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: Role of autophagy paradox and toxic aldehyde. Eur. Heart J. 2011, 32, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Muzio, G.; Maggiora, M.; Paiuzzi, E.; Oraldi, M.; Canuto, R.A. Aldehyde dehydrogenases and cell proliferation. Free Radic. Biol. Med. 2012, 52, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Townsend, A.J.; Leone-Kabler, S.; Haynes, R.L.; Wu, Y.; Szweda, L.; Bunting, K.D. Selective protection by stably transfected human ALDH3A1 (but not human ALDH1A1) against toxicity of aliphatic aldehydes in V79 cells. Chem. Biol. Interact. 2001, 130–132, 261–273. [Google Scholar] [CrossRef]
- Black, W.; Chen, Y.; Matsumoto, A.; Thompson, D.C.; Lassen, N.; Pappa, A.; Vasiliou, V. Molecular mechanisms of ALDH3A1-mediated cellular protection against 4-hydroxy-2-nonenal. Free Radic. Biol. Med. 2012, 52, 1937–1944. [Google Scholar] [CrossRef] [PubMed]
- Yoval-Sanchez, B.; Rodriguez-Zavala, J.S. Differences in susceptibility to inactivation of human aldehyde dehydrogenases by lipid peroxidation byproducts. Chem. Res. Toxicol. 2012, 25, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Ohsawa, I.; Kasahara, T.; Ohta, S. Cytoprotective role of mitochondrial amyloid beta peptide-binding alcohol dehydrogenase against a cytotoxic aldehyde. Neurobiol. Aging 2009, 30, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Vladykovskaya, E.; Barski, O.A.; Kaiserova, M.S.K.; Petrash, J.M.; Chung, S.S.; Hunt, G.; Dawn, B.; Bhatnagar, A. Aldose reductase protects against early atherosclerotic lesion formation in apolipoprotein e-null mice. Circ. Res. 2009, 105, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Keith, R.J.; Haberzettl, P.; Vladykovskaya, E.; Hill, B.G.; Kaiserova, K.; Srivastava, S.; Barski, O.; Bhatnagar, A. Aldose reductase decreases endoplasmic reticulum stress in ischemic hearts. Chem. Biol. Interact. 2009, 178, 242–249. [Google Scholar] [CrossRef]
- Li, D.; Ferrari, M.; Ellis, E.M. Human aldo-keto reductase AKR7A2 protects against the cytotoxicity and mutagenicity of reactive aldehydes and lowers intracellular reactive oxygen species in hamster V79-4 cells. Chem. Biol. Interact. 2012, 195, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.-A.; Kwak, M.-K. Enhanced 4-hydroxynonenal resistance in KEAP1 silenced human colon cancer cells. Oxid. Med. Cell. Longevity 2013. [Google Scholar] [CrossRef]
- Lyon, R.C.; Li, D.; McGarvie, G.; Ellis, E.M. Aldo-keto reductases mediate constitutive and inducible protection against aldehyde toxicity in human neuroblastoma SH-SY5Y cells. Neurochem. Int. 2013, 62, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Liu, Z.; Yan, R.; Johnson, S.; Zhao, Y.; Fang, X.; Cao, D. Aldo-keto reductase family 1 B10 protein detoxifies dietary and lipid-derived alpha, beta-unsaturated carbonyls at physiological levels. Biochem. Biophys. Res. Commun. 2009, 387, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.-J.; Maser, E. Role of human aldo-keto-reductase AKR1B10 in the protection against toxic aldehydes. Chem. Biol. Interact. 2009, 178, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Endo, S.; Matsunaga, T.; Kuragano, T.; Ohno, S.; Kitade, Y.; Tajima, K.; el-Kabbani, O.; Hara, A. Properties and tissue distribution of a novel aldo-keto reductase encoding in a rat gene (Akr1b10). Arch. Biochem. Biophys. 2010, 503, 230–237. [Google Scholar] [CrossRef]
- Matsunaga, T.; Shinoda, Y.; Inoue, Y.; Endo, S.; el-Kabbani, O.; Hara, A. Protective effect of rat aldo-keto reductase (AKR1C15) on endothelial cell damage elicited by 4-hydroxy-2-nonenal. Chem. Biol. Interact. 2011, 191, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Marchette, L.D.; Thompson, D.A.; Kravtsova, M.; Ngansop, T.N.; Mandal, M.N.A.; Kasus-Jacobi, A. Retinol dehydrogenase 12 detoxifies 4-hydroxynonenal in photoreceptor cells. Free Radic. Biol. Med. 2010, 48, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Amunom, I.; Dieter, L.J.; Tamasi, V.; Cai, J.; Conklin, D.J.; Srivastava, S.; Martin, M.V.; Guengerich, F.P.; Prough, R.A. Cytochromes p450 catalyze the reduction of α,β-unsaturated aldehydes. Chem. Res. Toxicol. 2011, 24, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-F.; Kombu, R.S.; Kasumov, T.; Han, Y.; Sadhukhan, S.; Zhang, J.; Sayre, L.M.; Ray, D.; Gibson, K.M.; Anderson, V.A.; et al. Catabolism of 4-hydroxyacids and 4-hydroxynonenal via 4-hydroxy-4-phosphoacyl-CoAs. J. Biol. Chem. 2009, 284, 33521–33534. [Google Scholar] [CrossRef] [PubMed]
- Berthiaume, J.M.; Li, Q.; Sadhukhan, S.; Henry, F.; Tochtrop, G.P.; Brunengraber, H.; Zhang, G. Catabolism of 4-hydroxy-2(E)-nonenal (HNE) via omega oxidation in perfused rat livers. FASEB J. 2013, 27. Abstract No. 794.15. [Google Scholar]
- Subramaniam, R.; Roediger, F.; Jordan, B.; Mattson, M.P.; Keller, J.N.; Waeg, G.; Butterfield, D.A. The lipid peroxidation product, 4-hydroxy-2-trans-nonenal, alters the conformation of cortical synaptosomal membrane proteins. J. Neurochem. 1997, 69, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Yu, B.P. Alterations in mitochondrial-membrane fluidity by lipid-peroxidation products. Free Radic. Biol. Med. 1994, 17, 411–418. [Google Scholar] [CrossRef]
- Fleuranceau-Morel, P.; Barrier, L.; Fauconneau, B.; Piriou, A.; Huguet, F. Origin of 4-hydroxynonenal incubation-induced inhibition of dopamine transporter and Na+/K+ adenosine triphosphate in rat striatal synaptosomes. Neurosci. Lett. 1999, 277, 91–94. [Google Scholar] [CrossRef]
- Liu, Q.; Ullery, J.; Zhu, J.; Liebler, D.C.; Marnett, L.J.; Zhang, B. RNA-seq data analysis at the gene and CDS levels provides a comprehensive view of transcriptome responses induced by 4-hydroxynonenal. Mol. Biosyst. 2013, 9, 3036–3046. [Google Scholar] [CrossRef] [PubMed]
- Schreier, S.M.; Muellner, M.K.; Steinkellner, H.; Hermann, M.; Esterbauer, H.; Exner, M.; Gmeiner, B.M.K.; Kapiotis, S.; Laggner, H. Hydrogen sulfide scavenges the cytotoxic lipid oxidation product 4-HNE. Neurotox. Res. 2010, 17, 249–256. [Google Scholar] [PubMed]
- Siems, W.; Grune, T. Intracellular metabolism of 4-hydroxynonenal. Mol. Asp. Med. 2003, 24, 167–175. [Google Scholar]
- Vila, A.; Tallman, K.A.; Jacobs, A.T.; Liebler, D.C.; Porter, N.A.; Marnett, L.J. Identification of protein targets of 4-hydroxynonenal using click chemistry for ex vivo biotinylation of azido and alkynyl derivatives. Chem. Res. Toxicol. 2008, 21, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Tallman, K.A.; Vila, A.; Porter, N.A.; Marnett, L.J. Measuring electrophile stress. Curr. Protoc. Toxicol. 2009, 11–17. [Google Scholar]
- Chavez, J.; Chung, W.-G.; Miranda, C.L.; Singhal, M.; Stevens, J.F.; Maier, C.S. Site-specific protein adducts of 4-hydroxy-2(E)-nonenal in human THP-1 monocytic cells: Protein carbonylation is diminished by ascorbic acid. Chem. Res. Toxicol. 2010, 23, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Codreanu, S.G.; Zhang, B.; Sobecki, S.M.; Billheimer, D.D.; Liebler, D.C. Global analysis of protein damage by the lipid electrophile 4-hydroxy-2-nonenal. Mol. Cell. Proteomics 2009, 8, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Petersen, D.R.; Carbone, D.; Doorn, J. Hepatocellular targets of 4-hydroxy-2-nonenal modification. In Proceedings of the 3rd International Meeting of the HNE-Club, Genova, Italy, 16–18 June 2006. Abstract No. 6.
- Hussain, S.N.A.; Matar, G.; Barreiro, E.; Florian, M.; Divangahi, M.; Vassilakopoulos, T. Modifications of proteins by 4-hydroxy-2-nonenal in the ventilatory muscles of rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L996–L1003. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Engels, M.; Kaiser, B.; Grune, T. Distribution of oxidized and HNE-modified proteins in U87 cells. Biofactors 2005, 24, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Carbone, D.L.; Doorn, J.A.; Kiebler, Z.; Ickes, B.R.; Petersen, D.R. Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. J. Pharmacol. Exp. Ther. 2005, 315, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Carbone, D.L.; Doorn, J.A.; Kiebler, Z.; Sampey, B.P.; Petersen, D.R. Inhibition of Hsp72-mediated protein refolding by 4-hydroxy-2-nonenal. Chem. Res. Toxicol. 2004, 17, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Legards, J.F.; des Rosiers, C. Assay of 4-hydroxynonenal (HNE) adducts with various polyaminoacids (PAA) using gas chromatography-mass spectrometry (GCMS). In Proceedings of the 3rd International Meeting of the HNE-Club, Genova, Italy, 16–18 June 2006.
- Doorn, J.A.; Petersen, D.R. Covalent adduction of nucleophilic amino acids by 4-hydroxynonenal and 4-oxononenal. Chem. Biol. Interact. 2003, 143, 93–100. [Google Scholar] [CrossRef]
- Isom, A.L.; Barnes, S.; Wilson, L.; Kirk, M.; Coward, L.; Darley-Usmar, V. Modification of cytochrome c by 4-hydroxy-2-nonenal: Evidence for histidine, lysine, and arginine-aldehyde adducts. J. Am. Soc. Mass Spectrom. 2004, 15, 1136–1147. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.F.; Minkler, P.E.; Sayre, L.A. Mass spectroscopic characterization of protein modification by 4-Hydroxy-2-(E)-nonenal and 4-Oxo-2-(E)-nonenal. Chem. Res. Toxicol. 2003, 16, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Aldini, G.; Dalle-Donne, I.; Vistoli, G.; Facino, R.M.; Carini, M. Covalent modification of actin by 4-hydroxy-trans-2-nonenal (HNE): LC-ESI-MS/MS evidence for Cys374 Michael adduction. J. Mass Spectrom. 2005, 40, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Bennaars-Eiden, A.; Higgins, L.A.; Hertzel, A.V.; Kapphahn, R.J.; Ferrington, D.; Bernlohr, D.A. Covalent modification of epithelial fatty acid binding protein by 4-hydroxynonenal in vitro and in vivo: Evidence for a role in antioxidant biology. FASEB J. 2003, 17. Abstract No. 845.847. [Google Scholar] [CrossRef]
- Carbone, D.L.; Doorn, J.A.; Kiebler, Z.; Petersen, D.R. Cysteine modification by lipid peroxidation products inhibits protein disulfide isomerase. Chem. Res. Toxicol. 2005, 18, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Asselin, C.; Bouchard, B.; Tardif, J.C.; des Rosiers, C. Circulating 4-hydroxynonenal-protein thioether adducts assessed by gas chromatography-mass spectrometry are increased with disease progression and aging in spontaneously hypertensive rats. Free Radic. Biol. Med. 2006, 41, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Moreau, R.; Nguyen, B.T.; Doneanu, C.E.; Hagen, T.M. Reversal by aminoguanidine of the age-related increase in glycoxidation and lipoxidation in the cardiovascular system of Fischer 344 rats. Biochem. Pharmacol. 2005, 69, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Zou, Y.N.; Kim, D.H.; Kim, N.D.; Yu, B.P.; Chung, H.Y. Proteomic analysis of nitrated and 4-hydroxy-2-nonenal-modified serum proteins during aging. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2006, 61, 332–338. [Google Scholar] [CrossRef]
- Petersen, D.R.; Doorn, J.A. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radic. Biol.Med. 2004, 37, 937–945. [Google Scholar] [CrossRef]
- Renner, A.; Sagstetter, M.R.; Harms, H.; Lange, V.; Gotz, M.E.; Elert, O. Formation of 4-hydroxy-2-nonenal protein adducts in the ischemic rat heart after transplantation. J. Heart Lung Transpl. 2005, 24, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Galligan, J.J.; Smathers, R.L.; Fritz, K.S.; Epperson, L.E.; Hunter, L.E.; Petersen, D.R. Protein carbonylation in a murine model for early alcoholic liver disease. Chem. Res. Toxicol. 2012, 25, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Matsubara, K.; Kobayashi, S. Aging and oxidative stress in progressive supranuclear palsy. Eur. J. Neurol. 2006, 13, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Grune, T. HNE-modified proteins: Formation, distribution and fate. In Proceedings of the 3rd International Meeting of the HNE-Club, Genova, Italy, 2006. Abstract No. 17.
- Carbone, D.L.; Doorn, J.A.; Petersen, D.R. 4-hydroxynonenal regulates 26S proteasomal degradation of alcohol dehydrogenase. Free Radic. Biol. Med. 2004, 37, 1430–1439. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Wangpoengtrakul, C.; Osawa, T.; Toyokuni, S.; Tanaka, K.; Uchida, K. 4-hydroxy-2-nonenal-mediated impairment of intracellular proteolysis during oxidative stress—Identification of proteasomes as target molecules. J. Biol. Chem. 1999, 274, 23787–23793. [Google Scholar] [CrossRef] [PubMed]
- Hyun, D.H.; Lee, M.H.; Halliwell, B.; Jenner, P. Proteasomal dysfunction induced by 4-hydroxy-2,3-trans-nonenal, an end-product of lipid peroxidation: A mechanism contributing to neurodegeneration? J. Neurochem. 2002, 83, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Bardag-Gorce, F.; Li, J.; French, B.A.; French, S.W. The effect of ethanol-induced CYP2E1 on proteasome activity: The role of 4-hydroxynonenal. Exp. Mol. Pathol. 2005, 78, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Kessova, I.G.; Cederbaum, A.I. The effect of CYP2E1-dependent oxidant stress on activity of proteasomes in HepG2 cells. J. Pharmacol. Exp. Ther. 2005, 315, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Kapphahn, R.J. Catalytic site-specific inhibition of the 20S proteasome by 4-hydroxynonenal. FEBS Lett. 2004, 578, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Yamaguchi, M.; Chikuma, T.; Hojo, H. Degradation of glyceraldehyde-3-phosphate dehydrogenase triggered by 4-hydroxy-2-nonenal and 4-hydroxy-2-hexenal. Arch. Biochem. Biophys. 2005, 438, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.Z.; Liu, Y.H.; Sayre, L.M. Independent synthesis, solution behavior, and studies on the mechanism of formation of a primary amine-derived fluorophore representing cross-linking of proteins by (E)-4-hydroxy-2-nonenal. J. Org. Chem. 1999, 64, 5732–5745. [Google Scholar] [CrossRef]
- Zarkovic, N. Protein-aldehydic adducts as biomarkers of oxidative stress, lipid peroxidation and oxidative homeostasis. Free Radic. Res. 2009, 43, 33. [Google Scholar]
- Wang, J.-F.; Tan, H. Oxidative protein modification of soluble N-ethylmaleimide-sensitive factor attachment protein receptors. FASEB J. 2013, 27, 890–897. [Google Scholar]
- Gallagher, E.P.; Gardner, J.L.; Barber, D.S. Several glutathione S-transferase isozymes that protect against oxidative injury are expressed in human liver mitochondria. Biochem. Pharmacol. 2006, 71, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Alary, J.; Fernandez, Y.; Debrauwer, L.; Perdu, E.; Gueraud, F. Identification of intermediate pathways of 4-hydroxynonenal metabolism in the rat. Chem. Res.Toxicol. 2003, 16, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.H.; Wheeler, D.G.; Guan, Z.; Suntres, Z.; Stokes, B.T. Effect of glutathione augmentation on lipid peroxidation after spinal cord injury. J. Neurotrauma 2002, 19, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.S.; Sharma, R.; Sharma, A.; Awasthi, S.; Awasthi, Y.C. Lipid peroxidation and cell cycle signaling: 4-Hydroxynonenal, a key molecule in stress mediated signaling. Acta Biochim. Polonica 2003, 50, 319–336. [Google Scholar]
- Patrick, B.; Li, J.; Jeyabal, P.V.S.; Reddy, P.; Yang, Y.S.; Sharma, R.; Sinha, M.; Luxon, B.; Zimniak, P.; Awasthi, S.; et al. Depletion of 4-hydroxynonenal in hGSTA4-transfected HLE B-3 cells results in profound changes in gene expression. Biochem. Biophys. Res. Commun. 2005, 334, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, S.; Sharma, R.; Sharma, A.; Zimniak, P.; Ceci, J.D.; Awasthi, Y.C.; Boor, P.J. The course of CCl4 induced hepatotoxicity is altered in mGSTA4-4 null (−/−) mice. Toxicology 2006, 218, 58–66. [Google Scholar] [CrossRef]
- Engle, M.R.; Singh, S.P.; Czernik, P.J.; Gadd, D.; Montague, D.C.; Ceci, J.D.; Yang, Y.S.; Awasthi, S.; Awasthi, Y.C.; Zimniak, P. Physiological role of mGSTA4-4, a glutathione S-transferase metabolizing 4-hydroxynonenal: Generation and analysis of mGSTA4 null mouse. Toxicol. Appl. Pharmacol. 2004, 194, 296–308. [Google Scholar] [CrossRef]
- Knoll, N.; Ruhe, C.; Veeriah, S.; Sauer, J.; Glei, M.; Gallagher, E.P.; Pool-Zobel, B.L. Genotoxicity of 4-hydroxy-2-nonenal in human colon tumor cells is associated with cellular levels of glutathione and the modulation of glutathione S-transferase A4 expression by butyrate. Toxicol. Sci. 2005, 86, 27–35. [Google Scholar] [CrossRef]
- Srivastava, S.; Ramana, K.V.; Bhatnagar, A.; Srivastava, S.K. Synthesis, quantification, characterization, and signaling properties of glutathionyl conjugates of enals. Methods Enzymol. 2010, 474, 297–313. [Google Scholar] [PubMed]
- Frohnert, B.I.; Long, E.K.; Hahn, W.S.; Bernlohr, D.A. Glutathionylated lipid aldehydes are products of adipocyte oxidative stress and activators of macrophage inflammation. Diabetes 2014, 63, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Spite, M.; Summers, L.; Porter, T.F.; Srivastava, S.; Bhatnagar, A.; Serhan, C.N. Resolvin D1 controls inflammation initiated by glutathione-lipid conjugates formed during oxidative stress. Br. J. Pharmacol. 2009, 158, 1062–1073. [Google Scholar] [CrossRef]
- Aldini, G.; Facino, R.M.; Beretta, G.; Carini, M. Carnosine and related dipeptides as quenchers of reactive carbonyl species: From structural studies to therapeutic perspectives. Biofactors 2005, 24, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Aldini, G.; Carini, M.; Beretta, G.; Bradamante, S.; Facino, R.M. Carnosine is a quencher of 4-hydroxy-nonenal: Through what mechanism of reaction? Biochem. Biophys. Res. Commun. 2002, 298, 699–706. [Google Scholar] [PubMed]
- Barski, O.A.; Xie, Z.; Baba, S.P.; Sithu, S.D.; Agarwal, A.; Cai, J.; Bhatnagar, A.; Srivastava, S. Dietary carnosine prevents early atherosclerotic lesion formation in apolipoprotein e-null mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Fang, H.; Han, X. Shotgun lipidomics analysis of 4-hydroxyalkenal species directly from lipid extracts after one-step in situ derivatization. Anal. Chem. 2012, 84, 4580–4586. [Google Scholar] [PubMed]
- Guiotto, A.; Calderan, A.; Ruzza, P.; Osler, A.; Rubini, C.; Jo, D.G.; Mattson, M.P.; Borin, G. Synthesis and evaluation of neuroprotective alpha,beta-unsaturated aldehyde scavenger histidyl-containing analogues of carnosine. J. Med. Chem. 2005, 48, 6156–6161. [Google Scholar] [CrossRef] [PubMed]
- Guiotto, A.; Calderan, A.; Ruzza, P.; Borin, G. Carnosine and carnosine-related antioxidants: A review. Curr. Med. Chem. 2005, 12, 2293–2315. [Google Scholar] [CrossRef] [PubMed]
- Vistoli, G.; Orioli, M.; Pedretti, A.; Regazzoni, L.; Canevotti, R.; Negrisoli, G.; Carini, M.; Aldini, G. Design, synthesis, and evaluation of carnosine derivatives as selective and efficient sequestering agents of cytotoxic reactive carbonyl species. Chemmedchem 2009, 4, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Bertinaria, M.; Rolando, B.; Giorgis, M.; Montanaro, G.; Guglielmo, S.; Buonsanti, M.F.; Carabelli, V.; Gavello, D.; Daniele, P.G.; Fruttero, R.; et al. Synthesis, physicochemical characterization, and biological activities of new carnosine derivatives stable in human serum as potential neuroprotective agents. J. Med. Chem. 2011, 54, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Sugano, E.; Isago, H.; Murayama, N.; Tamai, M.; Tomita, H. Different anti-oxidant effects of thioredoxin 1 and thioredoxin 2 in retinal epithelial cells. Cell Struct. Funct. 2013, 38, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Du, Y.; Zhang, X.; Lu, J.; Holmgren, A. Glutaredoxin 2 reduces both thioredoxin 2 and thioredoxin 1 and protects cells from apoptosis induced by auranofin and 4-hydroxynonenal. Antioxid. Redox Signal. 2014, 21, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Sayre, L.M.; Tochtrop, G.P. A mass spectrometric analysis of 4-hydroxy-2-(E)-nonenal modification of cytochrome c. J. Mass Spectrom. 2011, 46, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Gardner, H.W.; Deighton, N. Effect of 4-hydroxy-2(E)-nonenal on soybean lipoxygenase-1. Lipids 2001, 36, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Stadtman, E.R. Covalent attachment of 4-hydroxynonenal to glyceraldehyde-3-phosphate dehydrogenase—A possible involvement of intramolecular and intermolecular cross-linking reaction. J. Biol. Chem. 1993, 268, 6388–6393. [Google Scholar] [PubMed]
- Ishii, T.; Tatsuda, E.; Kumazawa, S.; Nakayama, T.; Uchida, K. Molecular basis of enzyme inactivation by an endogenous electrophile 4-hydroxy-2-nonenal: Identification of modification sites in glyceraldehyde-3-phosphate dehydrogenase. Biochemistry 2003, 42, 3474–3480. [Google Scholar] [CrossRef] [PubMed]
- Van Iersel, M.L.P.S.; Ploemen, J.-P.H.T.M.; lo Bello, M.; Federici, G.; van Bladeren, P.J. Interactions of α,β-unsaturated aldehydes and ketones with human glutathione S-transferase P1-1. Chem. Biol. Interact. 1997, 108, 67–78. [Google Scholar] [CrossRef]
- Bosch-Morell, F.; Flohe, L.; Marin, N.; Romero, F.J. 4-Hydroxynonenal inhibits glutathione peroxidase: Protection by glutathione. Free Radic. Biol. Med. 1999, 26, 1383–1387. [Google Scholar] [CrossRef]
- Korotchkina, L.G.; Yang, H.S.; Tirosh, O.; Packer, L.; Patel, M.S. Protection by thiols of the mitochondrial complexes from 4-hydroxy-2-nonenal. Free Radic. Biol. Med. 2001, 30, 992–999. [Google Scholar] [CrossRef]
- Del Corso, A.; dal Monte, M.; Vilardo, P.G.; Cecconi, I.; Moschini, R.; Banditelli, S.; Cappiello, M.; Tsai, L.; Mura, U. Site-specific inactivation of aldose reductase by 4-hydroxynonenal. Arch. Biochem. Biophys. 1998, 350, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.K.; Moos, P.J.; Cassidy, P.; Wade, M.; Fitzpatrick, F.A. Conditional expression of 15-lipoxygenase-1 inhibits the selenoenzyme thioredoxin reductase—Modulation of selenoproteins by lipoxygenase enzymes. J. Biol. Chem. 2004, 279, 28028–28035. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.G.; Holmgren, A. Inhibition of thioredoxin and thioredoxin reductase by 4-hydroxy-2-nonenal in vitro and in vivo. J. Am. Chem. Soc. 2006, 128, 1879–1885. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.H.; Saito, Y.; Yoshida, Y.; Sekine, A.; Noguchi, N.; Niki, E. 4-Hydroxynonenal induces adaptive response and enhances PC12 cell tolerance primarily through induction of thioredoxin reductase 1 via activation of Nrf2. J. Biol. Chem. 2005, 280, 41921–41927. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.H.; Yoshida, Y.; Saito, Y.; Noguchi, N.; Niki, E. Adaptive response induced by lipid peroxidation products in cell cultures. FEBS Lett. 2006, 580, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Morquette, B.; Shi, Q.; Lavigne, P.; Ranger, P.; Fernandes, J.C.; Benderdour, M. Production of lipid peroxidation products in osteoarthritic tissues—New evidence linking 4-hydroxynonenal to cartilage degradation. Arthritis Rheum. 2006, 54, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.; Mancini, R.A.; Suman, S.P.; Beach, C.M. Covalent binding of 4-hydroxy-2-nonenal to lactate dehydrogenase decreases NADH formation and metmyoglobin reducing activity. J. Agric. Food Chem. 2014, 62, 2112–2117. [Google Scholar] [CrossRef] [PubMed]
- Schlisser, A.E.; Yan, J.; Hales, B.F. Teratogen-induced oxidative stress targets glyceraldehyde-3-phosphate dehydrogenase in the organogenesis stage mouse embryo. Toxicol. Sci. 2010, 118, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Markesbery, W.R.; Carney, J.M. Oxidative alterations in Alzheimer’s disease. Brain Pathol. 1999, 9, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Lauderback, C.M. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress1,2. Free Radic. Biol. Med. 2002, 32, 1050–1060. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Lovell, M.A. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol. Aging 1998, 19, 33–36. [Google Scholar] [CrossRef]
- Sultana, R.; Butterfield, D.A. Oxidatively modified GST and MRP1 in Alzheimer’s disease brain: Implications for accumulation of reactive lipid peroxidation products. Neurochem. Res. 2004, 29, 2215–2220. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, W.; Kong, A.-N.T. Anti-oxidative stress regulator NF-E2-related factor 2 mediates the adaptive induction of antioxidant and detoxifying enzymes by lipid peroxidation metabolite 4-hydroxynonenal. Cell Biosci. 2012, 2, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Balogh, L.M.; le Trong, I.; Kripps, K.A.; Shireman, L.M.; Stenkamp, R.E.; Zhang, W.; Mannervik, B.; Atkins, W.M. Substrate specificity combined with stereopromiscuity in glutathione transferase A4-4-dependent metabolism of 4-hydroxynonenal. Biochemistry 2010, 49, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Calamaras, T.D.; Lee, C.; Lan, F.; Ido, Y.; Siwik, D.A.; Colucci, W.S. Post-translational modification of serine/threonine kinase LKB1 via adduction of the reactive lipid species 4-hydroxy-trans-2-nonenal (HNE) at lysine residue 97 directly inhibits kinase activity. J. Biol. Chem. 2012, 287, 42400–42406. [Google Scholar] [CrossRef] [PubMed]
- Shearn, C.T.; Backos, D.S.; Orlicky, D.J.; Smathers-McCullough, R.L.; Petersen, D.R. Identification of 5' AMP-activated kinase as a target of reactive aldehydes during chronic ingestion of high concentrations of ethanol. J. Biol. Chem. 2014, 289, 15449–15462. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Weerapana, E.; Blewett, M.M.; Cravatt, B.F. A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat. Methods 2014, 11, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Shearn, C.T.; Fritz, K.S.; Reigan, P.; Petersen, D.R. Modification of Akt2 by 4-hydroxynonenal inhibits insulin-dependent Akt signaling in HepG2 cells. Biochemistry 2011, 50, 3984–3996. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.J.; Langberg, E.C.; Tibell, A.; Sabat, G.; Kendrick, M.A.; Szweda, L.I.; Ostenson, C.G. Identification of ATP synthase as a lipid peroxide protein adduct in pancreatic islets from humans with and without type 2 Diabetes mellitus. J. Clin. Endocrinol. Metab. 2013, 98, E727–E731. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Prokai-Tatrai, K.; Vien, N.; Rauniyar, N.; Ughy, B.; Prokai, L. Protein targets for carbonylation by 4-hydroxy-2-nonenal in rat liver mitochondria. J. Proteomics 2011, 74, 2370–2379. [Google Scholar] [CrossRef] [PubMed]
- Shearn, C.T.; Smathers, R.L.; Stewart, B.J.; Fritz, K.S.; Galligan, J.J.; Hail, N., Jr.; Petersen, D.R. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) inhibition by 4-hydroxynonenal leads to increased Akt activation in hepatocytes. Mol. Pharmacol. 2011, 79, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Fu, Y.; Long, M.J.C.; Haegele, J.A.; Ge, E.J.; Parvez, S.; Aye, Y. Temporally controlled targeting of 4-hydroxynonenal to specific proteins in living cells. J. Am. Chem. Soc. 2013, 135, 14496–14499. [Google Scholar] [CrossRef] [PubMed]
- Los, G.V.; Encell, L.P.; McDougall, M.G.; Hartzell, D.D.; Karassina, N.; Zimprich, C.; Wood, M.G.; Learish, R.; Ohane, R.F.; Urh, M.; et al. HatoTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Fritz, K.S.; Galligan, J.J.; Smathers, R.L.; Roede, J.R.; Shearn, C.T.; Reigan, P.; Petersen, D.R. 4-hydroxynonenal inhibits SIRT3 via thiol-specific modification. Chem. Res. Toxicol. 2011, 24, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Krohne, T.U.; Kaemmerer, E.; Holz, F.G.; Kopitz, J. Lipid peroxidation products reduce lysosomal protease activities in human retinal pigment epithelial cells via two different mechanisms of action. Exp. Eye Res. 2010, 90, 261–266. [Google Scholar] [CrossRef]
- Wang, D.S.; Iwata, N.; Hama, E.; Saido, T.C.; Dickson, K. Oxidized neprilysin in aging and Alzheimer’s disease brains. Biochem. Biophys. Res. Commun. 2003, 310, 236–241. [Google Scholar]
- Wang, R.; Wang, S.; Malter, J.S.; Wang, D.-S. Effects of HNE-modification induced by A beta on neprilysin expression and activity in SH-SY5Y cells. J. Neurochem. 2009, 108, 1072–1082. [Google Scholar]
- Wang, R.; Malter, J.S.; Wang, D.-S. N-Acetylcysteine prevents 4-hydroxynonenal—And amyloid-β-induced modification and inactivation of neprilysin in SH-SY5Y cells. J. Alzheimers Dis. 2010, 19, 179–189. [Google Scholar] [PubMed]
- Liu, Q.; Simpson, D.C.; Gronert, S. Carbonylation of mitochondrial aconitase with 4-hydroxy-2-(E)-nonenal: Localization and relative reactivity of addition sites. Biochim. Biophys. Acta Proteins Proteomics 2013, 1834, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Miriyala, S.; Miao, L.; Mitov, M.; Schnell, D.; Dhar, S.K.; Cai, J.; Klein, J.B.; Sultana, R.; Butterfield, D.A.; et al. Redox proteomic identification of HNE-bound mitochondrial proteins in cardiac tissues reveals a systemic effect on energy metabolism after doxorubicin treatment. Free Radic. Biol. Med. 2014, 72, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Pizzimenti, S.; Arcaro, A.; Pettazzoni, P.; Minelli, R.; D’Angelo, D.; Mamone, G.; Ferranti, P.; Toaldo, C.; Cetrangolo, G.; et al. Exposure of HL-60 human leukaemic cells to 4-hydroxynonenal promotes the formation of adduct(s) with alpha-enolase devoid of plasminogen binding activity. Biochem. J. 2009, 422, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Reed, T.T.; Pierce, W.M.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of HNE-bound proteins in early Alzheimer disease: Insights into the role of lipid peroxidation in the progression of AD. Brain Res. 2009, 1274, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Bandemer, J.; Vindis, C.; Camare, C.; Mucher, E.; Gueraud, F.; Larroque-Cardoso, P.; Bernis, C.; Auge, N.; Salvayre, R.; et al. Protein disulfide isomerase modification and inhibition contribute to ER stress and apoptosis induced by oxidized low density lipoproteins. Antioxid. Redox Signal. 2013, 18, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Aluise, C.D.; Rose, K.; Boiani, M.; Reyzer, M.L.; Manna, J.D.; Tallman, K.; Porter, N.A.; Marnett, L.J. Peptidyl-prolyl cis/trans-isomerase A1 (Pin1) is a target for modification by lipid electrophiles. Chem. Res. Toxicol. 2013, 26, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Tanito, M.; Haniu, H.; Elliott, M.H.; Singh, A.K.; Matsumoto, H.; Anderson, R.E. Identification of 4-hydroxynonenal-modified retinal proteins induced by photooxidative stress prior to retinal degeneration. Free Radic. Biol. Med. 2006, 41, 1847–1859. [Google Scholar] [CrossRef] [PubMed]
- Toyokuni, S.; Yamada, S.; Kashima, M.; Ihara, Y.; Yamada, Y.; Tanaka, T.; Hiai, H.; Seino, Y.; Uchida, K. Serum 4-hydroxy-2-nonenal-modified albumin is elevated in patients with type 2 Diabetes Mellitus. Antioxid. Redox Signal. 2000, 2, 681–685. [Google Scholar] [CrossRef]
- Aldini, G.; Gamberoni, L.; Orioli, M.; Beretta, G.; Regazzoni, L.; Facino, R.M.; Carini, M. Mass spectrometric characterization of covalent modification of human serum albumin by 4-hydroxy-trans-2-nonenal. J. Mass Spectrom. 2006, 41, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Simpson, D.C.; Gronert, S. The reactivity of human serum albumin towards trans-4-hydroxy-2-nonenal. J. Mass Spectrom. 2012, 47, 411–424. [Google Scholar] [PubMed]
- Khatoon, F.; Alam, K.; Ali, A. Physicochemical and immunological studies on 4-hydroxynonenal modified HSA: Implications of protein damage by lipid peroxidation products in the etiopathogenesis of SLE. Hum. Immunol. 2012, 73, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Weisbach, V.; Sticht, H.; Seebahn, A.; Bussmann, J.; Zimmermann, R.; Becker, C.-M. Oxidative stress-induced posttranslational modifications of human hemoglobin in erythrocytes. Arch. Biochem. Biophys. 2013, 529, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Naveena, B.M.; Faustman, C.; Tatiyaborworntham, N.; Yin, S.; Ramanathan, R.; Mancini, R.A. Detection of 4-hydroxy-2-nonenal adducts of turkey and chicken myoglobins using mass spectrometry. Food Chem. 2010, 122, 836–840. [Google Scholar] [CrossRef]
- Smathers, R.L.; Fritz, K.S.; Galligan, J.J.; Shearn, C.T.; Reigan, P.; Marks, M.J.; Petersen, D.R. Characterization of 4-HNE modified L-FABP reveals alterations in structural and functional dynamics. PLoS ONE 2012, 7, e38459. [Google Scholar] [CrossRef] [PubMed]
- Hellberg, K.; Grimsrud, P.A.; Kruse, A.C.; Banaszak, L.J.; Ohlendorf, D.H.; Bernlohr, D.A. X-ray crystallographic analysis of adipocyte fatty acid binding protein (aP2) modified with 4-hydroxy-2-nonenal. Protein Sci. 2010, 19, 1480–1489. [Google Scholar] [CrossRef] [PubMed]
- Stewart, B.J.; Roede, J.R.; Doorn, J.A.; Petersen, D.R. Lipid aldehyde-mediated cross-linking of apolipoprotein B-100 inhibits secretion from HepG2 cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2009, 1791, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Pennathur, S.; Pagani, I.; Oda, M.N.; Witztum, J.L.; Oram, J.F.; Heinecke, J.W. Modifying apolipoprotein A–I by malondialdehyde, but not by an array of other reactive carbonyls, blocks cholesterol efflux by the ABCA1 pathway. J. Biol. Chem. 2010, 285, 18473–18484. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Tang, X.; Anderson, V.E.; Sayre, L.M. Mass spectrometric characterization of protein modification by the products of nonenzymatic oxidation of linoleic acid. Chem. Res. Toxicol. 2009, 22, 1386–1397. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.J.; Zhang, Y.L.; Montine, T.J. In vivo assessment of lipid peroxidation products associated with age-related neurodegenerative diseases. Exp. Neurol. 2002, 175, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Chan, S.L. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium 2003, 34, 385–397. [Google Scholar] [CrossRef]
- Pedersen, W.A.; Fu, W.; Keller, J.N.; Markesbery, W.R.; Appel, S.; Smith, R.G.; Kasarskis, E.; Mattson, M.P. Protein modification by the lipid peroxidation product 4-hydroxynonenal in the spinal cords of amyotrophic lateral sclerosis patients. Ann. Neurol. 1998, 44, 819–824. [Google Scholar] [CrossRef]
- Lovell, M.A.; Bradley, M.A.; Fister, S.X. 4-Hydroxyhexenal (HHE) impairs glutamate transport in astrocyte cultures. J. Alzheimers Dis. 2012, 32, 139–146. [Google Scholar] [PubMed]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [PubMed]
- Cabin, D.E.; Shimazu, K.; Murphy, D.; Cole, N.B.; Gottschalk, W.; McIlwain, K.L.; Orrison, B.; Chen, A.; Ellis, C.E.; Paylor, R.; et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci. 2002, 22, 8797–8807. [Google Scholar]
- Murphy, D.D.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 2000, 20, 3214–3220. [Google Scholar] [PubMed]
- Lotharius, J.; Brundin, P. Impaired dopamine storage resulting from α-synuclein mutations may contribute to the pathogenesis of Parkinson’s disease. Hum. Mol. Genet. 2002, 11, 2395–2407. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. 2010, 345, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Schlachetzki, J.C.M.; Helling, S.; Bussmann, J.C.; Berlinghof, M.; Schaeffer, T.E.; Marcus, K.; Winkler, J.; Klucken, J.; Becker, C.-M. Oxidative stress-induced posttranslational modifications of alpha-synuclein: Specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol. Cell. Neurosci. 2013, 54, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.-J.; Ho, D.-H.; Park, E.; Jung, J.W.; Cho, K.; Hong, J.H.; Lee, H.-J.; Kim, K.P.; Lee, S.-J. Lipid peroxidation product 4-hydroxy-2-nonenal promotes seeding-capable oligomer formation and cell-to-cell transfer of a-synuclein. Antioxid. Redox Signal. 2013, 18, 770–783. [Google Scholar] [CrossRef]
- Nasstrom, T.; Fagerqvist, T.; Barbu, M.; Karlsson, M.; Nikolajeff, F.; Kasrayan, A.; Ekberg, M.; Lannfelt, L.; Ingelsson, M.; Bergstrom, J. The lipid peroxidation products 4-oxo-2-nonenal and 4-hydroxy-2-nonenal promote the formation of α-synuclein oligomers with distinct biochemical, morphological, and functional properties. Free Radic. Biol. Med. 2011, 50, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Inose, Y.; Toi, S.; Hiroi, A.; Yamamoto, T.; Kobayashi, M. Involvement of 4-hydroxy-2-nonenal accumulation in multiple system atrophy. Acta Histochem. Cytochem. 2010, 43, 69–75. [Google Scholar] [CrossRef]
- Hortigon-Vinagre, M.P.; Chardonnet, S.; Montigny, C.; Gutierrez-Martin, Y.; Champeil, P.; Henao, F. Inhibition by 4-hydroxynonenal (HNE) of Ca2+ transport by SERCA1a: Low concentrations of HNE open protein-mediated leaks in the membrane. Free Radic. Biol. Med. 2011, 50, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Gunthorpe, M.J.; Benham, C.D.; Randall, A.; Davis, J.B. The diversity in the vanilloid (TRPV) receptor family of ion channels. Trends Pharmacol. Sci. 2002, 23, 183–191. [Google Scholar] [CrossRef]
- Kishimoto, E.; Naito, Y.; Handa, O.; Okada, H.; Mizushima, K.; Hirai, Y.; Nakabe, N.; Uchiyama, K.; Ishikawa, T.; Takagi, T.; et al. Oxidative stress-induced posttranslational modification of TRPV1 expressed in esophageal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G230–G238. [Google Scholar] [CrossRef] [PubMed]
- Vindis, C.; Escargueil-Blanc, I.; Elbaz, M.; Marcheix, B.; Grazide, M.H.; Uchida, K.; Salvayre, R.; Negre-Salvayre, A. Desensitization of platelet-derived growth factor receptor-beta by oxidized lipids in vascular cells and atherosclerotic lesions—Prevention by Aldehyde Scavengers. Circ. Res. 2006, 98, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Kumano-Kuramochi, M.; Shimozu, Y.; Wakita, C.; Ohnishi-Kameyama, M.; Shibata, T.; Matsunaga, S.; Takano-Ishikawa, Y.; Watanabe, J.; Goto, M.; Xie, Q.; et al. Identification of 4-hydroxy-2-nonenal-histidine adducts that serve as ligands for human lectin-like oxidized LDL receptor-1. Biochem. J. 2012, 442, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Park, Z.Y.; Kim, S.Y.; Jeong, E.; Lee, Y. Alteration of Toll-like receptor 4 activation by 4-hydroxy-2-nonenal mediated by the suppression of receptor homodimerization. Chem. Biol. Interact. 2009, 182, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Smith, M.A.; Avila, J.; DeBernardis, J.; Kansal, M.; Takeda, A.; Zhu, X.W.; Nunomura, A.; Honda, K.; Moreira, P.I.; et al. Alzheimer-specific epitopes of tau represent lipid peroxidation-induced conformations. Free Radic. Biol. Med. 2005, 38, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Santa-Maria, I.; Hernandez, F.; Martin, C.P.; Avila, J.; Moreno, F.J. Quinones facilitate the self-assembly of the phosphorylated tubulin binding region of tau into fibrillar polymers. Biochemistry 2004, 43, 2888–2897. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.; Hernaez, M.L.; Kamali, A.N.; Diez, A.; Puyet, A.; Bautista, J.M. Differential carbonylation of cytoskeletal proteins in blood group O erythrocytes: Potential role in protection against severe malaria. Infect. Genet. Evol. 2012, 12, 1780–1787. [Google Scholar] [CrossRef] [PubMed]
- Arashiki, N.; Otsuka, Y.; Ito, D.; Yang, M.; Komatsu, T.; Sato, K.; Inaba, M. The covalent modification of spectrin in red cell membranes by the lipid peroxidation product 4-hydroxy-2-nonenal. Biochem. Biophys. Res. Commun. 2010, 391, 1543–1547. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.; Hernaez, M.L.; Diez, A.; Puyet, A.; Bautista, J.M. Combined proteomic approaches for the identification of specific amino acid residues modified by 4-hydroxy-2-nonenal under physiological conditions. J. Proteome Res. 2010, 9, 5770–5781. [Google Scholar] [CrossRef] [PubMed]
- Yamashima, T. Hsp70.1 and related lysosomal factors for necrotic neuronal death. J. Neurochem. 2012, 120, 477–494. [Google Scholar] [CrossRef]
- Perluigi, M.; Poon, H.F.; Hensley, K.; Pierce, W.M.; Klein, J.B.; Calabrese, V.; de Marco, C.; Butterfield, D.A. Proteomic analysis of 4-hydroxy-2-nonenal-modified proteins in G93A-SOD1 transgenic mice—A model of familial amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2005, 38, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Sahara, S.; Yamashima, T. Calpain-mediated Hsp70.1 cleavage in hippocampal CA1 neuronal death. Biochem. Biophys. Res. Commun. 2010, 393, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Connor, R.E.; Marnett, L.J.; Liebler, D.C. Protein-selective capture to analyze electrophile adduction of Hsp90 by 4-hydroxynonenal. Chem. Res. Toxicol. 2011, 24, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Smathers, R.L.; Galligan, J.J.; Stewart, B.J.; Petersen, D.R. Overview of lipid peroxidation products and hepatic protein modification in alcoholic liver disease. Chem. Biol. Interact. 2011, 192, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, A.T.; Marnett, L.J. Systems analysis of protein modification and cellular responses induced by electrophile stress. Acc. Chem. Res. 2010, 43, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Galligan, J.J.; Fritz, K.S.; Backos, D.S.; Shearn, C.T.; Smathers, R.L.; Jiang, H.; MacLean, K.N.; Reigan, P.R.; Petersen, D.R. Oxidative stress-mediated aldehyde adduction of GRP78 in a mouse model of alcoholic liver disease: Functional independence of ATPase activity and chaperone function. Free Radic. Biol. Med. 2014, 73, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Buckingham, J.A.; Esteves, T.C.; Green, K.; Lambert, A.J.; Miwa, S.; Murphy, M.P.; Pakay, J.L.; Talbot, D.A.; Echtay, K.S. Mitochondrial superoxide and aging: Uncoupling-protein activity and superoxide production. Free Radic. 2004, 203–213. [Google Scholar] [CrossRef]
- Esteves, T.C.; Brand, M.D. The reactions catalysed by the mitochondrial uncoupling proteins UCP2 and UCP3. Biochim. Biophys. Acta Bioenerget. 2005, 1709, 35–44. [Google Scholar]
- Smith, A.M.O.; Ratcliffe, R.G.; Sweetlove, L.J. Activation and function of mitochondrial uncoupling protein in plants. J. Biol. Chem. 2004, 279, 51944–51952. [Google Scholar] [CrossRef] [PubMed]
- Echtay, K.S.; Esteves, T.C.; Pakay, J.L.; Jekabsons, M.B.; Lambert, A.J.; Portero-Otin, M.; Pamplona, R.; Vidal-Puig, A.J.; Wang, S.; Roebuck, S.J.; et al. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J. 2003, 22, 4103–4110. [Google Scholar] [CrossRef]
- Echtay, K.S.; Pakay, J.L.; Esteves, T.C.; Brand, M.D. Hydroxynonenal and uncoupling proteins: A model for protection against oxidative damage. Biofactors 2005, 24, 119–130. [Google Scholar] [CrossRef]
- Malingriaux, E.A.; Rupprecht, A.; Gille, L.; Jovanovic, O.; Jezek, P.; Jaburek, M.; Pohl, E.E. Fatty acids are key in 4-hydroxy-2-nonenal-mediated activation of uncoupling proteins 1 and 2. PLoS ONE 2013, 8, e77786. [Google Scholar] [PubMed]
- Woyda-Ploszczyca, A.; Jarmuszkiewicz, W. Hydroxynonenal-stimulated activity of the uncoupling protein in Acanthamoeba castellanii mitochondria under phosphorylating conditions. Biol. Chem. 2013, 394, 649–658. [Google Scholar] [CrossRef]
- Aguirre, E.; Cadenas, S. GDP and carboxyatractylate inhibit 4-hydroxynonenal-activated proton conductance to differing degrees in mitochondria from skeletal muscle and heart. Biochim. Biophys. Acta Bioenerget. 2010, 1797, 1716–1726. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Daly, D.S.; Marks, J.R.; Zangar, R.C. Oxidatively modified proteins as plasma biomarkers in breast cancer. Cancer Biomarkers 2013, 13, 193–200. [Google Scholar]
- Pillon, N.J.; Vella, R.E.; Soulere, L.; Becchi, M.; Lagarde, M.; Soulage, C.O. Structural and functional changes in human insulin induced by the lipid peroxidation byproducts 4-hydroxy-2-nonenal and 4-hydroxy-2-hexenal. Chem. Res.Toxicol. 2011, 24, 752–762. [Google Scholar] [CrossRef]
- Lee, S.H.; Takahashi, R.; Goto, T.; Oe, T. Mass spectrometric characterization of modifications to angiotensin II by lipid peroxidation products, 4-oxo-2(E)-nonenal and 4-hydroxy-2(E)-nonenal. Chem. Res. Toxicol. 2010, 23, 1771–1785. [Google Scholar] [CrossRef] [PubMed]
- El-Bikai, R.; Welman, M.; Margaron, Y.; Cote, J.-F.; Macqueen, L.; Buschmann, M.D.; Fahmi, H.; Shi, Q.; Maghni, K.; Fernandes, J.C.; et al. Perturbation of adhesion molecule-mediated chondrocyte-matrix interactions by 4-hydroxynonenal binding: Implication in osteoarthritis pathogenesis. Arthritis Res. Ther. 2010, 12. [Google Scholar] [CrossRef] [PubMed]
- Alzolibani, A.A.; al Robaee, A.A.; al-Shobaili, H.A.; Rasheed, Z. 4-Hydroxy-2-nonenal modified histone-H2A: A possible antigenic stimulus for systemic lupus erythematosus autoantibodies. Cell. Immunol. 2013, 284, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Guichardant, M.; Taibi-Tronche, P.; Fay, L.B.; Lagarde, M. Covalent modifications of aminophospholipids by 4-hydroxynonenal. Free Radic. Biol. Med. 1998, 25, 1049–1056. [Google Scholar] [CrossRef]
- Guo, L.; Chen, Z.; Amarnath, V.; Davies, S.S. Identification of novel bioactive aldehyde-modified phosphatidylethanolamines formed by lipid peroxidation. Free Radic. Biol. Med. 2012, 53, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Sowell, J.; Frei, B.; Stevens, J.F. Vitamin C conjugates of genotoxic lipid peroxidation products: Structural characterization and detection in human plasma. (Retracted article, see Proc. Natl. Acad. Sci. USA 2007, 104, 14543–14543). Proc. Natl. Acad. Sci. USA 2004, 101, 17964–17969. [Google Scholar] [CrossRef] [PubMed]
- Sowell, J.; Frei, B.; Stevens, J.F. Vitamin C conjugates of genotoxic lipid peroxidation products: Structural characterization and detection in human plasma. Proc. Natl. Acad. Sci. USA 2007, 104, 14543. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.L.; Reed, R.L.; Kuiper, H.C.; Alber, S.; Stevens, J.F. Ascorbic acid promotes detoxification and elimination of 4-hydroxy-2(E)-nonenal in human monocytic THP-1 cells. Chem. Res. Toxicol. 2009, 22, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Amarnath, V.; Amarnath, K.; Davies, S.; Roberts, L.J. Pyridoxamine: An extremely potent scavenger of 1,4-dicarbonyls. Chem. Res. Toxicol. 2004, 17, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Hardas, S.S.; Sultana, R.; Clark, A.M.; Beckett, T.L.; Szweda, L.I.; Murphy, M.P.; Butterfield, D.A. Oxidative modification of lipoic acid by HNE in Alzheimer disease brain. Redox Biol. 2013, 1, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Voulgaridou, G.-P.; Anestopoulos, I.; Franco, R.; Panayiotidis, M.I.; Pappa, A. DNA damage induced by endogenous aldehydes: Current state of knowledge. Mutat. Res. Fund. Mol. Mech. Mutagen. 2011, 711, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Minko, I.G.; Kozekov, I.D.; Harris, T.A.; Rizzo, C.J.; Lloyd, R.S.; Stone, M.P. Chemistry and biology of DNA containing 1,N2-deoxyguanosine adducts of the α,β-unsaturated aldehydes acrolein, crotonaldehyde, and 4-hydroxynonenal. Chem. Res. Toxicol. 2009, 22, 759–778. [Google Scholar] [CrossRef] [PubMed]
- Eckl, P.M.; Ortner, A.; Esterbauer, H. Genotoxic properties of 4-hydroxyalkenals and analogous aldehydes. Mutat. Res. Fund. Mol. Mech. Mutagen. 1993, 290, 183–192. [Google Scholar] [CrossRef]
- Karlhuber, G.M.; Bauer, H.C.; Eckl, P.M. Cytotoxic and genotoxic effects of 4-hydroxynonenal in cerebral endothelial cells. Mutat. Res. Fund. Mol. Mech. Mutagen. 1997, 381, 209–216. [Google Scholar] [CrossRef]
- Wang, H.; Kozekov, I.D.; Harris, T.M.; Rizzo, C.J. Site-specific synthesis and reactivity of oligonucleotides containing stereochemically defined 1,N2-deoxyguanosine adducts of the lipid peroxidation product trans-4-hydroxynonenal. J. Am. Chem. Soc. 2003, 125, 5687–5700. [Google Scholar] [PubMed]
- Wacker, M.; Wanek, P.; Eder, E. Detection of 1,N2-propanodeoxyguanosine adducts of trans-4-hydroxy-2-nonenal after gavage of trans-4-hydroxy-2-nonenal or induction of lipid peroxidation with carbon tetrachloride in F344 rats. Chem. Biol. Interact. 2001, 137, 269–283. [Google Scholar] [CrossRef]
- Kozekov, I.D.; Turesky, R.J.; Alas, G.R.; Harris, C.M.; Harris, T.M.; Rizzo, C.J. Formation of deoxyguanosine cross-links from calf thymus DNA treated with acrolein and 4-hydroxy-2-nonenal. Chem. Res. Toxicol. 2010, 23, 1701–1713. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Kozekov, I.D.; Kozekova, A.; Wang, H.; Lloyd, R.S.; Rizzo, C.J.; Stone, M.P. DNA Cross-link induced by trans-4-Hydroxynonenal. Environ. Mol. Mutagen. 2010, 51, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wang, H.; Kozekova, A.; Rizzo, C.J.; Stone, M.P. Formation of a N2-dG:N2-dG carbinolamine DNA cross-link by the trans-4-Hydroxynonenal-derived (6S,8R,11S) 1,N2-dG adduct. J. Am. Chem. Soc. 2011, 133, 16101–16110. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.; Dyba, M.; Pan, J.; Roy, R.; Chung, F.-L. Repair kinetics of acrolein- and (E)-4-hydroxy-2-nonenal-derived DNA adducts in human colon cell extracts. Mutat. Res. Fund. Mol. Mech. Mutagen. 2013, 751, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.; Pan, J.; Amin, S.; Chung, F.L.; Roy, R. Repair kinetics of trans-4-hydroxynonenal-induced cyclic 1,N2-propanodeoxyguanine DNA adducts by human cell nuclear extracts. Biochemistry 2004, 43, 7514–7521. [Google Scholar] [CrossRef]
- Winczura, A.; Zdzalik, D.; Tudek, B. Damage of DNA and proteins by major lipid peroxidation products in genome stability. Free Radic. Res. 2012, 46, 442–459. [Google Scholar] [PubMed]
- Janowska, B.; Komisarski, M.; Prorok, P.; Sokolowska, B.; Kusmierek, J.; Janion, C.; Tudek, B. Nucleotide excision repair and recombination are engaged in repair of trans-4-hydroxy-2-nonenal adducts to DNA bases in Escherichia coli. Int. J. Biol. Sci. 2009, 5, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Christov, P.P.; Kozekova, A.; Rizzo, C.J.; Egli, M.; Stone, M.P. Replication bypass of the trans-4-Hydroxynonenal-derived (6S,8R,11S)-1,N2-deoxyguanosine DNA adduct by the Sulfolobus solfataricus DNA polymerase IV. Chem. Res. Toxicol. 2012, 25, 422–435. [Google Scholar] [CrossRef]
- Janowska, B.; Kurpios-Piec, D.; Prorok, P.; Szparecki, G.; Komisarski, M.; Kowalczyk, P.; Janion, C.; Tudek, B. Role of damage-specific DNA polymerases in M13 phage mutagenesis induced by a major lipid peroxidation product trans-4-hydroxy-2-nonenal. Mutat. Res. Fund. Mol. Mech. Mutagen. 2012, 729, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.J.C.; Gonzalez, F.J.; Shou, M.G.; Chung, F.L. 2,3-Epoxy-4-hydroxynonanal, a potential lipid peroxidation product for etheno adduct formation, is not a substrate of human epoxide hydrolase. Carcinogenesis 1998, 19, 939–943. [Google Scholar] [CrossRef]
- Lee, S.H.; Arora, J.A.; Oe, T.; Blair, I.A. 4-Hydroperoxy-2-nonenal-induced formation of 1,N2-etheno-2'-deoxyguanosine adducts. Chem. Res. Toxicol. 2005, 18, 780–786. [Google Scholar] [CrossRef]
- Wang, Y.; Millonig, G.; Nair, J.; Patsenker, E.; Stickel, F.; Mueller, S.; Bartsch, H.; Seitz, H.K. Ethanol-induced cytochrome P4502E1 causes carcinogenic etheno-DNA lesions in alcoholic liver disease. Hepatology 2009, 50, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.; Srivatanakul, P.; Haas, C.; Jedpiyawongse, A.; Khuhaprema, T.; Seitz, H.K.; Bartsch, H. High urinary excretion of lipid peroxidation-derived DNA damage in patients with cancer-prone liver diseases. Mutat. Res. Fund. Mol. Mech. Mutagen. 2010, 683, 23–28. [Google Scholar]
- Grune, T.; Muller, K.; Zollner, S.; Haseloff, R.; Blasig, I.E.; David, H.; Siems, W. Evaluation of purine nucleotide loss, lipid peroxidation and ultrastructural alterations in post-hypoxic hepatocytes. J. Physiol. Lond. 1997, 498, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Grune, T.; Sommerburg, O.; Petras, T.; Siems, W.G. Postanoxic formation of aldehydic lipid-peroxidation products in human renal tubular cells. Free Radic. Biol. Med. 1995, 18, 21–27. [Google Scholar] [CrossRef]
- Blasig, I.E.; Grune, T.; Schonheit, K.; Rohde, E.; Jakstadt, M.; Haseloff, R.F.; Siems, W.G. 4-hydroxynonenal, a novel indicator of lipid-peroxidation for reperfusion injury of the myocardium. Am. J. Physiol. Heart Circ. Physiol. 1995, 269, H14–H22. [Google Scholar]
- Grune, T.; Siems, W.G.; Zollner, H.; Esterbauer, H. Metabolism of 4-hydroxynonenal, a cytotoxic lipid-peroxidation product, in ehrlich mouse ascites-cells at different proliferation stages. Cancer Res. 1994, 54, 5231–5235. [Google Scholar] [PubMed]
- Blasig, I.E.; Schoenheit, K.; Siems, W.G. Formation of 4-hydroxyalkenals by the reperfusion-injured rat-heart. Cell. Biochem. Mol. Asp. Reperfus. Injury 1994, 723, 462–465. [Google Scholar] [CrossRef]
- Siems, W.G.; Grune, T.; Esterbauer, H. 4-hydroxynonenal formation during ischemia and reperfusion of rat small-intestine. Life Sci. 1995, 57, 785–789. [Google Scholar] [CrossRef]
- Poli, G.; Dianzani, M.U.; Cheeseman, K.H.; Slater, T.F.; Lang, J.; Esterbauer, H. Separation and characterization of the aldehydic products of lipid-peroxidation stimulated by carbon-tetrachloride or adp iron in isolated rat hepatocytes and rat-liver microsomal suspensions. Biochem. J. 1985, 227, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.X.; Roberts, K.N.; Markesbery, W.R.; Scheff, S.W.; Lovell, M.A. Oxidative stress in head trauma in aging. Free Radic. Biol. Med. 2006, 41, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.I.; Lynn, B.C.; Markesbery, W.R.; Lovell, M.A. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer’s disease. Neurobiol. Aging 2006, 27, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Negre-Salvayre, A.; Auge, N.; Ayala, V.; Basaga, H.; Boada, J.; Brenke, R.; Chapple, S.; Cohen, G.; Feher, J.; Grune, T.; et al. Pathological aspects of lipid peroxidation. Free Radic. Res. 2010, 44, 1125–1171. [Google Scholar] [PubMed]
- Grune, T.; Siems, W.G.; Schneider, W. Accumulation of aldehydic lipid peroxidation products during postanoxic reoxygenation of isolated rat hepatocytes. Free Radic. Biol. Med. 1993, 15, 125–132. [Google Scholar] [CrossRef]
- Schwarzer, E.; Muller, O.; Arese, P.; Siems, W.G.; Grune, T. Increased levels of 4-hydroxynonenal in human monocytes fed with malarial pigment hemozoin—A possible clue for hemozoin toxicity. FEBS Lett. 1996, 388, 119–122. [Google Scholar] [CrossRef]
- Gil, L.; Siems, W.; Mazurek, B.; Gross, J.; Schroeder, P.; Voss, P.; Grune, T. Age-associated analysis of oxidative stress parameters in human plasma and erythrocytes. Free Radic. Res. 2006, 40, 495–505. [Google Scholar] [CrossRef]
- Selley, M.L.; Bartlett, M.R.; McGuiness, J.A.; Hapel, A.J.; Ardlie, N.G.; Lacey, M.J. Determination of the lipid-peroxidation product trans-4-hydroxy-2-nonenal in biological samples by high-performance liquid-chromatography and combined capillary column gas chromatography negative-ion chemical ionization mass-spectrometry. J. Chromatogr. Biomed. Appl. 1989, 488, 329–340. [Google Scholar] [CrossRef]
- Schmidt, H.; Grune, T.; Muller, R.; Siems, W.G.; Wauer, R.R. Increased levels of lipid peroxidation products malondialdehyde and 4-hydroxynonenal after perinatal hypoxia. Pediatr. Res. 1996, 40, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Michel, P.; Eggert, W.; AlbrechtNebe, H.; Grune, T. Increased lipid peroxidation in children with autoimmune diseases. Acta Paediatr. 1997, 86, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Itakura, K.; Kawakishi, S.; Hiai, H.; Toyokuni, S.; Stadtman, E.R. Characterization of epitopes recognized by 4-hydroxy-2-nonenal specific antibodies. Arch. Biochem. Biophys. 1995, 324, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Yoritaka, A.; Hattori, N.; Uchida, K.; Tanaka, M.; Stadtman, E.R.; Mizuno, Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc. Natl. Acad. Sci. USA 1996, 93, 2696–2701. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Toyokuni, S.; Uchida, K.; Ogawa, O.; Takenewa, J.; Kakehi, Y.; Kinoshita, H.; Hattorinakakuki, Y.; Hiai, H.; Yoshida, O. Formation of 8-hydroxy-2'-deoxyguanosine and 4-hydroxy-2-nonenal-modified proteins in human renal-cell carcinoma. Int. J. Cancer 1994, 58, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Nyhlin, N.; Suhr, O.; Holmgren, G.; Uchida, K.; ElSahly, M.; Yamashita, T.; Terasaki, H.; Nakamura, M.; Uchino, M.; et al. Oxidative stress is found in amyloid deposits in systemic amyloidosis. Biochem. Biophys. Res. Commun. 1997, 232, 497–502. [Google Scholar] [PubMed]
- Butterfield, D.A.; Reed, T.; Perluigi, M.; de Marco, C.; Coccia, R.; Cini, C.; Sultana, R. Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci. Lett. 2006, 397, 170–173. [Google Scholar] [CrossRef]
- Morikawa, S.; Kurauchi, O.; Tanaka, M.; Yoneda, M.; Uchida, K.; Itakura, A.; Furugori, K.; Mizutani, S.; Tomoda, Y. Increased mitochondrial damage by lipid peroxidation in trophoblast cells of preeclamptic placentas. Biochem. Mol. Biol. Int. 1997, 41, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, R.F.; Li, L.; Eschenbacher, W.L.; Szweda, L.; Holian, A. Potential involvement of 4-hydroxynonenal in the response of human lung cells to ozone. Am. J. Physiol. Lung Cell. Mol. Physiol. 1998, 274, L8–L16. [Google Scholar]
- Liu, X.L.; Lovell, M.A.; Lynn, B.C. Detection and quantification of endogenous cyclic DNA adducts derived from trans-4-hydroxy-2-nonenal in human brain tissue by isotope dilution capillary liquid chromatography nanoelectrospray tandem mass spectrometry. Chem. Res. Toxicol. 2006, 19, 710–718. [Google Scholar] [CrossRef]
- Surh, J.; Kwon, H. Estimation of daily exposure to 4-hydroxy-2-alkenals in Korean foods containing n-3 and n-6 polyunsaturated fatty acids. Food Addit. Contam. 2005, 22, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Kosuge, Y.; Sakikubo, T.; Horie, K.; Ishikawa, N.; Obokata, N.; Yokoyama, E.; Yamashina, K.; Yamamoto, M.; Saito, H.; et al. Protective effect of S-allyl-L-cysteine, a garlic compound, on amyloid beta-protein-induced cell death in nerve growth factor-differentiated PC12 cells. Neurosci. Res. 2003, 46, 119–125. [Google Scholar] [CrossRef]
- Glei, M.; Hofmann, T.; Kuster, K.; Hollmann, J.; Lindhauer, M.G.; Pool-Zobel, B.L. Both wheat (Triticum aestivum) bran arabinoxylans and gut flora-mediated fermentation products protect human colon cells from genotoxic activities of 4-hydroxynonenal and hydrogen peroxide. J. Agric. Food Chem. 2006, 54, 2088–2095. [Google Scholar] [CrossRef] [PubMed]
- Loguercio, C.; Federico, A.; Tuccillo, C.; Terracciano, F.; D’Auria, M.V.; de Simone, C.; Blanco, C.D. Beneficial effects of a probiotic VSL#3 on parameters of liver dysfunction in chronic liver diseases. J. Clin. Gastroenterol. 2005, 39, 540–543. [Google Scholar] [PubMed]
- Chowdhury, P.; Soulsby, M. Lipid peroxidation in rat brain is increased by simulated weightlessness and decreased by a soy-protein diet. Ann. Clin. Lab. Sci. 2002, 32, 188–192. [Google Scholar]
- Skrzydlewska, E.; Ostrowska, J.; Farbiszewski, R.; Michalak, K. Protective effect of green tea against lipid peroxidation in the rat liver, blood serum and the brain. Phytomedicine 2002, 9, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Leiphon, L.J.; Picklo, M.J., Sr. Inhibition of aldehyde detoxification in CNS mitochondria by fungicides. Neurotoxicology 2007, 28, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, O.; Grune, T.; Henke, W.; Esterbauer, H.; Siems, W.G. Identification of metabolic pathways of the lipid-peroxidation product 4-hydroxynonenal by mitochondria isolated from rat-kidney cortex. FEBS Lett. 1994, 352, 84–86. [Google Scholar] [CrossRef]
- Siems, W.G.; Zollner, H.; Grune, T.; Esterbauer, H. Metabolic fate of 4-hydroxynonenal in hepatocytes: 1,4-dihydroxynonene is not the main product. J. Lipid Res. 1997, 38, 612–622. [Google Scholar] [PubMed]
- Siems, W.G.; Grune, T. High capacity of secondary antioxidative pathways in thymocytes: Rapid detoxification of 4-hydroxynonenal. Int. J. Thymol. 1998, 6, 518–529. [Google Scholar]
- Ullrich, O.; Huser, H.; Ehrlich, W.; Grune, T. Intracellular metabolism of 4-hydroxynonenal in primary cultures of rabbit synovial fibroblasts. Free Radic. Biol. Med. 1997, 22, 1153–1157. [Google Scholar] [CrossRef]
- Grune, T.; Siems, W.G.; Petras, T. Identification of metabolic pathways of the lipid peroxidation product 4-hydroxynonenal in in situ perfused rat kidney. J. Lipid Res. 1997, 38, 1660–1665. [Google Scholar] [PubMed]
- Grune, T.; Siems, W.; Kowalewski, J.; Zollner, H.; Esterbauer, H. Identification of metabolic pathways of the lipid-peroxidation product 4-hydroxynonenal by enterocytes of rat small-intestine. Biochem. Int. 1991, 25, 963–971. [Google Scholar] [PubMed]
- Petras, T.; Siems, W.G.; Grune, T. 4-hydroxynonenal is degraded to mercapturic acid conjugate in rat-kidney. Free Radic. Biol. Med. 1995, 19, 685–688. [Google Scholar] [CrossRef]
- Kinter, M.; Roberts, R.J. Glutathione consumption and glutathione peroxidase inactivation in fibroblast cell lines by 4-hydroxy-2-nonenal. Free Radic. Biol. Med. 1996, 21, 457–462. [Google Scholar] [CrossRef]
- Esterbauer, H.; Zollner, H.; Lang, J. Metabolism of the lipid-peroxidation product 4-hydroxynonenal by isolated hepatocytes and by liver cytosolic fractions. Biochem. J. 1985, 228, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Fauler, G. Investigations on the Metabolism of 4-Hydroxy-Alkenals. Ph.D. Thesis, University of Graz, Graz, Austria, 1987. [Google Scholar]
- Siems, W.G.; Zollner, H.; Esterbauer, H. Metabolic pathways of lipid peroxidation product 4-hydroxynonenal in hepatocytes quantitative assessment of an antioxidative defense system. Free Radic. Biol. Med. 1990, 9, 110. [Google Scholar] [CrossRef]
- Schaur, R.J.; Zollner, H.; Esterbauer, H. Biological effects of aldehydes with particular attention to hydroxynonenal and malondialdehyde. In Membrane Lipid Peroxidation; Vigo-Pelfrey, C., Ed.; CRC Press: Boca Raton, FL, USA, 1991; Volume 3, pp. 141–163. [Google Scholar]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Siems, W.; Grune, T.; Zollner, H.; Esterbauer, H. Formation and metabolism of the lipid peroxidation product 4-hydroxynonenal in liver and small intestine. In Free Radicals: From Basic Science to Medicine; Poli, G., Albano, E., Dianzani, M.U., Eds.; Birkhäuser Verlag: Basel, Switzerland, 1993; pp. 89–101. [Google Scholar]
- Siems, W.G.; Zollner, H.; Grune, T.; Esterbauer, H. Qualitative and quantitative-determination of metabolites of the lipid-peroxidation product 4-hydroxynonenal from hepatocytes, enterocytes and tumor-cells. Fresenius J. Anal. Chem. 1992, 343, 75–76. [Google Scholar] [CrossRef]
- Canuto, R.A.; Ferro, M.; Muzio, G.; Bassi, A.M.; Leonarduzzi, G.; Maggiora, M.; Adamo, D.; Poli, G.; Lindahl, R. Effects of aldehyde products of lipid peroxidation on the activity of aldehyde metabolizing enzymes in hepatomas. Adv. Exp. Med. Biol. 1993, 328, 17–25. [Google Scholar]
- Canuto, R.A.; Muzio, G.; Maggiora, M.; Poli, G.; Biasi, F.; Dianzani, M.U.; Ferro, M.; Bassi, A.M.; Penco, S.; Marinari, U.M. Ability of different hepatoma-cells to metabolize 4-hydroxynonenal. Cell Biochem. Funct. 1993, 11, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Ferro, M.; Marinari, U.M.; Poli, G.; Dianzani, M.U.; Fauler, G.; Zollner, H.; Esterbauer, H. Metabolism of 4-hydroxynonenal by the rat hepatoma-cell line MH1C1. Cell Biochem. Funct. 1988, 6, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Canuto, R.A.; Muzio, G.; Bassi, A.M.; Biocca, M.E.; Poli, G.; Esterbauer, H.; Ferro, M. Metabolism of 4 hydroxynonenal in hepatoma cell lines. In Advances in Experimental Medicine and Biology, Vol. 284. Enzymology and Molecular Biology of Carbonyl Metabolism 3, Proceedings of the Fifth International Workshop, West Lafayette, IN, USA, 12–15 June 1990; Weiner, H., Wermuth, M.B., Crabb, D.W., Eds.; Plenum Press: New York, NY, USA, 1990; pp. 75–84. [Google Scholar]
- Srivastava, S.; Conklin, D.J.; Liu, S.Q.; Prakash, N.; Boor, P.J.; Srivastava, S.K.; Bhatnagar, A. Identification of biochemical pathways for the metabolism of oxidized low-density lipoprotein derived aldehyde-4-hydroxy trans-2-nonenal in vascular smooth muscle cells. Atherosclerosis 2001, 158, 339–350. [Google Scholar] [CrossRef]
- Srivastava, S.; Chandra, A.; Wang, L.F.; Seifert, W.E.; DaGue, B.B.; Ansari, N.H.; Srivastava, S.K.; Bhatnagar, A. Metabolism of the lipid peroxidation product, 4-hydroxy-trans-2-nonenal, in isolated perfused rat heart. J. Biol. Chem. 1998, 273, 10893–10900. [Google Scholar] [CrossRef] [PubMed]
- Spitz, D.R.; Malcolm, R.R.; Roberts, R.J. Cytotoxicity and metabolism of 4-hydroxy-2-nonenal and 2-nonenal in H2O2-resistant cell-lines—Do aldehydic by-products of lipid-peroxidation contribute to oxidative stress. Biochem. J. 1990, 267, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.J.; Mosely, D.E.; Amarnath, V.; Picklo, M.J. Metabohom of 4-hydroxy-trans-2-nonenal by central nervous system mitochondria is dependent on age and NAD(+) availability. Chem. Res. Toxicol. 2004, 17, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Canuto, R.A.; Muzio, G.; Biocca, M.E.; Dianzani, M.U. Oxidative-metabolism of 4-hydroxy-2,3-nonenal during diethyl-nitrosamine-induced carcinogenesis in rat-liver. Cancer Lett. 1989, 46, 7–13. [Google Scholar] [CrossRef]
- Chen, J.J.; Yu, B.P. Detoxification of reactive aldehydes in mitochondria: Effects of age and dietary restriction. Aging Clin. Exp. Res. 1996, 8, 334–340. [Google Scholar] [CrossRef]
- Honzatko, A.; Brichac, J.; Murphy, T.C.; Reberg, A.; Kubatova, A.; Smoliakova, I.P.; Picklo, M.J. Enantioselective metabolism of trans-4-hydroxy-2-nonenal by brain mitochondria. Free Radic. Biol. Med. 2005, 39, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Alary, J.; Gueraud, F.; Cravedi, J.-P. Fate of 4-hydroxynonenal in vivo: Disposition and metabolic pathways. Mol. Asp. Med. 2003, 24, 177–187. [Google Scholar] [CrossRef]
- Alary, J.; Bravais, F.; Cravedi, J.P.; Debrauwer, L.; Rao, D.; Bories, G. Mercapturic acid conjugates as urinary end metabolites of the lipid-peroxidation product 4-hydroxy-2-nonenal in the rat. Chem. Res. Toxicol. 1995, 8, 34–39. [Google Scholar] [CrossRef]
- Winter, C.K.; Segall, H.J.; Jones, A.D. Distribution of trans-4-hydroxy-2-hexenal and tandem mass-spectrometric detection of its urinary mercapturic acid in the rat. Drug Metab. Dispos. 1987, 15, 608–612. [Google Scholar] [PubMed]
- Alin, P.; Danielson, U.H.; Mannervik, B. 4-hydroxyalk-2-enals are substrates for glutathione transferase. FEBS Lett. 1985, 179, 267–270. [Google Scholar] [CrossRef]
- Danielson, U.H.; Esterbauer, H.; Mannervik, B. Structure-activity-relationships of 4-hydroxyalkenals in the conjugation catalyzed by mammalian glutathione transferases. Biochem. J. 1987, 247, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.Y.; Petersen, D.R. The oxidation of α,β-unsaturated aldehydic products of lipid-peroxidation by rat-liver aldehyde dehydrogenases. Toxicol. Appl. Pharmacol. 1987, 87, 403–410. [Google Scholar] [CrossRef]
- Mitchell, D.Y.; Petersen, D.R. Inhibition of rat hepatic mitochondrial aldehyde dehydrogenase mediated acetaldehyde oxidation by trans-4-hydroxy-2-nonenal. Hepatology 1991, 13, 728–734. [Google Scholar] [CrossRef]
- Hartley, D.P.; Ruth, J.A.; Petersen, D.R. The hepatocellular metabolism of 4-hydroxynonenal by alcohol-dehydrogenase, aldehyde dehydrogenase, and glutathione-S-transferase. Arch. Biochem. Biophys. 1995, 316, 197–205. [Google Scholar] [CrossRef]
- Singhal, S.S.; Zimniak, P.; Awasthi, S.; Piper, J.T.; He, N.G.; Teng, J.I.; Petersen, D.R.; Awasthi, Y.C. Several closely-related glutathione-S-transferase isozymes catalyzing conjugation of 4-hydroxynonenal are differentially expressed in human tissues. Arch. Biochem. Biophys. 1994, 311, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Berhane, K.; Widersten, M.; Engstrom, A.; Kozarich, J.W.; Mannervik, B. Detoxication of base propenals and other α,β-unsaturated aldehyde products of radical reactions and lipid-peroxidation by human glutathione transferases. Proc. Natl. Acad. Sci. USA 1994, 91, 1480–1484. [Google Scholar] [CrossRef]
- Fukuda, A.; Nakamura, Y.; Ohigashi, H.; Osawa, T.; Uchida, K. Cellular response to the redox active lipid peroxidation products: Induction of glutathione S-transferase P by 4-hydroxy-2-nonenal. Biochem. Biophys. Res. Commun. 1997, 236, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Z.; Yang, Y.S.; Trent, M.B.; He, N.G.; Lick, S.D.; Zimniake, P.; Awasthi, Y.C.; Boor, P.J. Glutathione-S-transferase A4-4 modulates oxidative stress in endothelium: Possible role in human atherosclerosis. Atherosclerosis 2004, 173, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Hardej, D.; Trombetta, L.D.; Li, Y. The role of chemically induced glutathione and glutathione S-transferase in protecting against 4-hydroxy-2-nonenal-mediated cytotoxicity in vascular smooth muscle cells. Cardiovas. Toxicol. 2003, 3, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Kaiserova, K.; Srivastava, S.; Hoetker, J.D.; Awe, S.O.; Tang, X.L.; Cai, J.; Bhatnagar, A. Redox activation of aldose reductase in the ischemic heart. J. Biol. Chem. 2006, 281, 15110–15120. [Google Scholar] [CrossRef] [PubMed]
- Bander Jagt, D.L.; Kolb, N.S.; bander Jagt, T.J.; Chino, J.; Martinez, F.J.; Hunsaker, L.A.; Royer, R.E. Substrate specificity of human aldose reductase: Identification of 4-hydroxynonenal as an endogenous substrate. Biochim. Biophys. Acta 1995, 1249, 117–126. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Ansari, N.H.; Hair, G.A.; Das, B. Aldose and aldehyde reductases in human-tissues. Biochim. Biophys. Acta 1984, 800, 220–227. [Google Scholar] [CrossRef]
- He, Q.; Khanna, P.; Srivastava, S.; van Kuijk, F.; Ansari, N.H. Reduction of 4-hydroxynonenal and 4-hydroxyhexenal by retinal aldose reductase. Biochem. Biophys. Res. Commun. 1998, 247, 719–722. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Yadav, U.C.S.; Reddy, A.B.M.; Saxena, A.; Tammali, R.; Shoeb, M.; Ansari, N.H.; Bhatnagar, A.; Petrash, M.J.; Srivastava, S.; et al. Aldose reductase inhibition suppresses oxidative stress-induced inflammatory disorders. Chem. Biol. Interact. 2011, 191, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.H.; Park, Y.S.; Takahashi, M.; Suzuki, K.; Taniguchi, N. Aldehyde reductase gene expression by lipid peroxidation end products, MDA and HNE. Free Radic. Res. 2000, 33, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Hoehn, A.; Koenig, J.; Grune, T. Protein oxidation in aging and the removal of oxidized proteins. J. Proteomics 2013, 92, 132–159. [Google Scholar] [CrossRef] [PubMed]
- Grune, T.; Davies, K.J.A. Breakdown of oxidized proteins as a part of secondary antioxidant defenses in mammalian cells. Biofactors 1997, 6, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Grune, T.; Reinheckel, T.; Davies, K.J.A. Degradation of oxidized proteins in mammalian cells. FASEB J. 1997, 11, 526–534. [Google Scholar] [PubMed]
- Laurent, A.; Perdu-Durand, E.; Alary, J.; Debrauwer, L.; Cravedi, J.P. Metabolism of 4-hydroxynonenal, a cytotoxic product of lipid peroxidation, in rat precision-cut liver slices. Toxicol. Lett. 2000, 114, 203–214. [Google Scholar] [CrossRef]
- Alary, J.; Debrauwer, L.; Fernandez, Y.; Paris, A.; Cravedi, J.P.; Dolo, L.; Rao, D.; Bories, G. Identification of novel urinary metabolites of the lipid peroxidation product 4-hydroxy-2-nonenal in rats. Chem. Res. Toxicol. 1998, 11, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Sadhukhan, S.; Berthiaume, J.M.; Ibarra, R.A.; Tang, H.; Deng, S.; Hamilton, E.; Nagy, L.E.; Tochtrop, G.P.; Zhang, G.-F. 4-Hydroxy-2(E)-nonenal (HNE) catabolism and formation of HNE adducts are modulated by beta oxidation of fatty acids in the isolated rat heart. Free Rad. Biol. Med. 2013, 58, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Kubatova, A.; Murphy, T.C.; Combs, C.; Picklo, M.J. Astrocytic biotransformation of trans-4-hydroxy-2-nonenal is dose-dependent. Chem. Res. Toxicol. 2006, 19, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Alary, J.; Debrauwer, L.; Fernandez, Y.; Cravedi, J.P.; Rao, D.; Bories, G. 1,4-dihydroxynonene mercapturic acid, the major end metabolite of exogenous 4-hydroxy-2-nonenal, is a physiological component of rat and human urine. Chem. Res. Toxicol. 1998, 11, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Laurent, A.; Alary, J.; Debrauwer, L.; Cravedi, J.P. Analysis in the rat of 4-hydroxynonenal metabolites excreted in bile: Evidence of enterohepatic circulation of these byproducts of lipid peroxidation. Chem. Res. Toxicol. 1999, 12, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.; Ito, K.; Suzuki, H.; Sugiyama, Y.; Horie, T. Multidrug resistance-associated protein2 (MRP2) plays an important role in the biliary excretion of glutathione conjugates of 4-hydroxynonenal. Free Rad. Biol. Med. 2002, 33, 370–378. [Google Scholar] [CrossRef]
- Esterbauer, H.; Ertl, A.; Scholz, N. Reaction of cysteine with α,β-unsaturated aldehydes. Tetrahedron 1976, 32, 285–289. [Google Scholar] [CrossRef]
- Grune, T.; Michel, P.; Sitte, N.; Eggert, W.; AlbrechtNebe, H.; Esterbauer, H.; Siems, W.G. Increased levels of 4-hydroxynonenal modified proteins in plasma of children with autoimmune diseases. Free Rad. Biol. Med. 1997, 23, 357–360. [Google Scholar] [CrossRef]
- Peiro, G.; Alary, J.; Cravedi, J.P.; Rathahao, E.; Steghens, J.P.; Gueraud, F. Dihydroxynonene mercapturic acid, a urinary metabolite of 4-hydroxynonenal, as a biomarker of lipid peroxidation. Biofactors 2005, 24, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Volkel, W.; Sicilia, T.; Pahler, A.; Gsell, W.; Tatschner, T.; Jellinger, K.; Leblhuber, F.; Riederer, P.; Lutz, W.K.; Gotz, M.E. Increased brain levels of 4-hydroxy-2-nonenal glutathione conjugates in severe Alzheimer’s disease. Neurochem. Int. 2006, 48, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Ramana, K.V.; Bhatnagar, A.; Srivastava, S.; Yadav, U.C.; Awasthi, S.; Awasthi, Y.C.; Srivastava, S.K. Mitogenic responses of vascular smooth muscle cells to lipid peroxidation-derived aldehyde 4-hydroxy-trans-2-nonenal (HNE)—Role of aldose reductase-catalyzed reduction of the HNE-glutathione conjugates in regulating cell growth. J. Biol. Chem. 2006, 281, 17652–17660. [Google Scholar] [CrossRef]
- Ramana, K.V.; Willis, M.S.; White, M.D.; Horton, J.W.; DiMaio, J.M.; Srivastava, D.; Bhatnagar, A.; Srivastava, S.K. Endotoxin-induced cardiomyopathy and systemic inflammation in mice is prevented by aldose reductase inhibition. Circulation 2006, 114, 1838–1846. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Ramana, K.V.; Tammali, R.; Srivastava, S.K.; Bhatnagar, A. Contribution of aldose reductase to diabetic hyperproliferation of vascular smooth muscle cells. Diabetes 2006, 55, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Tammali, R.; Ramana, K.V.; Singhal, S.S.; Awasthi, S.; Srivastava, S.K. Aldose reductase regulates growth factor-induced cyclooxygenase-2 expression and prostaglandin E2 production in human colon cancer cells. Cancer Res. 2006, 66, 9705–9713. [Google Scholar] [CrossRef] [PubMed]
- Ronis, M.J.J.; Butura, A.; Sampey, B.P.; Shankar, K.; Prior, R.L.; Korourian, S.; Albano, E.; Ingelman-Sundberg, M.; Petersen, D.R.; Badger, T.M. Effects of N-acetylcysteine on ethanol-induced hepatotoxicity in rats fed via total enteral nutrition. Free Rad. Biol. Med. 2005, 39, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, M.; Ushimaru, N.; Osada, N.; Oda, T.; Ishige, K.; Ito, Y. N-acetylcysteine selectively protects cerebellar granule cells from 4-hydroxynonenal-induced cell death. Neurosci. Res. 2006, 55, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.Y.; Mutcherson, R.; Helfand, S.L. Calorie restriction delays lipid oxidative damage in Drosophila melanogaster. Aging Cell 2005, 4, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Jung, K.J.; Kim, J.W.; Kim, H.J.; Yu, B.P.; Chung, H.Y. Suppression of apoptosis by calorie restriction in aged kidney. Exp. Gerontol. 2004, 39, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Zainal, T.A.; Oberley, T.D.; Allison, D.B.; Szweda, L.I.; Weindruch, R. Caloric restriction of rhesus monkeys lowers oxidative damage in skeletal muscle. FASEB J. 2000, 14, 1825–1836. [Google Scholar] [CrossRef]
- Shoeb, M.; Ansari, N.H.; Srivastava, S.K.; Ramana, K.V. 4-Hydroxynonenal in the pathogenesis and progression of human diseases. Curr. Med. Chem. 2014, 21, 230–237. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schaur, R.J.; Siems, W.; Bresgen, N.; Eckl, P.M. 4-Hydroxy-nonenal—A Bioactive Lipid Peroxidation Product. Biomolecules 2015, 5, 2247-2337. https://doi.org/10.3390/biom5042247
Schaur RJ, Siems W, Bresgen N, Eckl PM. 4-Hydroxy-nonenal—A Bioactive Lipid Peroxidation Product. Biomolecules. 2015; 5(4):2247-2337. https://doi.org/10.3390/biom5042247
Chicago/Turabian StyleSchaur, Rudolf J., Werner Siems, Nikolaus Bresgen, and Peter M. Eckl. 2015. "4-Hydroxy-nonenal—A Bioactive Lipid Peroxidation Product" Biomolecules 5, no. 4: 2247-2337. https://doi.org/10.3390/biom5042247