
Αlpha-Synuclein as a Mediator in the Interplay between Aging and Parkinson’s Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
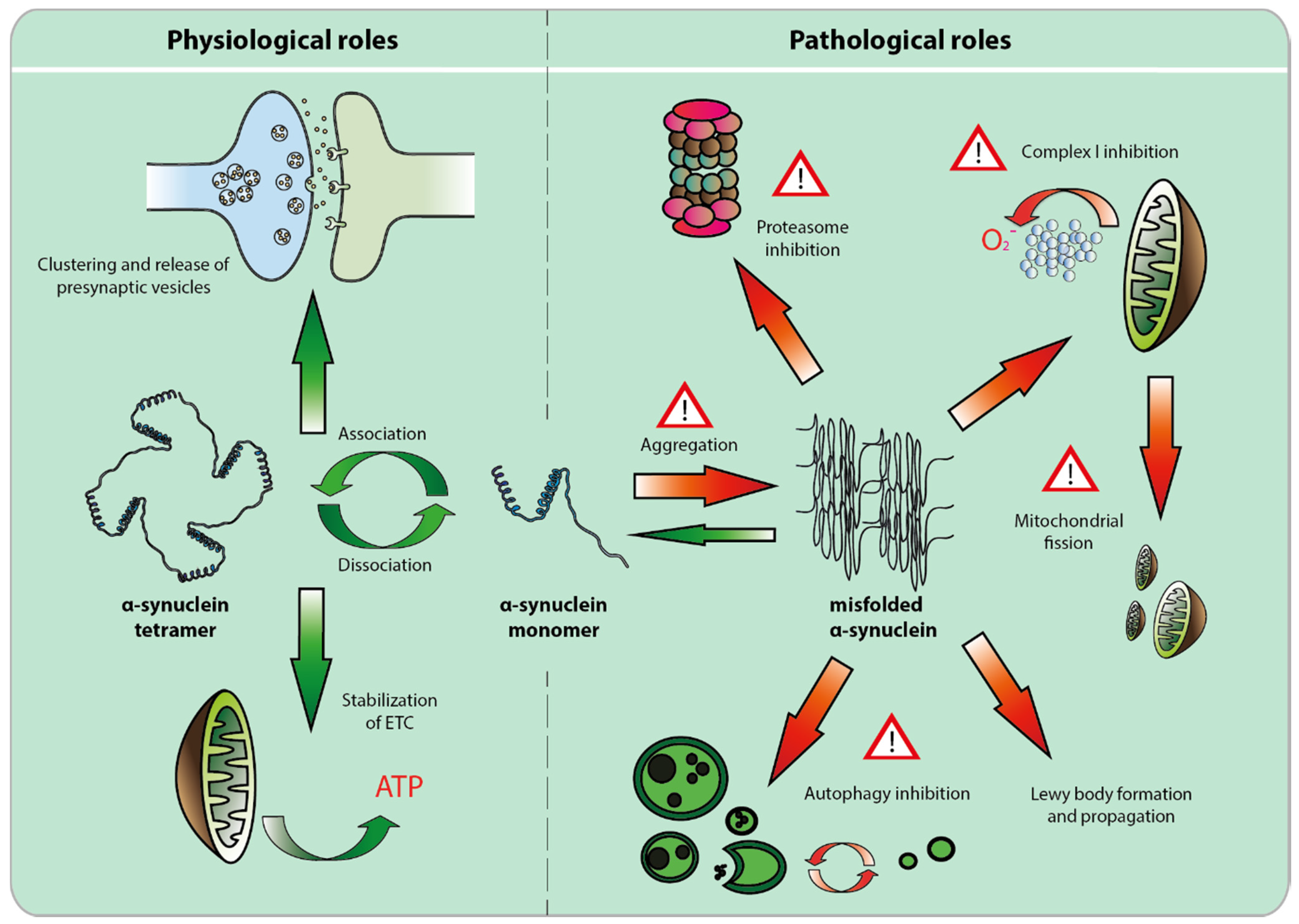
2. The Physiological Role of α-syn
2.1. Alpha-Synuclein and the Synapse
2.2. Alpha-Synuclein and the Mitochondria
3. Pathological Role of α-Syn
3.1. Alpha-Syn Misfolding, Aggregation and Propagation

3.2. Loss of Function Toxicity
3.3. Alpha-syn Toxicity Affects Multiple Compartments, but Mitochondrial Toxicity May Prevail
4. Aging as a Major Risk Factor for PD
4.1. Structural Changes in the Aging and Diseased Brain

4.2. Cellular Alterations in the Aging and Diseased Brain: Neuronal Pigments
4.3. Molecular Changes in the Aging and Diseased Brain: Proteostasis
4.4. Changes in Mitochondrial Activity
5. Dopaminergic Neurons of SNpc—Colossus with Feet Made of Clay
6. Animal Models of Aging
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Kiely, A.P.; Asi, Y.T.; Kara, E.; Limousin, P.; Ling, H.; Lewis, P.; Proukakis, C.; Quinn, N.; Lees, A.J.; Hardy, J.; et al. Alpha-synucleinopathy associated with G51D SNCA mutation: A link between Parkinson's disease and multiple system atrophy? Acta Neuropathol. 2013, 125, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science 2003, 302. PMID: 14593171. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Kagedal, K.; Halliday, G.M. Alpha-synuclein biology in Lewy body diseases. Alzheimers Res. Ther. 2014. [Google Scholar] [CrossRef] [PubMed]
- Farrer, M.; Maraganore, D.M.; Lockhart, P.; Singleton, A.; Lesnick, T.G.; de Andrade, M.; West, A.; de Silva, R.; Hardy, J.; Hernandez, D. Alpha-synuclein gene haplotypes are associated with Parkinson’s disease. Hum. Mol. Genet. 2001, 10, 1847–1851. [Google Scholar] [CrossRef] [PubMed]
- Pals, P.; Lincoln, S.; Manning, J.; Heckman, M.; Skipper, L.; Hulihan, M.; van den Broeck, M.; de Pooter, T.; Cras, P.; Crook, J.; et al. Alpha-synuclein promoter confers susceptibility to Parkinson’s disease. Ann. Neurol. 2004, 56, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.K.; Chai, A.; Teo, Y.Y.; Zhao, Y.; Tan, C.; Shen, H.; Chandran, V.R.; Teoh, M.L.; Yih, Y.; Pavanni, R.; et al. Alpha-synuclein haplotypes implicated in risk of Parkinson’s disease. Neurology 2004, 62, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of alpha-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Snead, D.; Eliezer, D. Alpha-synuclein function and dysfunction on cellular membranes. Exp. Neurobiol. 2014, 23, 292–313. [Google Scholar] [CrossRef] [PubMed]
- Kwakye, G.F.; Paoliello, M.M.; Mukhopadhyay, S.; Bowman, A.B.; Aschner, M. Manganese-induced parkinsonism and Parkinson’s disease: Shared and distinguishable features. Int. J. Environ. Res. Public Health 2015, 12, 7519–7540. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Ageing as a primary risk factor for Parkinson’s disease: Evidence from studies of non-human primates. Nat. Rev. Neurosci. 2011, 12, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Buchman, A.S.; Shulman, J.M.; Nag, S.; Leurgans, S.E.; Arnold, S.E.; Morris, M.C.; Schneider, J.A.; Bennett, D.A. Nigral pathology and parkinsonian signs in elders without Parkinson disease. Ann. Neurol. 2012, 71, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain 1991, 114, 2283–2301. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Bolam, J.P.; Pissadaki, E.K. Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov. Disord. 2012, 27, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.C.; Yu, J.T.; Tan, M.S.; Jiang, T.; Zhu, X.C.; Tan, L. Autophagy in aging and neurodegenerative diseases: Implications for pathogenesis and therapy. Neurobiol. Aging 2014, 35, 941–957. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Dimayuga, E.; Chen, Q.; Thorpe, J.; Gee, J.; Ding, Q. Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain. Int. J. Biochem. Cell Biol. 2004, 36, 2376–2391. [Google Scholar] [CrossRef] [PubMed]
- Lindner, A.B.; Demarez, A. Protein aggregation as a paradigm of aging. BBA-Gen Subjects 2009, 1790, 980–996. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I.; Cuervo, A.M. Proteostasis and the aging proteome in health and disease. J. Gerontol. A 2014, 69, S33–S38. [Google Scholar] [CrossRef] [PubMed]
- Cuanalo-Contreras, K.; Mukherjee, A.; Soto, C. Role of protein misfolding and proteostasis deficiency in protein misfolding diseases and aging. Int. J. Cell Biol. 2013, 2013, 638083. [Google Scholar] [CrossRef] [PubMed]
- Brunk, U.T.; Terman, A. The mitochondrial-lysosomal axis theory of aging—Accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur. J. Biochem. 2002, 269, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R. α-Synuclein and neuronal cell death. Mol. Neurodegener 2009. [Google Scholar] [CrossRef] [PubMed]
- Greten-Harrison, B.; Polydoro, M.; Morimoto-Tomita, M.; Diao, L.; Williams, A.M.; Nie, E.H.; Makani, S.; Tian, N.; Castillo, P.E.; Buchman, V.L.; et al. Alphabetagamma-synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. USA 2010, 107, 19573–19578. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Tan, L.; Yu, J.T. The link between the SNCA gene and parkinsonism. Neurobiol. Aging 2015, 36, 1505–1518. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Schmitz, Y.; Farinas, I.; Choi-Lundberg, D.; Ho, W.H.; Castillo, P.E.; Shinsky, N.; Verdugo, J.M.; Armanini, M.; Ryan, A.; et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000, 25, 239–252. [Google Scholar] [CrossRef]
- Cabin, D.E.; Shimazu, K.; Murphy, D.; Cole, N.B.; Gottschalk, W.; McIlwain, K.L.; Orrison, B.; Chen, A.; Ellis, C.E.; Paylor, R.; et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci. 2002, 22, 8797–8807. [Google Scholar] [PubMed]
- Steidl, J.V.; Gomez-Isla, T.; Mariash, A.; Ashe, K.H.; Boland, L.M. Altered short-term hippocampal synaptic plasticity in mutant α-synuclein transgenic mice. Neuroreport 2003, 14, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Clough, R.L.; Stefanis, L. A novel pathway for transcriptional regulation of alpha-synuclein. FASEB J. 2007, 21, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.; Olesen, O.F.; Mikkelsen, J.D. Developmental expression of α-synuclein in rat hippocampus and cerebral cortex. Neuroscience 1999, 91, 651–659. [Google Scholar] [CrossRef]
- Kholodilov, N.G.; Neystat, M.; Oo, T.F.; Lo, S.E.; Larsen, K.E.; Sulzer, D.; Burke, R.E. Increased expression of rat synuclein in the substantia nigra pars compacta identified by mRNA differential display in a model of developmental target injury. J. Neurochem. 1999, 73, 2586–2599. [Google Scholar] [CrossRef] [PubMed]
- Burre, J.; Sharma, M.; Sudhof, T.C. Definition of a molecular pathway mediating alpha-synuclein neurotoxicity. J. Neurosci. 2015, 35, 5221–5232. [Google Scholar] [CrossRef] [PubMed]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes snare-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Burre, J.; Sharma, M.; Sudhof, T.C. Alpha-synuclein assembles into higher-order multimers upon membrane binding to promote snare complex formation. Proc. Natl. Acad. Sci. USA 2014, 111, E4274–E4283. [Google Scholar] [CrossRef] [PubMed]
- Vargas, K.J.; Makani, S.; Davis, T.; Westphal, C.H.; Castillo, P.E.; Chandra, S.S. Synucleins regulate the kinetics of synaptic vesicle endocytosis. J. Neurosci. 2014, 34, 9364–9376. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [PubMed]
- Plaas, M.; Karis, A.; Innos, J.; Rebane, E.; Baekelandt, V.; Vaarmann, A.; Luuk, H.; Vasar, E.; Koks, S. Alpha-synuclein A30P point-mutation generates age-dependent nigrostriatal deficiency in mice. J. Physiol. Pharmacol. 2008, 59, 205–216. [Google Scholar] [PubMed]
- Janezic, S.; Threlfell, S.; Dodson, P.D.; Dowie, M.J.; Taylor, T.N.; Potgieter, D.; Parkkinen, L.; Senior, S.L.; Anwar, S.; Ryan, B.; et al. Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc. Natl. Acad. Sci. USA 2013, 110, E4016–E4025. [Google Scholar] [CrossRef] [PubMed]
- Guardia-Laguarta, C.; Area-Gomez, E.; Schon, E.A.; Przedborski, S. Novel subcellular localization for alpha-synuclein: Possible functional consequences. Front. Neuroanat. 2015. [Google Scholar] [CrossRef] [PubMed]
- Chicco, A.J.; Sparagna, G.C. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Cell Physiol. 2007, 292, C33–C44. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Qin, Z.J.; Hu, D.; Munishkina, L.A.; Fink, A.L. Alpha-synuclein can function as an antioxidant preventing oxidation of unsaturated lipid in vesicles. Biochemistry 2006, 45, 8135–8142. [Google Scholar] [CrossRef] [PubMed]
- Ai, S.X.; Xu, Q.; Hu, Y.C.; Song, C.Y.; Guo, J.F.; Shen, L.; Wang, C.R.; Yu, R.L.; Yan, X.X.; Tang, B.S. Hypomethylation of SNCA in blood of patients with sporadic Parkinson’s disease. J. Neurol. Sci. 2014, 337, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Jowaed, A.; Schmitt, I.; Kaut, O.; Wullner, U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J. Neurosci. 2010, 30, 6355–6359. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.Y.; Wu, L.; Zhao, Z.B.; Wang, Y.; Xiao, Q.; Liu, J.; Wang, G.; Ma, J.F.; Chen, S.D. Methylation of α-synuclein and leucine-rich repeat kinase 2 in leukocyte DNA of Parkinson’s disease patients. Parkinsonism Relat Disord 2014, 20, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L.; Larsen, K.E.; Rideout, H.J.; Sulzer, D.; Greene, L.A. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J. Neurosci. 2001, 21, 9549–9560. [Google Scholar] [PubMed]
- Tanaka, Y.; Engelender, S.; Igarashi, S.; Rao, R.K.; Wanner, T.; Tanzi, R.E.; Sawa, A.; Valina, L.D.; Dawson, T.M.; Ross, C.A. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum. Mol. Genet. 2001, 10, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, L.; O’Farrell, C.; Lockhart, P.J.; Baptista, M.; Kehoe, K.; Vink, L.; Choi, P.; Wolozin, B.; Farrer, M.; Hardy, J.; et al. Parkin protects against the toxicity associated with mutant α-synuclein: Proteasome dysfunction selectively affects catecholaminergic neurons. Neuron 2002, 36, 1007–1019. [Google Scholar] [CrossRef]
- Snyder, H.; Mensah, K.; Theisler, C.; Lee, J.; Matouschek, A.; Wolozin, B. Aggregated and monomeric alpha-synuclein bind to the S6' proteasomal protein and inhibit proteasomal function. J. Biol. Chem. 2003, 278, 11753–11759. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhang, C.; Yin, J.; Li, X.; Cheng, F.; Li, Y.; Yang, H.; Ueda, K.; Chan, P.; Yu, S. α-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex i activity. Neurosci. Lett. 2009, 454, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Mitochondrial pathology in Parkinson’s disease. Mt Sinai J. Med. 2011, 78, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [PubMed]
- Kamp, F.; Exner, N.; Lutz, A.K.; Wender, N.; Hegermann, J.; Brunner, B.; Nuscher, B.; Bartels, T.; Giese, A.; Beyer, K.; et al. Inhibition of mitochondrial fusion by alpha-synuclein is rescued by pink1, parkin and DJ-1. EMBO J. 2010, 29, 3571–3589. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. Alpha-synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Pfefferkorn, C.M.; Jiang, Z.; Lee, J.C. Biophysics of alpha-synuclein membrane interactions. Biochim. Biophys. Acta 2012, 1818, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Iwatsubo, T. Pathological biochemistry of alpha-synucleinopathy. Neuropathology 2007, 27, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Oueslati, A.; Paleologou, K.E.; Schneider, B.L.; Aebischer, P.; Lashuel, H.A. Mimicking phosphorylation at serine 87 inhibits the aggregation of human alpha-synuclein and protects against its toxicity in a rat model of Parkinson’s disease. J. Neurosci. 2012, 32, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.L.; Pham, C.L.; Galatis, D.; Fodero-Tavoletti, M.T.; Perez, K.; Hill, A.F.; Masters, C.L.; Ali, F.E.; Barnham, K.J.; Cappai, R. Formation of dopamine-mediated alpha-synuclein-soluble oligomers requires methionine oxidation. Free Radic Biol. Med. 2009, 46, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Orimo, S.; Uchihara, T.; Nakamura, A.; Mori, F.; Kakita, A.; Wakabayashi, K.; Takahashi, H. Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008, 131, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Fujishiro, H.; Frigerio, R.; Burnett, M.; Klos, K.J.; Josephs, K.A.; Delledonne, A.; Parisi, J.E.; Ahlskog, J.E.; Dickson, D.W. Cardiac sympathetic denervation correlates with clinical and pathologic stages of Parkinson’s disease. Mov. Disord. 2008, 23, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Shannon, K.M.; Keshavarzian, A.; Dodiya, H.B.; Jakate, S.; Kordower, J.H. Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Mov. Disord. 2012, 27, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Goldstein, D.S. Cardiovascular dysautonomia in Parkinson disease: From pathophysiology to pathogenesis. Neurobiol. Dis. 2012, 46, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Gelpi, E.; Navarro-Otano, J.; Tolosa, E.; Gaig, C.; Compta, Y.; Rey, M.J.; Marti, M.J.; Hernandez, I.; Valldeoriola, F.; Rene, R.; et al. Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov. Disord. 2014, 29, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Farrell, K.F.; Krishnamachari, S.; Villanueva, E.; Lou, H.; Alerte, T.N.; Peet, E.; Drolet, R.E.; Perez, R.G. Non-motor Parkinsonian pathology in aging A53T alpha-synuclein mice is associated with progressive synucleinopathy and altered enzymatic function. J. Neurochem. 2014, 128, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Shiba, M.; Bower, J.H.; Maraganore, D.M.; McDonnell, S.K.; Peterson, B.J.; Ahlskog, J.E.; Schaid, D.J.; Rocca, W.A. Anxiety disorders and depressive disorders preceding Parkinson’s disease: A case-control study. Mov. Disord. 2000, 15, 669–677. [Google Scholar] [CrossRef]
- Beach, T.G.; White, C.L., 3rd; Hladik, C.L.; Sabbagh, M.N.; Connor, D.J.; Shill, H.A.; Sue, L.I.; Sasse, J.; Bachalakuri, J.; Henry-Watson, J.; et al. Olfactory bulb alpha-synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol. 2009, 117, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Ghebremedhin, E.; Rub, U.; Bratzke, H.; del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Rajendran, L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009, 64, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Bjorklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Angot, E.; Steiner, J.A.; Lema Tome, C.M.; Ekstrom, P.; Mattsson, B.; Bjorklund, A.; Brundin, P. α-Synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS ONE 2012, 7, e39465. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, J.H.; Paik, S.R.; Lee, S.J. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int. J. Biochem. Cell Biol. 2008, 40, 1835–1849. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I. Dementia with Lewy bodies. Dialogues Clin. Neurosci. 2004, 6, 333–341. [Google Scholar] [CrossRef]
- Ueda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar] [CrossRef] [PubMed]
- Gai, W.P.; Power, J.H.; Blumbergs, P.C.; Blessing, W.W. Multiple-system atrophy: A new α-synuclein disease? Lancet 1998, 352, 547–548. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Jicha, G.A.; Liu, H.; Schmitt, F.A. Lewy body pathology in normal elderly subjects. J. Neuropathol. Exp. Neurol. 2009, 68, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. Alpha-synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, U.; Newman, A.J.; Soldner, F.; Luth, E.S.; Kim, N.C.; von Saucken, V.E.; Sanderson, J.B.; Jaenisch, R.; Bartels, T.; Selkoe, D. Parkinson-causing alpha-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat. Commun. 2015, 6, 7314. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyuk, O.S.; Li, S.; Nash, K.; Gorbatyuk, M.; Lewin, A.S.; Sullivan, L.F.; Mandel, R.J.; Chen, W.; Meyers, C.; Manfredsson, F.P.; et al. In vivo RNAi-mediated alpha-synuclein silencing induces nigrostriatal degeneration. Mol. Ther. 2010, 18, 1450–1457. [Google Scholar] [CrossRef] [PubMed]
- Manfredsson, F.P.; Redmond, D.E.; Malpass, R.M.; Lipton, J.W.; Collier , T.J. AAV-mediated knockdown of alpha-synuclein in the substantia nigra of the african green monkey results in nigral neurodegeneration. In Proceedings of the Alzheimer’s and Parkinson’s Congress, Nice, France, 18–22 March 2015.
- Mullin, S.; Schapira, A. Alpha-synuclein and mitochondrial dysfunction in Parkinson’s disease. Mol. Neurobiol. 2013, 47, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Colla, E.; Jensen, P.H.; Pletnikova, O.; Troncoso, J.C.; Glabe, C.; Lee, M.K. Accumulation of toxic alpha-synuclein oligomer within endoplasmic reticulum occurs in alpha-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3301–3305. [Google Scholar] [CrossRef] [PubMed]
- Coune, P.G.; Bensadoun, J.C.; Aebischer, P.; Schneider, B.L. RAB1A over-expression prevents golgi apparatus fragmentation and partially corrects motor deficits in an alpha-synuclein based rat model of Parkinson’s disease. J. Parkinsons Dis. 2011, 1, 373–387. [Google Scholar] [PubMed]
- Smith, W.W.; Jiang, H.; Pei, Z.; Tanaka, Y.; Morita, H.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 2005, 14, 3801–3811. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. Alpha-synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gehrke, S.; Imai, Y.; Huang, Z.; Ouyang, Y.; Wang, J.W.; Yang, L.; Beal, M.F.; Vogel, H.; Lu, B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of drosophila pink1 is rescued by parkin. Proc. Natl. Acad. Sci. USA 2006, 103, 10793–10798. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Dodson, M.W.; Huang, H.; Guo, M. The Parkinson’s disease genes PINK1 and parkin promote mitochondrial fission and/or inhibit fusion in drosophila. Proc. Natl. Acad. Sci. USA 2008, 105, 14503–14508. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. Paris (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci. 2015, 40, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.N.; Byers, B.; Cord, B.; Shcheglovitov, A.; Byrne, J.; Gujar, P.; Kee, K.; Schule, B.; Dolmetsch, R.E.; Langston, W.; et al. LRRK2 mutant IPSC-derived da neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 2011, 8, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Tabrizi, S.J.; Schapira, A.H.; Cooper, J.M. Alpha-synuclein expression in HEK293 cells enhances the mitochondrial sensitivity to rotenone. Neurosci. Lett. 2003, 351, 29–32. [Google Scholar] [CrossRef]
- Thomas, B.; Mandir, A.S.; West, N.; Liu, Y.; Andrabi, S.A.; Stirling, W.; Dawson, V.L.; Lee, M.K.; Dawson, T.M. Resistance to MPTP-neurotoxicity in alpha-synuclein knockout mice is complemented by human α-synuclein and associated with increased beta-synuclein and akt activation. PLoS ONE 2011, 6, e16706. [Google Scholar] [CrossRef] [PubMed]
- Brunk, U.T.; Terman, A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic Biol. Med. 2002, 33, 611–619. [Google Scholar] [CrossRef]
- Sousa, V.L.; Bellani, S.; Giannandrea, M.; Yousuf, M.; Valtorta, F.; Meldolesi, J.; Chieregatti, E. α-Synuclein and its A30P mutant affect actin cytoskeletal structure and dynamics. Mol. Biol. Cell 2009, 20, 3725–3739. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Y.; Koprich, J.B.; Siddiqi, H.; Isacson, O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J. Neurosci. 2009, 29, 3365–3373. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Kordower, J.H. Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: Is this the target for Parkinson’s disease? Neurobiol. Dis. 2007, 25, 134–149. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Kompoliti, K.; Cochran, E.J.; Mufson, E.J.; Kordower, J.H. Age-related decreases in NURR1 immunoreactivity in the human substantia nigra. J. Comp. Neurol. 2002, 450, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Zeighami, Y.; Ulla, M.; Iturria-Medina, Y.; Dadar, M.; Zhang, Y.; Larcher, K.M.; Fonov, V.; Evans, A.C.; Collins, D.L.; Dagher, A. Network structure of brain atrophy in de novo Parkinson’s disease. Elife 2015. [Google Scholar] [CrossRef] [PubMed]
- Double, K.L.; Dedov, V.N.; Fedorow, H.; Kettle, E.; Halliday, G.M.; Garner, B.; Brunk, U.T. The comparative biology of neuromelanin and lipofuscin in the human brain. Cell Mol. Life Sci. 2008, 65, 1669–1682. [Google Scholar] [CrossRef] [PubMed]
- Marsden, C.D. Parkinson’s disease. Lancet 1990, 335, 948–952. [Google Scholar] [CrossRef]
- Kordower, J.H.; Olanow, C.W.; Dodiya, H.B.; Chu, Y.; Beach, T.G.; Adler, C.H.; Halliday, G.M.; Bartus, R.T. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 2013, 136, 2419–2431. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Fariello, R.; Riederer, P.; Sulzer, D.; Gatti, A.; Tampellini, D. The absolute concentration of nigral neuromelanin, assayed by a new sensitive method, increases throughout the life and is dramatically decreased in Parkinson’s disease. FEBS Lett. 2002, 510, 216–220. [Google Scholar] [CrossRef]
- Zucca, F.A.; Basso, E.; Cupaioli, F.A.; Ferrari, E.; Sulzer, D.; Casella, L.; Zecca, L. Neuromelanin of the human substantia nigra: An update. Neurotox Res. 2014, 25, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.L.; Nelson, O.; Yazdani, U.; Pasbakhsh, P.; German, D.C. Inverse relationship between the contents of neuromelanin pigment and the vesicular monoamine transporter-2: Human midbrain dopamine neurons. J. Comp. Neurol. 2004, 473, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Stefani, M. Protein misfolding and aggregation: New examples in medicine and biology of the dark side of the protein world. Biochim. Biophys. Acta 2004, 1739, 5–25. [Google Scholar] [CrossRef] [PubMed]
- Lundmark, K.; Westermark, G.T.; Nystrom, S.; Murphy, C.L.; Solomon, A.; Westermark, P. Transmissibility of systemic amyloidosis by a prion-like mechanism. Proc. Natl. Acad. Sci. USA 2002, 99, 6979–6984. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- Sala, G.; Stefanoni, G.; Arosio, A.; Riva, C.; Melchionda, L.; Saracchi, E.; Fermi, S.; Brighina, L.; Ferrarese, C. Reduced expression of the chaperone-mediated autophagy carrier HSC70 protein in lymphomonocytes of patients with Parkinson’s disease. Brain Res. 2014, 1546, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Wang, X.; Feng, X.; Zhang, A.; Li, J.; Gu, K.; Huang, J.; Pang, S.; Dong, H.; Gao, H.; et al. Altered expression of autophagic genes in the peripheral leukocytes of patients with sporadic Parkinson’s disease. Brain Res. 2011, 1394, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.; Chen, D.; Zhang, A.; Qin, X.; Yan, B. Genetic analysis of the LAMP-2 gene promoter in patients with sporadic Parkinson’s disease. Neurosci. Lett. 2012, 526, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Ii, K.; Ito, H.; Tanaka, K.; Hirano, A. Immunocytochemical co-localization of the proteasome in ubiquitinated structures in neurodegenerative diseases and the elderly. J. Neuropathol. Exp. Neurol. 1997, 56, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [PubMed]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Bjorklund, A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef] [PubMed]
- Choubey, V.; Safiulina, D.; Vaarmann, A.; Cagalinec, M.; Wareski, P.; Kuum, M.; Zharkovsky, A.; Kaasik, A. Mutant A53T α-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem. 2011, 286, 10814–10824. [Google Scholar] [CrossRef] [PubMed]
- Sampaio-Marques, B.; Felgueiras, C.; Silva, A.; Rodrigues, M.; Tenreiro, S.; Franssens, V.; Reichert, A.S.; Outeiro, T.F.; Winderickx, J.; Ludovico, P. SNCA (α-synuclein)-induced toxicity in yeast cells is dependent on sirtuin 2 (SIR2)-mediated mitophagy. Autophagy 2012, 8, 1494–1509. [Google Scholar] [CrossRef] [PubMed]
- Koehler, N.K.; Stransky, E.; Meyer, M.; Gaertner, S.; Shing, M.; Schnaidt, M.; Celej, M.S.; Leyhe, T.; Jovin, T.M.; Laske, C.; et al. Alpha-synuclein levels in blood plasma decline with healthy aging. PLoS ONE 2015, 10, e0123444. [Google Scholar] [CrossRef] [PubMed]
- Mak, S.K.; McCormack, A.L.; Langston, J.W.; Kordower, J.H.; di Monte, D.A. Decreased α-synuclein expression in the aging mouse substantia nigra. Exp. Neurol. 2009, 220, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lesuisse, C.; Xu, Y.; Troncoso, J.C.; Price, D.L.; Lee, M.K. Stabilization of alpha-synuclein protein with aging and familial Parkinson’s disease-linked A53T mutation. J. Neurosci. 2004, 24, 7400–7409. [Google Scholar] [CrossRef] [PubMed]
- Pino, E.; Amamoto, R.; Zheng, L.; Cacquevel, M.; Sarria, J.C.; Knott, G.W.; Schneider, B.L. FOXO3 determines the accumulation of alpha-synuclein and controls the fate of dopaminergic neurons in the substantia nigra. Hum. Mol. Genet. 2014, 23, 1435–1452. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T. Genetic and functional changes in mitochondria associated with aging. Physiol. Rev. 1997, 77, 425–464. [Google Scholar] [PubMed]
- Richter, C. Oxidative damage to mitochondrial DNA and its relationship to ageing. Int. J. Biochem. Cell Biol. 1995, 27, 647–653. [Google Scholar] [CrossRef]
- Trimmer, P.A.; Swerdlow, R.H.; Parks, J.K.; Keeney, P.; Bennett, J.P., Jr.; Miller, S.W.; Davis, R.E.; Parker, W.D., Jr. Abnormal mitochondrial morphology in sporadic Parkinson’s and Alzheimer’s disease cybrid cell lines. Exp. Neurol. 2000, 162, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef] [PubMed]
- Ciron, C.; Zheng, L.; Bobela, W.; Knott, G.W.; Leone, T.C.; Kelly, D.P.; Schneider, B.L. PGC-1α activity in nigral dopamine neurons determines vulnerability to alpha-synuclein. Acta Neuropathol. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Brauer, R.; Bhaskaran, K.; Chaturvedi, N.; Dexter, D.T.; Smeeth, L.; Douglas, I. Glitazone treatment and incidence of Parkinson’s disease among people with diabetes: A retrospective cohort study. PLoS Med. 2015, 12, e1001854. [Google Scholar] [PubMed]
- Eschbach, J.; von Einem, B.; Muller, K.; Bayer, H.; Scheffold, A.; Morrison, B.E.; Rudolph, K.L.; Thal, D.R.; Witting, A.; Weydt, P.; et al. Mutual exacerbation of peroxisome proliferator-activated receptor gamma coactivator 1alpha deregulation and alpha-synuclein oligomerization. Ann. Neurol. 2015, 77, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.; Chinta, S.J.; Mallajosyula, J.K.; Rajagopolan, S.; Hanson, I.; Rane, A.; Melov, S.; Andersen, J.K. Selective binding of nuclear α-synuclein to the pgc1alpha promoter under conditions of oxidative stress may contribute to losses in mitochondrial function: Implications for Parkinson’s disease. Free Radic Biol. Med. 2012, 53, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Pujol, J.; Junque, C.; Vendrell, P.; Grau, J.M.; Marti-Vilalta, J.L.; Olive, C.; Gili, J. Biological significance of iron-related magnetic resonance imaging changes in the brain. Arch. Neurol. 1992, 49, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, J.; Panter, S.S.; Neema, M.; Arora, A.; Batt, C.E.; Bakshi, R. Iron in chronic brain disorders: Imaging and neurotherapeutic implications. Neurotherapeutics 2007, 4, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Asanuma, M. Dopaminergic neuron-specific oxidative stress caused by dopamine itself. Acta Med Okayama 2008, 62, 141–150. [Google Scholar] [PubMed]
- Matsuda, W.; Furuta, T.; Nakamura, K.C.; Hioki, H.; Fujiyama, F.; Arai, R.; Kaneko, T. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. 2009, 29, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Pissadaki, E.K.; Bolam, J.P. The energy cost of action potential propagation in dopamine neurons: Clues to susceptibility in Parkinson’s disease. Front. Comput. Neurosci. 2013. [Google Scholar] [CrossRef] [PubMed]
- Kim-Han, J.S.; Antenor-Dorsey, J.A.; O’Malley, K.L. The parkinsonian mimetic, MPP+, specifically impairs mitochondrial transport in dopamine axons. J. Neurosci. 2011, 31, 7212–7221. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.L.; Wang, T.T.; Luby-Phelps, K.; German, D.C. Mitochondria mass is low in mouse substantia nigra dopamine neurons: Implications for Parkinson’s disease. Exp. Neurol. 2007, 203, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Prots, I.; Veber, V.; Brey, S.; Campioni, S.; Buder, K.; Riek, R.; Bohm, K.J.; Winner, B. α-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J. Biol. Chem. 2013, 288, 21742–21754. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlof, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Kraytsberg, Y.; Simon, D.K.; Turnbull, D.M.; Khrapko, K. Do mtDNA deletions drive premature aging in mtDNA mutator mice? Aging Cell 2009, 8, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Hauser, D.N.; Dillman, A.A.; Ding, J.; Li, Y.; Cookson, M.R. Post-translational decrease in respiratory chain proteins in the POLG mutator mouse brain. PLoS ONE 2014, 9, e94646. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Pinto, M.; Hida, A.; Moraes, C.T. Striatal dysfunctions associated with mitochondrial DNA damage in dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neurosci. 2011, 31, 17649–17658. [Google Scholar] [CrossRef] [PubMed]
- Tyynismaa, H.; Mjosund, K.P.; Wanrooij, S.; Lappalainen, I.; Ylikallio, E.; Jalanko, A.; Paetau, A.; Spelbrink, J.N.; Suomalainen, A. Mutant mitochondrial helicase twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 17687–17692. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand, M.I.; Terzioglu, M.; Galter, D.; Zhu, S.; Hofstetter, C.; Lindqvist, E.; Thams, S.; Bergstrand, A.; Hansson, F.S.; Trifunovic, A.; et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bobela, W.; Aebischer, P.; Schneider, B.L. Αlpha-Synuclein as a Mediator in the Interplay between Aging and Parkinson’s Disease. Biomolecules 2015, 5, 2675-2700. https://doi.org/10.3390/biom5042675
Bobela W, Aebischer P, Schneider BL. Αlpha-Synuclein as a Mediator in the Interplay between Aging and Parkinson’s Disease. Biomolecules. 2015; 5(4):2675-2700. https://doi.org/10.3390/biom5042675
Chicago/Turabian StyleBobela, Wojciech, Patrick Aebischer, and Bernard Laurent Schneider. 2015. "Αlpha-Synuclein as a Mediator in the Interplay between Aging and Parkinson’s Disease" Biomolecules 5, no. 4: 2675-2700. https://doi.org/10.3390/biom5042675
APA StyleBobela, W., Aebischer, P., & Schneider, B. L. (2015). Αlpha-Synuclein as a Mediator in the Interplay between Aging and Parkinson’s Disease. Biomolecules, 5(4), 2675-2700. https://doi.org/10.3390/biom5042675