
The RNA Splicing Response to DNA Damage


Microbiologie et d’Infectiologie, Faculté de Médecine et des Sciences de la Santé, Université de Sherbrooke, Sherbrooke, QC J1E 4K8, Canada
*
Author to whom correspondence should be addressed.
Biomolecules 2015, 5(4), 2935-2977; https://doi.org/10.3390/biom5042935
Submission received: 12 August 2015
/
Revised: 20 September 2015
/
Accepted: 16 October 2015
/
Published: 29 October 2015
(This article belongs to the Special Issue DNA Damage Response)
Abstract
:The number of factors known to participate in the DNA damage response (DDR) has expanded considerably in recent years to include splicing and alternative splicing factors. While the binding of splicing proteins and ribonucleoprotein complexes to nascent transcripts prevents genomic instability by deterring the formation of RNA/DNA duplexes, splicing factors are also recruited to, or removed from, sites of DNA damage. The first steps of the DDR promote the post-translational modification of splicing factors to affect their localization and activity, while more downstream DDR events alter their expression. Although descriptions of molecular mechanisms remain limited, an emerging trend is that DNA damage disrupts the coupling of constitutive and alternative splicing with the transcription of genes involved in DNA repair, cell-cycle control and apoptosis. A better understanding of how changes in splice site selection are integrated into the DDR may provide new avenues to combat cancer and delay aging.
1. Introduction
Genomes are continuously bruised by products of normal cell metabolism and by errors in DNA replication. The frequency of DNA lesions increases when cells are exposed to UV light, gamma rays or toxic chemicals (Figure 1). While DNA damage impacts nearly every aspect of gene expression, including transcription and translation, more attention has recently been devoted to studying how DNA damage affects post-transcriptional events, with several excellent reviews published on this topic [1,2,3,4,5]. Here we review the interconnections that exist between DNA damage and pre-mRNA splicing. Following a brief overview of the basic principles of the DNA damage response (DDR), splicing and alternative splicing control, we outline how DNA damage affects the post-translational modification, localization, expression and activity of splicing factors. We then present examples showing that DNA damage often disrupts splicing by interfering with its coupling to transcription. Finally, we summarize a growing body of data that document the impact of DNA damage on the splicing and alternative splicing of genes intimately associated with the DDR and with cell fate.
1.1. The DNA Damage Response
DNA damage is a cause of cancer, and its accumulation is associated with organismal aging. Paradoxically, provoking DNA damage by irradiation, DNA intercalating drugs, crosslinking agents or topoisomerase inhibitors is a common strategy to treat patients suffering from cancer. This anti-cancer approach is based on the expectations that (1) an excessive number of lesions will overwhelm the DNA repair machinery of cancer cells and trigger apoptosis; and (2) that the normal and cancer cells that acquire a lesser load of mutations will not become cancerous or evolve toward more aggressive behavior, respectively.
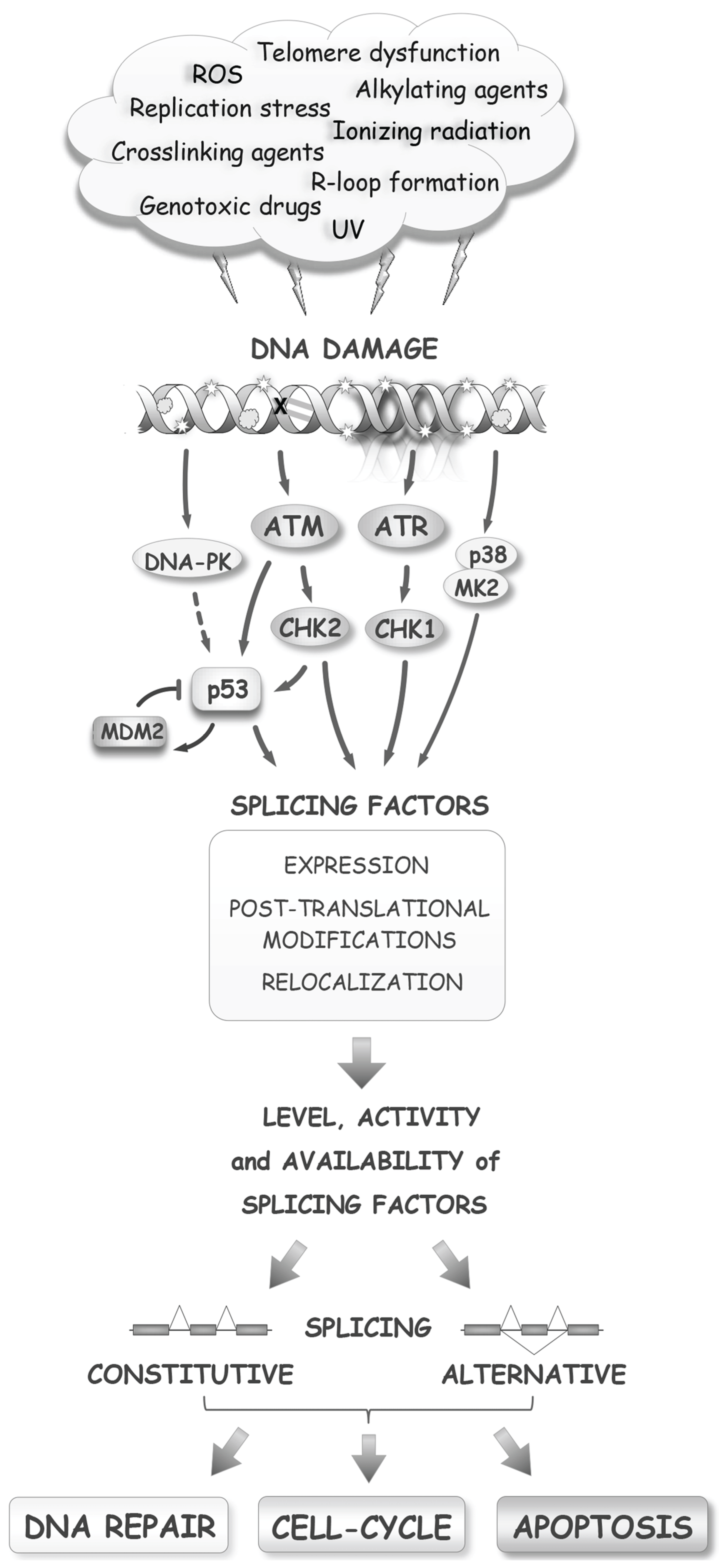
Distinct repair mechanisms are used to correct different types of DNA lesions. Mismatch repair and base-excision repair act on simple lesions, while nucleotide excision repair, non-homologous end-joining and homologous recombination (HR) deal with more complex lesions. DNA lesions, if not adequately repaired, impede transcription and replication and result in mutations or chromosomal rearrangements. To deal with DNA lesions, the cell must mount a response that is based first on sensing the DNA lesions themselves or their direct consequences, such as blocks in replication and transcription. DNA damage triggers the phosphorylation of the histone variant H2AX, the modifications of canonical histones, and the recruitment of poly(ADP-ribose) enzymes. Depending on the type of lesions, different axes of the DDR are activated. For example, double-strand breaks (DSBs) recruit the ATM and DNA-PK kinases, while single-stranded breaks (SSBs) recruit the ATR kinase. Following their activation, the ATM/ATR kinases activate other kinases, including CHK1 and CHK2 [6] (Figure 1). These early events ramify into a signal transduction cascade that mobilizes downstream pathways to mediate cell-cycle arrest, DNA repair, and apoptosis, if the intensity of the damage is excessive. Damage sensing and many steps of the DDR rely on a variety of post-translational modifications (e.g., phosphorylation, poly(ADP-ribosylation), acetylation, methylation, ubiquitylation, sumoylation) to guide the interactions between DDR factors and components of the repair and cell-cycle machinery [7]. In addition to these initial stages, the DDR initiates a slower route aimed at modulating transcription, of which the main contributor in the ATM/CHK2 axis is p53. The p53 protein regulates expression of cyclin-dependent inhibitor protein 1A (CDKN1A or p21), apoptotic proteins (e.g., BAX and PUMA) and DNA repair components. Importantly, the E3 ubiquitin ligase MDM2 represses p53 as part of a cyclic strategy that senses the progress of damage repair [8]. When p53 activity is defective, as is often the case in cancer cells, the cell-cycle checkpoint response to damage is rewired through the p38/MK2 pathway [9]. Thus, DNA damage elicits a complex signaling cascade that coordinates cell-cycle progression and DNA repair, and reconfigures gene expression at multiple levels.
Figure 1.
The RNA splicing response to DNA damage. Several early and late steps of the DNA damage response alter processes that impact the activity of splicing factors, ultimately affecting the production, through splicing, of components that maintain genome integrity and control cell fate.
Figure 1.
The RNA splicing response to DNA damage. Several early and late steps of the DNA damage response alter processes that impact the activity of splicing factors, ultimately affecting the production, through splicing, of components that maintain genome integrity and control cell fate.

While the expression of genes that encode sensing factors, and components of the DNA repair, cell-cycle and apoptotic machineries is controlled by the p53 activation branch of the DDR, these genes also produce splice variants that harbor potentially distinct, and sometimes, opposite activities. For example, the recruitment to chromatin of cyclin D1a, but not its splice variant cyclin D1b, is sufficient to activate the DDR [10], and Bcl-x produces variants with pro-survival or pro-death activities [11]. A change in splicing control elicited by the DDR therefore has the potential to provide feedback on every step of the DDR and regulate repair and cell fate.
1.2. Splicing and Alternative Splicing
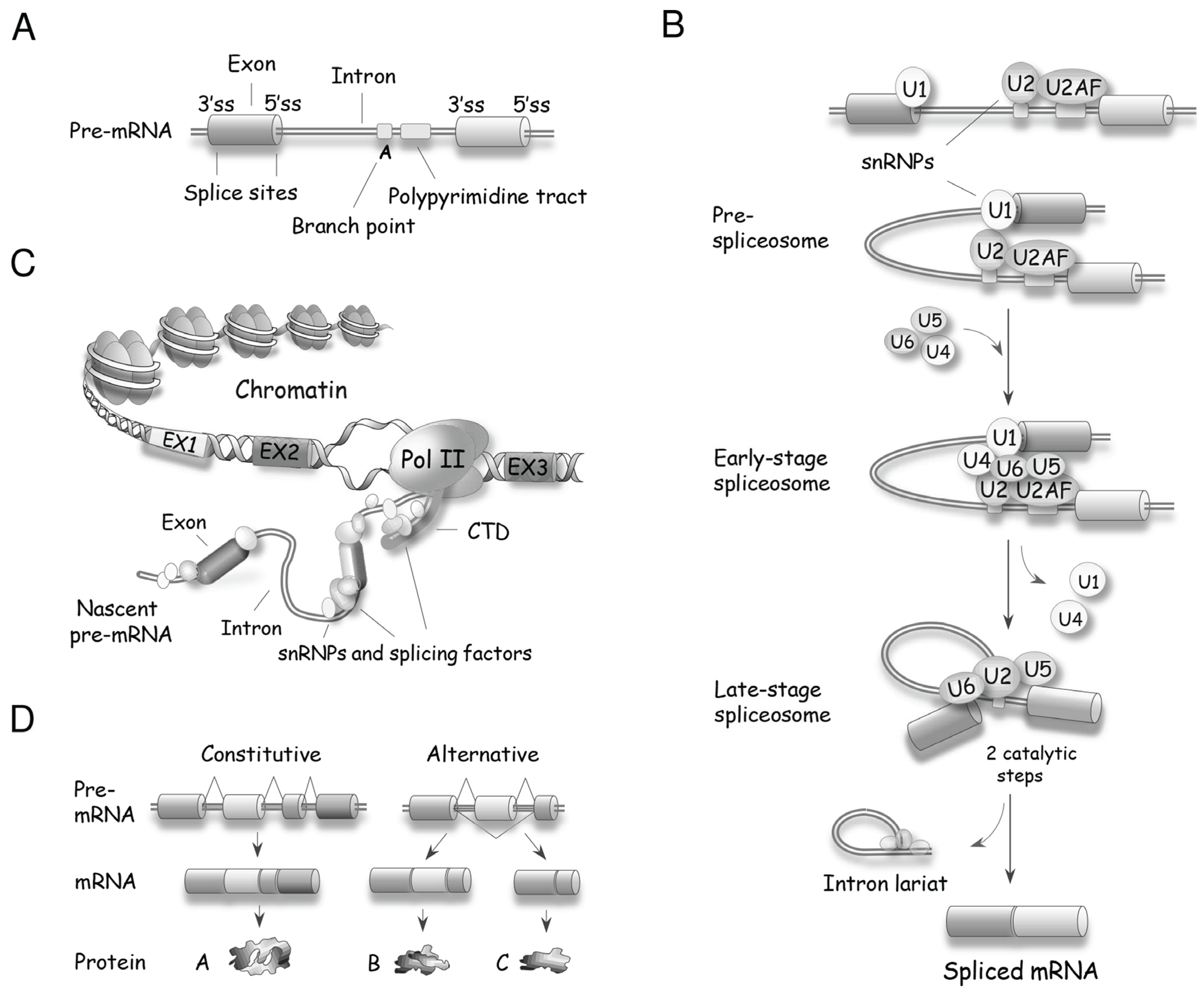
Precursor (pre)-mRNA splicing is the process by which introns are removed from a pre-mRNA and exons are joined to produce a mature mRNA. Removal of introns from pre-mRNAs occurs in eukaryotes from yeast to human. The majority of introns in the budding yeast Saccharomyces cerevisiae are found in ribosomal protein genes, which produce approximately 90% of the pre-mRNAs in growing cells [12]. In mammals, except for histones and a few other genes, nearly all RNA polymerase II-transcribed genes contain introns. Splicing is performed by the spliceosome, a large nuclear macromolecular complex that contains five small nuclear ribonucleoproteins (snRNPs) (U1, U2, U4, U5 and U6) and more than 150 accessory proteins [13,14,15,16,17]. Fewer than 0.5% of human introns are processed by a minor form of spliceosome that uses the functionally homologous U11, U12, U4atac and U6atac snRNPs. The U5 snRNP is used in both spliceosome types [18]. Spliceosome assembly is a dynamic process initiated by the recognition of splice sites (Figure 2A,B); the U1 snRNP recognizes the 5' splice site, while the U2AF proteins and U2 snRNP interact with the 3' splice site and the branch site, respectively [14]. Once the borders of the intron are defined, the pre-assembled U4/U6.U5 tri-snRNP is recruited and, with the help of auxiliary proteins, the U1 and U4 snRNPs are displaced to allow U6 and U2 snRNPs to form a catalytically competent core that positions the branch point adenosine for the first of two cleavage steps. The first step produces a free upstream exon and a lariat intron covalently linked to the downstream exon. Following further spliceosome rearrangements, the second step of splicing leads to the excision of the lariat intron and the ligation of both exons. The efficiency of spliceosome assembly is increased when it is coupled to transcription [19], at least in part because the CTD of RNA polymerase II recruits spliceosome components to facilitate their deposition on the nascent pre-mRNA [20] (Figure 2C).
A single type of pre-mRNA can be spliced in different ways (i.e., by inclusion of specific exons in the final spliced product) to generate distinct mRNAs (Figure 2D). This process is named alternative splicing, and is a major contributor to transcriptomic and proteomic diversity in higher eukaryotes. In humans, nearly all multi-exon primary transcripts are alternatively spliced [21,22]. On average, a human gene is made up of 8–10 exons [23], and examples of the diversifying power of alternative splicing range from two to several thousand variants from a single gene (e.g., the Drosophila DSCAM gene produces over 38,000 splice variants [24]). Although much remains to be done to document the remarkable diversity of functions resulting from alternative splicing, examples of functionally relevant splice variants are continuously being reported, and are found in all cellular processes [25]. The production of proteins displaying different functions is expected to be tightly controlled. Indeed, profiles of alternative splicing vary in a tissue-specific manner [26], and are often altered in diseases, including cancer [27,28,29].
Figure 2.
Basic principles of pre-mRNA splicing. (A) Schematic structure of a pre-mRNA with the position of core signal sequences that define exons and introns. ss: splice site; (B) A snRNP-biased view of spliceosome assembly leading to two catalytic steps that produce the mRNA and the excised intron. U2AF is a heterodimer made of the U2AF2 (U2AF65) and U2AF1 (U2AF35) proteins that respectively recognize the polypyrimidine tract and the AG dinucleotide at the 3' splice site [15]; (C) Spliceosome assembly is often coupled with transcription, with the carboxyl-terminal domain (CTD) of RNA polymerase II recruiting splicing components that are deposited on the nascent pre-mRNA; (D) In contrast to constitutive splicing, alternative splicing produces different mRNAs from a single kind of pre-mRNA.
Figure 2.
Basic principles of pre-mRNA splicing. (A) Schematic structure of a pre-mRNA with the position of core signal sequences that define exons and introns. ss: splice site; (B) A snRNP-biased view of spliceosome assembly leading to two catalytic steps that produce the mRNA and the excised intron. U2AF is a heterodimer made of the U2AF2 (U2AF65) and U2AF1 (U2AF35) proteins that respectively recognize the polypyrimidine tract and the AG dinucleotide at the 3' splice site [15]; (C) Spliceosome assembly is often coupled with transcription, with the carboxyl-terminal domain (CTD) of RNA polymerase II recruiting splicing components that are deposited on the nascent pre-mRNA; (D) In contrast to constitutive splicing, alternative splicing produces different mRNAs from a single kind of pre-mRNA.

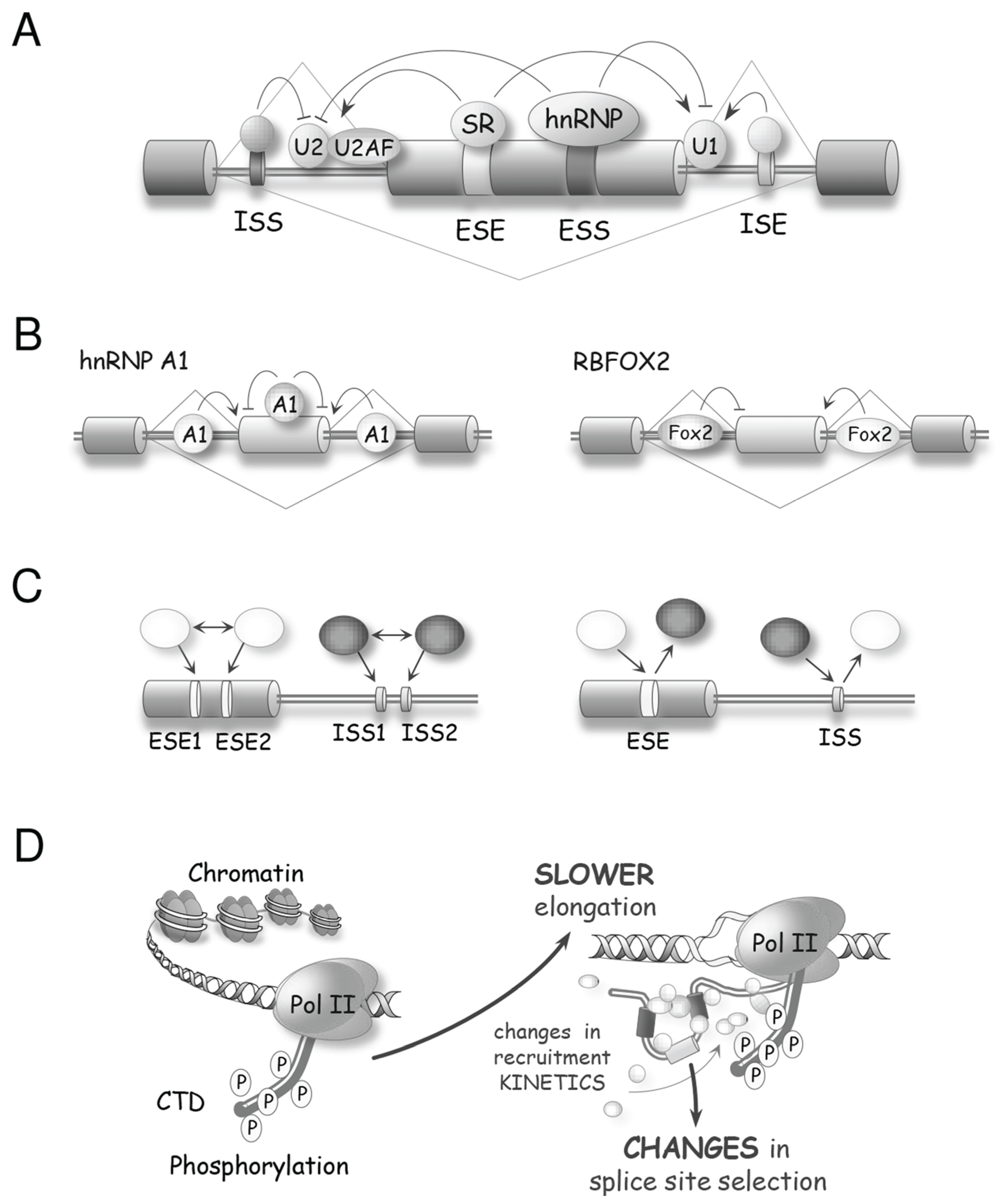
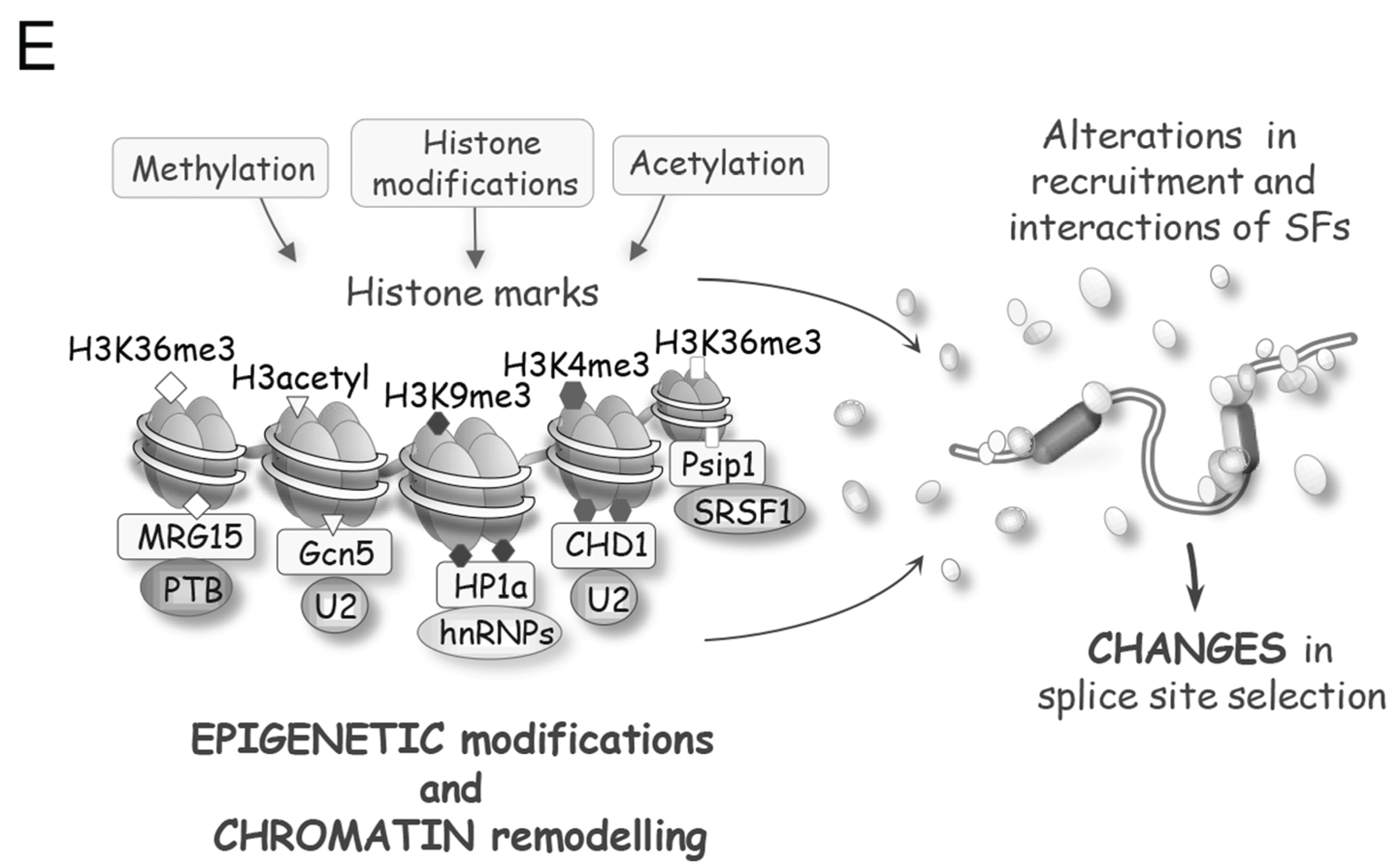
Sophisticated mechanisms that regulate alternative splicing profiles are also emerging (Figure 3). Alternative splicing units usually have weak splice sites whose utilization is controlled by sequence elements recognized by RNA binding proteins (RBPs) that act positively or negatively to recruit spliceosome components or prevent spliceosome assembly [30,31]. For example, SR proteins can interact with exonic enhancer sequences to antagonize the activity of a nearby splicing silencer element [32]. The activity of exonic silencers is often mediated by hnRNP proteins of which A1, L and PTBP1 have received most of the attention [33,34,35,36]. The respectively positive and negative functions of SR and hnRNP proteins bound to exons are often reversed when they bind to introns. For example, the binding of an SR protein near the branch site prevents U2 snRNP binding [37], while the binding of hnRNP A1 and H in introns stimulates splicing [38]. Transcript-specific studies and global analyses of alternative splicing indicate that positive and negative interactions between splicing factors with a wide range of sequence specificities play an important role in splicing regulation [30] (Figure 3A–C). In addition, since most introns are removed in a cotranscriptional manner, the control of alternative splicing is often coupled to the local state of the chromatin. Histone marks and chromatin remodeling factors impact the speed of transcription elongation and the recruitment of splicing regulators to alter splice site selection (Figure 3D,E) [39]. Thus, while great progress has been achieved in documenting the function of individual regulatory proteins, we need to better understand how these splicing factors combine their activity to control specific splicing decisions, how these activities are integrated with transcription and chromatin structure, and how they are affected by various cellular inputs and environmental insults. As is often the case in biology, insight can be provided by disrupting homeostasis. Below we will present examples of how DNA damage, by modifying the expression, localization and activity of spliceosomal proteins and splicing regulators, is providing precious information on the rules and mechanisms that control distinct steps of splice site selection.
Figure 3.
Molecular mechanisms controlling splice site selection. (A) A variety of splicing regulators, including hnRNP and SR proteins, bind to exon or intron splicing enhancers (ESE or ISE, respectively) and to exon or intron splicing silencers (ESS or ISS, respectively) to control splice site recognition and utilization; (B) The activity of splicing regulators is often position-dependent. For example, hnRNP A1 often acts as a repressor when bound to exons but can enhance splicing when bound to introns. RBFOX2 is associated with exon skipping when bound upstream of that exon, but triggers exon inclusion when bound downstream of the exon; (C) Depending on the identity of the factors that recognize them, the combinatorial configuration of regulatory elements leads either to synergy (left) or to antagonism (right) [30]. Inhibitory and stimulatory factors are shown as black and white, respectively; (D) The structure of the chromatin and the phosphorylation status of the CTD of RNA polymerase II affect the speed of transcription, which in turn impacts the time given for the binding and assembly of regulatory complexes, hence affecting splice site selection [39]; (E) Post-translational modifications of chromatin components and chromatin remodeling activities alter the recruitment of adaptor proteins and splicing regulators to modulate alternative splicing.
Figure 3.
Molecular mechanisms controlling splice site selection. (A) A variety of splicing regulators, including hnRNP and SR proteins, bind to exon or intron splicing enhancers (ESE or ISE, respectively) and to exon or intron splicing silencers (ESS or ISS, respectively) to control splice site recognition and utilization; (B) The activity of splicing regulators is often position-dependent. For example, hnRNP A1 often acts as a repressor when bound to exons but can enhance splicing when bound to introns. RBFOX2 is associated with exon skipping when bound upstream of that exon, but triggers exon inclusion when bound downstream of the exon; (C) Depending on the identity of the factors that recognize them, the combinatorial configuration of regulatory elements leads either to synergy (left) or to antagonism (right) [30]. Inhibitory and stimulatory factors are shown as black and white, respectively; (D) The structure of the chromatin and the phosphorylation status of the CTD of RNA polymerase II affect the speed of transcription, which in turn impacts the time given for the binding and assembly of regulatory complexes, hence affecting splice site selection [39]; (E) Post-translational modifications of chromatin components and chromatin remodeling activities alter the recruitment of adaptor proteins and splicing regulators to modulate alternative splicing.


2. DNA Damage Modifies Splicing Proteins
The post-translational covalent modification of DDR proteins plays a critical role in mounting the response to DNA damage [7]; it is therefore not surprising that many of the changes that affect splicing factors following DNA damage also occur at the post-translational level. Such modifications alter the steric or electrostatic profile of proteins, and provide ways to modulate their activity by modifying their interaction with other proteins or with RNA. Below, we review cases where DNA damage has been associated with the post-translational modifications of splicing factors to affect their localization, stability and activity. The emerging concept is that many of these modifications represent the initial steps of a concerted mechanism that coordinates the RNA splicing response to DNA damage.
2.1. PARylation
Poly(ADP-ribose) polymerases (PARPs) utilize NAD to synthesize poly(ADP-ribose) polymer (PAR) varying from 2 to 200 ADP-ribose units [40]. The activity of PARPs is counteracted by PARG, which hydrolyzes PAR. The addition of PAR, or PARylation, by the PARP1, PARP2 and PARP3 enzymes is an early event associated with DNA damage repair [41]. The PARylation of chromatin components is used to recruit DNA repair factors [6,42]. PAR is also bound by the splicing factors NONO (aka p54nrb), hnRNP A1 and RBMX (aka hnRNP G) [43,44], suggesting that PARylation mobilizes splicing factors at sites of damage. The binding of SRSF1 to PAR inhibits its phosphorylation [45]. Splicing factors and regulators are also substrates for PARP1 [46,47], and the PARylation of hnRNP proteins inhibits RNA binding to alter alternative splicing [48]. After treatment with methylmethane sulfonate (MMS), peroxide, ultraviolet (UV) or ionizing radiation (IR), PARylation occurs on many splicing factors (see Figure 3) [49]. While the PARylation of THRAP3 promotes its relocalization to nuclear speckles [49], it is unclear if this modification contributes to the observed exclusion of THRAP from DNA damage sites [50]. Overall, the recruitment of splicing factors by PAR and their subsequent PARylation likely represent important early events associated with the detection of DNA lesions. However, the consequence of these interactions and their impact on the splicing of DDR-related genes need to be investigated in more detail.
2.2. Arginine Methylation
Many splicing factors contain arginine residues that are methylated by arginine methyl transferases [51], and some factors, such as Sam68, RBM15, EWS, hnRNP A1/A2 and hnRNP K, are implicated in the DDR. Interestingly, the methylation of two arginines in hnRNP K prevents the PKCδ-mediated phosphorylation of a flanking serine, and mutating these two arginines increases the genotoxic effect of the topoisomerase II (TOP2) inhibitor etoposide, suggesting that arginine methylation of hnRNP K has anti-apoptotic function [52]. While hnRNP K normally represses the production of pro-apoptotic Bcl-xS [53], the role of arginine methylation on the splicing function of hnRNP K remains to be evaluated.
2.3. Acetylation
Proteins implicated in splicing are often acetylated (e.g., addition of the chemical group COCH3) [54], and this modification has been associated with their activity [55]. Following treatment of U2OS cells with the TOP2 inhibitor etoposide, changes in acetylation have been observed on many splicing factors (Figure 3) [50]. Likewise, cisplatin decreases the hyperacetylation of SRSF2 by TIP60 (aka KAT5) in SAOS2 cells, contributing to its stabilization [56]. The impact of cisplatin on TIP60 also favors the nuclear translocation of SR protein kinases SRPK1/SRPK2 that leads to an increase in the phosphorylation of SRSF2 [56]. BCLAF1 (a protein that interacts with BRCA1 to recruit spliceosome components following DNA damage) is rapidly deacetylated following the treatment of U2OS cells with IR [57]. Thus, the fact that acetylation can alter the activity of splicing factors warrants a broader assessment of the role of DNA damage-induced acetylation in splicing.
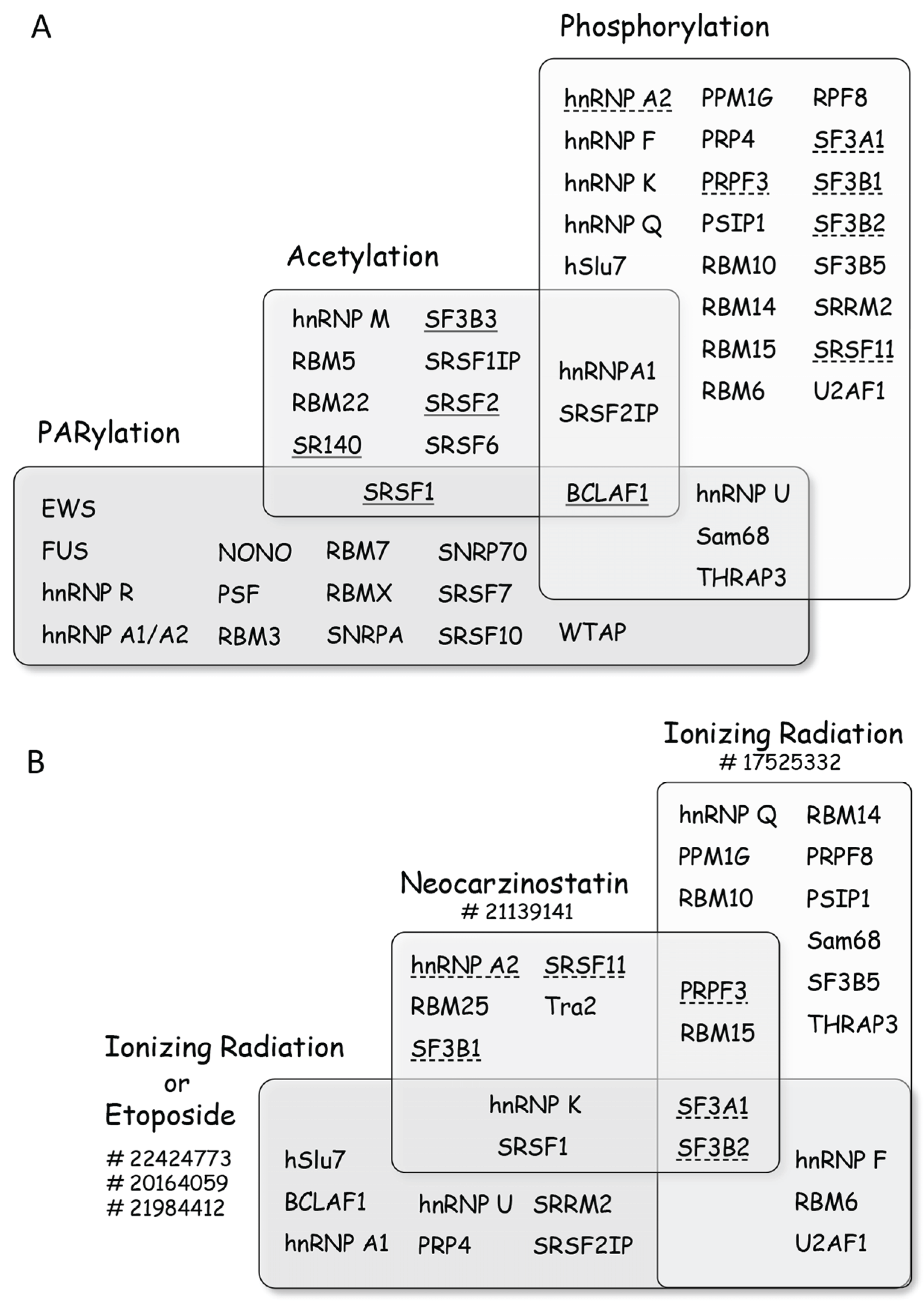
Figure 4.
Post-translational modifications of splicing factors promoted by DNA damage. (A) Diagram representing splicing factors that are modified upon treatment of cells with genotoxic agents. The list is non-exhaustive and more factors can be found in the references given in the text. Splicing factors are grouped according to the type of modifications they sustain, with BCLAF1 being subjected to all three types of modifications. When DNA damage elicits deacetylation the name of the splicing factor is underlined, whereas a dashed underline indicates dephosphorylation; (B) Diagram representing the impact of selected treatments on the phosphorylation/dephosphorylation of splicing factors (a dashed underline indicates dephosphorylation), with SF3A1 and SF3B2 being dephosphorylated in all groups of treatments. PubMed Identification (PMID) numbers for the different studies are indicated next to treatments. The impact of other agents on specific factors is discussed in the text.
Figure 4.
Post-translational modifications of splicing factors promoted by DNA damage. (A) Diagram representing splicing factors that are modified upon treatment of cells with genotoxic agents. The list is non-exhaustive and more factors can be found in the references given in the text. Splicing factors are grouped according to the type of modifications they sustain, with BCLAF1 being subjected to all three types of modifications. When DNA damage elicits deacetylation the name of the splicing factor is underlined, whereas a dashed underline indicates dephosphorylation; (B) Diagram representing the impact of selected treatments on the phosphorylation/dephosphorylation of splicing factors (a dashed underline indicates dephosphorylation), with SF3A1 and SF3B2 being dephosphorylated in all groups of treatments. PubMed Identification (PMID) numbers for the different studies are indicated next to treatments. The impact of other agents on specific factors is discussed in the text.

2.4. Ubiquitylation and Sumoylation
Sumoylation, or conjugation with a small ubiquitin-like modifier (SUMO), has been associated with DNA damage [58]. For example, treatment with the topoisomerase I (TOP1) inhibitor camptothecin delocalizes TOP1 from the nucleolus and promotes its sumoylation [59]. Many RBPs, including hnRNP proteins, are sumoylated [60]. Notably, SRSF1 is a cofactor for the SUMO E2 conjugating enzyme UBC9, and it interacts with the SUMO E3 ligase PIAS1 [61]. hnRNP K is a substrate for the ubiquitin E3 ligase MDM2 and is de-ubiquitylated upon DNA damage [62], and also sumoylated to affect its p53 transcriptional co-activator function [63]. Although SRSF1 stimulates the sumoylation of Sam68 [61], it is not known whether DNA damage affects the sumoylation of Sam68, and whether the splicing functions of Sam68 and hnRNP K are affected by sumoylation.
2.5. Phosphorylation
Phosphorylation is tightly associated with the DDR since the sensing of DNA lesions rapidly activates ATM, ATR and downstream kinases. A large-scale proteomics study identified more than 700 proteins that become phosphorylated at ATM/ATR consensus sites following treatment with ionizing radiation (IR) [64]. The list contains several splicing factors (Figure 4). A similar study in yeast identified PRP19 as a target for Mec1 and Tel1 (orthologs of the human ATR and ATM kinases, respectively) [65]. While PRP19 (aka Pso4) is a bona fide splicing factor in yeast, in mammals it is implicated in transcription elongation through the recruitment of TREX components [66]. Other proteomic studies focusing on the ATM pathway identified targets for more downstream kinases and phosphatases, or occurring at sites that are modified later during the response [50,67]. IR or etoposide affects the phosphorylation of many splicing factors (Figure 4). Another large-scale proteomic study using the radiomimetic agent neocarzinostatin (NCS) identified hundreds of phosphorylation and dephosphorylation events (40% of them independently of the ATM pathway (Figure 4) [68]. Hyperphosphorylation of SRSF1 also occurs when cells are treated with UV light or etoposide [69]. While the ATM-dependent increase in the phosphorylation of hnRNP K co-activates p53 transcription [70], it is not yet known if its function in alternative splicing is also altered. Splicing factors that are substrates of CDK1 (which is activated by ATR) include hnRNP M, MATR3, RBM7, RBM14 and SRRM2 [71].
Global proteomic studies have therefore clearly established splicing factors as targets for the DDR kinases. While there is considerable overlap in the identity of proteins that get modified by different types of agents, these studies have also revealed targets that are specific to the type of treatment or to the cell line used [50,67,71]. Global studies have rarely addressed the impact of phosphorylation events on splicing. Nevertheless, the impact of specific kinases that are activated by DNA damage on the splicing or the alternative splicing of specific genes has been examined in a few cases. Cisplatin promotes the nuclear translocation of SRPK1/SRPK2, which increases the phosphorylation of a hypoacetylated form of SRSF2 to regulate Casp8 splicing [56]. In Drosophila, DNA damage by IR or camptothecin alters the alternative splicing of Taf1 in a CHK2-dependent manner [72]. The activation of CHK2 by IR in human cells in turn activates CDK11 to stimulate the splicing of a reporter gene [73]. Oxaliplatin and cisplatin also elicit an ATM/CHK2-dependent response that alters the alternative splicing of Bcl-x [74]. Camptothecin reduces the level of Tra2 in Drosophila cells in an ATR-dependent manner [75]. CHK2 has also been linked with the phosphorylation of HuR, which affects hTra2β splicing and decreases the level of the hTra2β protein [76]. In human cells, Tra2 proteins favors the production of the long splice variant of CHK1, and a drop in Tra2 increases the level of phosphorylated γH2AX and reduces cell viability [77]. UV irradiation, by changing the localization of the splicing factor EWS, promotes the expression of a splice mRNA variant of Chek2 that lacks the initiation codon, hence reducing the level of CHK2 kinase [78]. Thus, while DNA damage activates kinases that impact the alternative splicing of downstream DDR genes, splicing regulation may feedback on the signaling genes themselves, possibly as part of mechanisms to amplify the response or facilitate cell recovery.
Splicing decisions are often taken while the pre-mRNA is still being transcribed [31,39]. Post-translational modifications elicited by DNA damage occur on protein components of the transcription machinery that alter speed or pausing to impact splice site selection. For example and as discussed in more details below, exposure to UV alters the level of phosphorylation of RNA polymerase II to affect transcription elongation and splicing [79]. In addition to changes in the RNA polymerase complex, DNA damage also leads to the modification of histones and histone-binding proteins to alter nucleosome positioning and global chromatin structure [80]. These changes may in turn modulate the speed of transcription and the interaction with chromatin components of the splicing machinery to affect splicing [39,81].
3. The Depletion of Splicing Factors Causes DNA Damage
While irradiation and genotoxic compounds are primary causes of DNA damage, DNA lesions also occur when basic processes are deficient. A genome-wide siRNA screen demonstrated that the depletion of many splicing factors increases DNA damage, as monitored by the phosphorylation of H2AX [82]. One explanation for this result is based on the impact of depleting or mutating components of the yeast THO-TREX complex that normally coats nascent polymerase II transcripts to couple transcription and pre-mRNA maturation with mRNA export [83]. Nascent RNA sequences that are not bound by RNA binding proteins (RBPs) hybridize to the template strand of the melted DNA to form R-loops, a structure that slows transcription and promotes mutations and hyper-recombination [84,85]. Because THO-TREX components have equivalents in higher eukaryotes, with a similar impact on genomic instability when depleted [86], their depletion or that of additional splicing factors, such as SR and hnRNP proteins, may create similar problems with similar impact on genome stability [87,88]. Indeed, SR proteins are important participants in the maintenance of genomic stability. The inactivation of SRSF1 provokes the accumulation of R-loops, which lead to DNA breaks, mutations and chromosomal rearrangements that activate ATM [89,90]. The depletion of SRSF2 also promotes genomic instability [88], but the mechanism remains unclear since overexpression of SRSF2, in contrast to that of RNPS1 [91], cannot rescue defects caused by the loss of SRSF1. TOP1 also collaborates with SRSF1 to prevent R-loop formation [92]. The observations that TOP1 phosphorylates SR proteins [93] and other splicing factors, including PSF and NONO [94], and that the kinase activity of TOP1 is inhibited by the topoisomerase inhibitor camptothecin [93], suggest that camptothecin may promote genomic instability not only by blocking TOP1 function in DNA topology but also possibly by modulating the activity of splicing factors.
The R-loop-mediated genomic instability caused by reducing the level of THO-TREX components and SR proteins may also occur when the activity of other RBPs are affected, potentially amplifying DDR signaling and splicing changes, as when late-stage spliceosomes are displaced after RNA polymerase II encounters DNA lesions (Figure 5 and see below). Consistent with this view, both the use of the splicing inhibitor pladienolide B, which targets components of the SF3B complex, and the depletion of BUGZ and BUB3, which interact with U2AF and SF3A3, promote R-loop formation and activate the DDR [95]. The splicing regulator hnRNP A1 is implicated in telomere biogenesis [96]. Although hnRNP A1 displays affinity for telomerase RNA and single-stranded DNA telomeric repeats [97,98], it also interacts with TERRA transcripts synthesized from telomeric repeats to contribute to telomere capping and genome integrity [99]. However, whether the depletion of hnRNP A1 activates the DDR through R-loop formation and telomere dysfunction remains to be evaluated.
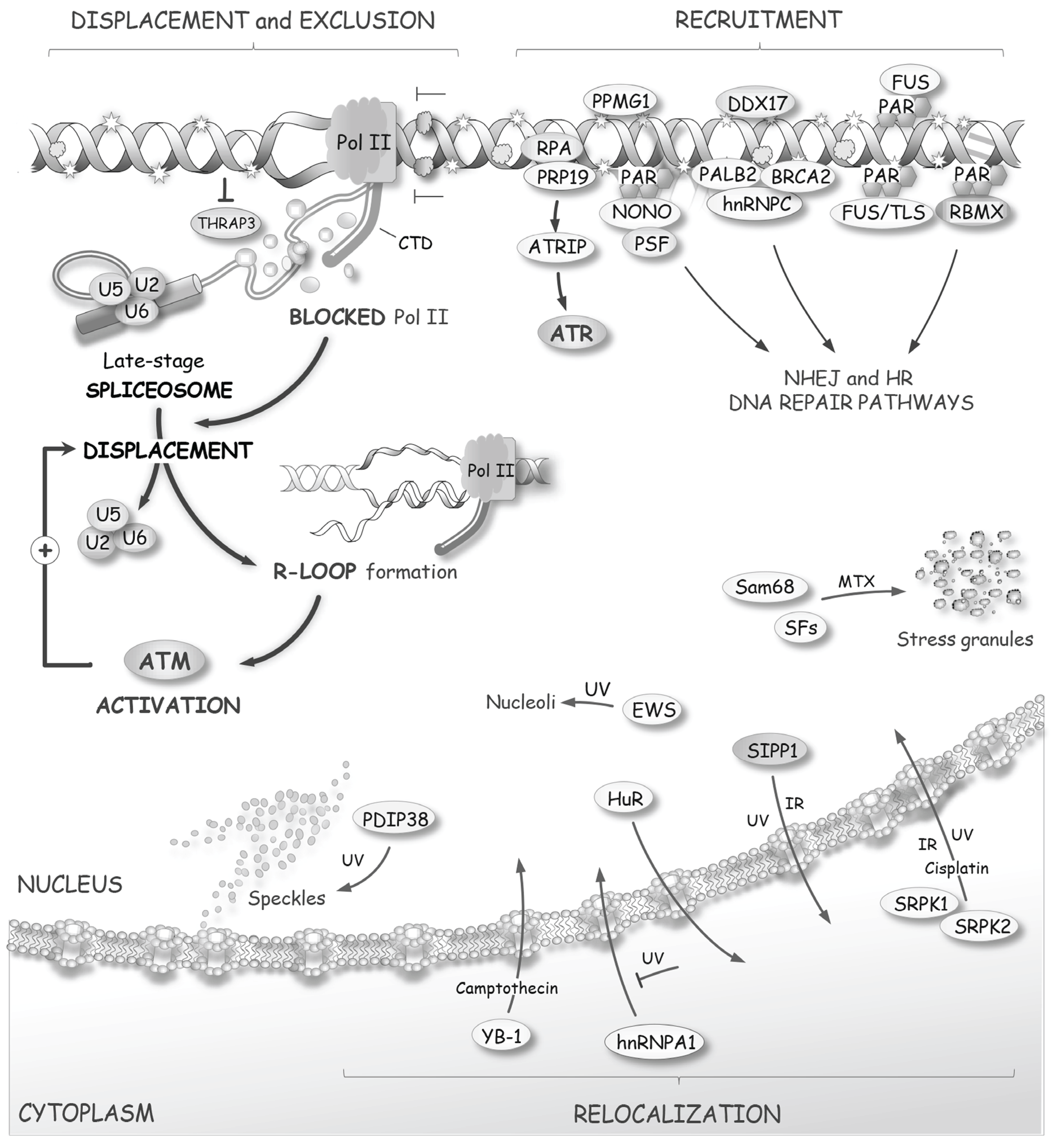
Figure 5.
DNA damage affects the distribution of splicing factors. As described in the text, some splicing factors are recruited at sites of damage to participate in sensing or DNA repair. On the other hand, nascent pre-mRNAs that are associated with transcription complexes blocked at lesions will be stripped of late-spliceosome components to form RNA/DNA duplexes (R-loops) that activate ATM. DNA damaging agents also affect the subcellular localization of several splicing factors.
Figure 5.
DNA damage affects the distribution of splicing factors. As described in the text, some splicing factors are recruited at sites of damage to participate in sensing or DNA repair. On the other hand, nascent pre-mRNAs that are associated with transcription complexes blocked at lesions will be stripped of late-spliceosome components to form RNA/DNA duplexes (R-loops) that activate ATM. DNA damaging agents also affect the subcellular localization of several splicing factors.

4. Exclusion and Recruitment of Splicing Factors at Sites of Damage
DNA lesions are deleterious if they allow the misincorporation of nucleotides that lead to protein malfunction. If the affected protein is involved in DNA repair directly or in cell-cycle control, this may lead to more instability and damage. Transcriptional stalling at sites of lesions may also generate replication stress. It is therefore crucial for the cell to recognize sites of damage, to limit local transcription and to provide access to the repair machinery. Given that transcription is silenced at sites of DNA damage [100], the association of splicing factors at these sites is expected to decrease. Consistent with this view, THRAP3, a factor that interacts with splicing factors to couple transcription with splicing [101,102], is excluded from DNA damage sites (Figure 5), correlating with loss of transcription and decreased mRNA processing [50]. Interestingly, DNA lesions that block transcription (elicited by UV irradiation, but not by oxidative damage, DSBs or DNA inter-strand crosslinks) rapidly decrease the localization of U2 and U5/U6 snRNPs at irradiated sites (Figure 5). Because the U1 and U4 snRNPs are not similarly displaced, this form of damage suggests the specific loss of late-stage spliceosomes [103]. Notably, spliceosome displacement elicits R-loop formation, which in turn activates ATM, further enhancing the displacement of splicing components, and providing the necessary signaling output to reconfigure splicing and alternative splicing globally [103]. The mechanism that triggers the dissociation of late-stage spliceosomes is unknown. Since it is known that the phosphatase PPM1G (aka PP2Cγ) regulates splicing positively and is recruited at DNA damage sites [50,104,105], it would be of interest to test whether PPM1G contributes to the DNA damage-induced delocalization of THRAP3 and the disengagement of late-stage spliceosomes.
While DNA damage can prevent the association of some splicing components, other observations indicate that splicing factors are recruited at sites of damage. However, it is important to indicate that sites of damage are often experimentally defined as large chromatin regions that recruit massive amounts of proteins such as γ-H2AX to form nuclear foci visible by light microscopy, and that proteins recruited to foci are unlikely to be all in the immediate vicinity of the DNA lesion itself [80]. Thus, recruitment of splicing factors does not necessarily imply a function in repair but may be part of a strategy to coordinate repair with splicing decisions.
An early step associated with the recruitment of repair proteins is the transient polymerization of PAR. Because several hnRNP proteins and some SR proteins display affinity for PAR [46,106], these proteins may be rapidly recruited at sites of DNA damage. The splicing regulator RBMX associates with DSBs in a PAR-dependent manner [107,108] (Figure 5). The siRNA-mediated depletion of RBMX sensitizes cells to DNA-damaging agents that use homologous recombination (HR) for repair (e.g., IR, camptothecin, mitomycin C, chlorambucil, oxaliplatin and carboplatin) but also to UV and tert-Butyl hydroperoxide (tBHP), all of which cause lesions not primarily repaired by HR [108]. The mechanism by which RBMX contributes to HR remains intriguing since the localization of RBMX at sites of damage is not required for repair [108]. However, the depletion of RBMX, as well as the depletion of several other pre-mRNA processing factors, affect BRCA2 expression [108], suggesting that RBMX may be post-translationally modified when recruited at sites of damage, and then released to affect the alternative splicing of target genes [109,110,111,112,113,114,115,116]. The screen that identified RBMX as contributing to HR also identified other RNA splicing factors involved in HR, including U2 snRNP proteins SF3A3 and SF3B1, and U5 snRNP protein PRP8 [108]. Depleting some of these proteins reduced BRCA2 expression, but the impact of these factors and of RBMX on BRCA2 splicing was not tested [108]. DDX17 also localizes at sites of DSB repair, and is known to regulate the alternative splicing of genes encoding DNA- and chromatin-binding factors [117], as well as components of the regulatory network of androgen and estrogen receptors [118,119].
The splicing regulator FUS [120] is also recruited to DSBs in a PAR-dependent manner [121] (Figure 5). Likewise, the fused oncogenic version FUS/TLS is mobilized at lesions created by oxidative DNA damage in a PARP1-dependent manner [122]. A role for FUS in DSB repair is suggested by the observation that ablation of FUS in mice confers high sensitivity to IR [123]. Familial mutations in FUS linked to amyotrophic lateral sclerosis (ALS) decrease an interaction with the histone deacetylase HDAC1 and produce a FUS protein defective in DNA repair and DDR [124]. Many ALS-causing mutations in FUS result in mislocalization of FUS to the cytoplasm in interaction with U1 snRNP [125], supporting the view that ALS cells are deficient in their ability to repair DNA. The ablation of the related protein EWS also confers high sensitivity to IR [123]. Importantly, EWS plays a critical role in regulating the changes in alternative splicing induced by UV light, and partially relocalizes to the nucleolus upon UV irradiation, an event not seen when cells are treated with NCS or etoposide [78]. The EWS-interacting factor YB-1 has also been implicated in DNA repair [126,127,128].
Other splicing factors are multifunctional proteins that play a direct role in DNA repair. For example, hnRNP C partially localizes to sites of DNA damage following IR, and this interaction occurs in association with PALB2 and BRCA2 [129] (Figure 5). hnRNP C antagonizes the activity of U2AF65 (U2AF2) to prevent portions of Alu elements to be recognized as exons [130], and its depletion has a broad impact on alternative splicing [131]. Since hnRNP C associates with the SWI/SNF chromatin remodeling complex that controls alternative splicing [132,133], hnRNP C may mediate some of its splicing effects cotranscriptionally. hnRNP C therefore contributes to the repair of DNA lesions directly through interaction with the repair proteins PALB2 and BRCA2, and more globally by regulating the splicing of genes that encode components of the HR repair machinery (BRCA1, BRCA2, RAD51 and BRIP1) [129]. RBM14, a protein structurally related to hnRNP A1, may also play a direct role in DNA repair by controlling the efficiency of NHEJ through an interaction with Ku80 that is stimulated by IR [134].
Another multifunctional protein is the splicing and ubiquitin E3 ligase protein PRP19. PRP19 is essential for DNA repair in yeast [135], and in mammals it interacts with CDC5L and the repair helicase WRN [136]. DNA damage promotes the ubiquitylation of PRP19, which then dissociates from CDC5L [137]. PRP19 recognizes DNA damage through RPA bound to ssDNA [138,139]. PRP19 then ubiquitylates RPA to recruit ATRIP and activate ATR [138,139] (Figure 5).
The pleiotropic PSF protein is mobilized to lesions after laser-induced DNA damage [140] (Figure 5). PSF displays in vitro DSB end-rejoining activity [141], and is implicated in NHEJ and HR repair pathways [140,142,143]. PSF forms a complex with NONO, a protein that binds to PAR and is implicated in NHEJ and HR [43,144]. PSF also associates with the nuclear matrix and splicing regulator MATR3, which itself interacts with PTBP1 [140,145]. The depletion of MATR3 increases the retention of PSF at DNA damage sites [140].
Thus, the available data suggest that the localization of splicing factors at sites of damage is affected in different ways. Some factors are expelled to block splicing, allowing formation of R-loops and amplification of the DDR signaling pathway; other splicing factors are recruited to act directly in DNA repair, while others are recruited in the vicinity of lesions to be modified, and released to execute more downstream portions of the DDR.
5. DNA Damage Relocalizes Splicing Factors
Although DNA damage impacts the association of splicing factors at sites of lesions, it also affects their subcellular localization (Figure 5). For example, UV irradiation triggers the cytoplasmic retention of hnRNP A1, with an impact on alternative splicing [146,147]. Notably, doses of UV light that do not trigger this A1 relocalization also have an impact on splicing, most likely by affecting the phosphorylation and elongation speed of RNA polymerase II [79]. UV irradiation and IR also redistribute the putative splicing factor SIPP1 to the cytoplasm [148].
The TOP2 inhibitor mitoxantrone (MTX) redistributes Sam68, along with other splicing regulators, to nuclear stress granules in PC-3 cells and promotes splicing changes in genes regulated by Sam68 [149]. However, this pathway is independent of the DDR signaling cascade. The siRNA-mediated depletion of Sam68 sensitizes LNCaP cells to cisplatin-induced apoptosis [150]. Cisplatin and the depletion of Sam68 in LNCaP cells shift splicing in favor of pro-apoptotic Bcl-xS [74,150,151]. It is unclear whether DNA damage affects the cellular distribution of Sam68 to reduce its recruitment at the Bcl-x gene locus. The activity of Sam68 in Bcl-x splicing requires hnRNP A1 [152,153], whose localization is also affected by genotoxic stresses [146,147].
In MRC-5 cells, PDIP38 helps install specialized polymerases at UV-damaged sites to mediate repair [154]. In HeLa and A549 cells however, UV irradiation translocates PDIP38 not to UV repair foci but to nuclear speckles where its association with splicing components may help control the alternative splicing of Mdm2 [155].
Cisplatin provokes the nuclear accumulation of SRPK1 and SRPK2 kinases to increase the phosphorylation of SR proteins [56]. IR and reactive oxygen species (ROS) also elicit the nuclear accumulation of SRPK2 [156]. On the other hand, UV irradiation induces a dynamic redistribution of SRSF1, SRSF9, SRSF7, U1-70K, hTra2β and NONO to areas around nucleolar fibrillar components [157].
HuR has been implicated in splicing control [158], and DNA damage triggers its export to the cytoplasm through the phosphorylation of CDK1 by the CHK1 and CHK2 pathways [159,160,161]. While exposure of HCT116 cells to ROS-generating sodium arsenate stimulates the CHK2- and p38-mediated phosphorylation of HuR, in this case it stimulates the binding of HuR to the nuclear pre-mRNA encoding the splicing regulator hTra2β to alter its alternative splicing [76].
Camptothecin relocalizes YB-1 from the cytoplasm to the nucleus where it loses its ability to interact with EWS and participate in cotranscriptional splicing [2,162]. On the other hand, UV, but not etoposide, elicits the partial relocalization of EWS to the nucleolar compartment [78].
Thus, while DNA damage affects the localization of several splicing factors, the nature of the lesions and the cell line used may determine which splicing factors are affected.
6. DNA Damage Alters the Expression of Splicing Factors
Early studies revealed the negative impact that DNA damage has on global transcription [163,164]. However, further analyses indicated that the steady-state levels of specific transcripts or their products were increased [165], possibly reflecting mRNA stabilization and/or stimulation of translation [166]. The impact of DNA damage-induced changes in transcription, mRNA stability and translation on the expression of splicing factors is difficult to predict because splicing factors are often drafted to accomplish a variety of functions, sometimes in different cellular compartments [167]. Nevertheless, adjusting the level of spliceosome components and splicing regulators may be important after DNA damage. For example, the overexpression of RBM17 in the A2780 ovarian carcinoma cell line is associated with broad resistance to DNA damage-inducing drugs through the splicing of genes transcriptionally regulated by ESR2 [168]. Increases in the mRNA and protein levels of hnRNP proteins have been noted to occur following DNA damage [169,170,171,172,173]. Mitomycin C increases the expression of SRSF6, SRSF1 and SRSF2 in U2OS-derived cells [174]. UV also increases the level of SRSF1 in MCF-7 cells to modulate Mdm2 splicing; however this increase in SRSF1 is not due to increased mRNA levels but by a change in the alternative splicing of SRSF1 that affects protein abundance [175,176]. The treatment of HCT116 cells with camptothecin also affects the alternative splicing of genes encoding splicing factors including RBM8A (aka Y14), the branch site protein SF1, SF3A1, U2AF1, hnRNP A2, SIP1 and SRSF8 to produce transcripts lacking important functional domains [177]. Global proteomics studies have noted only a few changes in the level of splicing factors. For example, the treatment of U2OS cells with etoposide leads to a two-fold decrease in levels of QKI [50].
Interestingly, a recent study has revealed an intimate relationship between DNA damage at telomeres and the expression of splicing factors [178]. Persistent telomere dysfunction in mice elicits a hematopoietic defect that mimics human myelodysplastic syndromes [178]. Defective hematopoietic stem cells display ATR-dependent DNA damage signaling that reduces the expression of U2AF2, SRSF2, SRSF10, SF3B2 and SF3A3, and globally alters the alternative splicing of genes involved in maintaining genome stability, DDR, chromatin remodeling and histone modifications. The functional impact of these changes is supported by the fact that nearly one-third of them should produce non-functional proteins, mainly because of premature stop codons. Notably, downregulating the expression of SRSF2 in mice hematopoietic cells alters the alternative splicing of DNA repair and telomere maintenance genes and induces telomere dysfunction that may exacerbate splicing defects [178].
Translational control may represent a more efficient way than transcription to control the production of splicing factors, particularly when only a short-term adaptation is needed before lesions are repaired. Another strategy to control the level of splicing factors is through their alternative splicing. SR and hnRNP proteins are exquisitely designed to respond in this manner since each group contains alternative exons with premature stop codons (PTC), allowing rapid fine-tuning of protein levels through nonsense-mediated RNA decay (NMD) [179]. Consistent with this view, the alternative splicing changes induced by DNA damage often target genes encoding splicing factors with PTC-containing exons [180].
7. Impact of DNA Damage on Constitutive Splicing
Although DNA damage was initially associated with global drops in gene expression and RNA processing activities, both positive and negative impacts have now been documented. While it may be important to decrease transcription and splicing at sites of lesions to facilitate sensing and access by the DNA repair machinery, broader splicing alterations may be required to optimize the production of DNA repair enzymes, to implement more stable adjustments in the cell-cycle or to initiate the apoptotic program. Notably, UV irradiation reduces the splicing efficiency of individual introns in a dozen genes including Akt1, Fancg, Atr, Atm, Aurka and Aurkb in an ATM-dependent manner [103], although the functional impact of these events is unclear. On the other hand, etoposide stimulates the removal of introns in genes encoding the DNA repair components ATRIP, BACH and EXO1 [181]. The molecular pathway at work in this case involves the ATM/ATR-dependent phosphorylation of serine-1423 in chromatin-bound BRCA1 to promote its interaction with BCLAF1, itself in association with several spliceosomal proteins including U2AF, SF3B1 and PRP8, to stimulate splicing and increase the production of repair factors [182]. Consistent with the importance of this route for mounting an appropriate DDR response, phosphorylation of serine-1423 in BRCA1 confers resistance to IR and is associated with cell-cycle arrest [183]. Moreover, the siRNA-mediated depletion of BRCA1, BCLAF1 or U2AF increases sensitivity to IR and etoposide, and results in defective DNA repair and genomic instability in MCF-7 and 293T cells [181]. Thus, the noted increase in the mRNA level of DNA repair genes associated with genotoxic stresses [184] may be the consequence, at least in part, of more efficient pre-mRNA splicing.
A similar outcome was reported for two genes involved in cell-cycle control. First, resistance to DNA damage and improved splicing of Cdkn1a (encoding the CDK inhibitor p21) are stimulated by SKIP, a protein that interacts with SNIP1, THRAP3, BCLAF1, U2AF and PRP19, and also associates with the Ccnd1 gene to increase the levels of cyclin D1 [101,185]. Thus, the association of BCLAF1 with spliceosome components [186] may coordinate the splicing of both DNA repair and cell-cycle genes following DNA damage. The fact that BCLAF1 is a target for several types of post-translational modifications elicited by DNA damage (Figure 4A) is consistent with this central regulatory position.
8. DNA Damage Alters Transcription-Coupled Splicing Decisions
As discussed above, several features of transcription affect splice site selection in different ways (Figure 3D,E) [31,39]. The elongation speed and pauses of a transcribing RNA polymerase determine how much time is given for enhancer or repressor complexes to assemble on a nascent pre-mRNA and influence the commitment of competing pairs of splice sites. This speed of elongation is altered by (1) the phosphorylation the carboxyl-terminal domain (CTD) of the large polymerase II subunit; (2) the association of elongation factors like P-TEFb; and (3) the modification of chromatin components including histones. In addition, interactions between components of the RNA polymerase complex and spliceosomal factors increase splicing efficiency and alter splice site selection. Finally, chromatin remodeling components and epigenetic changes on histones modulate the interaction with adaptors that recruit splicing regulators. Below we present DDR-relevant examples in these categories.
- •
- UV treatment of Hep3B and HCT116 cells changes the phosphorylation of the CTD of RNA polymerase II to affect transcription elongation and alternative splicing in an ATM/ATR-independent manner [79]. On the other hand, and as discussed above, lesions created by UV block transcription leading to the dissociation of late-stage spliceosomes [103].
- •
- SR proteins regulate splicing decisions by interacting directly with exon and intron sequence elements on pre-mRNAs [30,31,187]. More recently however, SRSF2 was implicated in the release of the transcription elongation factor P-TEFb from the repressor 7SK RNA at paused transcription sites [188]. A similar function for hnRNP A1/A2 in the release of P-TEFb and transcription elongation has been reported [189]. Since DNA damage promotes changes in the level of SRSF2 and in the localization of hnRNP A1 [56,146], splicing alterations may possibly occur through P-TEFb-mediated effects on transcription elongation.
- •
- The RNA polymerase II-associated protein EWS confers resistance to IR and UV light [78,190], and mice lacking EWS are hypersensitive to IR [123]. These phenotypes may be due, in part, by the fact that a deficiency in EWS affects the alternative splicing of cyclin D1 [191], Fas [192], Mdm2 [162], and the DNA repair genes Abl1, Chek2 and Map4k2 [78]. EWS interacts with spliceosome components, including the U1 snRNP protein U1C [193], the branch site protein SF1 [194] and YB-1 [162,195]. UV decreases the association of EWS with target RNAs [78]. Camptothecin impairs the interaction between EWS and YB-1, possibly affecting spliceosome assembly to provoke exon skipping in Mdm2 and other genes [162]. Although camptothecin treatment leads to hyperphosphorylation of the CTD of RNA polymerase II [162,196], the transcription elongation inhibitor DRB prevents camptothecin-mediated polymerase II phosphorylation, but not the impact of camptothecin on splicing, suggesting that a change in transcription elongation is not responsible for the observed shift in Mdm2 splicing. While the impact of camptothecin on Mdm2 splicing is independent of p53 [162], it is unclear how it promotes a loss of interaction between EWS and YB-1. UV irradiation partially relocalizes EWS to the nucleolus, but this effect is not seen with the TOP2 inhibitor etoposide [78].
- •
- The epigenetic histone mark H3K36me3 is required to recruit the mismatch repair machinery and for HR-mediated repair [197,198]. H3K36me3 is elevated in nucleosomes residing on exons relative to those found in introns [199,200,201,202], and a splicing inhibitor or splice site mutations alters the deposition of the H3K36me3 mark [203]. The preferential association of H3K36me3 with coding sequences may therefore be used to prioritize the repair of coding portions of the genome. Notably, the chromatin-associated protein PSIP1 interacts with H3K36me3 and with several splicing regulators including SRSF1, SRSF2, SRSF10, hnRNP proteins and snRNP helicases [204]. Moreover, silencing the expression of the short splice variant of PSIP1 changes the localization of SRSF1 and impacts alternative splicing [204]. DNA damage leads to the phosphorylation of PSIP1 [64], but it is not known if this alters the interaction of PSIP with H3K36me3 or with splicing regulators to affect splicing. Interestingly, the splicing factor protein SF3B1, whose expression is altered by DNA damage and whose activity is linked to DNA repair, preferentially associates with nucleosomes residing on exons to modulate splicing [205]. If DNA damage promotes the dissociation of splicing components from exon-specific nucleosomes, this could represent a strategy for the unobstructed sensing of local damage and the recruitment of the repair machinery.
- •
- Several large non-coding RNAs (lncRNAs) provide binding platforms for splicing regulators [206,207] and factors that modify chromatin to alter splice site selection [208]. DNA damage affects the transcription of the lncRNAs TUG1, Panda and lincRNA-p21 [209,210]. Panda sequesters transcription factors induced by DNA damage and prevents apoptosis when cells are treated with doxorubicin [211]. LincRNA-p21 recruits hnRNP K to control the transcription of p53-dependent genes [212], and enhances sensitivity to radiotherapy [210]. Whether the interaction of hnRNP K with lincRNA-p21 is affected by DNA damage to impact hnRNP K-mediated splicing events is not yet known.
9. DNA Damage Modulates the Alternative Splicing of Genes Involved in the DDR
Genes involved in DNA repair, cell-cycle control and apoptosis use alternative splicing to expand their functional diversity. In the DNA repair category, splice variants of BRCA1 sensitize cells to DNA damage and alter repair mechanisms [213]. Ercc1 produces a splice variant that increases sensitivity to cisplatin [214]. In cell-cycle genes, splice variants of CHK2 and the CDC25B phosphatase [215,216] display dominant-negative effects, while variants of cyclin D1 differentially regulate the DDR [10]. At least a dozen genes involved in apoptosis produce splice variants with distinct and sometimes opposite activities, including Fas, Bcl-x, Mcl1, casp8 and casp9 [217]. Mdm2 produces splice variants that differentially control the activity of p53 and, hence, have a major impact on cell fate [218]. Coordination between repair, cell-cycle control and apoptosis is important to insure homeostasis and an efficient response to genotoxic stresses; thus we may anticipate common regulatory principles as well as cross-talks between molecular processes that are controlling splicing in these three functional categories. Apoptosis and cell-cycle control are already linked at the transcriptional level, for example, through the activity p53 and FOXO [219,220]. Signal transduction also serves to link cell-cycle control with the alternative splicing of apoptotic genes. For example, the AURKA kinase, which controls mitosis, converges on SRSF1 to prevent the production of pro-apoptotic variants of Bcl-x, Mcl1 and Casp9 [221]. Although this aspect has not yet been explored in detail, coordination between these functional categories likely includes the contribution of splicing factors and regulators. As mentioned above, EWS controls the alternative splicing of cyclin D1, of the apoptotic genes Fas and Mdm2, and of the DNA repair genes Abl1 and Chk2. SRSF1 and Sam68 both control the alternative splicing of Bcl-x and cyclin D1 [152,222,223,224]. HnRNP C controls the alternative splicing of Bcl2l12, Bard1 and Wrn [130,131], while hnRNP A1 modulates the splicing of Lrdd and Bard1 [131]. More complex regulatory strategies can arise since many splicing regulators lead alternate lives in the cytoplasm. For example, PTBP1 controls the splicing of Fas, but also promotes the translation of the CDK inhibitor p27Kip [34,225]. Based on these examples and the fact that several splicing regulators are modified or relocalized in response to DNA damage, many of the functional adjustments implemented to deal with DNA damage may occur through control of splicing.
Studies that have focused on a few genes or that have interrogated entire transcriptomes indicate that DNA damage has a broad impact on alternative splicing (Table 1 and Table 2). While the functional impact of these shifts is lacking in most cases, many of the genes affected are involved in DNA repair, cell-cycle control and apoptosis. In the DNA damage repair category, DNA lesions shift alternative splicing in Atrip [103], and improve the production of a splice variant encoding NBS1, a protein that interacts with the break-sensing MRE11/RAD50 complex [226]. In cell-cycle control genes, UV irradiation promotes splicing shifts in Chek2, Mapk2 and Abl1 [78]. UV and camptothecin also alter the alternative splicing of several genes involved in sensing/repair (e.g., Atm, Atr, Chek1, Chek2, Parp2, Ddb1, Mlh1, Msh6) and cell-cycle control (e.g., Ccnb2, Aurka, Aurkb, Ccnt2) [79,177] (Table 2).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Alternative splicing events validated by RT-PCR that are affected by DNA damaging agents. The reference for each study is provided as a PubMed Identification (PMID) number.
| Treatment | Cell Line | Affected Gene | PMID Number |
|---|---|---|---|
| 5-Aza dC | MCF-7 | FN1, SYNE2 | 25313066 |
| Aclarubicin | SMA fibroblasts | SMN2 | 11734549 |
| Amsacrine | U-937 | CASP2 | 14757846 |
| Arsenic (III) chloride | BEAS-2B | GADD45 | 18942077 |
| Arsenite | AGS | CD44, SFRS10 (TRA2B) | 19439532 |
| Bleomycin (BLM) | HLE, HLF | FIR | 24811221 |
| BN80927 (TOP1, TOP2 inhibitor) | U-937 | CASP2 | 14757846 |
| Cadmium dichloride | RKO, EB-1 | PA26 | 9926927 |
| Camptothecin | HCT116 | AASDHPPT, APTX, BAT1, CASP2, CCT2, DDX17, EIF2S2, PNN, PPFIA1, PSMD12, RBM8A, RIOK1, RTN4, SF1, TCP1, WHSC1, ZRANB2 | 20817775 |
| HeLa, U-937 | CASP2 | 14757846, 18166155 | |
| MCF-7 | CDC25C, FN1, SYNE2, HRAS, CHD2, EED, KIAA0232, MDM2, PAPOLG, RC3H2, THUMPD2, ZCCHC8, VEGF-A, RBM8A, SF1 | 22871320, 25313066, 17709397, 20972445, 18086921, 20817775 | |
| A431 | FOS (generic splicing) | 16921380 | |
| Jurkat T lymphoma | HNRPDL, IVNS1ABP, SF3B3, RUNX1, HMGXb4, SNRPB, SRSF2 | 21163941 | |
| HaCat, MDA-MB-231 | VEGF-A | 18086921 | |
| Capecitabine | MCF-7, HeLa S3, PA-1 | BCL2L1 (Bcl-x) | 18566212 |
| Carboplatin | RTC | BCL2L1 (Bcl-x) | 21198546 |
| Chlorambucil | EcR293 | BCL2L1 (Bcl-x) | 18566212 |
| Cisplatin | SH-SY5Y | APAF1, H-RAS | 23613995 |
| Hep3B | BCL2L1 (Bcl-x) | 19450518 | |
| EcR293, MCF-7, HeLa S3, PC3, PA-1, SKOV-3 | BCL2L1 (Bcl-x) | 18566212 | |
| HT1080 | BCL2L1 (Bcl-x), PIG3, Smac/DIABLO, MDM2 | 21327085, 25884497 | |
| H358 | CASP8 | 21157427 | |
| MCF-7 | CDC25C, MDM4, MAGOH, AMZ2, CSDE1, EIF4A2, MDM2, MTA1, NFE2L1, STRAP, TMPO, VEGF, MDM2 | 22871320, 18711402, 25884497, 25845590, 17018606 | |
| AT5BIVA, MO59J | HNRNPDL | 25884497 | |
| HeLa S3, BT549, HDF1, MDA-MB-231, MG-63, MSU, RD, U2OS | MDM2 | 25845590, 25884497, 17018606 | |
| H1299 | MDM2, MDM4 | 17018606, 18711402 | |
| Ishikawa | MDM2, VEGF | 25884497 | |
| HCT116, IMR90 | MDM4 | 18711402 | |
| Cyclohexamide | U937 | CASP2, FAS | 15746654, 16131458 |
| Cyclophosphamide | SKOV3 | BCL2L1 (Bcl-x) | 18566212 |
| H358 | BCL2L1 (Bcl-x), CASP9 | 18806759 | |
| Cytarabine | PC3 | BCL2L1 (Bcl-x) | 18566212 |
| Dacarbazine | EcR293 | BCL2L1 (Bcl-x) | 18566212 |
| Dactinomycin | EcR293, MCF-7, HeLa S3, PC3 | BCL2L1 (Bcl-x) | 18566212 |
| NIH3T3 | MDM2 | 18469520 | |
| Daunorubicin | MCF-7, PC3, PA1 | BCL2L1 (Bcl-x) | 18566212 |
| Diflomotecan | U-937 | CASP2 | 14757846 |
| Docetacel | HeLa S3 | BCL2L1 (Bcl-x) | 18566212 |
| Doxorubicin | EU-3 | BIRC5 | 15334064 |
| U-937, HCT116 | CASP2 | 14757846, 20817775 | |
| MDA-MB-231 | CDC25C | 22871320 | |
| MCF-7 | CDC25C, PPM1D | 2287132, 18845566 | |
| NIH3T3 | MDM2 | 18469520 | |
| EB-1, RKO | PA26 | 9926927 | |
| T47D | PPM1D | 18845566 | |
| Epirubicin | EcR293, MCF-7, HeLa S3, PC3 | BCL2L1 (Bcl-x) | 18566212 |
| HeLa, HL-60 | CASP2 | 14757846, 12169392 | |
| U937 | CASP2, FAS | 15746654, 16131458, 12169392, 14757846 | |
| MCF-7 | CDC25C | 22871320 | |
| U2OS | NOXA, GADD45 (generic splicing) | 21460037 | |
| Fluorouracil (5FU) | EcR293 | BCL2L1 (Bcl-x) | 18566212 |
| Gemcitabine | A549 | BCL2L1 (Bcl-x), CASP9 | 11801602 |
| EcR293, MCF-7, PA-1, SKOV3, MiaPaCa2, PT45P1 | BCL2L1 (Bcl-x), MKNK2 | 18566212, 22797067 | |
| H2O2 | Saos2 | ATF-3 | 12034827 |
| HCT116 | SRSF3 | 24284797 | |
| IDC92 (Indole derivative) | MDA-MB-435S | RON | 20864806 |
| Indolocarbazole derivative (NB-506) | P388 | BCL2L1 (Bcl-x), CD44, SC35, STY | 11559564 |
| Ionizing radiation (IR) | MCF-7 | BCL2L1 (Bcl-x), CLU | 16465415, 15530543 |
| SH-SY5Y | APAF1, H-RAS | 23613995 | |
| LCL lymphoblastoid | ASPM, FBXW7, GADD45G, MDM2, VWCE | 22039421 | |
| Ionizing radiation (IR) | Primary fibroblasts | ATF-2 | 12833146 |
| PBMCs | NBS1 | 18582154 | |
| NF AG1519 | RAD17 | 11602352 | |
| Irinotecan | U-937 | CASP2 | 14757846 |
| L-mimosin | MCF-7 | VEGF-A | 18086921 |
| Methotrexate | EcR293, MCF-7, HeLa S3 | BCL2L1 (Bcl-x) | 18566212 |
| Mitomycin C | PC3, U2OS, HTC116 | CD44, KSR1, IL24 | 20110258, 17699766 |
| U2OS | FAS | 18571879 | |
| MCF-7, OVCAR3, SKOV3 | VEGF-A | 18086921, 25990504 | |
| Mitoxantrone | U-937 | CASP2 | 14757846 |
| Oxaliplatin | EcR293, MCF-7, HeLa S3, PC3, PA-1, SKOV-3 | BCL2L1 (Bcl-x) | 18566212, 20980256 |
| MCF-7 | MDM2 | 25884497 | |
| Paclitaxel | U937 | CASP2, FAS | 15746654, 16131458 |
| Paraquat | SH-SY5Y | APAF1, BIN1, CASP9, CHN1, ERRC1, GNAO1, H-RAS, LMO3, NRG1, SKP2, SMN1, RPRD1A | 23613995, 21120952 |
| Sodium arsenite | HeLa | ABCG2, MGP, NCAM2 | 25879800 |
| HCT116 | SFRS10, SRSF3 | 24865968, 24284797 | |
| TAS-103 (TOP1, TOP2 inhibitor) | U-937 | CASP2 | 14757846 |
| Topotecan | EcR293, PC3 | BCL2L1 (Bcl-x) | 18566212 |
| UV irradiation | HeLa | ABL1, CHEK2, MAP4K2, MDM2, PIG3 | 21816343, 25845590, 18801469 |
| HT1080 | BCL2L1 (Bcl-x), PIG3, Smac/DIABLO | 21327085 | |
| Human skin | ELN | 19054052 | |
| H1299 | MDM2, MDM4 | 17018606 | |
| MCF-7 | MDM2, MDM4, VEGF-A, PIG3 | 25845590, 17018606, 18086921, 18801469 | |
| MRC-5V1 | SRSF1 | 21984412 | |
| UV-B irradiation | HaCat | VEGF-A | 18086921 |
| MDA-MB-231 | VEGF-A | 18086921 | |
| UV-C irradiation | HeLa | ADAR2, DDO | 15728250 |
| Hep3B | BCL2L1 (Bcl-x), CASP9 | 19450518 |
Many splicing shifts caused by DNA damage occur in apoptotic genes. Cisplatin favors the production of pro-apoptotic splice variants of c-flip, casp8, casp9 and Bcl-x [227]. While cisplatin and oxaliplatin modify Bcl-x splicing in different cells lines (Table 2), not all DNA damaging drugs (e.g., topotecan, etoposide, 5FU) have a similar impact on Bcl-x splicing, and these effects vary in different cell lines [151], Likewise, DNA damaging drugs alter the alternative splicing of other apoptotic genes but the sets of targets in different cell lines display little overlap [151] (Table 2). Cisplatin and oxaliplatin shift Bcl-x splicing in 293 cells in an ATM/CHK2-dependent manner [74]. UV irradiation promotes an ATM-independent shift in Bcl-x and Mdm2 splicing in Hep3B cells and MCF-7 cells, respectively [79,228]. UV irradiation of human dermal fibroblasts fosters ATM-dependent changes in alternative splicing of genes that include Atrip, Dnmt3a and Sirt3 [103]. Hundreds of alternative splicing changes occur when MCF-7 cells are treated with cisplatin [229], but these changes rely on the activation of the PI3K P110β and not the typical ATM and ATR signaling pathways. Overall, these results reinforce the notion that different treatments and different doses activate distinct pathways. Since the operational status of these pathways likely differs between cell lines, these variations will also be reflected in the identity of genes whose splicing and alternative splicing are affected by the treatments.
Table 2.
Alternative splicing events affected by DNA damaging agents and identified by high-throughput screening. A web link address is provided with the source file. The reference for each study is provided as a PubMed Identification (PMID) number.
| Treatment | Cell Line | Reference for Web Link | Source | PMID Number |
|---|---|---|---|---|
| Twenty chemotherapeutic drugs | EcR293, MCF-7, HeLa, PC3, PA-1, SKOV-3 | [230] | Supplementary Information | 18566212 |
| Camptothecin | HCT116 | [231] | Table S3 | 20817775 |
| Camptothecin | Jurkat T lymphoma | [232] | Table S2 | 21163941 |
| Camptothecin | MCF-7 | [233] | Table S1 | 20972445 |
| Cisplatin | MCF-7 | [234] | File S4 | 25884497 |
| Ionizing radiation (IR) | Lymphoblastoid cell lines, Primary fibroblasts | [235] | Table S8, Table S11 | 22039421 |
| Sodium arsenite | HeLa | [236] | File S12, Table 6 | 25879800 |
| UV-C irradiation | Hep3B | [237] | Table S2 | 19450518 |
| UV-irradiation | Human dermal fibroblasts | [238] | Table S2 | 26106861 |
| UV-B irradiation | Several | [239] | Table 2 | 18086921 |
ROS production also affects the alternative splicing of genes involved in the DDR (e.g., Ercc1 for DNA repair, Hras and Skp2 for cell-cycle control, and Apaf1 and Bin1 for apoptosis) [156]. Finally, bleomycin reduces the expression of SF3B1 and increases the production of a splice variant of FIR (aka PUF60, a U2AF2-related protein) that is deficient in its ability to confer transcriptional repression of c-myc [240]. The FIR proteins form a complex with SF3B1, which controls the splicing of Fir itself [240,241], of the cell-cycle gene p27kip1 [240] and of Bcl-x [242]. The bleomycin-mediated drop in the expression of SF3B1 may therefore coordinate alternative splicing decisions to affect cell-cycle and apoptosis.
To determine if the impact of DNA damage on alternative splicing control preferentially affects the production of DDR components, we carried out an analysis using the PANTHER classification system [243]. We included in the analysis the splicing changes of six studies that used camptothecin, UV irradiation and sodium arsenate in different cell lines (Table 2). Notably, DNA damage elicited alternative splicing changes in genes that are preferentially associated with DNA repair, cell-cycle control and apoptotic signaling (Table 3), suggesting a concerted reprogramming of alternative splicing. Interestingly, an overrepresentation of splicing changes occurred in genes involved in splicing control, as previously noted by the group of Blencowe [180], and in genes involved in chromatin organization and modification. This last observation is interesting but not totally surprising given that chromatin coordinates DNA repair activities, and is now known to be intimately associated with the control of splice site selection.
Table 3.
DNA damage-induced changes in alternative splicing occur preferentially in genes implicated in DNA repair, cell-cycle control and apoptosis. We compiled alternative splicing changes occurring in 2214 genes from six studies that used camptothecin, UV and sodium arsenate (PMID number 20817775, 21163941, 20972445, 25879800, 19450518 and 26106861; see Table 3). The annotation to biological processes was carried out using the PANTHER bioinformatics platform [243]. Relative to the distribution of 20,814 human genes in each process, PANTHER identified processes that were enriched in genes whose splicing is affected by DNA damage (the number of genes affected is indicated). The statistical significance of the enrichment, expressed as a p value (Bonferroni corrected), is indicated for each process. p Values inferior to 0.01 were observed for 3.4% (268/7812) of processes. Of these, 36 processes displayed a p value of less than 0.001 and a gene enrichment greater than 2-fold. Nineteen processes from this set are listed below.
| GO Biological Process Complete | Number of Genes | Fold Enrichment | p Value |
|---|---|---|---|
| DNA repair | 94 | 2.29 | 4.04E−09 |
| cell cycle checkpoint | 58 | 2.4 | 2.09E−05 |
| cell cycle phase transition | 64 | 2.18 | 1.19E−04 |
| mitotic cell cycle phase transition | 63 | 2.18 | 1.70E−04 |
| mitotic cell cycle phase | 57 | 2.05 | 5.09E−04 |
| mitotic cell cycle process | 138 | 2.04 | 1.00E−10 |
| cell cycle phase | 57 | 2.04 | 6.26E−04 |
| mitotic cell cycle | 51 | 2.03 | 6.87E−12 |
| intrinsic apoptotic signaling pathway | 41 | 2.49 | 1.72E−04 |
| apoptotic signaling pathway | 75 | 2.25 | 1.85E−06 |
| RNA splicing | 93 | 2.92 | 3.69E−15 |
| RNA splicing, via transesterification reactions | 68 | 3.19 | 3.00E−12 |
| mRNA splicing, via spliceosome | 67 | 3.19 | 5.26E−12 |
| chromatin modification | 117 | 2.42 | 1.29E−13 |
| covalent chromatin modification | 75 | 2.55 | 6.99E−09 |
| chromatin organization | 123 | 2.1 | 4.28E−10 |
| chromosome organization | 167 | 2.04 | 1.31E−13 |
| histone modification | 74 | 2.54 | 1.18E−08 |
| regulation of gene expression, epigenetic | 49 | 2.39 | 4.28E−04 |
10. Conclusions
Recent efforts have helped establish the multiple ways through which DNA damage alters the activity of splicing factors and modulates constitutive and alternative splicing. DNA lesions attract splicing factors that participate in mounting the first steps of the DDR. Occasionally, some of these factors (e.g., BRCA1, BCLAF1, PSF and hnRNP C) play an even more direct role in damage repair. Other splicing proteins are recruited at sites of DNA damage, only to be tagged with post-translational modifications that affect their localization and/or activity. Yet other splicing factors are modified by enzymes that dissociate from sites of damage, such as the CHK1 and CHK2 kinases. These alterations affect splicing in different ways. For example, DDR-specific splicing complexes can be formed to improve splicing, or specific interactions can be disrupted to prevent the normal coupling of transcription with splicing. The splicing changes that often occur in genes involved in damage sensing, DNA repair, cell-cycle control and apoptosis therefore has the potential to feedback on every step of the DDR. However, despite the increasing number of examples documenting the global impact of DNA damage on splicing and alternative splicing, the functional impact of most of these changes remains to be evaluated. The variety of DNA damaging agents, their doses and the identity of cell lines selected to carry out studies also limit the usefulness of comparing the splicing response in different systems. Different types of damage require the contribution of distinct repair machineries, whose operational status may vary between cell lines. The baseline expression of DDR components may also vary considerably between genomically unstable cancer cell lines (often lacking p53) and immortalized ones. The complexity created by the various combinations of treatment and cell lines will hopefully be mitigated by the continuous development of high-throughput procedures and bioinformatics tools to help distill the common and specific rules that guide the interconnections between DNA damage and the splicing response.
Striving to obtain a complete description of the splicing-relevant alterations elicited by DNA damage has the potential to improve anti-cancer strategies. Anti-cancer treatments often aim to overwhelm the DNA repair machinery as a means of triggering apoptosis. While the intrinsically high genomic instability of cancer cells makes them more dependent on enhanced DNA repair activities, hence contributing to anti-cancer drug resistance, this reliance also provides a therapeutic opportunity because inactivating one or several DDR components may sensitize cancer cells to DNA damaging agents [244,245]. This potential was first illustrated by BRCA1, a DDR factor that also directly contributes to splicing [182]. Some BRCA1 mutations increase sensitivity to DNA damaging agents [246], and the inactivation of both BRCA1 and PARP1 is lethal [247]. Many splicing factors implicated in the DDR are also mutated or aberrantly expressed in cancer. For example, genes that encode U2AF, SF3B1 and SRSF2 are mutated in myelodysplastic syndromes [248,249], while others promote cancer or provide a cancer-specific metabolic signature [250,251]. It is revealing that the depletion of RBMX, EWS, FUS, SKIP and Tra2 all augment DNA damage-induced apoptosis [77,108,123,185], and that knocking down PTBP1 abolishes resistance to genotoxic drugs in pancreatic cancer cells [252]. Thus, small molecules that target generic spliceosomal components [253] or specific splicing regulators [254] may be offering new strategies to combat cancer and chemoresistance.
The stochastic accumulation of DNA damage caused by replication errors, intrinsic metabolic and external mutagens has also been associated with organismal and stem cell aging [255]. The persistence of unrepaired DNA lesions and the resulting chronic DDR activation cause a permanent cell-cycle arrest that defines cellular senescence [256,257]. Aging and senescence are associated with changes in chromatin structure, in the expression of splicing factors and in alternative splicing [257,258,259,260]. The diversity of factors that affect the splicing of DDR genes and the activity of the splice variant itself are providing a large selection of novel targets that can potentially be used to prevent stem cell senescence and impair cancer development in aging individuals.
Acknowledgments
We thank Nancy Greenbaum for helpful comments on the manuscript. This work was supported by grants from the Canadian Institutes of Health Research and the Natural Sciences and Engineering Research Council of Canada. Benoit Chabot held the Canada Research Chair in Functional Genomics, and is now the Pierre C. Fournier Chair in Functional Genomics.
Author Contributions
Lulzim Shkreta and Benoit Chabot wrote the manuscript and prepared the figures and the tables.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lenzken, S.C.; Loffreda, A.; Barabino, S.M. RNA splicing: A new player in the DNA damage response. Int. J. Cell Biol. 2013. [Google Scholar] [CrossRef]
- Dutertre, M.; Lambert, S.; Carreira, A.; Amor-Gueret, M.; Vagner, S. DNA damage: RNA-binding proteins protect from near and far. Trends Biochem. Sci. 2014, 39, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Busa, R.; Sette, C. An emerging role for nuclear RNA-mediated responses to genotoxic stress. RNA Biol. 2010, 7, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Naro, C.; Bielli, P.; Pagliarini, V.; Sette, C. The interplay between DNA damage response and RNA processing: The unexpected role of splicing factors as gatekeepers of genome stability. Front. Genet. 2015. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, M.; Sanchez, G.; Barbier, J.; Corcos, L.; Auboeuf, D. The emerging role of pre-messenger RNA splicing in stress responses: Sending alternative messages and silent messengers. RNA Biol. 2011, 8, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, E.; Loewer, A.; Lahav, G. The ups and downs of p53: Understanding protein dynamics in single cells. Nat. Rev. Cancer 2009, 9, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Boucas, J.; Riabinska, A.; Jokic, M.; Herter-Sprie, G.S.; Chen, S.; Hopker, K.; Reinhardt, H.C. Posttranscriptional regulation of gene expression-adding another layer of complexity to the DNA damage response. Front. Genet. 2012. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jiao, X.; Wang, C.; Shirley, L.A.; Elsaleh, H.; Dahl, O.; Wang, M.; Soutoglou, E.; Knudsen, E.S.; Pestell, R.G. Alternative cyclin D1 splice forms differentially regulate the DNA damage response. Cancer Res. 2010, 70, 8802–8811. [Google Scholar] [CrossRef] [PubMed]
- Boise, L.H.; Gonzalez-Garcia, M.; Postema, C.E.; Ding, L.; Lindsten, T.; Turka, L.A.; Mao, X.; Nunez, G.; Thompson, C.B. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993, 74, 597–608. [Google Scholar] [CrossRef]
- Munding, E.M.; Shiue, L.; Katzman, S.; Donohue, J.P.; Ares, M., Jr. Competition between pre-mRNAs for the splicing machinery drives global regulation of splicing. Mol. Cell 2013, 51, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Jurica, M.S.; Moore, M.J. Pre-mRNA splicing: Awash in a sea of proteins. Mol. Cell 2003, 12, 5–14. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Luhrmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed]
- Matera, A.G.; Wang, Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Wahl, M.C.; Luhrmann, R. SnapShot: Spliceosome dynamics II. Cell 2015, 162. [Google Scholar] [CrossRef] [PubMed]
- Wahl, M.C.; Luhrmann, R. SnapShot: Spliceosome Dynamics I. Cell 2015, 161. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.A.; Steitz, J.A. Splicing double: Insights from the second spliceosome. Nat. Rev. Mol. Cell Biol. 2003, 4, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Dufu, K.; Romney, B.; Feldt, M.; Elenko, M.; Reed, R. Functional coupling of RNAP II transcription to spliceosome assembly. Genes Dev. 2006, 20, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.L. Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet. 2014, 15, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, M.; Long, M. Intron-exon structures of eukaryotic model organisms. Nucleic Acids Res. 1999, 27, 3219–3228. [Google Scholar] [PubMed]
- Schmucker, D.; Clemens, J.C.; Shu, H.; Worby, C.A.; Xiao, J.; Muda, M.; Dixon, J.E.; Zipursky, S.L. Drosophila Dscam is an axon guidance receptor exhibiting extraordinary molecular diversity. Cell 2000, 101, 671–684. [Google Scholar] [CrossRef]
- Kelemen, O.; Convertini, P.; Zhang, Z.; Wen, Y.; Shen, M.; Falaleeva, M.; Stamm, S. Function of alternative splicing. Gene 2013, 514, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Blencowe, B.J. Alternative splicing: New insights from global analyses. Cell 2006, 126, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Shkreta, L.; Bell, B.; Revil, T.; Venables, J.P.; Prinos, P.; Elela, S.A.; Chabot, B. Cancer-associated perturbations in alternative pre-messenger RNA splicing. In RNA and Cancer; Wu, J., Ed.; Springer Berlin: Heidelberg, Germany, 2013; pp. 41–94. [Google Scholar]
- Zhang, J.; Manley, J.L. Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov. 2013, 3, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Tazi, J.; Bakkour, N.; Stamm, S. Alternative splicing and disease. Biochim. Biophys. Acta 2009, 1792, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.D.; Ares, M., Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Rio, D.C. Mechanisms and regulation of alternative pre-mRNA splicing. Ann. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [PubMed]
- Kan, J.L.; Green, M.R. Pre-mRNA splicing of IgM exons M1 and M2 is directed by a juxtaposed splicing enhancer and inhibitor. Genes Dev. 1999, 13, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Mayeda, A.; Krainer, A.R. Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Mol. Cell 2001, 8, 1351–1361. [Google Scholar] [CrossRef]
- Izquierdo, J.M.; Majos, N.; Bonnal, S.; Martinez, C.; Castelo, R.; Guigo, R.; Bilbao, D.; Valcarcel, J. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol. Cell 2005, 19, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Sebestyen, E.; Zawisza, M.; Eyras, E. Detection of recurrent alternative splicing switches in tumor samples reveals novel signatures of cancer. Nucleic Acids Res. 2015, 43, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Chiou, N.T.; Shankarling, G.; Lynch, K.W. hnRNP L and hnRNP A1 induce extended U1 snRNA interactions with an exon to repress spliceosome assembly. Mol. Cell 2013, 49, 972–982. [Google Scholar] [CrossRef] [PubMed]
- Kanopka, A.; Muhlemann, O.; Akusjarvi, G. Inhibition by SR proteins of splicing of a regulated adenovirus pre-mRNA. Nature 1996, 381, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Contreras, R.; Fisette, J.F.; Nasim, F.U.; Madden, R.; Cordeau, M.; Chabot, B. Intronic binding sites for hnRNP A/B and hnRNP F/H proteins stimulate pre-mRNA splicing. PLoS Biol. 2006, 4, e21. [Google Scholar] [CrossRef] [PubMed]
- Naftelberg, S.; Schor, I.E.; Ast, G.; Kornblihtt, A.R. Regulation of alternative splicing through coupling with transcription and chromatin structure. Ann. Rev. Biochem. 2015, 84, 165–198. [Google Scholar] [CrossRef] [PubMed]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Kraus, W.L. On PAR with PARP: Cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012, 26, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Dery, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Krietsch, J.; Caron, M.C.; Gagne, J.P.; Ethier, C.; Vignard, J.; Vincent, M.; Rouleau, M.; Hendzel, M.J.; Poirier, G.G.; Masson, J.Y. PARP activation regulates the RNA-binding protein NONO in the DNA damage response to DNA double-strand breaks. Nucleic Acids Res. 2012, 40, 10287–10301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hottiger, M.O. SnapShot: ADP-ribosylation signaling. Mol. Cell 2015. [Google Scholar] [CrossRef] [PubMed]
- Malanga, M.; Czubaty, A.; Girstun, A.; Staron, K.; Althaus, F.R. Poly(ADP-ribose) binds to the splicing factor ASF/SF2 and regulates its phosphorylation by DNA topoisomerase I. J. Biol. Chem. 2008, 283, 19991–19998. [Google Scholar] [CrossRef] [PubMed]
- Gagne, J.P.; Hunter, J.M.; Labrecque, B.; Chabot, B.; Poirier, G.G. A proteomic approach to the identification of heterogeneous nuclear ribonucleoproteins as a new family of poly(ADP-ribose)-binding proteins. Biochem. J. 2003, 371, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Tulin, A.V. Post-transcriptional regulation by poly(ADP-ribosyl)ation of the RNA-binding proteins. Int. J. Mol. Sci. 2013, 14, 16168–16183. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Tulin, A.V. Poly(ADP-ribosyl)ation of heterogeneous nuclear ribonucleoproteins modulates splicing. Nucleic Acids Res. 2009, 37, 3501–3513. [Google Scholar] [CrossRef] [PubMed]
- Jungmichel, S.; Rosenthal, F.; Altmeyer, M.; Lukas, J.; Hottiger, M.O.; Nielsen, M.L. Proteome-wide identification of poly(ADP-Ribosyl)ation targets in different genotoxic stress responses. Mol. Cell 2013, 52, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Beli, P.; Lukashchuk, N.; Wagner, S.A.; Weinert, B.T.; Olsen, J.V.; Baskcomb, L.; Mann, M.; Jackson, S.P.; Choudhary, C. Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 2012, 46, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Bedford, M.T.; Richard, S. Arginine methylation an emerging regulator of protein function. Mol. Cell 2005, 18, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Chiou, Y.Y.; Fu, S.L.; Shih, I.Y.; Weng, T.H.; Lin, W.J.; Lin, C.H. Arginine methylation of hnRNPK negatively modulates apoptosis upon DNA damage through local regulation of phosphorylation. Nucleic Acids Res. 2014, 42, 9908–9924. [Google Scholar] [CrossRef] [PubMed]
- Revil, T.; Pelletier, J.; Toutant, J.; Cloutier, A.; Chabot, B. Heterogeneous nuclear ribonucleoprotein K represses the production of pro-apoptotic Bcl-xS splice isoform. J. Biol. Chem. 2009, 284, 21458–21467. [Google Scholar] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Nakka, K.K.; Chaudhary, N.; Joshi, S.; Bhat, J.; Singh, K.; Chatterjee, S.; Malhotra, R.; De, A.; Santra, M.K.; Dilworth, F.J.; et al. Nuclear matrix-associated protein SMAR1 regulates alternative splicing via HDAC6-mediated deacetylation of Sam68. Proc. Natl. Acad. Sci. USA 2015, 112, E3374–E3383. [Google Scholar] [CrossRef] [PubMed]
- Edmond, V.; Moysan, E.; Khochbin, S.; Matthias, P.; Brambilla, C.; Brambilla, E.; Gazzeri, S.; Eymin, B. Acetylation and phosphorylation of SRSF2 control cell fate decision in response to cisplatin. EMBO J. 2011, 30, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Bennetzen, M.V.; Larsen, D.H.; Dinant, C.; Watanabe, S.; Bartek, J.; Lukas, J.; Andersen, J.S. Acetylation dynamics of human nuclear proteins during the ionizing radiation-induced DNA damage response. Cell Cycle 2013, 12, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Durocher, D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell 2013, 49, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.Y.; Yu, Y.; Shen, Z.; Beck, W.T. Nucleolar delocalization of human topoisomerase I in response to topotecan correlates with sumoylation of the protein. J. Biol. Chem. 2002, 277, 2958–2964. [Google Scholar] [CrossRef] [PubMed]
- Vassileva, M.T.; Matunis, M.J. SUMO modification of heterogeneous nuclear ribonucleoproteins. Mol. Cell. Biol. 2004, 24, 3623–3632. [Google Scholar] [CrossRef] [PubMed]
- Pelisch, F.; Gerez, J.; Druker, J.; Schor, I.E.; Munoz, M.J.; Risso, G.; Petrillo, E.; Westman, B.J.; Lamond, A.I.; Arzt, E.; et al. The serine/arginine-rich protein SF2/ASF regulates protein sumoylation. Proc. Natl. Acad. Sci. USA 2010, 107, 16119–16124. [Google Scholar] [CrossRef] [PubMed]
- Moumen, A.; Masterson, P.; O’Connor, M.J.; Jackson, S.P. hnRNP K: An HDM2 target and transcriptional coactivator of p53 in response to DNA damage. Cell 2005, 123, 1065–1078. [Google Scholar] [CrossRef] [PubMed]
- Pelisch, F.; Pozzi, B.; Risso, G.; Munoz, M.J.; Srebrow, A. DNA damage-induced heterogeneous nuclear ribonucleoprotein K sumoylation regulates p53 transcriptional activation. J. Biol. Chem. 2012, 287, 30789–30799. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Smolka, M.B.; Albuquerque, C.P.; Chen, S.H.; Zhou, H. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc. Natl. Acad. Sci. USA 2007, 104, 10364–10369. [Google Scholar] [CrossRef] [PubMed]
- Chanarat, S.; Burkert-Kautzsch, C.; Meinel, D.M.; Strasser, K. Prp19C and TREX: Interacting to promote transcription elongationand mRNA export. Transcription 2012, 3, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Bennetzen, M.V.; Larsen, D.H.; Bunkenborg, J.; Bartek, J.; Lukas, J.; Andersen, J.S. Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol. Cell. Proteomics 2010, 9, 1314–1323. [Google Scholar] [CrossRef] [PubMed]
- Bensimon, A.; Schmidt, A.; Ziv, Y.; Elkon, R.; Wang, S.Y.; Chen, D.J.; Aebersold, R.; Shiloh, Y. ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage. Sci. Signal. 2010. [Google Scholar] [CrossRef] [PubMed]
- Leva, V.; Giuliano, S.; Bardoni, A.; Camerini, S.; Crescenzi, M.; Lisa, A.; Biamonti, G.; Montecucco, A. Phosphorylation of SRSF1 is modulated by replicational stress. Nucleic Acids Res. 2012, 40, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Moumen, A.; Magill, C.; Dry, K.L.; Jackson, S.P. ATM-dependent phosphorylation of heterogeneous nuclear ribonucleoprotein K promotes p53 transcriptional activation in response to DNA damage. Cell Cycle 2013, 12, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Blasius, M.; Forment, J.V.; Thakkar, N.; Wagner, S.A.; Choudhary, C.; Jackson, S.P. A phospho-proteomic screen identifies substrates of the checkpoint kinase Chk1. Genome Biol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Katzenberger, R.J.; Marengo, M.S.; Wassarman, D.A. ATM and ATR pathways signal alternative splicing of Drosophila TAF1 pre-mRNA in response to DNA damage. Mol. Cell. Biol. 2006, 26, 9256–9267. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.H.; Choi, H.K.; Jung, S.Y.; Hyle, J.; Kim, B.J.; Yoon, K.; Cho, E.J.; Youn, H.D.; Lahti, J.M.; Qin, J.; et al. CHK2 kinase promotes pre-mRNA splicing via phosphorylating CDK11(p110). Oncogene 2014, 33, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Shkreta, L.; Michelle, L.; Toutant, J.; Tremblay, M.L.; Chabot, B. The DNA damage response pathway regulates the alternative splicing of the apoptotic mediator Bcl-x. J. Biol. Chem. 2011, 286, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Katzenberger, R.J.; Marengo, M.S.; Wassarman, D.A. Control of alternative splicing by signal-dependent degradation of splicing-regulatory proteins. J. Biol. Chem. 2009, 284, 10737–10746. [Google Scholar] [CrossRef] [PubMed]
- Akaike, Y.; Masuda, K.; Kuwano, Y.; Nishida, K.; Kajita, K.; Kurokawa, K.; Satake, Y.; Shoda, K.; Imoto, I.; Rokutan, K. HuR regulates alternative splicing of the TRA2beta gene in human colon cancer cells under oxidative stress. Mol. Cell. Biol. 2014, 34, 2857–2873. [Google Scholar] [CrossRef] [PubMed]
- Best, A.; James, K.; Dalgliesh, C.; Hong, E.; Kheirolahi-Kouhestani, M.; Curk, T.; Xu, Y.; Danilenko, M.; Hussain, R.; Keavney, B.; et al. Human Tra2 proteins jointly control a CHEK1 splicing switch among alternative and constitutive target exons. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Minana, B.; Valcarcel, J. The Ewing sarcoma protein regulates DNA damage-induced alternative splicing. Mol. Cell 2011, 43, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Munoz, M.J.; Perez Santangelo, M.S.; Paronetto, M.P.; de la Mata, M.; Pelisch, F.; Boireau, S.; Glover-Cutter, K.; Ben-Dov, C.; Blaustein, M.; Lozano, J.J.; et al. DNA damage regulates alternative splicing through inhibition of RNA polymerase II elongation. Cell 2009, 137, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Lukas, C.; Bartek, J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011, 13, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Gómez Acuña, L.I.; Fiszbein, A.; Allo, M.; Schor, I.E.; Kornblihtt, A.R. Connections between chromatin signatures and splicing. Wiley Interdiscip. Rev. RNA 2013, 4, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, R.D.; Soni, D.V.; Wollman, R.; Hahn, A.T.; Yee, M.C.; Guan, A.; Hesley, J.A.; Miller, S.C.; Cromwell, E.F.; Solow-Cordero, D.E.; et al. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol. Cell 2009, 35, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, S.; Rondon, A.G.; Luna, R.; Aguilera, A. The yeast THO complex and mRNA export factors link RNA metabolism with transcription and genome instability. EMBO J. 2002, 21, 3526–3535. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Manley, J.L. Cotranscriptional processes and their influence on genome stability. Genes Dev. 2006, 20, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Garcia-Muse, T. R loops: From transcription byproducts to threats to genome stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Sanchez, M.S.; Barroso, S.; Gomez-Gonzalez, B.; Luna, R.; Aguilera, A. Genome instability and transcription elongation impairment in human cells depleted of THO/TREX. PLoS Genet. 2011, 7, e1002386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Manley, J.L. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 2005, 122, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Sun, Y.; Ding, J.H.; Lin, S.; Rose, D.W.; Rosenfeld, M.G.; Fu, X.D.; Li, X. Splicing regulator SC35 is essential for genomic stability and cell proliferation during mammalian organogenesis. Mol. Cell. Biol. 2007, 27, 5393–5402. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Manley, J.L. Alternative splicing and control of apoptotic DNA fragmentation. Cell Cycle 2006, 5, 1286–1288. [Google Scholar] [CrossRef] [PubMed]
- Sordet, O.; Redon, C.E.; Guirouilh-Barbat, J.; Smith, S.; Solier, S.; Douarre, C.; Conti, C.; Nakamura, A.J.; Das, B.B.; Nicolas, E.; et al. Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks. EMBO Rep. 2009, 10, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Niu, T.; Manley, J.L. The RNA binding protein RNPS1 alleviates ASF/SF2 depletion-induced genomic instability. RNA 2007, 13, 2108–2115. [Google Scholar] [CrossRef] [PubMed]
- Tuduri, S.; Crabbe, L.; Conti, C.; Tourriere, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; de Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009, 11, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Labourier, E.; Forne, T.; Divita, G.; Derancourt, J.; Riou, J.F.; Antoine, E.; Cathala, G.; Brunel, C.; Tazi, J. Specific phosphorylation of SR proteins by mammalian DNA topoisomerase I. Nature 1996, 381, 80–82. [Google Scholar] [CrossRef] [PubMed]
- Czubaty, A.; Girstun, A.; Kowalska-Loth, B.; Trzcinska, A.M.; Purta, E.; Winczura, A.; Grajkowski, W.; Staron, K. Proteomic analysis of complexes formed by human topoisomerase I. Biochim. Biophys. Acta 2005, 1749, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Zheng, X.; Chen, H.; Guo, Y.; Jiang, H.; He, X.; Zhu, X.; Zheng, Y. Splicing function of mitotic regulators links R-loop-mediated DNA damage to tumor cell killing. J. Cell Biol. 2015, 209, 235–246. [Google Scholar] [CrossRef] [PubMed]
- LaBranche, H.; Dupuis, S.; Ben-David, Y.; Bani, M.R.; Wellinger, R.J.; Chabot, B. Telomere elongation by hnRNP A1 and a derivative that interacts with telomeric repeats and telomerase. Nat. Genet. 1998, 19, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Fiset, S.; Chabot, B. hnRNP A1 may interact simultaneously with telomeric DNA and the human telomerase RNA in vitro. Nucleic Acids Res. 2001, 29, 2268–2275. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.S.; Manche, L.; Xu, R.M.; Krainer, A.R. hnRNP A1 associates with telomere ends and stimulates telomerase activity. RNA 2006, 12, 1116–1128. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.L.; Centore, R.C.; O’Sullivan, R.J.; Rai, R.; Tse, A.; Songyang, Z.; Chang, S.; Karlseder, J.; Zou, L. TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature 2011, 471, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Bracken, C.P.; Wall, S.J.; Barre, B.; Panov, K.I.; Ajuh, P.M.; Perkins, N.D. Regulation of cyclin D1 RNA stability by SNIP1. Cancer Res. 2008, 68, 7621–7628. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Hsu, I.W.; Tarn, W.Y. TRAP150 activates pre-mRNA splicing and promotes nuclear mRNA degradation. Nucleic Acids Res. 2010, 38, 3340–3350. [Google Scholar] [CrossRef] [PubMed]
- Tresini, M.; Warmerdam, D.O.; Kolovos, P.; Snijder, L.; Vrouwe, M.G.; Demmers, J.A.; van IJcken, W.F.; Grosveld, F.G.; Medema, R.H.; Hoeijmakers, J.H.; et al. The core spliceosome as target and effector of non-canonical ATM signalling. Nature 2015, 523, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Allemand, E.; Hastings, M.L.; Murray, M.V.; Myers, M.P.; Krainer, A.R. Alternative splicing regulation by interaction of phosphatase PP2Cgamma with nucleic acid-binding protein YB-1. Nat. Struct. Mol. Biol. 2007, 14, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.V.; Kobayashi, R.; Krainer, A.R. The type 2C Ser/Thr phosphatase PP2Cgamma is a pre-mRNA splicing factor. Genes Dev. 1999, 13, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Mandusic, V.; Nikolic-Vukosavljevic, D.; Tanic, N.; Kanjer, K.; Neskovic-Konstantinovic, Z.; Celeketic, D.; Dimitrijevic, B. Expression of estrogen receptor beta wt isoform (ERbeta1) and ERbetaDelta5 splice variant mRNAs in sporadic breast cancer. J. Cancer Res. Clin. Oncol. 2007, 133, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.H.; Kim, R.H.; Kang, M.K.; Kim, R.H.; Kim, S.G.; Lim, P.K.; Yochim, J.M.; Baluda, M.A.; Park, N.H. p53 promotes the fidelity of DNA end-joining activity by, in part, enhancing the expression of heterogeneous nuclear ribonucleoprotein G. DNA Repair 2007, 6, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Adamson, B.; Smogorzewska, A.; Sigoillot, F.D.; King, R.W.; Elledge, S.J. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol. 2012, 14, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, B.; Zhang, Z.; Raitskin, O.; Hiller, M.; Benderska, N.; Hartmann, A.M.; Bracco, L.; Elliott, D.; Ben-Ari, S.; Soreq, H.; et al. Heterogeneous nuclear ribonucleoprotein G regulates splice site selection by binding to CC(A/C)-rich regions in pre-mRNA. J. Biol. Chem. 2009, 284, 14303–14315. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, Y.; Lorson, C.L.; Stamm, S.; Androphy, E.J.; Wirth, B. Htra2-beta 1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2). Proc. Natl. Acad. Sci. USA 2000, 97, 9618–9623. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, Y.; Wirth, B. hnRNP-G promotes exon 7 inclusion of survival motor neuron (SMN) via direct interaction with Htra2-β1. Hum. Mol. Genet. 2002, 11, 2037–2049. [Google Scholar] [CrossRef] [PubMed]
- Venables, J.P.; Elliott, D.J.; Makarova, O.V.; Makarov, E.M.; Cooke, H.J.; Eperon, I.C. RBMY, a probable human spermatogenesis factor, and other hnRNP G proteins interact with Tra2β and affect splicing. Hum. Mol. Genet. 2000, 9, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Nasim, M.T.; Chernova, T.K.; Chowdhury, H.M.; Yue, B.G.; Eperon, I.C. HnRNP G and Tra2β: Opposite effects on splicing matched by antagonism in RNA binding. Hum. Mol. Genet. 2003, 12, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Barnard, D.C.; Patton, J.G. Identification and characterization of a novel serine-arginine-rich splicing regulatory protein. Mol. Cell. Biol. 2000, 20, 3049–3057. [Google Scholar] [CrossRef] [PubMed]
- Barnard, D.C.; Li, J.; Peng, R.; Patton, J.G. Regulation of alternative splicing by SRrp86 through coactivation and repression of specific SR proteins. RNA 2002, 8, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Barnard, D.C.; Patton, J.G. A unique glutamic acid-lysine (EK) domain acts as a splicing inhibitor. J. Biol. Chem. 2002, 277, 39485–39492. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, E.; Pierredon, S.; Driouch, K.; Gratadou, L.; Lacroix-Triki, M.; Espinoza, M.P.; Zonta, E.; Germann, S.; Mortada, H.; Villemin, J.P.; et al. Splicing switch of an epigenetic regulator by RNA helicases promotes tumor-cell invasiveness. Nat. Struct. Mol. Biol. 2012, 19, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, E.; Espinoza, M.P.; Fattet, L.; Germann, S.; Lambert, M.P.; Neil, H.; Zonta, E.; Mortada, H.; Gratadou, L.; Deygas, M.; et al. RNA helicases DDX5 and DDX17 dynamically orchestrate transcription, miRNA, and splicing programs in cell differentiation. Cell Rep. 2014, 7, 1900–1913. [Google Scholar] [CrossRef] [PubMed]
- Samaan, S.; Tranchevent, L.C.; Dardenne, E.; Espinoza, M.P.; Zonta, E.; Germann, S.; Gratadou, L.; Dutertre, M.; Auboeuf, D. The Ddx5 and Ddx17 RNA helicases are cornerstones in the complex regulatory array of steroid hormone-signaling pathways. Nucleic Acids Res. 2014, 42, 2197–2207. [Google Scholar] [CrossRef] [PubMed]
- Rogelj, B.; Easton, L.E.; Bogu, G.K.; Stanton, L.W.; Rot, G.; Curk, T.; Zupan, B.; Sugimoto, Y.; Modic, M.; Haberman, N.; et al. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci. Rep. 2012. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, A.S.; Kim, S.H.; Trinh, A.T.; Rodenkirch, L.A.; Tibbetts, R.S. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J. Biol. Chem. 2013, 288, 24731–24741. [Google Scholar] [CrossRef] [PubMed]
- Rulten, S.L.; Rotheray, A.; Green, R.L.; Grundy, G.J.; Moore, D.A.; Gomez-Herreros, F.; Hafezparast, M.; Caldecott, K.W. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 2014, 42, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Watford, W.; Li, C.; Parmelee, A.; Bryant, M.A.; Deng, C.; O’Shea, J.; Lee, S.B. Ewing sarcoma gene EWS is essential for meiosis and B lymphocyte development. J. Clin. Investig. 2007, 117, 1314–1323. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.; Huang, E.J.; Tsai, L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Chi, B.; Xia, W.; Gangopadhyay, J.; Yamazaki, T.; Winkelbauer-Hurt, M.E.; Yin, S.; Eliasse, Y.; Adams, E.; Shaw, C.E.; et al. U1 snRNP is mislocalized in ALS patient fibroblasts bearing NLS mutations in FUS and is required for motor neuron outgrowth in zebrafish. Nucleic Acids Res. 2015, 43, 3208–3218. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.W.; Mai, R.T.; Fang, W.H.; Lin, C.C.; Chiu, C.C.; Wu Lee, Y.H. YB-1 disrupts mismatch repair complex formation, interferes with MutSalpha recruitment on mismatch and inhibits mismatch repair through interacting with PCNA. Oncogene 2014, 33, 5065–5077. [Google Scholar] [CrossRef] [PubMed]
- Gaudreault, I.; Guay, D.; Lebel, M. YB-1 promotes strand separation in vitro of duplex DNA containing either mispaired bases or cisplatin modifications, exhibits endonucleolytic activities and binds several DNA repair proteins. Nucleic Acids Res. 2004, 32, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.R.; Selyutina, A.A.; Buldakov, I.A.; Evdokimova, V.; Ovchinnikov, L.P.; Sorokin, A.V. The proteolytic YB-1 fragment interacts with DNA repair machinery and enhances survival during DNA damaging stress. Cell Cycle 2013, 12, 3791–3803. [Google Scholar] [CrossRef] [PubMed]
- Anantha, R.W.; Alcivar, A.L.; Ma, J.; Cai, H.; Simhadri, S.; Ule, J.; Konig, J.; Xia, B. Requirement of heterogeneous nuclear ribonucleoprotein C for BRCA gene expression and homologous recombination. PLoS ONE 2013, 8, e61368. [Google Scholar] [CrossRef] [PubMed]
- Zarnack, K.; Konig, J.; Tajnik, M.; Martincorena, I.; Eustermann, S.; Stevant, I.; Reyes, A.; Anders, S.; Luscombe, N.M.; Ule, J. Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell 2013, 152, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Venables, J.P.; Koh, C.S.; Froehlich, U.; Lapointe, E.; Couture, S.; Inkel, L.; Bramard, A.; Paquet, E.R.; Watier, V.; Durand, M.; et al. Multiple and specific mRNA processing targets for the major human hnRNP proteins. Mol. Cell. Biol. 2008, 28, 6033–6043. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, M.C.; Narlikar, G.J.; Boyapaty, G.; Kingston, R.E.; Weissman, S.M. Heterogeneous nuclear ribonucleoprotein C1/C2, MeCP1, and SWI/SNF form a chromatin remodeling complex at the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 2005, 102, 15012–15017. [Google Scholar] [CrossRef] [PubMed]
- Batsche, E.; Yaniv, M.; Muchardt, C. The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nat. Struct. Mol. Biol. 2006, 13, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Eberhart, C.G.; Kai, M. RNA binding protein RBM14 promotes radio-resistance in glioblastoma by regulating DNA repair and cell differentiation. Oncotarget 2014, 5, 2820–2826. [Google Scholar] [CrossRef] [PubMed]
- Grey, M.; Dusterhoft, A.; Henriques, J.A.; Brendel, M. Allelism of PSO4 and PRP19 links pre-mRNA processing with recombination and error-prone DNA repair in Saccharomyces cerevisiae. Nucleic Acids Res. 1996, 24, 4009–4014. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Kaur, R.; Lu, X.; Shen, X.; Li, L.; Legerski, R.J. The Pso4 mRNA splicing and DNA repair complex interacts with WRN for processing of DNA interstrand cross-links. J. Biol. Chem. 2005, 280, 40559–40567. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Legerski, R.J. The Prp19/Pso4 core complex undergoes ubiquitylation and structural alterations in response to DNA damage. Biochem. Biophys. Res. Commun. 2007, 354, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Li, J.M.; Ji, X.Y.; Wu, C.S.; Yazinski, S.A.; Nguyen, H.D.; Liu, S.; Jimenez, A.E.; Jin, J.; Zou, L. PRP19 transforms into a sensor of RPA-ssDNA after DNA damage and drives ATR activation via a ubiquitin-mediated circuitry. Mol. Cell 2014, 53, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Huang, J. The PSO4 protein complex associates with replication protein A (RPA) and modulates the activation of ataxia telangiectasia-mutated and Rad3-related (ATR). J. Biol. Chem. 2014, 289, 6619–6626. [Google Scholar] [CrossRef] [PubMed]
- Salton, M.; Lerenthal, Y.; Wang, S.Y.; Chen, D.J.; Shiloh, Y. Involvement of Matrin 3 and SFPQ/NONO in the DNA damage response. Cell Cycle 2010, 9, 1568–1576. [Google Scholar] [CrossRef] [PubMed]
- Bladen, C.L.; Udayakumar, D.; Takeda, Y.; Dynan, W.S. Identification of the polypyrimidine tract binding protein-associated splicing factor.p54(nrb) complex as a candidate DNA double-strand break rejoining factor. J. Biol. Chem. 2005, 280, 5205–5210. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert, A.; Schmidt, U.; Monajembashi, S.; Franke, C.; Schlott, B.; Grosse, F.; Greulich, K.O.; Saluz, H.P.; Hanel, F. Proteomic identification of PSF and p54(nrb) as TopBP1-interacting proteins. J. Cell. Biochem. 2012, 113, 1744–1753. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, C.; Baker, D.K.; Pierce, A.J.; Pittman, D.L. The splicing-factor related protein SFPQ/PSF interacts with RAD51D and is necessary for homology-directed repair and sister chromatid cohesion. Nucleic Acids Res. 2011, 39, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Kuhne, W.W.; Kulharya, A.; Hudson, F.Z.; Ha, K.; Cao, Z.; Dynan, W.S. Involvement of p54(nrb), a PSF partner protein, in DNA double-strand break repair and radioresistance. Nucleic Acids Res. 2009, 37, 6746–6753. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.B.; Attig, J.; Bellora, N.; Konig, J.; Hallegger, M.; Kayikci, M.; Eyras, E.; Ule, J.; Smith, C.W. Nuclear matrix protein Matrin3 regulates alternative splicing and forms overlapping regulatory networks with PTB. EMBO J. 2015, 34, 653–668. [Google Scholar] [CrossRef] [PubMed]
- Van Oordt, W.H.; Diaz-Meco, M.T.; Lozano, J.; Krainer, A.R.; Moscat, J.; Caceres, J.F. The MKK(3/6)-p38-signaling cascade alters the subcellular distribution of hnRNP A1 and modulates alternative splicing regulation. J. Cell Biol. 2000, 149, 307–316. [Google Scholar] [CrossRef]
- Guil, S.; Long, J.C.; Caceres, J.F. hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol. Cell. Biol. 2006, 26, 5744–5758. [Google Scholar] [CrossRef] [PubMed]
- Llorian, M.; Beullens, M.; Lesage, B.; Nicolaescu, E.; Beke, L.; Landuyt, W.; Ortiz, J.M.; Bollen, M. Nucleocytoplasmic shuttling of the splicing factor SIPP1. J. Biol. Chem. 2005, 280, 38862–38869. [Google Scholar] [CrossRef] [PubMed]
- Busa, R.; Geremia, R.; Sette, C. Genotoxic stress causes the accumulation of the splicing regulator Sam68 in nuclear foci of transcriptionally active chromatin. Nucleic Acids Res. 2010, 38, 3005–3018. [Google Scholar] [CrossRef] [PubMed]
- Busa, R.; Paronetto, M.P.; Farini, D.; Pierantozzi, E.; Botti, F.; Angelini, D.F.; Attisani, F.; Vespasiani, G.; Sette, C. The RNA-binding protein Sam68 contributes to proliferation and survival of human prostate cancer cells. Oncogene 2007, 26, 4372–4382. [Google Scholar] [CrossRef] [PubMed]
- Shkreta, L.; Froehlich, U.; Paquet, E.R.; Toutant, J.; Elela, S.A.; Chabot, B. Anticancer drugs affect the alternative splicing of Bcl-x and other human apoptotic genes. Mol. Cancer Ther. 2008, 7, 1398–1409. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Achsel, T.; Massiello, A.; Chalfant, C.E.; Sette, C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J. Cell Biol. 2007, 176, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Cai, L.; Zhu, J.; Chen, M.; Chen, J.; Li, Z.H.; Liu, X.D.; Wang, S.G.; Bie, P.; Jiang, P.; et al. Fyn requires HnRNPA2B1 and Sam68 to synergistically regulate apoptosis in pancreatic cancer. Carcinogenesis 2011, 32, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Tissier, A.; Janel-Bintz, R.; Coulon, S.; Klaile, E.; Kannouche, P.; Fuchs, R.P.; Cordonnier, A.M. Crosstalk between replicative and translesional DNA polymerases: PDIP38 interacts directly with Poleta. DNA Repair 2010, 9, 922–928. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Zhang, S.; Mordue, D.; Wu, J.M.; Zhang, Z.; Darzynkiewicz, Z.; Lee, E.Y.; Lee, M.Y. PDIP38 is translocated to the spliceosomes/nuclear speckles in response to UV-induced DNA damage and is required for UV-induced alternative splicing of MDM2. Cell Cycle 2013, 12, 3184–3193. [Google Scholar] [CrossRef] [PubMed]
- Vivarelli, S.; Lenzken, S.C.; Ruepp, M.D.; Ranzini, F.; Maffioletti, A.; Alvarez, R.; Muhlemann, O.; Barabino, S.M. Paraquat modulates alternative pre-mRNA splicing by modifying the intracellular distribution of SRPK2. PLoS ONE 2013, 8, e61980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakashita, E.; Endo, H. SR and SR-related proteins redistribute to segregated fibrillar components of nucleoli in a response to DNA damage. Nucleus 2010, 1, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, J.M. Hu antigen R (HuR) functions as an alternative pre-mRNA splicing regulator of Fas apoptosis-promoting receptor on exon definition. J. Biol. Chem. 2008, 283, 19077–19084. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.H.; Abdelmohsen, K.; Gorospe, M. Regulation of HuR by DNA damage response kinases. J. Nucleic Acids 2010. [Google Scholar] [CrossRef] [PubMed]
- Masuda, K.; Abdelmohsen, K.; Kim, M.M.; Srikantan, S.; Lee, E.K.; Tominaga, K.; Selimyan, R.; Martindale, J.L.; Yang, X.; Lehrmann, E.; et al. Global dissociation of HuR-mRNA complexes promotes cell survival after ionizing radiation. EMBO J. 2011, 30, 1040–1053. [Google Scholar] [CrossRef] [PubMed]
- Mazan-Mamczarz, K.; Hagner, P.R.; Zhang, Y.; Dai, B.; Lehrmann, E.; Becker, K.G.; Keene, J.D.; Gorospe, M.; Liu, Z.; Gartenhaus, R.B. ATM regulates a DNA damage response posttranscriptional RNA operon in lymphocytes. Blood 2011, 117, 2441–2450. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, M.; Sanchez, G.; de Cian, M.C.; Barbier, J.; Dardenne, E.; Gratadou, L.; Dujardin, G.; le Jossic-Corcos, C.; Corcos, L.; Auboeuf, D. Cotranscriptional exon skipping in the genotoxic stress response. Nat. Struct. Mol. Biol. 2010, 17, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Vichi, P.; Coin, F.; Renaud, J.P.; Vermeulen, W.; Hoeijmakers, J.H.; Moras, D.; Egly, J.M. Cisplatin- and UV-damaged DNA lure the basal transcription factor TFIID/TBP. EMBO J. 1997, 16, 7444–7456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rockx, D.A.; Mason, R.; van Hoffen, A.; Barton, M.C.; Citterio, E.; Bregman, D.B.; van Zeeland, A.A.; Vrieling, H.; Mullenders, L.H. UV-induced inhibition of transcription involves repression of transcription initiation and phosphorylation of RNA polymerase II. Proc. Natl. Acad. Sci. USA 2000, 97, 10503–10508. [Google Scholar] [CrossRef] [PubMed]
- Rieger, K.E.; Chu, G. Portrait of transcriptional responses to ultraviolet and ionizing radiation in human cells. Nucleic Acids Res. 2004, 32, 4786–4803. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Cannell, I.G.; Morandell, S.; Yaffe, M.B. Is post-transcriptional stabilization, splicing and translation of selective mRNAs a key to the DNA damage response? Cell Cycle 2011, 10, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Long, J.C.; Caceres, J.F. The SR protein family of splicing factors: Master regulators of gene expression. Biochem. J. 2009, 417, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Perry, W.L., 3rd; Shepard, R.L.; Sampath, J.; Yaden, B.; Chin, W.W.; Iversen, P.W.; Jin, S.; Lesoon, A.; O’Brien, K.A.; Peek, V.L.; et al. Human splicing factor SPF45 (RBM17) confers broad multidrug resistance to anticancer drugs when overexpressed—A phenotype partially reversed by selective estrogen receptor modulators. Cancer Res. 2005, 65, 6593–6600. [Google Scholar] [PubMed]
- Haley, B.; Paunesku, T.; Protic, M.; Woloschak, G.E. Response of heterogeneous ribonuclear proteins (hnRNP) to ionising radiation and their involvement in DNA damage repair. Int. J. Radiat. Biol. 2009, 85, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Yan, C.; Gan, T.; Chen, Z.; Lu, X.; Duerksen-Hughes, P.J.; Zhu, X.; Yang, J. Nuclear proteome analysis of cisplatin-treated HeLa cells. Mutat. Res. 2010, 691, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Decorsiere, A.; Cayrel, A.; Vagner, S.; Millevoi, S. Essential role for the interaction between hnRNP H/F and a G quadruplex in maintaining p53 pre-mRNA 3'-end processing and function during DNA damage. Genes Dev. 2011, 25, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.T.; Shin, W.K.; Caudill, M.A.; Stover, P.J. A UV-responsive internal ribosome entry site enhances serine hydroxymethyltransferase 1 expression for DNA damage repair. J. Biol. Chem. 2009, 284, 31097–31108. [Google Scholar] [CrossRef] [PubMed]
- Decker, E.D.; Zhang, Y.; Cocklin, R.R.; Witzmann, F.A.; Wang, M. Proteomic analysis of differential protein expression induced by ultraviolet light radiation in HeLa cells. Proteomics 2003, 3, 2019–2027. [Google Scholar] [CrossRef] [PubMed]
- Filippov, V.; Filippova, M.; Duerksen-Hughes, P.J. The early response to DNA damage can lead to activation of alternative splicing activity resulting in CD44 splice pattern changes. Cancer Res. 2007, 67, 7621–7630. [Google Scholar] [CrossRef] [PubMed]
- Comiskey, D.F., Jr.; Jacob, A.G.; Singh, R.K.; Tapia-Santos, A.S.; Chandler, D.S. Splicing factor SRSF1 negatively regulates alternative splicing of MDM2 under damage. Nucleic Acids Res. 2015, 43, 4202–4218. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhang, Z.; Sinha, R.; Karni, R.; Krainer, A.R. SF2/ASF autoregulation involves multiple layers of post-transcriptional and translational control. Nat. Struct. Mol. Biol. 2010, 17, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Solier, S.; Barb, J.; Zeeberg, B.R.; Varma, S.; Ryan, M.C.; Kohn, K.W.; Weinstein, J.N.; Munson, P.J.; Pommier, Y. Genome-wide analysis of novel splice variants induced by topoisomerase I poisoning shows preferential occurrence in genes encoding splicing factors. Cancer Res. 2010, 70, 8055–8065. [Google Scholar] [CrossRef] [PubMed]
- Colla, S.; Ong, D.S.; Ogoti, Y.; Marchesini, M.; Mistry, N.A.; Clise-Dwyer, K.; Ang, S.A.; Storti, P.; Viale, A.; Giuliani, N.; et al. Telomere dysfunction drives aberrant hematopoietic differentiation and myelodysplastic syndrome. Cancer Cell. 2015, 27, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.Z.; Grate, L.; Donohue, J.P.; Preston, C.; Nobida, N.; O’Brien, G.; Shiue, L.; Clark, T.A.; Blume, J.E.; Ares, M., Jr. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev. 2007, 21, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Ip, J.Y.; Schmidt, D.; Pan, Q.; Ramani, A.K.; Fraser, A.G.; Odom, D.T.; Blencowe, B.J. Global impact of RNA polymerase II elongation inhibition on alternative splicing regulation. Genome Res. 2011, 21, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.I.; Gorski, J.J.; Barros, E.M.; Irwin, G.W.; Manti, L.; Powell, A.J.; Pellagatti, A.; Lukashchuk, N.; McCance, D.J.; McCluggage, W.G.; et al. Identification of a BRCA1-mRNA splicing complex required for efficient DNA repair and maintenance of genomic stability. Mol. Cell 2014, 54, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.I.; Harkin, D.P. BRCA1, a “complex” protein involved in the maintenance of genomic stability. FEBS J. 2015, 282, 630–646. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D.; Wang, Y.; Qin, J.; Elledge, S.J. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science 1999, 286, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Otomo, T.; Hishii, M.; Arai, H.; Sato, K.; Sasai, K. Microarray analysis of temporal gene responses to ionizing radiation in two glioblastoma cell lines: Up-regulation of DNA repair genes. J. Radiat. Res. 2004, 45, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, L.; Jones, K.A. SKIP counteracts p53-mediated apoptosis via selective regulation of p21Cip1 mRNA splicing. Genes Dev. 2011, 25, 701–716. [Google Scholar] [CrossRef] [PubMed]
- Merz, C.; Urlaub, H.; Will, C.L.; Luhrmann, R. Protein composition of human mRNPs spliced in vitro and differential requirements for mRNP protein recruitment. RNA 2007, 13, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Fu, X.D. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 2013, 122, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Zhou, Y.; Pandit, S.; Huang, J.; Li, H.; Lin, C.Y.; Xiao, R.; Burge, C.B.; Fu, X.D. SR proteins collaborate with 7SK and promoter-associated nascent RNA to release paused polymerase. Cell 2013, 153, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, B.; Blanchette, M.; Monette, A.; Mouland, A.J.; Wellinger, R.J.; Chabot, B. A Function for the hnRNP A1/A2 Proteins in Transcription Elongation. PLoS ONE 2015, 10, e0126654. [Google Scholar] [CrossRef] [PubMed]
- Hurov, K.E.; Cotta-Ramusino, C.; Elledge, S.J. A genetic screen identifies the Triple T complex required for DNA damage signaling and ATM and ATR stability. Genes Dev. 2010, 24, 1939–1950. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, G.; Bittencourt, D.; Laud, K.; Barbier, J.; Delattre, O.; Auboeuf, D.; Dutertre, M. Alteration of cyclin D1 transcript elongation by a mutated transcription factor up-regulates the oncogenic D1b splice isoform in cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 6004–6009. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Bernardis, I.; Volpe, E.; Bechara, E.; Sebestyen, E.; Eyras, E.; Valcarcel, J. Regulation of FAS exon definition and apoptosis by the Ewing sarcoma protein. Cell Rep. 2014, 7, 1211–1226. [Google Scholar] [CrossRef] [PubMed]
- Knoop, L.L.; Baker, S.J. The splicing factor U1C represses EWS/FLI-mediated transactivation. J. Biol. Chem. 2000, 275, 24865–24871. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Paley, A.J.; Childs, G. The transcriptional repressor ZFM1 interacts with and modulates the ability of EWS to activate transcription. J. Biol. Chem. 1998, 273, 18086–18091. [Google Scholar] [CrossRef] [PubMed]
- Chansky, H.A.; Hu, M.; Hickstein, D.D.; Yang, L. Oncogenic TLS/ERG and EWS/Fli-1 fusion proteins inhibit RNA splicing mediated by YB-1 protein. Cancer Res. 2001, 61, 3586–3590. [Google Scholar] [PubMed]
- Sordet, O.; Larochelle, S.; Nicolas, E.; Stevens, E.V.; Zhang, C.; Shokat, K.M.; Fisher, R.P.; Pommier, Y. Hyperphosphorylation of RNA polymerase II in response to topoisomerase I cleavage complexes and its association with transcription- and BRCA1-dependent degradation of topoisomerase I. J. Mol. Biol. 2008, 381, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Mao, G.; Tong, D.; Huang, J.; Gu, L.; Yang, W.; Li, G.M. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell 2013, 153, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Pfister, S.X.; Ahrabi, S.; Zalmas, L.P.; Sarkar, S.; Aymard, F.; Bachrati, C.Z.; Helleday, T.; Legube, G.; La Thangue, N.B.; Porter, A.C.; et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014, 7, 2006–2018. [Google Scholar] [CrossRef] [PubMed]
- Dhami, P.; Saffrey, P.; Bruce, A.W.; Dillon, S.C.; Chiang, K.; Bonhoure, N.; Koch, C.M.; Bye, J.; James, K.; Foad, N.S.; et al. Complex exon-intron marking by histone modifications is not determined solely by nucleosome distribution. PLoS ONE 2010, 5, e12339. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Enroth, S.; Rada-Iglesias, A.; Wadelius, C.; Komorowski, J. Nucleosomes are well positioned in exons and carry characteristic histone modifications. Genome Res. 2009, 19, 1732–1741. [Google Scholar] [CrossRef]
- Spies, N.; Nielsen, C.B.; Padgett, R.A.; Burge, C.B. Biased chromatin signatures around polyadenylation sites and exons. Mol. Cell 2009, 36, 245–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolasinska-Zwierz, P.; Down, T.; Latorre, I.; Liu, T.; Liu, X.S.; Ahringer, J. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat. Genet. 2009, 41, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, H.; Fong, N.; Erickson, B.; Bentley, D.L. Pre-mRNA splicing is a determinant of histone H3K36 methylation. Proc. Natl. Acad. Sci. USA 2011, 108, 13564–13569. [Google Scholar] [CrossRef] [PubMed]
- Pradeepa, M.M.; Sutherland, H.G.; Ule, J.; Grimes, G.R.; Bickmore, W.A. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012, 8, e1002717. [Google Scholar] [CrossRef] [PubMed]
- Kfir, N.; Lev-Maor, G.; Glaich, O.; Alajem, A.; Datta, A.; Sze, S.K.; Meshorer, E.; Ast, G. SF3B1 association with chromatin determines splicing outcomes. Cell Rep. 2015, 11, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Zhang, Q.C.; da Rocha, S.T.; Flynn, R.A.; Bharadwaj, M.; Calabrese, J.M.; Magnuson, T.; Heard, E.; Chang, H.Y. Systematic discovery of Xist RNA binding proteins. Cell 2015, 161, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, I.; Munita, R.; Agirre, E.; Dittmer, T.A.; Gysling, K.; Misteli, T.; Luco, R.F. A lncRNA regulates alternative splicing via establishment of a splicing-specific chromatin signature. Nat. Struct. Mol. Biol. 2015, 22, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Peng, G. Non-coding RNAs: An emerging player in DNA damage response. Mutat. Res. Rev. Mutat. Res. 2015, 763, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, Z.; Zhao, Q.; Zhu, Y.; Zhao, C.; Li, X.; Ma, Z.; Li, X.; Zhang, Y. LincRNA-p21 enhances the sensitivity of radiotherapy for human colorectal cancer by targeting the Wnt/beta-catenin signaling pathway. Oncol. Rep. 2014, 31, 1839–1845. [Google Scholar] [PubMed]
- Hung, T.; Wang, Y.; Lin, M.F.; Koegel, A.K.; Kotake, Y.; Grant, G.D.; Horlings, H.M.; Shah, N.; Umbricht, C.; Wang, P.; et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat. Genet. 2011, 43, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, N.; Zamudio, J.R.; Jong, R.M.; Soukup, D.; Resnick, R.; Sarma, K.; Ward, A.J.; Raj, A.; Lee, J.T.; Sharp, P.A.; et al. LincRNA-p21 activates p21 in cis to promote Polycomb target gene expression and to enforce the G1/S checkpoint. Mol. Cell 2014, 54, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Sevcik, J.; Falk, M.; Macurek, L.; Kleiblova, P.; Lhota, F.; Hojny, J.; Stefancikova, L.; Janatova, M.; Bartek, J.; Stribrna, J.; et al. Expression of human BRCA1Delta17–19 alternative splicing variant with a truncated BRCT domain in MCF-7 cells results in impaired assembly of DNA repair complexes and aberrant DNA damage response. Cell. Signal. 2013, 25, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, T.; Ma, K.; Tian, Z.; Zhu, Y.; Chen, F.; Hu, G. The impacts of ERCC1 gene exon VIII alternative splicing on cisplatin-resistance in ovarian cancer cells. Cancer Investig. 2009, 27, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Berge, E.O.; Staalesen, V.; Straume, A.H.; Lillehaug, J.R.; Lonning, P.E. Chk2 splice variants express a dominant-negative effect on the wild-type Chk2 kinase activity. Biochim. Biophys. Acta 2010, 1803, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Baldin, V.; Cans, C.; Knibiehler, M.; Ducommun, B. Phosphorylation of human CDC25B phosphatase by CDK1-cyclin A triggers its proteasome-dependent degradation. J. Biol. Chem. 1997, 272, 32731–32734. [Google Scholar] [CrossRef] [PubMed]
- Schwerk, C.; Schulze-Osthoff, K. Regulation of apoptosis by alternative pre-mRNA splicing. Mol. Cell 2005, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sigalas, I.; Calvert, A.H.; Anderson, J.J.; Neal, D.E.; Lunec, J. Alternatively spliced mdm2 transcripts with loss of p53 binding domain sequences: Transforming ability and frequent detection in human cancer. Nat. Med. 1996, 2, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Eijkelenboom, A.; Burgering, B.M. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012, 28, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Wang, Q.; Kennedy, C.J.; Silver, P.A. An alternative splicing network links cell-cycle control to apoptosis. Cell 2010, 142, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Anczukow, O.; Rosenberg, A.Z.; Akerman, M.; Das, S.; Zhan, L.; Karni, R.; Muthuswamy, S.K.; Krainer, A.R. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol. 2012, 19, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Olshavsky, N.A.; Comstock, C.E.; Schiewer, M.J.; Augello, M.A.; Hyslop, T.; Sette, C.; Zhang, J.; Parysek, L.M.; Knudsen, K.E. Identification of ASF/SF2 as a critical, allele-specific effector of the cyclin D1b oncogene. Cancer Res. 2010, 70, 3975–3984. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Cappellari, M.; Busa, R.; Pedrotti, S.; Vitali, R.; Comstock, C.; Hyslop, T.; Knudsen, K.E.; Sette, C. Alternative splicing of the cyclin D1 proto-oncogene is regulated by the RNA-binding protein Sam68. Cancer Res. 2010, 70, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Kim, J.H.; Back, S.H.; Jang, S.K. Polypyrimidine tract-binding protein enhances the internal ribosomal entry site-dependent translation of p27Kip1 mRNA and modulates transition from G1 to S phase. Mol. Cell. Biol. 2005, 25, 1283–1297. [Google Scholar] [CrossRef] [PubMed]
- Takai, K.; Sakamoto, S.; Sakai, T.; Yasunaga, J.; Komatsu, K.; Matsuoka, M. A potential link between alternative splicing of the NBS1 gene and DNA damage/environmental stress. Radiat. Res. 2008, 170, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Merdzhanova, G.; Edmond, V.; de Seranno, S.; van den Broeck, A.; Corcos, L.; Brambilla, C.; Brambilla, E.; Gazzeri, S.; Eymin, B. E2F1 controls alternative splicing pattern of genes involved in apoptosis through upregulation of the splicing factor SC35. Cell. Death Differ. 2008, 15, 1815–1823. [Google Scholar] [CrossRef] [PubMed]
- Chandler, D.S.; Singh, R.K.; Caldwell, L.C.; Bitler, J.L.; Lozano, G. Genotoxic stress induces coordinately regulated alternative splicing of the p53 modulators MDM2 and MDM4. Cancer Res. 2006, 66, 9502–9508. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, M.; Delforge, Y.; Deward, A.; Habraken, Y.; Hennuy, B.; Piette, J.; Klinck, R.; Chabot, B.; Colige, A.; Lambert, C. Role of the splicing factor SRSF4 in cisplatin-induced modifications of pre-mRNA splicing and apoptosis. BMC Cancer 2015. [Google Scholar] [CrossRef] [PubMed]
- Index of /annotate/luli. Available online: http://lgfus.ca/annotate/luli/ (accessed on 23 October 2015).
- Genome-wide analysis of novel splice variants induced by topoisomerase I poisoning shows preferential occurrence in genes encoding splicing factors. Supplemental Table 3. Supplementary Material. Available online: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2992871/#SD1 (accessed on 23 October 2015).
- Global impact of RNA polymerase II elongation inhibition on alternative splicing regulation. Supplemental Material. Supp. Table S3.xls. Available online: http://genome.cshlp.org/content/21/3/390/suppl/DC1 (accessed on 23 October 2015).
- Cotranscriptional exon skipping in the genotoxic stress response. Supplementary Table 1. Available online: http://www.nature.com/nsmb/journal/v17/n11/extref/nsmb.1912-S1.pdf (accessed on 23 October 2015).
- Additional file 4. Post-transcriptional events, from RNA-Seq in control and cisplatin-treated MCF7 cells. Available online: http://www.biomedcentral.com/1471-2407/15/227/suppl/S4 (accessed on 23 October 2015).
- PLOS ONE: Alternative Transcript Initiation and Splicing as a Response to DNA Damage. Supporting Information. Table S8. Genes predicted to be alternatively spliced in LCLs at 4 hours post 10 Gy IR based on SI. Table S11. Genes predicted to be alternatively spliced in fibroblasts at 4 hours post 10 Gy IR based on SI. Available online: http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0025758#pone.0025758.s010 (accessed on 23 October 2015).
- Additional file 12: Table S6.Genes with iAs-mediated alternatively spliced events. Available online: http://www.biomedcentral.com/1471-2164/16/212/suppl/S12 (accessed on 23 October 2015).
- DNA Damage Regulates Alternative Splicing through Inhibition of RNA Polymerase II Elongation: Supplemental Data. Document S2. Supplemental Spreadsheet. Available online: http://www.sciencedirect.com/science/article/pii/S0092867409002700 (accessed on 23 October 2015).
- Nature14512-s1.pdf. Supplementary Information. SI Table 2: UV-triggered and ATM-dependent alternative splicing events. Available online: http://www.nature.com/nature/journal/v523/n7558/extref/nature14512-s1.pdf (accessed on 23 October 2015).
- Newly identified biologically active and proteolysis-resistant VEGF-A isoform VEGF111 is induced by genotoxic agents. Table II. Induction of VEGF111 and Bcl-Xs/Bcl-Xl ratio by UV-B. Available online: http://jcb.rupress.org/content/179/6/1261.full (accessed on 23 October 2015).
- Rahmutulla, B.; Matsushita, K.; Satoh, M.; Seimiya, M.; Tsuchida, S.; Kubo, S.; Shimada, H.; Ohtsuka, M.; Miyazaki, M.; Nomura, F. Alternative splicing of FBP-interacting repressor coordinates c-Myc, P27Kip1/cyclinE and Ku86/XRCC5 expression as a molecular sensor for bleomycin-induced DNA damage pathway. Oncotarget 2014, 5, 2404–2417. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Kajiwara, T.; Tamura, M.; Satoh, M.; Tanaka, N.; Tomonaga, T.; Matsubara, H.; Shimada, H.; Yoshimoto, R.; Ito, A.; et al. SAP155-mediated splicing of FUSE-binding protein-interacting repressor serves as a molecular switch for c-myc gene expression. Mol. Cancer Res. 2012, 10, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Massiello, A.; Roesser, J.R.; Chalfant, C.E. SAP155 Binds to ceramide-responsive RNA cis-element 1 and regulates the alternative 5' splice site selection of Bcl-x pre-mRNA. J. Fed. Am. Soc. Exp. Biol. 2006, 20, 1680–1682. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protocols 2013, 8, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nature Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.E.; Kennedy, R.D.; Mullan, P.B.; Gilmore, P.M.; Carty, M.; Johnston, P.G.; Harkin, D.P. BRCA1 functions as a differential modulator of chemotherapy-induced apoptosis. Cancer Res. 2003, 63, 6221–6228. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Damm, F.; Kosmider, O.; Gelsi-Boyer, V.; Renneville, A.; Carbuccia, N.; Hidalgo-Curtis, C.; Valle, V.D.; Couronne, L.; Scourzic, L.; Chesnais, V.; et al. Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood 2012, 119, 3211–3218. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H.; Visconte, V.; Sakaguchi, H.; Jankowska, A.M.; Abu Kar, S.; Jerez, A.; Przychodzen, B.; Bupathi, M.; Guinta, K.; Afable, M.G.; et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 2012, 119, 3203–3210. [Google Scholar] [CrossRef] [PubMed]
- Karni, R.; de Stanchina, E.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 2007, 14, 185–193. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2009, 463, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Calabretta, S.; Bielli, P.; Passacantilli, I.; Pilozzi, E.; Fendrich, V.; Capurso, G.; Fave, G.D.; Sette, C. Modulation of PKM alternative splicing by PTBP1 promotes gemcitabine resistance in pancreatic cancer cells. Oncogene 2015. [Google Scholar] [CrossRef] [PubMed]
- Bonnal, S.; Vigevani, L.; Valcarcel, J. The spliceosome as a target of novel antitumour drugs. Nat. Rev. Drug Discov. 2012, 11, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Bakkour, N.; Lin, Y.L.; Maire, S.; Ayadi, L.; Mahuteau-Betzer, F.; Nguyen, C.H.; Mettling, C.; Portales, P.; Grierson, D.; Chabot, B.; et al. Small-molecule inhibition of HIV pre-mRNA splicing as a novel antiretroviral therapy to overcome drug resistance. PLoS Pathog. 2007, 3, 1530–1539. [Google Scholar] [CrossRef] [PubMed]
- Sperka, T.; Wang, J.; Rudolph, K.L. DNA damage checkpoints in stem cells, ageing and cancer. Nat. Rev. Mol. Cell Biol. 2012, 13, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Yankner, B.A. The aging stress response. Mol. Cell 2010, 40, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Sulli, G.; di Micco, R.; d’Adda di Fagagna, F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat. Rev. Cancer 2012, 12, 709–720. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Karlseder, J. The great unravelling: Chromatin as a modulator of the aging process. Trends Biochem. Sci. 2012, 37, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Holly, A.C.; Melzer, D.; Pilling, L.C.; Fellows, A.C.; Tanaka, T.; Ferrucci, L.; Harries, L.W. Changes in splicing factor expression are associated with advancing age in man. Mech. Ageing Dev. 2013, 134, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Blair, C.D.; Faddah, D.A.; Kieckhaefer, J.E.; Olive, M.; Erdos, M.R.; Nabel, E.G.; Collins, F.S. Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. J. Clin. Investig. 2011, 121, 2833–2844. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shkreta, L.; Chabot, B. The RNA Splicing Response to DNA Damage. Biomolecules 2015, 5, 2935-2977. https://doi.org/10.3390/biom5042935
AMA Style
Shkreta L, Chabot B. The RNA Splicing Response to DNA Damage. Biomolecules. 2015; 5(4):2935-2977. https://doi.org/10.3390/biom5042935
Chicago/Turabian StyleShkreta, Lulzim, and Benoit Chabot. 2015. "The RNA Splicing Response to DNA Damage" Biomolecules 5, no. 4: 2935-2977. https://doi.org/10.3390/biom5042935