
Glutaminase Increases in Rat Dorsal Root Ganglion Neurons after Unilateral Adjuvant-Induced Hind Paw Inflammation


Abstract
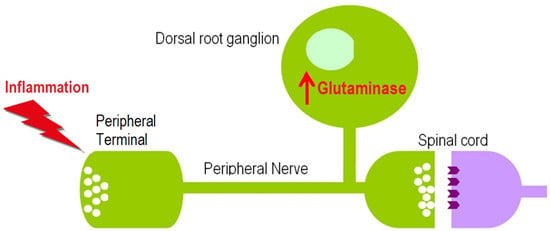
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Evoked Pain Behavioral Responses
2.3. Hind Paw Edema
2.4. Immunohistochemistry (IHC)
2.5. Quantitative Image Analysis
2.6. Western blot
2.7. Statistical Analysis
3. Results
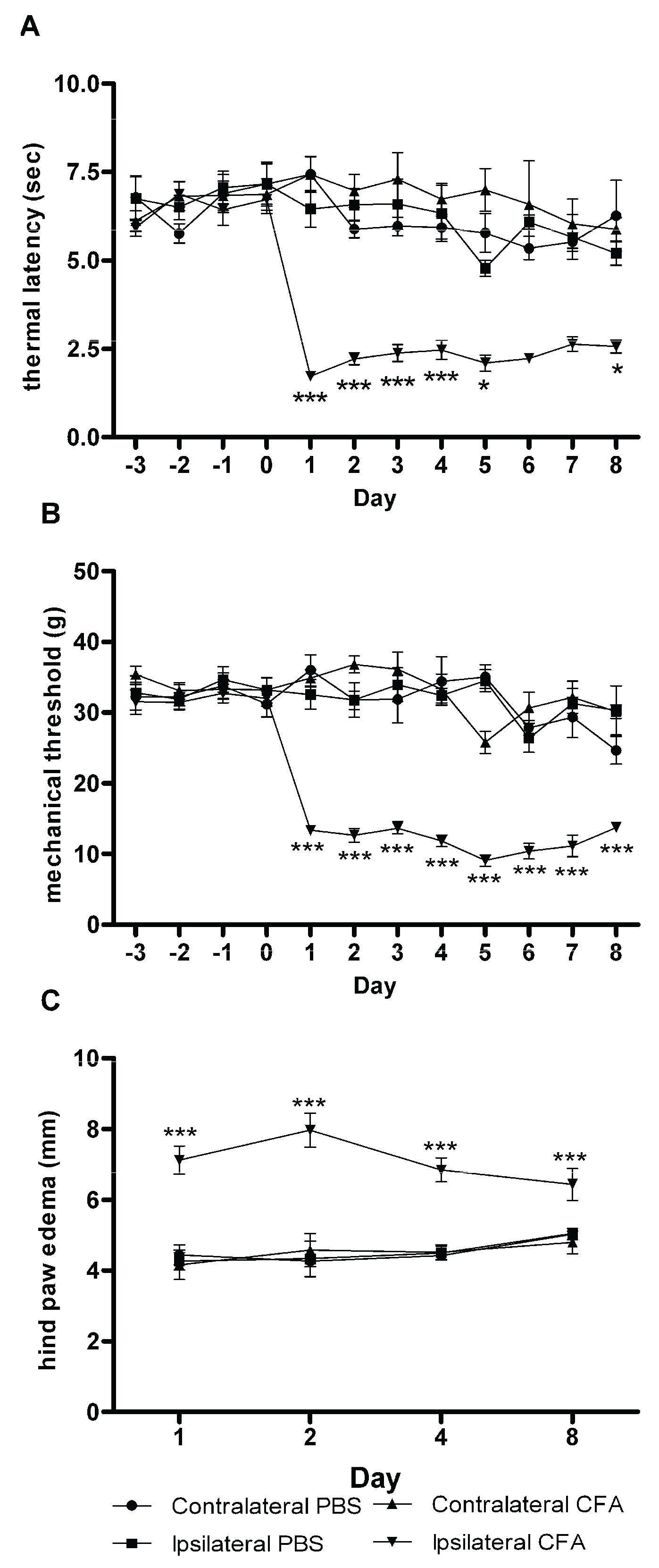
3.1. Evoked Pain Behavioral Responses

3.2. Hind Paw Edema
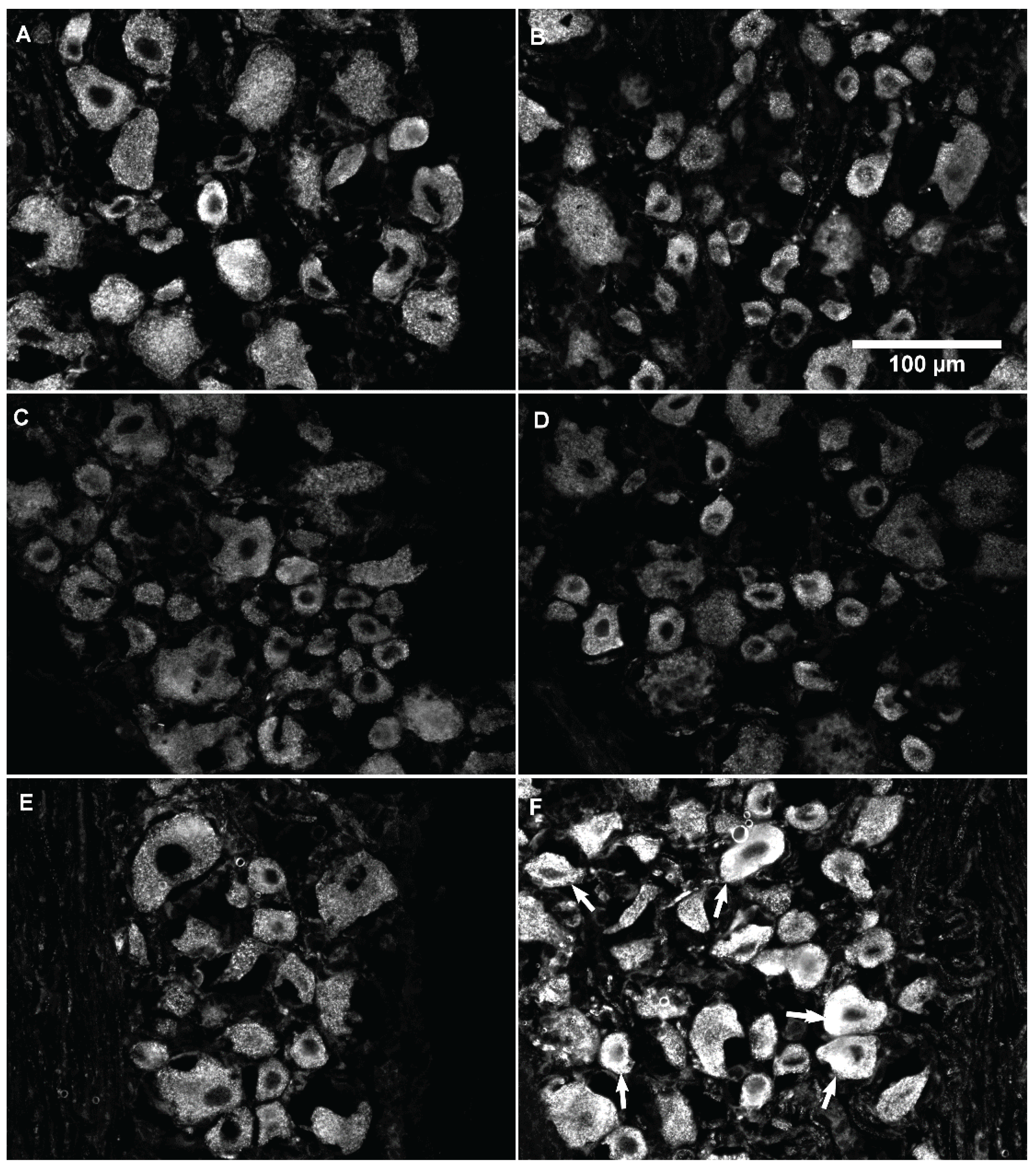
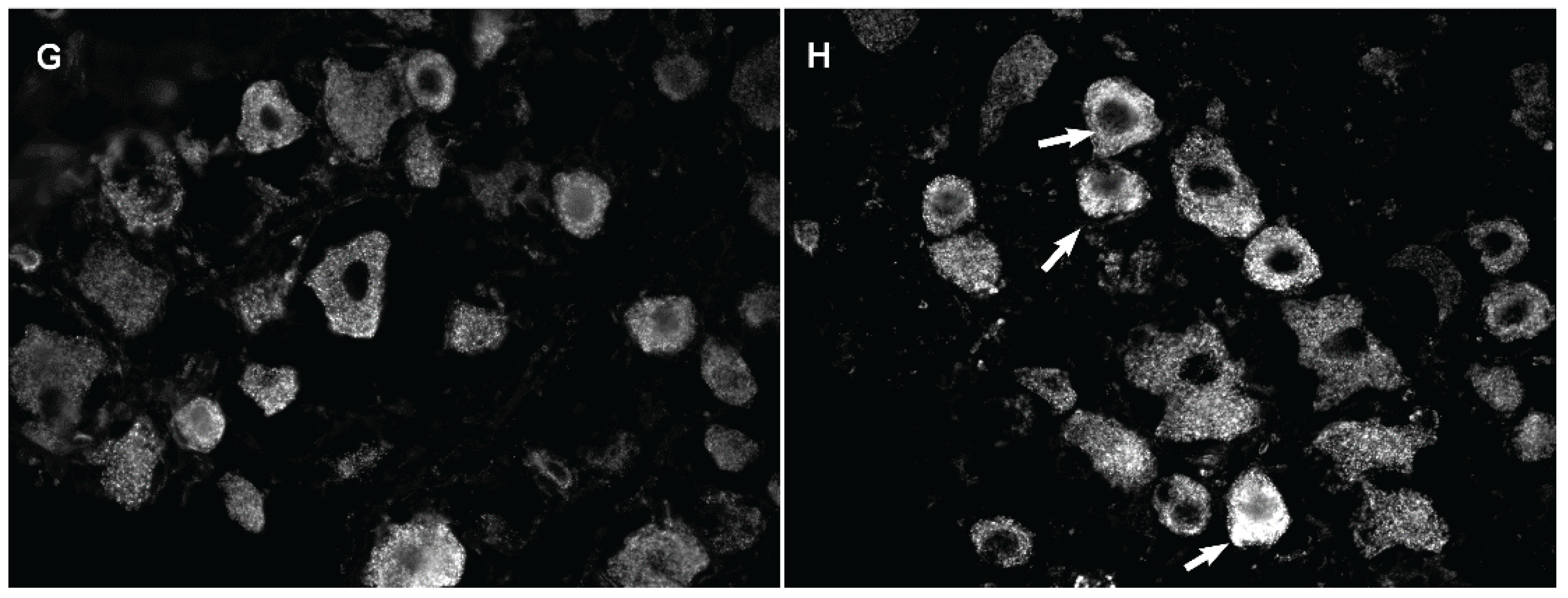
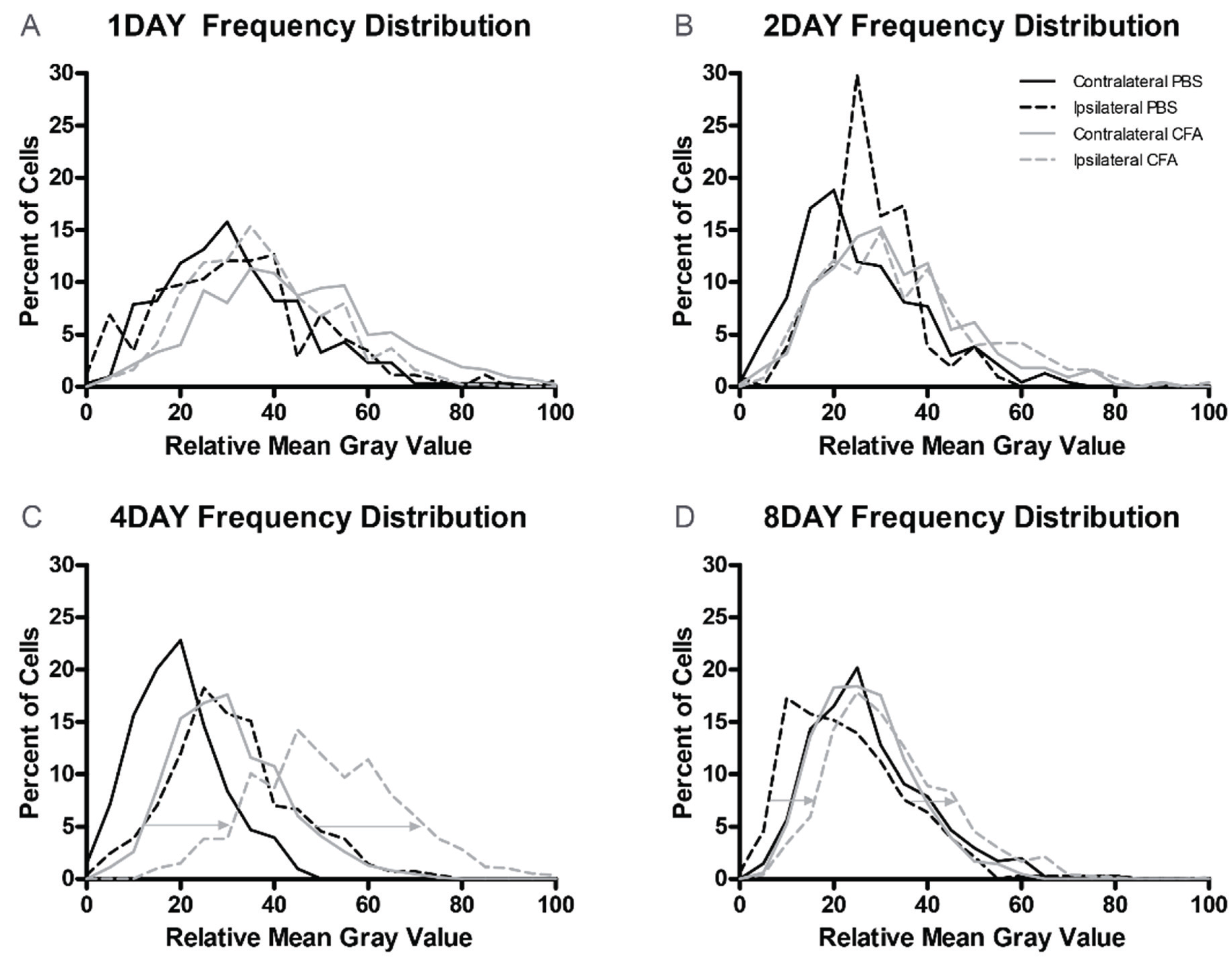
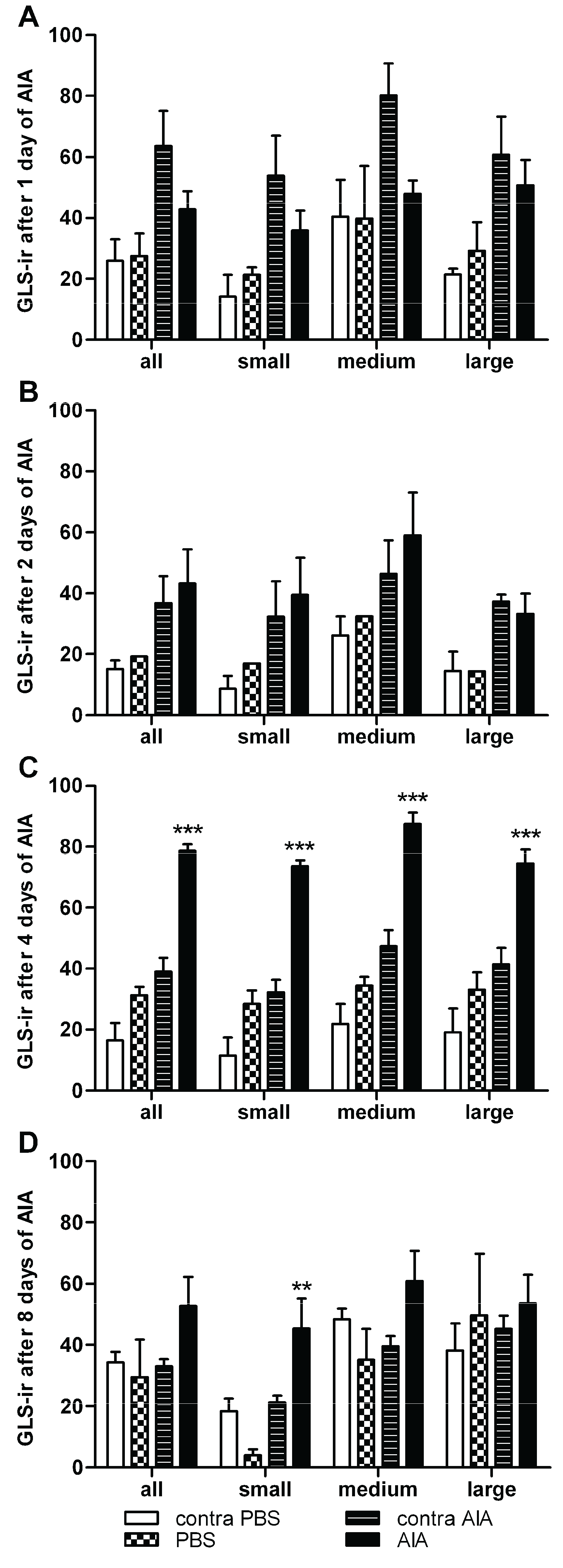
3.3. Changes in GLS-ir During Inflammation




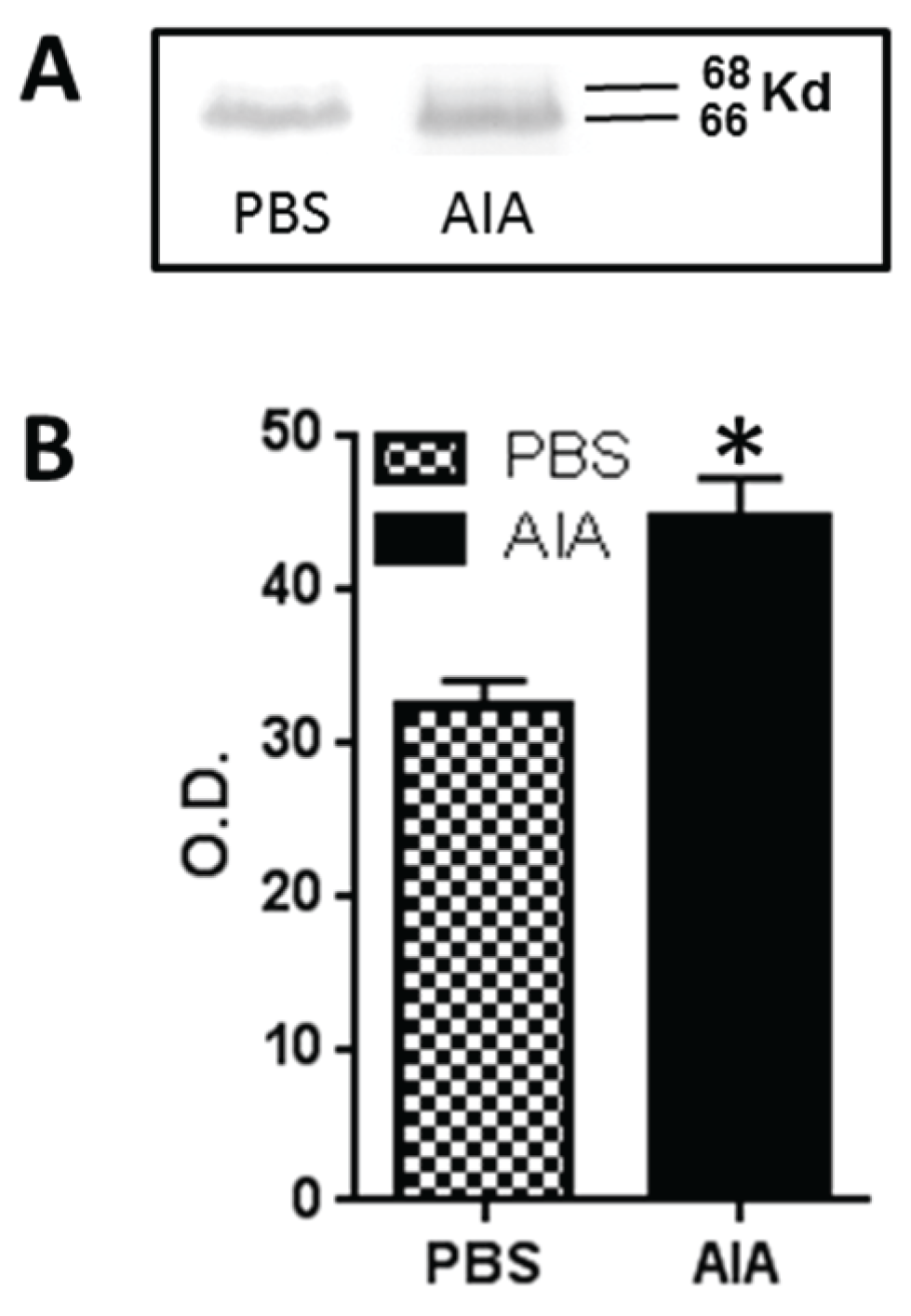
3.4. GLS Western Blots

4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Martin, J.H. Spinal Somatic Sensory Systems, Neuroanatomy: Text and Atlas; McGraw Hill: New York, NY, USA, 2003; pp. 107–133. [Google Scholar]
- De Biasi, S.; Rustioni, A. Ultrastructural immunocytochemical localization of excitatory amino acids in the somatosensory system. J. Histochem. Cytochem. 1990, 38, 1745–1754. [Google Scholar] [CrossRef] [PubMed]
- Dmitrieva, N.; Rodriguez-Malaver, A.J.; Perez, J.; Hernandez, L. Differential release of neurotransmitters from superficial and deep layers of the dorsal horn in response to acute noxious stimulation and inflammation of the rat paw. Eur. J. Pain 2004, 8, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Skilling, S.R.; Smullin, D.H.; Beitz, A.J.; Larson, A.A. Extracellular amino acid concentrations in the dorsal spinal cord of freely moving rats following veratridine and nociceptive stimulation. J. Neurochem. 1988, 51, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Zahn, P.K.; Sluka, K.A.; Brennan, T.J. Excitatory amino acid release in the spinal cord caused by plantar incision in the rat. Pain 2002, 100, 65–76. [Google Scholar] [CrossRef]
- Sann, H.; Pierau, F.K. Efferent functions of C-fiber nociceptors. Z. Rheumatol. 1998, 57, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.E.; Hoffman, E.M.; Sutharshan, M.; Schechter, R. Glutamate pharmacology and metabolism in peripheral primary afferents: Physiological and pathophysiological mechanisms. Pharmacol. Ther. 2011, 130, 283–309. [Google Scholar] [CrossRef] [PubMed]
- DeGroot, J.; Zhou, S.; Carlton, S.M. Peripheral glutamate release in the hindpaw following low and high intensity sciatic stimulation. Neuroreport 2000, 11, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.H.; Nishioka, H.; Wakabayashi, K.; Fujita, T.; Yonehara, N. Effect of morphine on the release of excitatory amino acids in the rat hind instep: Pain is modulated by the interaction between the peripheral opioid and glutamate systems. Neuroscience 2006, 138, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Omote, K.; Kawamata, T.; Kawamata, M.; Namiki, A. Formalin-induced release of excitatory amino acids in the skin of the rat hindpaw. Brain Res. 1998, 787, 161–164. [Google Scholar] [CrossRef]
- Woolf, C.J.; Ma, Q. Nociceptors—Noxious stimulus detectors. Neuron 2007, 55, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Woolf, C.J.; Salter, M.W. Neuronal plasticity: Increasing the gain in pain. Science 2000, 288, 1765–1769. [Google Scholar] [CrossRef] [PubMed]
- Merighi, A.; Polak, J.M.; Theodosis, D.T. Ultrastructural visualization of glutamate and aspartate immunoreactivities in the rat dorsal horn, with special reference to the co-localization of glutamate, substance P and calcitonin-gene related peptide. Neuroscience 1991, 40, 67–80. [Google Scholar] [CrossRef]
- Miller, K.E.; Douglas, V.D.; Kaneko, T. Glutaminase immunoreactive neurons in the rat dorsal root ganglion contain calcitonin gene-related peptide (CGRP). Neurosci. Lett. 1993, 160, 113–116. [Google Scholar] [CrossRef]
- Miller, K.E.; Balbás, J.C.; Benton, R.L.; Lam, T.S.; Edwards, K.M.; Kriebel, R.M.; Schechter, R. Glutaminase immunoreactivity and enzyme activity is increased in the rat dorsal root ganglion following peripheral inflammation. Pain Res. Treat. 2012. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.C. The glutamate-glutamine cycle is not stoichiometric: Fates of glutamate in brain. J. Neurosci. Res. 2007, 85, 3347–3358. [Google Scholar] [CrossRef] [PubMed]
- Berger, U.V.; Hediger, M.A. Distribution of the glutamate transporters GLAST and GLT-1 in rat circumventricular organs, meninges, and dorsal root ganglia. J. Comp. Neurol. 2000, 421, 385–399. [Google Scholar] [CrossRef]
- Hoffman, E.M.; Schechter, R.; Miller, K.E. Fixative composition alters distributions of immunoreactivity for glutaminase and two markers of nociceptive neurons, Nav1.8 and TRPV1, in the rat dorsal root ganglion. J. Histochem. Cytochem. 2010, 58, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Rajasekhar, P.; Poole, D.P.; Liedtke, W.; Bunnett, N.W.; Veldhuis, N.A. P2Y1 Receptor activation of the TRPV4 ion channel enhances purinergic signaling in satellite glial cells. J. Biol. Chem. 2015, 290, 29051–29062. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.E.; Richards, B.A.; Kriebel, R.M. Glutamine-, glutamine synthetase-, glutamate dehydrogenase- and pyruvate carboxylase-immunoreactivities in the rat dorsal root ganglion and peripheral nerve. Brain Res. 2002, 945, 202–211. [Google Scholar] [CrossRef]
- Tao, F.; Liaw, W.J.; Zhang, B.; Yaster, M.; Rothstein, J.D.; Johns, R.A.; Tao, Y.X. Evidence of neuronal excitatory amino acid carrier 1 expression in rat dorsal root ganglion neurons and their central terminals. Neuroscience 2004, 123, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain 1983, 16, 109–110. [Google Scholar] [CrossRef]
- National Institutes of Health, N.R.C. Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research; The National Academic Press: Washington, DC, USA, 2003; p. 223. [Google Scholar]
- Kaneko, T.; Itoh, K.; Shigemoto, R.; Mizuno, N. Glutaminase-like immunoreactivity in the lower brainstem and cerebellum of the adult rat. Neuroscience 1989, 32, 79–98. [Google Scholar] [CrossRef]
- Hoffman, E.M.; Zhang, Z.; Anderson, M.B.; Edwards, K.M.; Schechter, R.; Miller, K.E. Potential mechanisms for hypoalgesia induced by anti-nerve growth factor immunoglobulin are identified using autoimmune nerve growth factor deprivation. Neuroscience 2011, 193, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Miller, K.E. Characterization of glutamatergic neurons in the rat atrial intrinsic cardiac ganglia that project to the cardiac ventricular wall. Neuroscience 2016. in revision. [Google Scholar]
- Fang, X.; Djouhri, L.; McMullan, S.; Berry, C.; Waxman, S.G.; Okuse, K.; Lawson, S.N. Intense isolectin-B4 binding in rat dorsal root ganglion neurons distinguishes C-fiber nociceptors with broad action potentials and high Nav1.9 expression. J. Neurosci. 2006, 26, 7281–7292. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; McMullan, S.; Lawson, S.N.; Djouhri, L. Electrophysiological differences between nociceptive and non-nociceptive dorsal root ganglion neurones in the rat in vivo. J. Physiol. 2005, 565, 927–943. [Google Scholar] [CrossRef] [PubMed]
- Schechter, R.; Beju, D.; Miller, K.E. The effect of insulin deficiency on tau and neurofilament in the insulin knockout mouse. Biochem. Biophys. Res. Commun. 2005, 334, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, T.; Taylor, L.; Trohkimoinen, J.; Curthoys, N.P. Isolation, characterization and expression of a human brain mitochondrial glutaminase cDNA. Brain Res. Mol. Brain Res. 2000, 76, 56–63. [Google Scholar] [CrossRef]
- Schechter, R.; Yanovitch, T.; Abboud, M.; Johnson, G., 3rd; Gaskins, J. Effects of brain endogenous insulin on neurofilament and MAPK in fetal rat neuron cell cultures. Brain Res. 1998, 808, 270–278. [Google Scholar] [CrossRef]
- Kvamme, E.; Torgner, I.A.; Roberg, B. Kinetics and localization of brain phosphate activated glutaminase. J. Neurosci. Res. 2001, 66, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Altier, C.; Zamponi, G.W. Targeting Ca2+ channels to treat pain: T-type versus N-type. Trends Pharmacol. Sci. 2004, 25, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Bautista, D.M.; Jordt, S.E.; Nikai, T.; Tsuruda, P.R.; Read, A.J.; Poblete, J.; Yamoah, E.N.; Basbaum, A.I.; Julius, D. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 2006, 124, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Westlund, K.N.; Sun, Y.C.; Sluka, K.A.; Dougherty, P.M.; Sorkin, L.S.; Willis, W.D. Neural changes in acute arthritis in monkeys. II. Increased glutamate immunoreactivity in the medial articular nerve. Brain Res. Brain Res. Rev. 1992, 17, 15–27. [Google Scholar] [CrossRef]
- Messersmith, W.; Oppenheimer, D.; Peralba, J.; Sebastiani, V.; Amador, M.; Jimeno, A.; Embuscado, E.; Hidalgo, M.; Iacobuzio-Donahue, C. Assessment of epidermal growth factor receptor (EGFR) signaling in paired colorectal cancer and normal colon tissue samples using computer-aided immunohistochemical analysis. Cancer Biol. Ther. 2005, 4, 1381–1386. [Google Scholar] [CrossRef] [PubMed]
- Chada, S.R.; Hollenbeck, P.J. Mitochondrial movement and positioning in axons: The role of growth factor signaling. J. Exp. Biol. 2003, 206, 1985–1992. [Google Scholar] [CrossRef] [PubMed]
- Chada, S.R.; Hollenbeck, P.J. Nerve growth factor signaling regulates motility and docking of axonal mitochondria. Curr. Biol. 2004, 14, 1272–1276. [Google Scholar] [CrossRef] [PubMed]
- Pezet, S.; McMahon, S.B. Neurotrophins: Mediators and modulators of pain. Annu. Rev. Neurosci. 2006, 29, 507–538. [Google Scholar] [CrossRef] [PubMed]
- McDougal, D.B., Jr.; Yu, M.J.; Gorin, P.D.; Johnson, E.M., Jr. Transported enzymes in sciatic nerve and sensory ganglia of rats exposed to maternal antibodies against nerve growth factor. J. Neurochem. 1981, 36, 1847–1852. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.E.; Akesson, E.; Seiger, A. Nerve growth factor-induced stimulation of dorsal root ganglion/spinal cord co-grafts in oculo: Enhanced survival and growth of CGRP-immunoreactive sensory neurons. Cell Tissue Res. 1999, 298, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.E.; Caire, S.R.; Dennis, R.W.; Chandler, M.J.; Foreman, R.D.; Kriebel, R.M. Effects of nerve growth factor (NGF) on glutamate metabolism in primary sensory neurons. In Proceedings of the Society for Neuroscience, San Diego, CA, USA, 10–15 November 2001.
- Tomita, H.; Ishiguro, S.; Abe, T.; Tamai, M. Administration of nerve growth factor, brain-derived neurotrophic factor and insulin-like growth factor-II protects phosphate-activated glutaminase in the ischemic and reperfused rat retinas. Tohoku J. Exp. Med. 1999, 187, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Koltzenburg, M.; Carlton, S.M. Glutamate-induced excitation and sensitization of nociceptors in rat glabrous skin. Pain 2001, 89, 187–198. [Google Scholar] [CrossRef]
- Bhave, G.; Karim, F.; Carlton, S.M.; Gereau, R.W. Peripheral group I metabotropic glutamate receptors modulate nociception in mice. Nat. Neurosci. 2001, 4, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Carlton, S.M.; Hargett, G.L.; Coggeshall, R.E. Localization and activation of glutamate receptors in unmyelinated axons of rat glabrous skin. Neurosci. Lett. 1995, 197, 25–28. [Google Scholar] [CrossRef]
- Carlton, S.M.; Zhou, S.; Coggeshall, R.E. Localization and activation of substance P receptors in unmyelinated axons of rat glabrous skin. Brain Res. 1996, 734, 103–108. [Google Scholar] [CrossRef]
- Carlton, S.M.; Zhou, S.; Coggeshall, R.E. Evidence for the interaction of glutamate and NK1 receptors in the periphery. Brain Res. 1998, 790, 160–169. [Google Scholar] [CrossRef]
- Carlton, S.M.; Coggeshall, R.E. Inflammation-induced changes in peripheral glutamate receptor populations. Brain Res. 1999, 820, 63–70. [Google Scholar] [CrossRef]
- Davidson, E.M.; Coggeshall, R.E.; Carlton, S.M. Peripheral NMDA and non-NMDA glutamate receptors contribute to nociceptive behaviors in the rat formalin test. Neuroreport 1997, 8, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Giovengo, S.L.; Kitto, K.F.; Kurtz, H.J.; Velazquez, R.A.; Larson, A.A. Parenterally administered kainic acid induces a persistent hyperalgesia in the mouse and rat. Pain 1999, 83, 347–358. [Google Scholar] [CrossRef]
- Jackson, D.L.; Graff, C.B.; Richardson, J.D.; Hargreaves, K.M. Glutamate participates in the peripheral modulation of thermal hyperalgesia in rats. Eur. J. Pharmacol. 1995, 284, 321–325. [Google Scholar] [CrossRef]
- Lawand, N.B.; Willis, W.D.; Westlund, K.N. Excitatory amino acid receptor involvement in peripheral nociceptive transmission in rats. Eur. J. Pharmacol. 1997, 324, 169–177. [Google Scholar] [CrossRef]
- Zhou, S.; Bonasera, L.; Carlton, S.M. Peripheral administration of NMDA, AMPA or KA results in pain behaviors in rats. Neuroreport 1996, 7, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Gazerani, P.; Wang, K.; Cairns, B.E.; Svensson, P.; Arendt-Nielsen, L. Effects of subcutaneous administration of glutamate on pain, sensitization and vasomotor responses in healthy men and women. Pain 2006, 124, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Lawand, N.B.; McNearney, T.; Westlund, K.N. Amino acid release into the knee joint: Key role in nociception and inflammation. Pain 2000, 86, 69–74. [Google Scholar] [CrossRef]
- Nordlind, K.; Johansson, O.; Liden, S.; Hokfelt, T. Glutamate- and aspartate-like immunoreactivities in human normal and inflamed skin. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1993, 64, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhou, S.; Coggeshall, R.E.; Carlton, S.M. N-methyl-d-aspartate-induced excitation and sensitization of normal and inflamed nociceptors. Neuroscience 2003, 118, 547–562. [Google Scholar] [CrossRef]
- Warncke, T.; Jorum, E.; Stubhaug, A. Local treatment with the N-methyl-d-aspartate receptor antagonist ketamine, inhibit development of secondary hyperalgesia in man by a peripheral action. Neurosci. Lett. 1997, 227, 1–4. [Google Scholar] [CrossRef]
- Hoffman, E.M.; Miller, K.E. Peripheral inhibition of glutaminase reduces carrageenan-induced Fos expression in the superficial dorsal horn of the rat. Neurosci. Lett. 2010, 472, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.M.; Sessle, B.J.; Haas, D.A.; Izzo, A.; Vernon, H.; Hu, J.W. Involvement of NMDA receptor mechanisms in jaw electromyographic activity and plasma extravasation induced by inflammatory irritant application to temporomandibular joint region of rats. Pain 1996, 68, 169–178. [Google Scholar] [CrossRef]
- Carlton, S.M. Peripheral excitatory amino acids. Curr. Opin. Pharmacol. 2001, 1, 52–56. [Google Scholar] [CrossRef]
- Dougherty, P.M.; Willis, W.D. Enhancement of spinothalamic neuron responses to chemical and mechanical stimuli following combined micro-iontophoretic application of N-methyl-d-aspartic acid and substance P. Pain 1991, 47, 85–93. [Google Scholar] [CrossRef]
- Dougherty, P.M.; Palecek, J.; Zorn, S.; Willis, W.D. Combined application of excitatory amino acids and substance P produces long-lasting changes in responses of primate spinothalamic tract neurons. Brain Res. Brain Res. Rev. 1993, 18, 227–246. [Google Scholar] [CrossRef]
- Hu, H.J.; Alter, B.J.; Carrasquillo, Y.; Qiu, C.S.; Gereau, R.W.T. Metabotropic glutamate receptor 5 modulates nociceptive plasticity via extracellular signal-regulated kinase-Kv4.2 signaling in spinal cord dorsal horn neurons. J. Neurosci. 2007, 27, 13181–13191. [Google Scholar] [CrossRef] [PubMed]
- Pezet, S.; Marchand, F.; D’Mello, R.; Grist, J.; Clark, A.K.; Malcangio, M.; Dickenson, A.H.; Williams, R.J.; McMahon, S.B. Phosphatidylinositol 3-kinase is a key mediator of central sensitization in painful inflammatory conditions. J. Neurosci. 2008, 28, 4261–4270. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, H.; Sheng, M.; Jan, L.Y.; Jan, Y.N.; Basbaum, A.I. Evidence for presynaptic N-methyl-d-aspartate autoreceptors in the spinal cord dorsal horn. Proc. Natl. Acad. Sci. USA 1994, 91, 8383–8387. [Google Scholar] [CrossRef] [PubMed]
- Hanani, M. Satellite glial cells in sensory ganglia: From form to function. Brain Res. Brain Res. Rev. 2005, 48, 457–476. [Google Scholar] [CrossRef] [PubMed]
- Weick, M.; Cherkas, P.S.; Hartig, W.; Pannicke, T.; Uckermann, O.; Bringmann, A.; Tal, M.; Reichenbach, A.; Hanani, M. P2 receptors in satellite glial cells in trigeminal ganglia of mice. Neuroscience 2003, 120, 969–977. [Google Scholar] [CrossRef]
- Miller, K.E.; Kriebel, R.M. Glutamine and glutamine synthetase levels are increased in rat DRG satellite cells following peripheral inflammation. In Proceedings of the Society for Neuroscience, New Orleans, LA, USA, 8–12 November 2003.
- Miller, K.E.; Kriebel, R.M.; Edwards, K.M.; Bartley, E.; Varoqui, H.; Erickson, J.D.; Schechter, R. Localization of neutral amino acid transporters (SNAT1, ASCT1, ASCT2) in rat DRG and spinal dorsal horn. In Proceedings of the Society for Neuroscience, Washington, DC, USA, 12–16 November 2005.
- Miller, K.E.; Edwards, K.M.; Schechter, R. Localization of neutral amino acid transporter SNAT2 in rat DRG. Oklahoma State University: Tulsa, OK, USA, Unpublished work. 2008. [Google Scholar]
- Brumovsky, P.; Watanabe, M.; Hokfelt, T. Expression of the vesicular glutamate transporters-1 and -2 in adult mouse dorsal root ganglia and spinal cord and their regulation by nerve injury. Neuroscience 2007, 147, 469–490. [Google Scholar] [CrossRef] [PubMed]
- Li, J.L.; Xiong, K.H.; Dong, Y.L.; Fujiyama, F.; Kaneko, T.; Mizuno, N. Vesicular glutamate transporters, VGluT1 and VGluT2, in the trigeminal ganglion neurons of the rat, with special reference to coexpression. J. Comp. Neurol. 2003, 463, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.L.; Konig, P.; Shimizu, T.; Jobling, P.; Gibbins, I.L. Most peptide-containing sensory neurons lack proteins for exocytotic release and vesicular transport of glutamate. J. Comp. Neurol. 2005, 483, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Nunzi, M.G.; Pisarek, A.; Mugnaini, E. Merkel cells, corpuscular nerve endings and free nerve endings in the mouse palatine mucosa express three subtypes of vesicular glutamate transporters. J. Neurocytol. 2004, 33, 359–376. [Google Scholar] [CrossRef] [PubMed]
- Li, J.L.; Fujiyama, F.; Kaneko, T.; Mizuno, N. Expression of vesicular glutamate transporters, VGluT1 and VGluT2, in axon terminals of nociceptive primary afferent fibers in the superficial layers of the medullary and spinal dorsal horns of the rat. J. Comp. Neurol. 2003, 457, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Alothman, S.; Zhang, Z.; Bolt, B.; Miller, K.E. Vesicular glutamate transporter 2 expression is altered in dorsal root ganglion neurons during the regenerative phase following sciatic nerve crush. In Proceedings of the 14th International Symposium on Neural Regeneration, P-8, Pacific Grove, CA, USA, 7 December 2011.
- Alothman, S.; Zhang, Z.; Bolt, B.; Anderson, M.B.; Miller, K.E. Expression of vesicular glutamate transporter 2 in dorsal root ganglion neurons during the acute phase following sciatic nerve transection. In Proceedings of the Society for Neuroscience, New Orleans, LA, USA, 13–17 October 2012.
- Moechars, D.; Weston, M.C.; Leo, S.; Callaerts-Vegh, Z.; Goris, I.; Daneels, G.; Buist, A.; Cik, M.; van der Spek, P.; Kass, S.; et al. Vesicular glutamate transporter VGLUT2 expression levels control quantal size and neuropathic pain. J. Neurosci. 2006, 26, 12055–12066. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.E.; Herzog, B.H.; Sutharshan, M. 6-Diazo-5-oxo-l-norleucine: A glutaminase inhibitor with local analgesic properties. In Proceedings of the 13th World Congress on Pain, PT 421, Montreal, Canada, 31 August 2010.
- Zhang, Z. The Role of Dorsal Root Ganglion Glutaminase in Acute and Chronic Inflammatory Pain. Ph.D. Dissertation, Oklahoma State University, Stillwater, OK, USA, 2013. [Google Scholar]
- Chiang, C.Y.; Wang, J.; Xie, Y.F.; Zhang, S.; Hu, J.W.; Dostrovsky, J.O.; Sessle, B.J. Astroglial glutamate-glutamine shuttle is involved in central sensitization of nociceptive neurons in rat medullary dorsal horn. J. Neurosci. 2007, 27, 9068–9076. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.Y.; Li, Z.; Dostrovsky, J.O.; Hu, J.W.; Sessle, B.J. Glutamine uptake contributes to central sensitization in the medullary dorsal horn. Neuroreport 2008, 19, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffman, E.M.; Zhang, Z.; Schechter, R.; Miller, K.E. Glutaminase Increases in Rat Dorsal Root Ganglion Neurons after Unilateral Adjuvant-Induced Hind Paw Inflammation. Biomolecules 2016, 6, 10. https://doi.org/10.3390/biom6010010
Hoffman EM, Zhang Z, Schechter R, Miller KE. Glutaminase Increases in Rat Dorsal Root Ganglion Neurons after Unilateral Adjuvant-Induced Hind Paw Inflammation. Biomolecules. 2016; 6(1):10. https://doi.org/10.3390/biom6010010
Chicago/Turabian StyleHoffman, E. Matthew, Zijia Zhang, Ruben Schechter, and Kenneth E. Miller. 2016. "Glutaminase Increases in Rat Dorsal Root Ganglion Neurons after Unilateral Adjuvant-Induced Hind Paw Inflammation" Biomolecules 6, no. 1: 10. https://doi.org/10.3390/biom6010010