
Epigenetic Impact on EBV Associated B-Cell Lymphomagenesis


Abstract
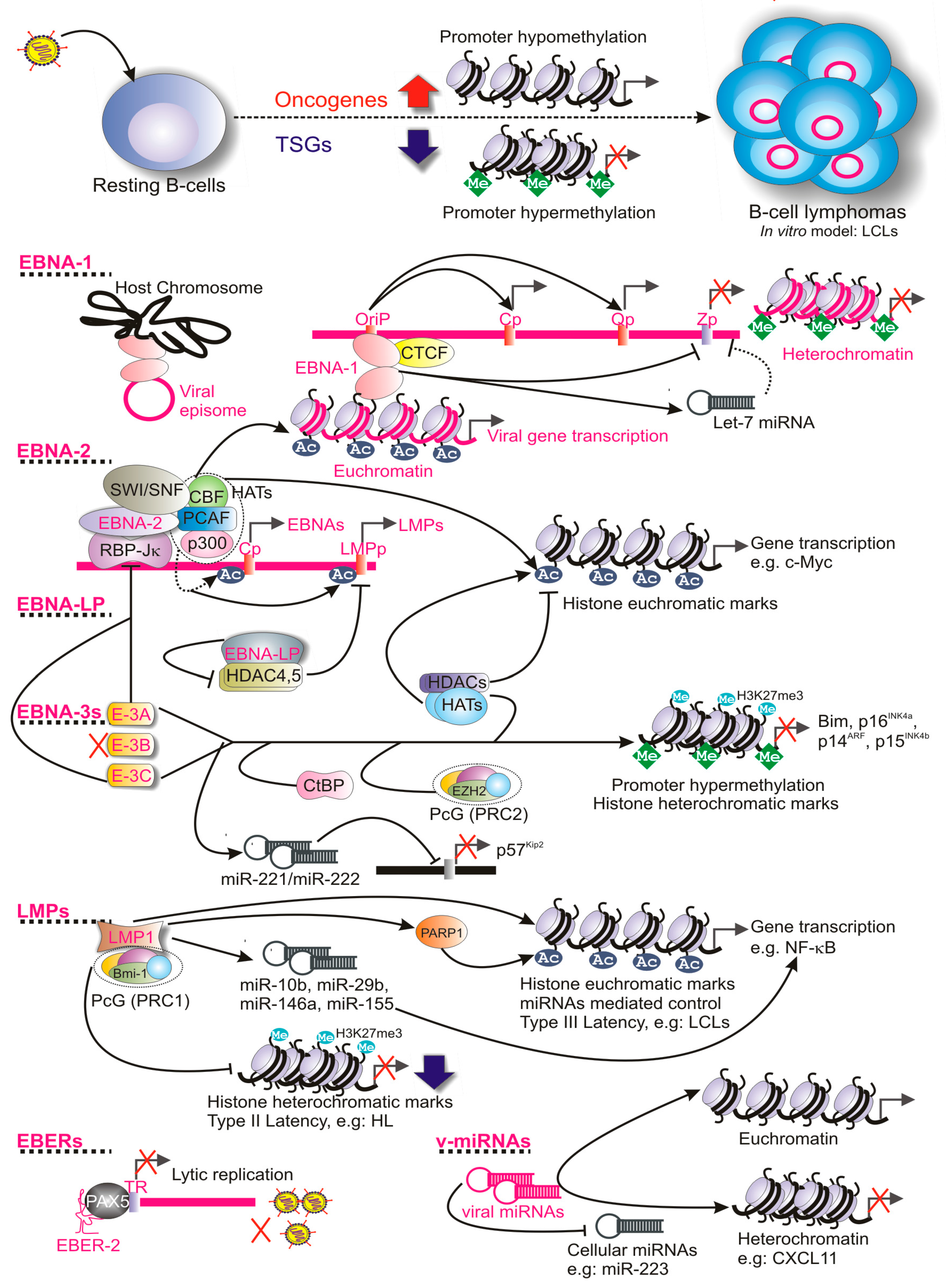
:1. Introduction
2. Epigenetic Regulation during EBV Lytic Replication
3. Epigenetic Regulation during EBV Latency
3.1. Epigenetic Regulation of EBV Latency Promoters
3.2. Epigenetic Regulation by the Viral Oncoproteins
3.2.1. Role of EBNAs
3.2.2. Role of LMPs
3.2.3. Role of EBERs
3.2.4. Role of Viral miRNAs
4. Epigenetic Profiles of Cellular Genomes during EBV-Induced B-Cell Lymphomagenesis
4.1. Early Infection
4.2. Lymphoblastoid Cell Lines
4.2.1. Host miRNAs
4.2.2. Super-Enhancers
4.3. Burkitt’s Lymphoma (BL)
4.4. Hodgkin’s Lymphoma (HL)
5. Future Perspective
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Bartova, E.; Krejci, J.; Harnicarova, A.; Galiova, G.; Kozubek, S. Histone modifications and nuclear architecture: A review. J. Histochem. Cytochem. 2008, 56, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.D.; Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Denis, H.; Ndlovu, M.N.; Fuks, F. Regulation of mammalian DNA methyltransferases: A route to new mechanisms. EMBO Rep. 2011, 12, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, J.; Esteller, M. Cancer epigenomics: Beyond genomics. Curr. Opin. Genet. Dev. 2012, 22, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Kaul, R.; Murakami, M.; Robertson, E.S. Tumor viruses and cancer biology: Modulating signaling pathways for therapeutic intervention. Cancer Biol. Ther. 2010, 10, 961–978. [Google Scholar] [CrossRef] [PubMed]
- Casiraghi, C.; Dorovini-Zis, K.; Horwitz, M.S. Epstein-Barr virus infection of human brain microvessel endothelial cells: A novel role in multiple sclerosis. J. Neuroimmunol. 2011, 230, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.; Rivera, C.; Sgadari, C.; Franklin, J.; Max, E.E.; Bhatia, K.; Tosato, G. Infection of human endothelial cells with Epstein-Barr virus. J. Exp. Med. 1995, 182, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, C.A.; Dreyfus, D.H.; Jones, J.F.; Gelfand, E.W. EBV infection of T cells: Potential role in malignant transformation. Semin. Cancer Biol. 1996, 7, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Coleman, C.B.; Wohlford, E.M.; Smith, N.A.; King, C.A.; Ritchie, J.A.; Baresel, P.C.; Kimura, H.; Rochford, R. Epstein-Barr virus type 2 latently infects T cells, inducing an atypical activation characterized by expression of lymphotactic cytokines. J. Virol. 2014, 89, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Robertson, E.S. Epstein-Barr virus-associated B-cell lymphomas: Pathogenesis and clinical outcomes. Clin. Cancer Res. 2011, 17, 3056–3063. [Google Scholar] [CrossRef] [PubMed]
- Allday, M.J. EBV finds a polycomb-mediated, epigenetic solution to the problem of oncogenic stress responses triggered by infection. Front. Genet. 2013, 4, 212. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Ghosh Roy, S.; Bose, P.; Saha, A. Role of EBNA-3 family proteins in EBV associated B-cell lymphomagenesis. Front. Microbiol. 2016, 7, 457. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.; Robertson, K.D.; Manns, A.; Hildesheim, A.; Ambinder, R.F. The Epstein-Barr virus major latent promoter QP is constitutively active, hypomethylated, and methylation sensitive. J. Virol. 1998, 72, 7075–7083. [Google Scholar] [PubMed]
- Tempera, I.; Wiedmer, A.; Dheekollu, J.; Lieberman, P.M. CTCF prevents the epigenetic drift of EBV latency promoter QP. PLoS Pathog. 2010, 6, e1001048. [Google Scholar] [CrossRef] [PubMed]
- Deacon, E.M.; Pallesen, G.; Niedobitek, G.; Crocker, J.; Brooks, L.; Rickinson, A.B.; Young, L.S. Epstein-Barr virus and Hodgkin’s disease: Transcriptional analysis of virus latency in the malignant cells. J. Exp. Med. 1993, 177, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Brink, A.A.; Meijer, C.J.; Nicholls, J.M.; Middeldorp, J.M.; van den Brule, A.J. Activity of the EBNA1 promoter associated with lytic replication (FP) in Epstein-Barr virus associated disorders. Mol. Pathol. 2001, 54, 98–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamson, A.L.; Darr, D.; Holley-Guthrie, E.; Johnson, R.A.; Mauser, A.; Swenson, J.; Kenney, S. Epstein-Barr virus immediate-early proteins BZLF1 and BRLF1 activate the ATF2 transcription factor by increasing the levels of phosphorylated p38 and c-Jun N-terminal kinases. J. Virol. 2000, 74, 1224–1233. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Su, X.; Choi, G.C.; Cao, Y.; Ambinder, R.F.; Tao, Q. Methylation profiling of Epstein-Barr virus immediate-early gene promoters, BZLF1 and BRLF1 in tumors of epithelial, NK- and B-cell origins. BMC Cancer 2012, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Tsurumi, T. Switching of EBV cycles between latent and lytic states. Rev. Med. Virol. 2013, 24, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Woellmer, A.; Arteaga-Salas, J.M.; Hammerschmidt, W. BZLF1 governs CPG-methylated chromatin of Epstein-Barr virus reversing epigenetic repression. PLoS Pathog. 2012, 8, e1002902. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Kondo, Y.; Sugimoto, A.; Kawashima, D.; Saito, S.; Isomura, H.; Kanda, T.; Tsurumi, T. Epigenetic histone modification of Epstein-Barr virus BZLF1 promoter during latency and reactivation in raji cells. J. Virol. 2012, 86, 4752–4761. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.S.; Kieff, E. Epstein-Barr virus latent genes. Exp. Mol. Med. 2015, 47, e131. [Google Scholar] [CrossRef] [PubMed]
- Takacs, M.; Banati, F.; Koroknai, A.; Segesdi, J.; Salamon, D.; Wolf, H.; Niller, H.H.; Minarovits, J. Epigenetic regulation of latent Epstein-Barr virus promoters. Biochim. Biophys. Acta 2010, 1799, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Kempkes, B.; Ling, P.D. EBNA2 and its coactivator EBNA-LP. Curr. Top. Microbiol. Immunol. 2015, 391, 35–59. [Google Scholar] [PubMed]
- Chen, H.S.; Martin, K.A.; Lu, F.; Lupey, L.N.; Mueller, J.M.; Lieberman, P.M.; Tempera, I. Epigenetic deregulation of the LMP1/LMP2 locus of Epstein-Barr virus by mutation of a single CTCF-cohesin binding site. J. Virol. 2014, 88, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.D.; Veenstra, H.; Khasnis, S.; Gunnell, A.; Webb, H.M.; Shannon-Lowe, C.; Andrews, S.; Osborne, C.S.; West, M.J. MYC activation and BCL2L11 silencing by a tumour virus through the large-scale reconfiguration of enhancer-promoter hubs. eLife 2016, 5, e18270. [Google Scholar] [CrossRef] [PubMed]
- Paschos, K.; Smith, P.; Anderton, E.; Middeldorp, J.M.; White, R.E.; Allday, M.J. Epstein-Barr virus latency in B cells leads to epigenetic repression and CPG methylation of the tumour suppressor gene Bim. PLoS Pathog. 2009, 5, e1000492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotter, M.A., 2nd; Robertson, E.S. Modulation of histone acetyltransferase activity through interaction of Epstein-Barr nuclear antigen 3C with prothymosin alpha. Mol. Cell. Biol. 2000, 20, 5722–5735. [Google Scholar] [CrossRef] [PubMed]
- Anderton, E.; Yee, J.; Smith, P.; Crook, T.; White, R.E.; Allday, M.J. Two Epstein-Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor Bim: Clues to the pathogenesis of Burkitt’s lymphoma. Oncogene 2008, 27, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Portal, D.; Rosendorff, A.; Kieff, E. Epstein-Barr nuclear antigen leader protein coactivates transcription through interaction with histone deacetylase 4. Proc. Natl. Acad. Sci. USA 2006, 103, 19278–19283. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.A.; Lupey, L.N.; Tempera, I. Epstein-Barr virus oncoprotein LMP1 mediates epigenetic changes in host gene expression through PARP1. J. Virol. 2016, 90, 8520–8530. [Google Scholar] [CrossRef] [PubMed]
- Motsch, N.; Pfuhl, T.; Mrazek, J.; Barth, S.; Grasser, F.A. Epstein-Barr virus-encoded latent membrane protein 1 (LMP1) induces the expression of the cellular microrna miR-146a. RNA Biol. 2007, 4, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; McBride, J.; Fewell, C.; Lacey, M.; Wang, X.; Lin, Z.; Cameron, J.; Flemington, E.K. MicroRNA-155 is an Epstein-Barr virus-induced gene that modulates Epstein-Barr virus-regulated gene expression pathways. J. Virol. 2008, 82, 5295–5306. [Google Scholar] [CrossRef] [PubMed]
- Elia, A.; Vyas, J.; Laing, K.G.; Clemens, M.J. Ribosomal protein L22 inhibits regulation of cellular activities by the Epstein-Barr virus small RNA EBER-1. Eur. J. Biochem. 2004, 271, 1895–1905. [Google Scholar] [CrossRef] [PubMed]
- Fok, V.; Friend, K.; Steitz, J.A. Epstein-Barr virus noncoding RNAs are confined to the nucleus, whereas their partner, the human La protein, undergoes nucleocytoplasmic shuttling. J. Cell Biol. 2006, 173, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Pimienta, G.; Steitz, J.A. AUF1/hnRNP D is a novel protein partner of the EBER1 noncoding RNA of Epstein-Barr virus. RNA 2012, 18, 2073–2082. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Moss, W.N.; Yario, T.A.; Steitz, J.A. EBV noncoding RNA binds nascent RNA to drive host PAX5 to viral DNA. Cell 2015, 160, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; O’Hara, A.; Araujo, I.; Barreto, J.; Carvalho, E.; Sapucaia, J.B.; Ramos, J.C.; Luz, E.; Pedroso, C.; Manrique, M.; et al. EBV microRNAs in primary lymphomas and targeting of CXCL-11 by EBV-mir-BHRF1-3. Cancer Res. 2008, 68, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.J.; Rabinowitz, G.S.; Yario, T.A.; Luna, J.M.; Darnell, R.B.; Steitz, J.A. EBV and human microRNAs co-target oncogenic and apoptotic viral and human genes during latency. EMBO J. 2012, 31, 2207–2221. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Wang, S.; Nguyen, T.; Shire, K.; Frappier, L. The EBNA1 protein of Epstein-Barr virus functionally interacts with Brd4. J. Virol. 2008, 82, 12009–12019. [Google Scholar] [CrossRef] [PubMed]
- Sarkari, F.; Sanchez-Alcaraz, T.; Wang, S.; Holowaty, M.N.; Sheng, Y.; Frappier, L. EBNA1-mediated recruitment of a histone H2B deubiquitylating complex to the Epstein-Barr virus latent origin of DNA replication. PLoS Pathog. 2009, 5, e1000624. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, S.; Pan, Q.; Blencowe, B.J.; Claycomb, J.M.; Frappier, L. Epstein-Barr virus EBNA1 protein regulates viral latency through effects on let-7 microRNA and dicer. J. Virol. 2014, 88, 11166–11177. [Google Scholar] [CrossRef] [PubMed]
- Zimber-Strobl, U.; Strobl, L.J. EBNA2 and Notch signalling in Epstein-Barr virus mediated immortalization of B lymphocytes. Semin. Cancer Biol. 2001, 11, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Grossman, S.R.; Kieff, E. Epstein-Barr virus nuclear protein 2 interacts with p300, CBP, and PCAF histone acetyltransferases in activation of the LMP1 promoter. Proc. Natl. Acad. Sci. USA 2000, 97, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.Y.; Krumm, A.; Schubach, W.H. Promoter-specific targeting of human SWI-SNF complex by Epstein-Barr virus nuclear protein 2. J. Virol. 2000, 74, 8893–8903. [Google Scholar] [CrossRef] [PubMed]
- White, R.E.; Ramer, P.C.; Naresh, K.N.; Meixlsperger, S.; Pinaud, L.; Rooney, C.; Savoldo, B.; Coutinho, R.; Bodor, C.; Gribben, J.; et al. EBNA3B-deficient EBV promotes B cell lymphomagenesis in humanized mice and is found in human tumors. J. Clin. Investig. 2012, 122, 1487–1502. [Google Scholar] [CrossRef] [PubMed]
- Maruo, S.; Johannsen, E.; Illanes, D.; Cooper, A.; Zhao, B.; Kieff, E. Epstein-Barr virus nuclear protein 3A domains essential for growth of lymphoblasts: Transcriptional regulation through RBP-Jkappa/CBF1 is critical. J. Virol. 2005, 79, 10171–10179. [Google Scholar] [CrossRef] [PubMed]
- Maruo, S.; Wu, Y.; Ishikawa, S.; Kanda, T.; Iwakiri, D.; Takada, K. Epstein-Barr virus nuclear protein EBNA3C is required for cell cycle progression and growth maintenance of lymphoblastoid cells. Proc. Natl. Acad. Sci. USA 2006, 103, 19500–19505. [Google Scholar] [CrossRef] [PubMed]
- Maruo, S.; Wu, Y.; Ito, T.; Kanda, T.; Kieff, E.D.; Takada, K. Epstein-Barr virus nuclear protein EBNA3C residues critical for maintaining lymphoblastoid cell growth. Proc. Natl. Acad. Sci. USA 2009, 106, 4419–4424. [Google Scholar] [CrossRef] [PubMed]
- Maruo, S.; Zhao, B.; Johannsen, E.; Kieff, E.; Zou, J.; Takada, K. Epstein-Barr virus nuclear antigens 3C and 3A maintain lymphoblastoid cell growth by repressing p16INK4A and p14ARF expression. Proc. Natl. Acad. Sci. USA 2011, 108, 1919–1924. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, C.; Hasan, S.; Rowe, M.; Hottiger, M.; Orre, R.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C and prothymosin alpha interact with the p300 transcriptional coactivator at the CH1 and CH3/HAT domains and cooperate in regulation of transcription and histone acetylation. J. Virol. 2002, 76, 4699–4708. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Lan, K.; Subramanian, C.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C recruits histone deacetylase activity and associates with the corepressors mSin3A and NCoR in human B-cell lines. J. Virol. 2003, 77, 4261–4272. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Mar, J.C.; Maruo, S.; Lee, S.; Gewurz, B.E.; Johannsen, E.; Holton, K.; Rubio, R.; Takada, K.; Quackenbush, J.; et al. Epstein-Barr virus nuclear antigen 3C regulated genes in lymphoblastoid cell lines. Proc. Natl. Acad. Sci. USA 2011, 108, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Schmidt, S.C.; Jiang, S.; Willox, B.; Bernhardt, K.; Liang, J.; Johannsen, E.C.; Kharchenko, P.; Gewurz, B.E.; Kieff, E.; et al. Epstein-Barr virus oncoprotein super-enhancers control B cell growth. Cell Host Microbe 2015, 17, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Harth-Hertle, M.L.; Scholz, B.A.; Erhard, F.; Glaser, L.V.; Dolken, L.; Zimmer, R.; Kempkes, B. Inactivation of intergenic enhancers by EBNA3A initiates and maintains polycomb signatures across a chromatin domain encoding CXCL10 and CXCL9. PLoS Pathog. 2013, 9, e1003638. [Google Scholar] [CrossRef] [PubMed]
- Allday, M.J.; Bazot, Q.; White, R.E. The EBNA3 family: Two oncoproteins and a tumour suppressor that are central to the biology of EBV in B cells. Curr. Top. Microbiol. Immunol. 2015, 391, 61–117. [Google Scholar] [PubMed]
- Skalska, L.; White, R.E.; Parker, G.A.; Turro, E.; Sinclair, A.J.; Paschos, K.; Allday, M.J. Induction of p16(INK4a) is the major barrier to proliferation when Epstein-Barr virus (EBV) transforms primary B cells into lymphoblastoid cell lines. PLoS Pathog. 2013, 9, e1003187. [Google Scholar] [CrossRef]
- Skalska, L.; White, R.E.; Franz, M.; Ruhmann, M.; Allday, M.J. Epigenetic repression of p16(INK4A) by latent Epstein-Barr virus requires the interaction of EBNA3A and EBNA3C with CtBP. PLoS Pathog. 2010, 6, e1000951. [Google Scholar] [CrossRef] [PubMed]
- Bazot, Q.; Deschamps, T.; Tafforeau, L.; Siouda, M.; Leblanc, P.; Harth-Hertle, M.L.; Rabourdin-Combe, C.; Lotteau, V.; Kempkes, B.; Tommasino, M.; et al. Epstein-Barr virus nuclear antigen 3A protein regulates CDKN2B transcription via interaction with MIZ-1. Nucleic Acids Res. 2014, 42, 9700–9716. [Google Scholar] [CrossRef] [PubMed]
- Bazot, Q.; Paschos, K.; Skalska, L.; Kalchschmidt, J.S.; Parker, G.A.; Allday, M.J. Epstein-Barr Virus Proteins EBNA3A and EBNA3C Together Induce Expression of the Oncogenic MicroRNA Cluster miR-221/miR-222 and Ablate Expression of Its Target p57KIP2. PLoS Pathog. 2015, 11, e1005031. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, J.; Homig-Holzel, C.; Seagal, J.; Muller, W.; Hermann, A.C.; Rajewsky, K.; Zimber-Strobl, U. LMP1 signaling can replace CD40 signaling in B cells in vivo and has unique features of inducing class-switch recombination to IgG1. Blood 2008, 111, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.J.; Longnecker, R. EBV LMP2A provides a surrogate pre-B cell receptor signal through constitutive activation of the ERK/MAPK pathway. J. Gen. Virol. 2008, 89, 1563–1568. [Google Scholar] [CrossRef] [PubMed]
- Siegler, G.; Kremmer, E.; Gonnella, R.; Niedobitek, G. Epstein-Barr virus encoded latent membrane protein 1 (LMP1) and TNF receptor associated factors (TRAF): Colocalisation of LMP1 and TRAF1 in primary EBV infection and in EBV associated Hodgkin lymphoma. Mol. Pathol. 2003, 56, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Dutton, A.; Woodman, C.B.; Chukwuma, M.B.; Last, J.I.; Wei, W.; Vockerodt, M.; Baumforth, K.R.; Flavell, J.R.; Rowe, M.; Taylor, A.M.; et al. Bmi-1 is induced by the Epstein-Barr virus oncogene LMP1 and regulates the expression of viral target genes in Hodgkin lymphoma cells. Blood 2007, 109, 2597–2603. [Google Scholar] [CrossRef] [PubMed]
- Anderton, J.A.; Bose, S.; Vockerodt, M.; Vrzalikova, K.; Wei, W.; Kuo, M.; Helin, K.; Christensen, J.; Rowe, M.; Murray, P.G.; et al. The H3K27ME3 demethylase, KDM6B, is induced by Epstein-Barr virus and over-expressed in Hodgkin’s lymphoma. Oncogene 2011, 30, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.L.; Li, H.P.; Lu, Y.J.; Hsueh, C.; Liang, Y.; Chen, C.L.; Tsao, S.W.; Tse, K.P.; Yu, J.S.; Chang, Y.S. Activation of DNA methyltransferase 1 by EBV LMP1 involves c-Jun NH(2)-terminal kinase signaling. Cancer Res. 2006, 66, 11668–11676. [Google Scholar] [CrossRef] [PubMed]
- Leonard, S.; Wei, W.; Anderton, J.; Vockerodt, M.; Rowe, M.; Murray, P.G.; Woodman, C.B. Epigenetic and transcriptional changes which follow Epstein-Barr virus infection of germinal center B cells and their relevance to the pathogenesis of Hodgkin’s lymphoma. J. Virol. 2011, 85, 9568–9577. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, D.; Takada, K. Role of EBERs in the pathogenesis of EBV infection. Adv. Cancer Res. 2010, 107, 119–136. [Google Scholar] [PubMed]
- Medvedovic, J.; Ebert, A.; Tagoh, H.; Busslinger, M. Pax5: A master regulator of B cell development and leukemogenesis. Adv. Immunol. 2011, 111, 179–206. [Google Scholar] [PubMed]
- Pfeffer, S.; Zavolan, M.; Grasser, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identification of virus-encoded microRNAs. Science 2004, 304, 734–736. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Kieff, E. Epstein-Barr virus BHRF1 micro- and stable RNAs during latency III and after induction of replication. J. Virol. 2007, 81, 9967–9975. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Huang, J.; Wu, F.Y.; Liao, G.; Hutt-Fletcher, L.; Hayward, S.D. Regulation of expression of the Epstein-Barr virus BamHI-A rightward transcripts. J. Virol. 2005, 79, 1724–1733. [Google Scholar] [CrossRef] [PubMed]
- Kuzembayeva, M.; Hayes, M.; Sugden, B. Multiple functions are mediated by the miRNAs of Epstein-Barr virus. Curr. Opin. Virol. 2014, 7, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Navari, M.; Etebari, M.; De Falco, G.; Ambrosio, M.R.; Gibellini, D.; Leoncini, L.; Piccaluga, P.P. The presence of Epstein-Barr virus significantly impacts the transcriptional profile in immunodeficiency-associated Burkitt lymphoma. Front. Microbiol. 2015, 6, 556. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Ai, J.; Xie, Z.; Zhou, C.; Liu, C.; Zhang, H.; Shen, K. Dynamic expression of viral and cellular microRNAs in infectious mononucleosis caused by primary Epstein-Barr virus infection in children. Virol. J. 2015, 12, 208. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Li, W.; Wu, Y.; Wei, F.; Gong, Z.; Bo, H.; Wang, Y.; Li, X.; Xiang, B.; Guo, C.; et al. Epstein-Barr virus-encoded miR-BART6-3p inhibits cancer cell metastasis and invasion by targeting long non-coding RNA LOC553103. Cell Death Dis. 2016, 7, e2353. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Lee, H.; Kim, S.R.; Gho, Y.S.; Lee, S.K. Epstein-Barr virus-encoded microrna BART15-3p promotes cell apoptosis partially by targeting BRUCE. J. Virol. 2013, 87, 8135–8144. [Google Scholar] [CrossRef] [PubMed]
- Babu, S.G.; Ponia, S.S.; Kumar, D.; Saxena, S. Cellular oncomiR orthologue in EBV oncogenesis. Comput. Biol. Med. 2011, 41, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Haneklaus, M.; Gerlic, M.; Kurowska-Stolarska, M.; Rainey, A.A.; Pich, D.; McInnes, I.B.; Hammerschmidt, W.; O’Neill, L.A.; Masters, S.L. Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1β production. J. Immunol. 2012, 189, 3795–3799. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. CPG island hypermethylation and tumor suppressor genes: A booming present, a brighter future. Oncogene 2002, 21, 5427–5440. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Jha, H.C.; Upadhyay, S.K.; Robertson, E.S. Epigenetic silencing of tumor suppressor genes during in vitro Epstein-Barr virus infection. Proc. Natl. Acad. Sci. USA 2015, 112, E5199–E5207. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, P.A.; Yan, C.M.; Forte, E.; Bocedi, A.; Tourigny, J.P.; White, R.E.; Allday, M.J.; Patel, A.; Dave, S.S.; Kim, W.; et al. An ATM/CHK2-mediated DNA damage-responsive signaling pathway suppresses Epstein-Barr virus transformation of primary human B cells. Cell Host Microbe 2010, 8, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Bornkamm, G.W. Epstein-Barr virus and the pathogenesis of Burkitt’s lymphoma: More questions than answers. Int. J. Cancer 2009, 124, 1745–1755. [Google Scholar] [CrossRef] [PubMed]
- Kretzmer, H.; Bernhart, S.H.; Wang, W.; Haake, A.; Weniger, M.A.; Bergmann, A.K.; Betts, M.J.; Carrillo-de-Santa-Pau, E.; Doose, G.; Gutwein, J.; et al. DNA methylome analysis in Burkitt and follicular lymphomas identifies differentially methylated regions linked to somatic mutation and transcriptional control. Nat. Genet. 2015, 47, 1316–1325. [Google Scholar] [CrossRef] [PubMed]
- Kreck, B.; Richter, J.; Ammerpohl, O.; Barann, M.; Esser, D.; Petersen, B.S.; Vater, I.; Murga Penas, E.M.; Bormann Chung, C.A.; Seisenberger, S.; et al. Base-pair resolution DNA methylome of the EBV-positive Endemic Burkitt lymphoma cell line DAUDI determined by SOLiD bisulfite-sequencing. Leukemia 2013, 27, 1751–1753. [Google Scholar] [CrossRef] [PubMed]
- Love, C.; Sun, Z.; Jima, D.; Li, G.; Zhang, J.; Miles, R.; Richards, K.L.; Dunphy, C.H.; Choi, W.W.; Srivastava, G.; et al. The genetic landscape of mutations in Burkitt lymphoma. Nat. Genet. 2012, 44, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Au Yeung, C.L.; Tsang, W.P.; Tsang, T.Y.; Co, N.N.; Yau, P.L.; Kwok, T.T. HPV-16 E6 upregulation of DNMT1 through repression of tumor suppressor p53. Oncol. Rep. 2010, 24, 1599–1604. [Google Scholar] [PubMed]
- Qiu, X.; Zhang, L.; Lu, S.; Song, Y.; Lao, Y.; Hu, J.; Fan, H. Upregulation of DNMT1 mediated by HBx suppresses RASSF1A expression independent of DNA methylation. Oncol. Rep. 2014, 31, 202–208. [Google Scholar] [PubMed]


{kind=link}
| B-Cell Lymphomas | Activated Promoters | Latency Programs | Latent Transcripts | Refs. |
|---|---|---|---|---|
| Burkitt’s lymphoma (BL) | Qp, Cp | I | EBNA-1, EBERs | [14,15] |
| Hodgkin’s lymphoma (HL) | Cp, LMP-1p | II | EBNA-1, LMP-1/2, EBERs | [16,17] |
| AIDS-associated B-cell lymphomas | Wp, Cp, LMP-1p/2Ap | III | All EBNAs, LMPs, EBERs and miRNAs | [15] |
| Post-transplant lymphoproliferative disorder (PTLD) | Wp, Cp, LMP-1p/2Ap | III | All EBNAs, LMPs, EBERs and miRNAs | [15] |
| Diffuse large B-cell lymphomas (DLBCLs) | Wp, Cp, LMP-1p/2Ap | III | All EBNAs, LMPs, EBERs and miRNAs | [15] |
| Latent Transcripts | Proposed Functions | Refs. |
|---|---|---|
| EBNA-1 |
| [15] |
| EBNA-2 |
| [27] |
| EBNA-3A |
| [27,28] |
| EBNA-3C |
| [27,28,29,30] |
| EBNA-LP |
| [31] |
| LMP-1 |
| [32,33] |
| LMP-2A |
| [34] |
| EBERs |
| [35,36,37,38] |
| miRNAs |
| [39,40] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghosh Roy, S.; Robertson, E.S.; Saha, A. Epigenetic Impact on EBV Associated B-Cell Lymphomagenesis. Biomolecules 2016, 6, 46. https://doi.org/10.3390/biom6040046
Ghosh Roy S, Robertson ES, Saha A. Epigenetic Impact on EBV Associated B-Cell Lymphomagenesis. Biomolecules. 2016; 6(4):46. https://doi.org/10.3390/biom6040046
Chicago/Turabian StyleGhosh Roy, Shatadru, Erle S. Robertson, and Abhik Saha. 2016. "Epigenetic Impact on EBV Associated B-Cell Lymphomagenesis" Biomolecules 6, no. 4: 46. https://doi.org/10.3390/biom6040046