Overview of Bile Acids Signaling and Perspective on the Signal of Ursodeoxycholic Acid, the Most Hydrophilic Bile Acid, in the Heart

 , and
, and
Abstract
:
1. Introduction
2. Bile Acid as a Signaling Molecule
2.1. Nuclear Receptor-Mediated Response
2.2. Takeda G-Protein-Coupled Receptor 5
2.3. Muscarinic Receptor
2.4. Sphingosine-1-Phosphate Receptor
2.5. Large Conductance Voltage- and Ca2+-Activated Potassium (K+) (BK) Channels
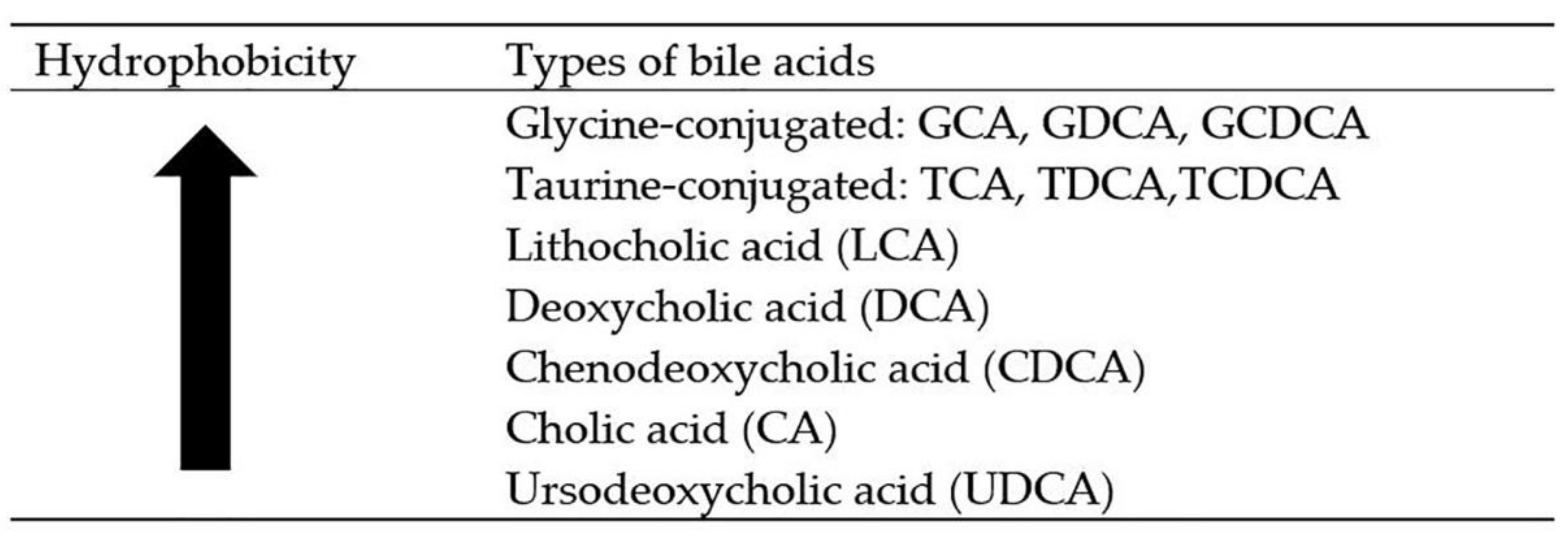
3. The Most Hydrophilic Bile Acids—Ursodeoxycholic Acid
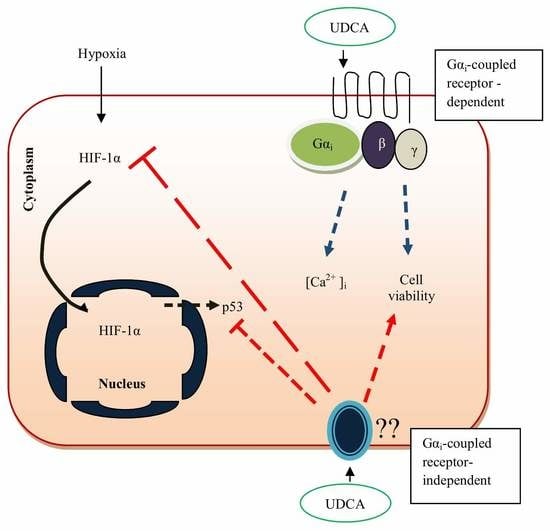
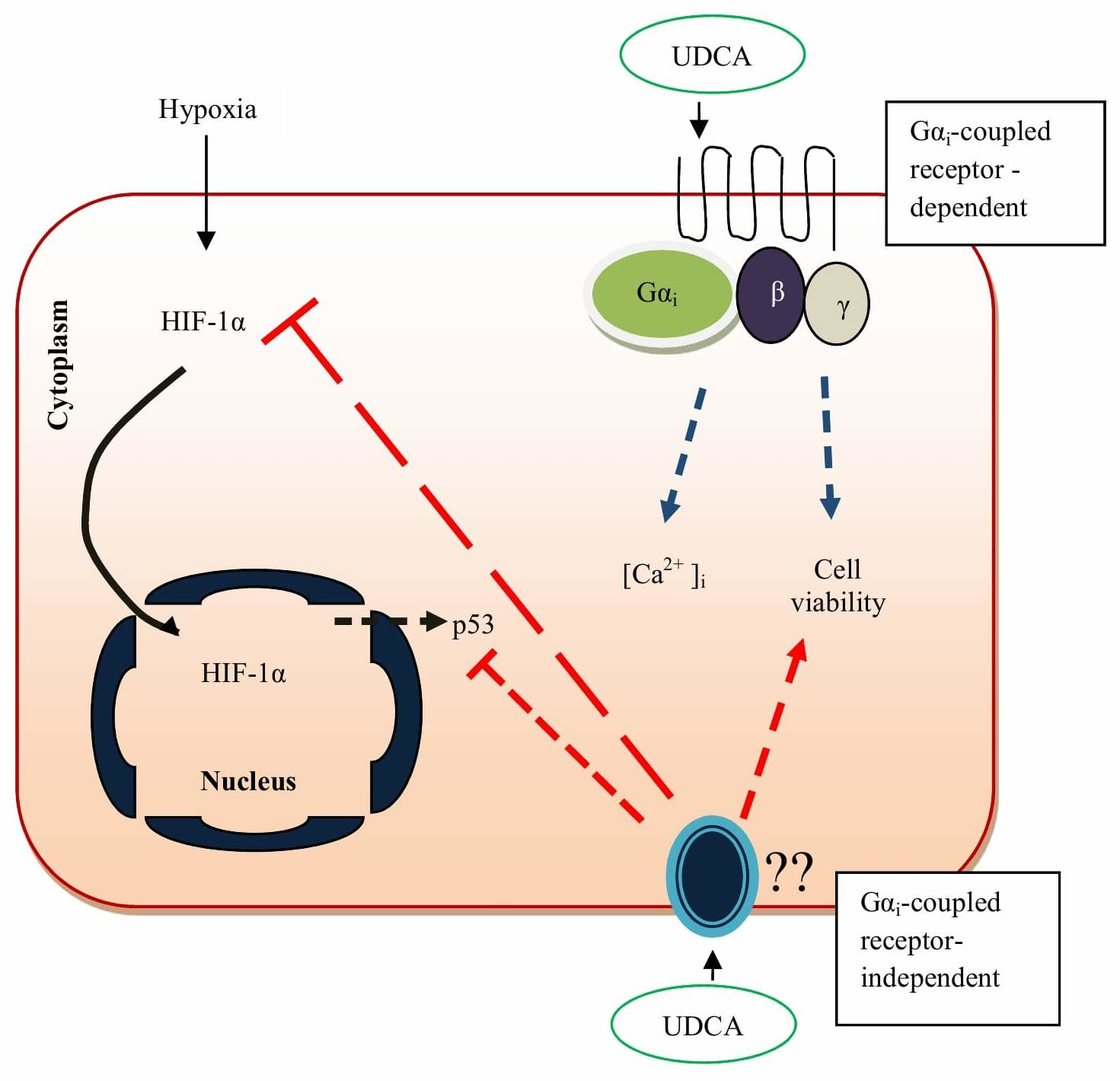
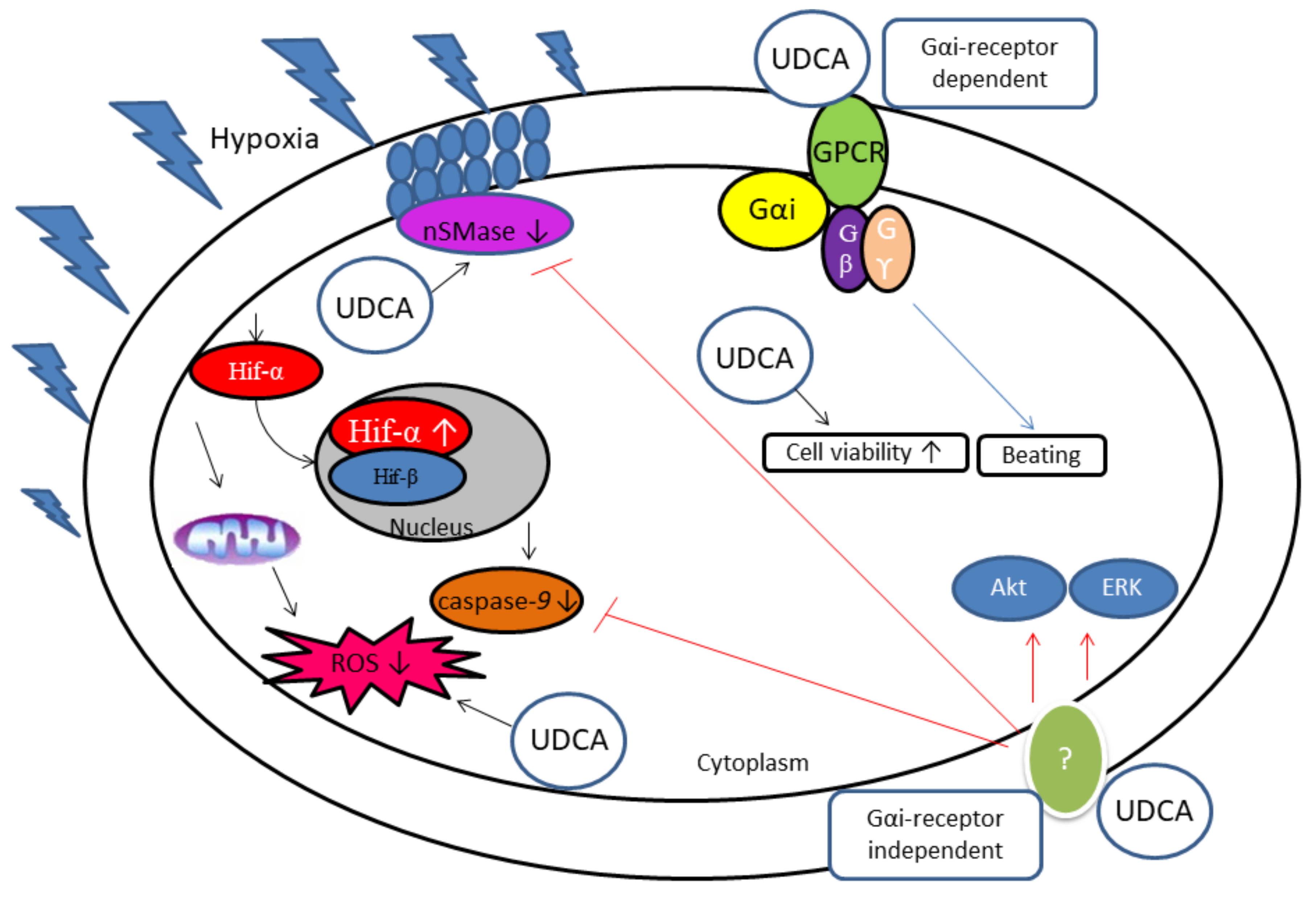
4. Mechanism of Action by Ursodeoxycholic Acid
5. Ursodeoxycholic Acid Protects the Heart against More Hydrophobic Bile Acids
6. Past, Current, and Future Perspectives on Role of Ursodeoxycholic Acid in Cardiovascular Diseases
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rutgeerts, P.; Ghoos, Y.; Vantrappen, G. The enterohepatic circulation of bile acids during continuous liquid formula perfusion of the duodenum. J. Lipid Res. 1983, 24, 614–619. [Google Scholar] [PubMed]
- Staels, B.; Fonseca, V.A. Bile acids and metabolic regulation: Mechanisms and clinical responses to bile acid sequestration. Diabetes Care 2009, 32 (Suppl. 2), S237–S245. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.-J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of Bile Acids and Bile Acid Receptors in Metabolic Regulation. Physiol. Rev. 2009, 147–191. [Google Scholar] [CrossRef] [PubMed]
- Kakiyama, G.; Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Heuman, D.M.; Kang, D.J.; Takel, H.; Nittono, H.; Ridlon, J.; Fuchs, M.; et al. Colonic inflammation and secondary bile acids in alcoholic cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Batta, A.K.; Salen, G.; Rapole, K.R.; Batta, M.; Batta, P.; Alberts, D.; Earnest, D. Highly simplified method for gas-liquid chromatographic quantitation of bile acids and sterols in human stool. J. Lipid Res. 1999, 40, 1148–1154. [Google Scholar] [PubMed]
- Hofmann, A.F. The continuing importance of bile acids in liver and intestinal disease. Arch. Intern. Med. 1999, 159, 2647–2658. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chiang, J.Y.L. Bile Acid signaling in liver metabolism and diseases. J. Lipids 2012, 2012, 754067. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.; Bouscarel, B. Bile acids and signal transduction: Role in glucose homeostasis. Cell. Signal. 2008, 20, 2180–2197. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.C.; Duong, H.Q.; Parajuli, K.R.; Han, S.I. Pro-apoptotic role of the MEK/ERK pathway in ursodeoxycholic acid-induced apoptosis in SNU601 gastric cancer cells. Oncol. Rep. 2012, 28, 1429–1434. [Google Scholar] [CrossRef] [PubMed]
- Degirolamo, C.; Modica, S.; Palasciano, G.; Moschetta, A. Bile acids and colon cancer: Solving the puzzle with nuclear receptors. Trends Mol. Med. 2011, 17, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Yang, L.; Wang, Z.; Huang, W. Bile acid nuclear receptor FXR and digestive system diseases. Acta Pharm. Sin. B 2015, 5, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duboc, H.; Taché, Y.; Hofmann, A.F. The bile acid TGR5 membrane receptor: From basic research to clinical application. Dig. Liver Dis. 2014, 46, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Beuers, U.; Trauner, M.; Jansen, P.; Poupon, R. New paradigms in the treatment of hepatic cholestasis: From UDCA to FXR, PXR and beyond. J. Hepatol. 2015, 62 (Suppl. 1), S25–S37. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki-Anzai, S.; Masuda, M.; Levi, M.; Keenan, A.L.; Miyazaki, M. Dual activation of the bile acid nuclear receptor FXR and G-Protein-Coupled receptor TGR5 protects mice against atherosclerosis. PLoS ONE 2014, 9, e108270. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous Bile Acids Are Ligands for the Nuclear Receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Xu, G.; Li, H.; Pan, L.-X.; Shang, Q.; Honda, A.; Ananthanarayanan, M.; Erickson, S.; Shneider, B.; Shefer, S.; Bollineni, J.; et al. FXR-mediated down-regulation of CYP7A1 dominates LXRα in long-term cholesterol-fed NZW rabbits. J. Lipid Res. 2003, 44, 1956–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagahashi, M.; Takabe, K.; Liu, R.; Peng, K.; Wang, X.; Wang, Y.; Hait, N.C.; Wang, X.; Allegood, J.C.; Yamada, A.; et al. Conjugated bile acid-activated S1P receptor 2 is a key regulator of sphingosine kinase 2 and hepatic gene expression. Hepatology 2015, 61, 1216–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Radominska-Pandya, A.; Shi, Y.; Simon, C.M.; Nelson, M.C.; Ong, E.S.; Waxmann, D.J.; Evans, R.M. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc. Natl. Acad. Sci. USA 2001, 98, 3375–3380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peet, D.J.; Janowski, B.A.; Mangelsdorf, D.J. The LXRs: A new class of oxysterol receptors. Curr. Opin. Genet. Dev. 1998, 8, 571–575. [Google Scholar] [CrossRef]
- Matsubara, T.; Li, F.; Gonzalez, F.J. FXR signaling in the enterohepatic system. Mol. Cell. Endocrinol. 2013, 368, 17–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Péan, N.; Doignon, I.; Garcin, I.; Besnard, A.; Julien, B.; Liu, B.; Branchereau, S.; Spraul, A.; Guettier, C.; Humbert, L.; et al. The receptor TGR5 protects the liver from bile acid overload during liver regeneration in mice. Hepatology 2013, 58, 1451–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichikawa, R.; Takayama, T.; Yoneno, K.; Kamada, N.; Kitazume, M.T.; Higuchi, H.; Matsuoka, K.; Watanabe, M.; Itoh, H.; Kanai, T.; et al. Bile acids induce monocyte differentiation toward interleukin-12 hypo-producing dendritic cells via a TGR5-dependent pathway. Immunology 2012, 136, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Raufman, J.-P.; Chen, Y.; Cheng, K.; Compadre, C.; Compadre, L.; Zimniak, P. Selective interaction of bile acids with muscarinic receptors: A case of molecular mimicry. Eur. J. Pharmacol. 2002, 457, 77–84. [Google Scholar] [CrossRef]
- Zhu, X.; Zhou, W.; Cui, Y.; Zhu, L.; Li, J.; Feng, X.; Shao, B.; Qi, H.; Zheng, J.; Wang, H.; et al. Pilocarpine protects cobalt chloride-induced apoptosis of RGC-5 cells: Involvement of muscarinic receptors and HIF-1α pathway. Cell. Mol. Neurobiol. 2010, 30, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Serriere-Lanneau, V.; Teixeira-Clerc, F.; Li, L.; Schippers, M.; de Wries, W.; Julien, B.; Tran-Van-Nhieu, J.; Manin, S.; Poelstra, K.; Chun, J.; et al. The sphingosine 1-phosphate receptor S1P2 triggers hepatic wound healing. FASEB J. 2007, 21, 2005–2013. [Google Scholar] [CrossRef] [PubMed]
- Studer, E.; Zhou, X.; Zhao, R.; Wang, Y.; Takabe, K.; Nagahashi, M.; Pandak, W.M.; Dent, P.; Spiegel, S.; Shi, R.; et al. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology (Balt. Md.) 2012, 55, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, X.; Luo, L.; Qiang, X.; Hylemon, P.B.; Jiang, Z.; Zhang, L.; Zhou, H. Taurocholate Induces Cyclooxygenase-2 Expression via the Sphingosine 1-phosphate Receptor 2 in a Human Cholangiocarcinoma Cell Line. J. Biol. Chem. 2015, 290, 30988–31002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Means, C.K.; Brown, J.H. Sphingosine-1-phosphate receptor signaling in the heart. Cardiovasc. Res. 2009, 82, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Lenzig, P.; Oslender-Bujotzek, A.; Kusch, J.; Lucas, S.D.; Gründer, S.; Wiemuth, D. The Bile Acid-Sensitive Ion Channel (BASIC) Is activated by alterations of its membrane environment. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Mozos, I. Arrhythmia risk in liver cirrhosis. World J. Hepatol. 2015, 7, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Gui, T.; Gai, Z. Genome-wide profiling to analyze the effects of FXR activation on mouse renal proximal tubular cells. Genom. Data 2015, 6, 31–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop-Bailey, D.; Walsh, D.T.; Warner, T.D. Expression and activation of the farnesoid X receptor in the vasculature. Proc. Natl. Acad. Sci. USA 2004, 101, 3668–3773. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Lo, J.L.; Huang, L.; Zhao, A.; Metzger, E.; Adams, A.; Meinket, P.T.; Wright, S.D.; Cui, J. Lithocholic acid decreases expression of bile salt 139 export pump through farnesoid X receptor antagonist activity. J. Biol. Chem. 2002, 277, 31441–31447. [Google Scholar] [CrossRef] [PubMed]
- Tu, H. FXR, a Bile Acid Receptor and Biological Sensor. Trends Cardiovasc. Med. 2000, 10, 30–35. [Google Scholar] [CrossRef]
- Fiorucci, S.; Mencarelli, A.; Palladino, G.; Cipriani, S. Bile-acid-activated receptors: Targeting TGR5 and farnesoid-X-receptor in lipid and glucose disorders. Trends Pharmacol. Sci. 2009, 30, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Jiang, C.; Cheng, J.; Krausz, K.W.; Li, T.; Ferrell, J.M.; Gonzalez, F.J.; Chiang, J.Y.L. Bile acid signaling in lipid metabolism: Metabolomic and lipidomic analysis of lipid and bile acid markers linked to anti-obesity and anti-diabetes in mice. Biochim. Biophys. Acta (BBA) 2015, 1851, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Wang, H.; Shi, Y.; Dong, Y.; Zhang, Y.; Wang, J. Impact of bile acids on the growth of human cholangiocarcinoma via FXR. J. Hematol. Oncol. 2011, 4, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, M.S.; Mathur, B.; Eblimit, Z.; Vasquez, H.; Taegtmeyer, H.; Karpen, S.J.; Penny, D.J.; Moore, D.D.; Anakk, S. Bile acid excess induces cardiomyopathy and metabolic dysfunctions in the heart. Hepatology 2017, 65, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Jonker, J.W.; Liddle, C.; Downes, M. FXR and PXR: Potential therapeutic targets in cholestasis. J. Steroid Biochem. Mol. Biol. 2012, 130, 147–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, S.; Piquette-Miller, M. Hepatoprotective role of PXR activation and MRP3 in cholic acid-induced cholestasis. Br. J. Pharmacol. 2007, 151, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Mangelsdorf, D.J.; Meyer, U.A. Pregnane X receptor is a target of farnesoid X receptor. J. Biol. Chem. 2006, 281, 19081–19091. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Liu, D. Activation of pregnane X receptor by pregnenolone 16 alpha-carbonitrile prevents high-fat diet-induced obesity in AKR/J mice. PLoS ONE 2012, 7, e38734. [Google Scholar] [CrossRef]
- Han, S.; Li, T.; Ellis, E.; Strom, S.; Chiang, J.Y.L. A Novel Bile Acid-Activated Vitamin D Receptor Signaling in Human Hepatocytes. Mol. Endocrinol. 2010, 24, 1151–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizwa, M.; Akagi, D.; Makishima, M. Lithocholic acid is a vitamin D receptor ligand that acts preferentially in the ileum. Int. J. Mol. Sci. 2018, 19, 1975. [Google Scholar] [CrossRef] [PubMed]
- Bouillon, R.; Carmeliet, G.; Verlinden, L.; Van Etten, E.; Verstuyf, A.; Luderer, H.F.; Lieben, L.; Mathieu, C.; Demay, M. Vitamin D and human health: Lessons from vitamin D receptor null mice. Endocr. Rev. 2008, 29, 726–776. [Google Scholar] [CrossRef] [PubMed]
- Adachi, R.; Honma, Y.; Masuno, H.; Kawana, K.; Shimomura, I.; Yamada, S.; Makishima, M. Selective activation of vitamin D receptor by lithocholic acid acetate, a bile acid derivative. J. Lipid Res. 2005, 46, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Polly, P.; Tan, T.C. The role of vitamin D in skeletal and cardiac muscle function. Front. Physiol. 2014, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mozos, I.; Marginean, O. Links between Vitamin D Deficiency and Cardiovascular Diseases. Biomed. Res. Int. 2005, 2015, 109275. [Google Scholar] [CrossRef] [PubMed]
- Mozos, I.; Stoian, D.; Luca, C.T. Crosstalk between Vitamins A, B12, D, K, C, and E Status and Arterial Stiffness. Dis. Markers 2017, 2017, 8784971. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Hiipakka, R.A.; Liao, S. Selective activation of liver X receptor α by 6α-hydroxy bile acids and analogs. Steroids 2000, 65, 423–427. [Google Scholar] [CrossRef]
- Li, T.; Matozel, M.; Boehme, S.; Kong, B.; Nilsson, L.M.; Guo, G.; Ellis, E.; Chiang, J.Y. Overexpression of cholesterol 7α-hydroxylase promotes hepatic bile acid synthesis and secretion and maintains cholesterol homeostasis. Hepatology 2011, 53, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukumsumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Keitel, V.; Donner, M.; Winandy, S.; Kubitz, R.; Häussinger, D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem. Biophys. Res. Commun. 2008, 372, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Pols, T.W.H.; Nomura, M.; Stein, S.; Pellicciari, R.; Schoonjans, K. TGR5 reduces macrophage migration through mTOR-induced C/EBPβ differential translation. J. Clin. Investig. 2014, 124, 5424–5436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kida, T.; Tsubosaka, Y.; Hori, M.; Ozaki, H.; Murata, T. Bile acid receptor TGR5 agonism induces no production and reduces monocyte adhesion in vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Ehlert, F.J.; Roeske, W.R.; Yamamura, H.I. Molecular Biology, Pharmacology, and Brain Distribution of Subtypes the Muscarinic Receptor. Distribution 1950, 64, 1–13. [Google Scholar]
- Mitchelson, F. Muscarinic Receptors; Fryer, D.A., Christopoulos, A., Nathanson, M.N., Eds.; Springer: Berlin/Heidelberg, Germnay, 2012; pp. 263–298. [Google Scholar]
- Dhein, S.; van Koppen, C.J.; Brodde, O.E. Muscarinic receptors in the mammalian heart. Pharmacol. Res. 2001, 44, 161–182. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Khurana, S.; Chen, Y.; Kennedy, R.H.; Zimniak, P.; Raufman, J.-P. Lithocholylcholine, a bile acid/acetylcholine hybrid, is a muscarinic receptor antagonist. J. Pharmacol. Exp. Ther. 2002, 303, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Sheikh Abdul Kadir, S.H.; Miragoli, M.; Abu-Hayyeh, S.; Moshkov, A.V.; Xie, Q.; Keitel, V.; Nikolaev, V.O.; Williamson, C.; Gorelik, J. Bile acid-induced arrhythmia is mediated by muscarinic M2 receptors in neonatal rat cardiomyocytes. PLoS ONE 2010, 5, e9689. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, E.; Diakonov, I.; Arunthavarajah, D.; Swift, T.; Goodwin, M.; Mcllyride, S.; Nikolova, V.; Williamson, C.; Gorelik, J. Bile acids and their respective conjugates elicit different responses in neonatal cardiomyocytes: Role of Gi protein, muscarinic receptors and TGR5. Sci. Rep. 2018, 8, 7110–7122. [Google Scholar] [CrossRef] [PubMed]
- Kwong, E.; Li, Y.; Hylemon, P.B.; Zhou, H. Bile acids and sphingosine-1-phosphate receptor 2 in hepatic lipid metabolism. Acta Pharm. Sin. B 2015, 5, 151–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukiya, A.N.; McMillan, J.; Parrill, A.L.; Dopico, A.M. Structural determinants of monohydroxylated bile acids to activate beta 1 subunit-containing BK channels. J. Lipid Res. 2008, 49, 2441–2451. [Google Scholar] [CrossRef] [PubMed]
- Bukiya, A.N.; McMillan, J.; Liu, J.; Shivakumar, B.; Parrill, A.L.; Dopico, A.M. Activation of calcium- and voltage-gated potassium channels of large conductance by leukotriene B4. J. Biol. Chem. 2014, 289, 35314–35325. [Google Scholar] [CrossRef] [PubMed]
- Binah, O.; Rubinstein, I.; Bomzon, A.; Better, O.S. Effects of bile acids on ventricular muscle contraction and electrophysiological properties: Studies in rat papillary muscle and isolated ventricular myocytes. Naunyn Schmiedebergs. Arch. Pharmacol. 1987, 335, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Laifer, S.A.; Stiller, R.J.; Siddiqui, D.S.; Dunston-Boone, G.; Whetham, J.C. Ursodeoxycholic acid for the treatment of intrahepatic cholestasis of pregnancy. J. Matern.-Fetal Med. 2001, 10, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Sokolovic, D.; Nikolic, I.; Kocic, G.; Ievtovic-Stoimenov, T.; Veljkovic, A.; Stojanovic, M.; Stanojkovic, Z.; Sokolovic, D.M.; Jelic, M. The effect of ursodeoxycholic acid on oxidative stress level and DNase activity in rat liver after bile duct ligation. Drug Chem. Toxicol. 2012, 36, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Yacoub, A.; Studer, E.; Gupta, S.; Pei, X.Y.; Grant, S.; Hylemon, P.B.; Dent, P. Inhibition of the MAPK and PI3K pathways enhances UDCA-induced apoptosis in primary rodent hepatocytes. Hepatology (Balt. MD) 2002, 35, 779–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Im, E.; Martinez, J.D. UDCA can inhibit DCA-induced apoptosis via modulation of EGFR/Raf-1/ERK signaling in human colon cancer cells. J. Nutr. 2004, 134, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Alberts, D.S.; Martínez, M.E.; Hess, L.M.; Einspahr, J.G.; Green, S.B.; Bhattacharyya, A.K.; Guillen, J.; Krutzsch, M.; Batta, A.K.; Salen, G.; et al. Phase III trial of ursodeoxycholic acid to prevent colorectal adenoma recurrence. J. Natl. Cancer Inst. 2005, 97, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Laukens, D.; Devisscher, L.; Van den Bossche, L.; Hindryckx, P.; Vandenbroucke, R.E.; Vandewynckel, Y.P.; Cuvelier, C.; Brinkman, B.M.; Libert, C.; Vandenabeele, P.; et al. Tauroursodeoxycholic acid inhibits experimental colitis by preventing early intestinal epithelial cell death. Lab. Investig. 2014, 94, 1419–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, E.; Haller, D. Structure-function analysis of the tertiary bile acid TUDCA for the resolution of endoplasmic reticulum stress in intestinal epithelial cells. Biochem. Biophys. Res. Commun. 2011, 409, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Fekaj, E.; Gjata, A.; Maxhuni, M. The effect of ursodeoxycholic acid in liver functional restoration of patients with obstructive jaundice after endoscopic treatment: A prospective, randomized, and controlled study. BMC Surg. 2013, 13, 38. [Google Scholar] [CrossRef] [PubMed]
- Geenes, V.; Williamson, C. Intrahepatic cholestasis of pregnancy. World J. Gastroenterol. 2009, 15, 1–16. [Google Scholar] [CrossRef]
- Beuers, U. Drug insight: Mechanisms and sites of action of ursodeoxycholic acid in cholestasis. Nature Clinical Practice. Gastroenterol. Hepatol. 2006, 3, 318–328. [Google Scholar] [CrossRef]
- Thistle, J.L.; Larusso, N.F.; Hofmann, A.F.; Turcotte, J.; Carlson, G.L.; Ott, B.J. Differing effects of ursodeoxycholic or chenodeoxycholic acid on biliary cholesterol saturation and bile acid metabolism in man. A dose-response study. Dig. Dis. Sci. 1982, 27, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Mas, M.R.; Comert, B.; Mas, N.; Yamanel, L.; Ozotuk, H.; Tasci, I.; Jasrawi, R.P. Effects of long-term hydrophilic bile acid therapy on in vitro containing of gallbladder muscle strips in patients with cholesterol gallstones. World J. Gastroenterol. 2007, 13, 4336–4339. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.M.P.; Fan, G.; Ma, X.; Kren, B.T.; Steer, C.J. A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. J. Clin. Investig. 1998, 101, 2790–2799. [Google Scholar] [CrossRef] [PubMed]
- Amaral, J.D.; Viana, R.J.S.; Ramalho, R.M.; Steer, C.J.; Rodrigues, C.M.P. Bile acids: Regulation of apoptosis by ursodeoxycholic acid. J. Lipid Res. 2009, 50, 1721–1734. [Google Scholar] [CrossRef] [PubMed]
- Halilbasic, E.; Claudel, T.; Trauner, M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J. Hepatol. 2013, 58, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Palmela, I.; Correia, L.; Silva, R.F.M.; Sasaki, H.; Kim, K.S.; Brites, D.; Brito, M.A. Hydrophilic bile acids protect human blood-brain barrier endothelial cells from disruption by unconjugated bilirubin: An in vitro study. Front. Neurosci. 2015, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Boatright, J.H.; Nickerson, J.M.; Moring, A.G.; Pardue, M.T. Bile acids in treatment of ocular disease. J. Ocul. Biol. Dis. Inf. 2009, 2, 149–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, M.; Une, N.; Nishizawa, M.; Suzuki, S.; Ito, H.; Horiuchi, T. Incretin secretion stimulated by ursodeoxycholic acid in healthy subjects. SpringerPlus 2013, 2, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Guarino, M.P.; Carotti, S.; Morini, S.; Perrone, G.; Behar, J.; Altomare, A.; Alloni, R.; Caviglia, R.; Emerenziani, S.; Rabitti, C.; et al. Decreased number of activated macrophages in gallbladder muscle layer of cholesterol gallstone patients following ursodeoxycholic acid. Gut 2008, 57, 1740–1741. [Google Scholar] [CrossRef] [PubMed]
- Mortiboys, H.; Aasly, J.; Bandmann, O. Ursocholanic acid rescues mitochondrial function in common forms of familial Parkinson’s disease. Brain 2013, 136, 3038–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Gaspá, S.; Dubreuil, M.; Guaňabens, N.; Combalia, A.; Peris, P.; Monegal, A.; Parés, A. Ursodeoxycholic acid decreases bilirubin-induced osteoblast apoptosis. Eur. J. Clin. Investig. 2014, 44, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Williamson, C.; Gorelik, J.; Eaton, B.M.; Lab, M.; de Swiet, M.; Korchev, Y. The bile acid taurocholate impairs rat cardiomyocyte function: A proposed mechanism for intra-uterine fetal death in obstetric cholestasis. Clin. Sci. (Lond. Engl.) 2001, 100, 363–369. [Google Scholar] [CrossRef]
- Gorelik, J.; Harding, S.E.; Shevchuk, A.I.; Koralage, D.; Swiet, M.D.E.; Korchev, Y.; Williamson, C. Taurocholate induces changes in rat cardiomyocyte contraction and calcium dynamics. Clin. Sci. (Lond.) 2002, 200, 191–200. [Google Scholar] [CrossRef]
- Miragoli, M.; Kadir, S.H.S.A.; Sheppard, M.N.; Salvarani, N.; Virta, M.; Wells, S.; Lab, M.J.; Nikolaev, V.O.; Moshkov, A.; Hague, W.M.; et al. A protective antiarrhythmic role of ursodeoxycholic acid in an in vitro rat model of the cholestatic fetal heart. Hepatology (Balt. MD) 2011, 54, 1282–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh Abdul Kadir, S.H.; Ali, N.N.; Abu-Hayyeh, S.; Harding, S.E.; Williamson, C.; Gorelik, J. The use of embryonic stem cell-derived cardiomyocytes as a model to study fetal arrhythmia related to maternal disease. J. Cell. Mol. Med. 2009, 13, 3730–3741. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.S.; Hanafi, N.I.; Siran, R.; Md Nor, J.; Abdul Hamid Hasani, N.; Ab Rahim, S.; Sheikh Abdul Kadir, S.H. Ursodeoxycholic acid protects cardiomyocytes against cobalt chloride induced hypoxia by regulating transcriptional mediator of cells stress hypoxia inducible factor 1α and p53 protein. Cell Biochem. Funct. 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, J. Dexamethasone and ursodeoxycholic acid protect against the arrhythmogenic effect of taurocholate in an in vitro study of rat cardiomyocytes. BJOG Int. J. Obstet. Gynaecol. 2003, 110, 424–429. [Google Scholar] [CrossRef]
- Anwer, M.S.; Engelking, L.R.; Nolan, K.; Sullivan, D.; Zimniak, P.; Lester, R. Hepatotoxic bile acids increase cytosolic Ca++ activity of isolated rat hepatocytes. Hepatology 1988, 8, 887–891. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Francis, H.; Glaser, S.; Alpini, G.; LeSage, G. Bile Acid Interactions with Cholangiocytes. World J. Gastroenterol. 2006, 12, 3553–3563. [Google Scholar] [CrossRef] [PubMed]
- Combettes, L.; Berthon, B.; Doucet, E.; Erlinger, S.; Claret, M. Characteristics of bile acid-mediated Ca release from permeabilized liver cells and liver microsomes. J. Biol. Chem. 1989, 264, 157–167. [Google Scholar] [PubMed]
- Takashi, S.; Kuboyama, A.; Mita, M.; Murata, S.; Shimizu, M.; Inoue, J.; Mori, K.; Sato, R. The Exercise-Inducible Bile Acid Receptor Tgr5 Improves Skeletal Muscle Function in Mice. J. Biol. Chem. 2018, 293, 10322–10332. [Google Scholar] [CrossRef]
- Coquil, J.; Berthon, B.; Chomiki, N.; Combettes, L.; Jourdon, P.; Schteingart, C.; Erlinger, S.; Claret, M. Effects of taurolithocholate, a Ca2(+)-mobilizing agent, on cell Ca2(+) in rat hepatocytes, human platelets and neuroblastoma NG108-15 cell line. Biochem. J. 1991, 273, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Bradley, M.N.; Hong, C.; Chen, M.; Joseph, S.B.; Wilpitz, D.C.; Wang, X.; Lusis, A.J.; Collins, A.; Hseuh, W.A.; Collins, J.L.; et al. Ligand activation of LXRβ reverses atherosclerosis and cellular cholesterol overload in mice lacking LXRα and apoE. J. Clin. Investig. 2007, 117, 2337–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Lee, W.; Han, S.; Cho, T. Effect of Ursodeoxycholic Acid Injury in Isolated Rat Heart Ischemia/Reperfusion. Arch. Pharm. Res. 1999, 22, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, K.G.; Suzuki, R.; Maeda, H.; Yamamoto, M.; Yutong, X.; Sasaguri, S. Hydrophilic bile salt ursodeoxycholic acid protects myocardium against reperfusion injury in a PI3K/Akt dependent pathway. J. Mol. Cell. Cardiol. 2005, 39, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.; Elshazly, S.M. Ursodeoxycholic Acid ameliorates fructose-induced metabolic syndrome in rats. PLoS ONE 2014, 9, e106993. [Google Scholar] [CrossRef] [PubMed]
- Hanafi, N.I.; Mohamed, A.S.; Md Noor, J.; Abdul Hamid Hasani, N.; Siran, R.; Osman, N.J.; Ab Rahim, S.; Sheikh Abdul Kadir, S.H. Ursodeoxycholic acid upregulates ERK and Akt in the protection of cardiomyocytes against CoCl2. Genet. Mol. Res. GMR 2016, 15. [Google Scholar] [CrossRef]
- Hanafi, N.I.; Sheikh Abdul Kadir, S.H.; Mohamed, A.S.; Md Noor, J.; Osman, N.J.; Osman, N.J.; Ab Rahim, S.; Abdul Hamid Hasani, N. Ursodeoxycholic Acid Regulates Caspase-9 and ROS Production in Protecting Cardiomyocytes Against Hypoxia. J. Teknol. (Sci. Eng.) UTM 2017, 79, 1–8. [Google Scholar]
- Chung, J.; An, S.H.; Kang, S.W.; Kwon, K. Ursodeoxycholic Acid (UDCA) Exerts Anti-Atherogenic Effects by Inhibiting RAGE Signaling in Diabetic Atherosclerosis. PLoS ONE 2016, 11, e0147839. [Google Scholar] [CrossRef] [PubMed]
- Sinisalo, J.; Vanhanen, H.; Pajunen, P.; Vapaatalo, H.; Nieminen, M.S. Ursodeoxycholic acid and endothelial-dependent, nitric oxide-independent vasodilatation of forearm resistance arteries in patients with coronary heart disease. Br. J. Clin. Pharmacol. 1999, 47, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Von Haehling, S.; Schefold, J.C.; Jankowska, E.A.; Springer, J.; Vazir, A.; Kalra, P.R.; Sandek, A.; Fauler, G.; Stojakovic, T.; Trauner, M.; et al. Ursodeoxycholic acid in patients with chronic heart failure: A double-blind, randomized, placebo-controlled, crossover trial. J. Am. Coll. Cardiol. 2012, 59, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Bährle, S.; Szabó, G.; Stiehl, A.; Theilmann, L.; Tj, D.; Zimmermann, R.; Kübler, W. Adjuvant treatment with ursodeoxycholic acid may reduce the incidence of acute cardiac allograft rejection. J. Heart Lung Transplant. 1998, 17, 592–598. [Google Scholar] [PubMed]
- Liu, X.; Fassett, J.; Wei, Y.; Chen, Y. Regulation of DDAH1 as a Potential Therapeutic Target for Treating Cardiovascular Diseases. Évid.-Based Complement. Altern. Med. ECAM 2013, 2013, 619207. [Google Scholar] [CrossRef] [PubMed]
- Vasavan, T.; Ferraro, E.; Ibrahim, E.; Dixon, P.; Gorelik, J.; Williamson, C. Heart and bile acids–Clinical consequences of altered bile acid metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864 (4 Pt B.), 1345–1355. [Google Scholar] [CrossRef]
- Voiosu, A.M.; Wiese, S.; Voiosu, T.A.; Hove, J.; Bendtsen, F.; Møller, S. Total bile acid levels are associated with left atrial volume and cardiac output in patients with cirrhosis. Eur. J. Gastroenterol. Hepatol. 2018, 30, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Macrophage Apoptosis in Atherosclerosis: Consequences on Plaque Progression and the Role of Endoplasmic Reticulum. Antioxid Redox Signal 2009, 11, 2333–2339. [Google Scholar] [CrossRef] [PubMed]
- Granados, D.P.; Tanguay, P.-L.; Hardy, M.-P., Caron; de Verteuil, D.; Meloche, S.; Perreault, C. ER stress processing of MHC class I-associated peptides. BMC Immunology 2009, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, L.; Hindryckx, P.D.; Welden, L.D.V.; Holvoet, S.; Vilchez-Vargas, T.; Vital, R.; Pieper, M.; Bussche, D.H.; Vanhaecke, J.V.; Vanhaecke, L.; et al. Ursodeoxycholic Acid and Its Taurine- or Colitogenic Dysbiosis and Equally Suppress Experimental Colitis in Mice. Appl. Environ. Microbiol. 2017, 83, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, B.M.; Hildebrand, F.; Kubica, M.; Goosens, D.; Del Favero, J.; Declercq, W.; Raes, J.; Vandenabeele, P. Caspase deficiency alters the murine gut microbiome. Cell Death Dis. 2011, 2, e220. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, L.E.; Sturino, J.M.; Carroll, R.J.; Rooney, L.W.; Azcarate-Peril, M.A.; Turner, N.D. Polyphenol-rich sorghum brans alter colon microbiota and impact species diversity and species richness after multiple bouts of dextran sodium sulfate-induced colitis. FEMS Microbiol. Ecol. 2015, 91, fiv008. [Google Scholar] [CrossRef] [PubMed]
- Sadeghzadeh, J.; Vakili, A.; Sameni, H.R.; Shadnoush, M.; Bandegi, A.R.; Khorasani, M.Z. The effect of oral consumption of probiotics in prevention of heart injury in a rat myocardial infarction model: A. histopathological, hemodynamic and biochemical evaluation. Iran. Biomed. J. 2017, 21, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.T.; Ettinger, G.; Huang, C.X.; Burton, J.P.; Haist, J.V.; Rajapurohitam, V.; Sidaway, J.E.; Martin, G.; Gloor, G.B.; Swann, J.R.; et al. Probiotic administration attenuates myocardial hypertrophy and heart failure after myocardial infarction in the rat. Circ. Heart Fail. 2014, 7, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Costanza, A.C.; Moscavitch, S.D.; Faria Neto, H.C.; Mesquita, E.T. Probiotic therapy with Saccharomyces boulardii for heart failure patients: A. randomized, double-blind, placebo-controlled pilot trial. Int. J. Cardiol. 2015, 179, 348–350. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Cheng, Y.; Wu, J.; Tauschel, H.-D.; Duan, R.-D. Ursodeoxycholic acid differentially affects three types of sphingomyelinase in human colon cancer Caco 2 cells. Cancer Lett. 2006, 235, 141–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Empinado, H.M.; Deevska, G.M.; Nikolova-Karakashian, M.; Yoo, J.-K.; Christou, D.D.; Ferreira, L.F. Diaphragm dysfunction in heart failure is accompanied by increases in neutral sphingomyelinase activity and ceramide content. Eur. J. Heart Fail. 2014, 16, 519–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavoine, C.; Pecker, F. Sphingomyelinases: Their regulation and roles in cardiovascular pathophysiology. Cardiovasc. Res. 2009, 82, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, O.M.; Discher, D.J.; Bishopric, N.H.; Webster, K.A. Rapid Activation of Neutral Sphingomyelinase by Hypoxia-Reoxygenation of Cardiac Myocytes. Circ. Res. 2000, 86, 198–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Pan, W.; Yang, T.; Zhang, G.; Shi, R. Relationship Between Sphingomyelinase, Ceramide and Clinical Presentation, Extent and Severity of Atherosclerotic Coronary Artery Disease. Heart 2012, 98 (Suppl. 2), 177–181. [Google Scholar] [CrossRef]
- Klevstig, M.; Ståhlman, M.; Lundqvist, A.; Scharin Täng, M.; Fogelstrand, P.; Adiels, M.; Anderson, L.; Kolesnick, R.; Jeppsson, A.; Borén, J.; et al. Targeting acid sphingomyelinase reduces cardiac ceramide accumulation in the post-ischemic heart. J. Mol. Cell. Cardiol. 2016, 93, 69–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Means, C.K.; Xiao, C.; Li, Z.; Zhang, T.; Omens, J.H.; Ishii, I.; Chun, J.; Brown, J.H. Sphingosine 1-phosphate S1P2 and S1P3 receptor-mediated Akt activation protects against in vivo myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, 2944–2951. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | Tissue | Bile Acid | Summary of Implications of High BA Pool | References |
|---|---|---|---|---|
| FXR | Liver, cholangiocytes, colonocytes, small intestine, heart | CDCA, DCA, LCA | Glucose metabolism and cholesterol metabolism are altered, which result in downregulation of LDL-R expression, increase in LDL-C levels, and upregulation of transcriptional activity of bile acids. | [14,15,16,17] |
| Others; PXR, LXR, VDR, S1P | Liver and heart | LCA | The receptors regulate hepatic lipid metabolism, activate ERK 1/2, and Akt and then lead to regulation of lipid and glucose metabolism. | [18,19,20,21] |
| TGR5 | Liver, heart, dendritic cells, | TLCA, LCA, DCA, CDCA, CA, UDCA, | Modulates insulin signaling pathway and aids in the regulation hepatic glucose metabolism and inhibition of LPS-induced cytokine expression. | [13,22,23] |
| Muscarinic | Liver, brain, eyes, heart, and colon carcinoma | Lithocholyltaurine (LCT), TCA | Modulate glucose homeostasis, thermogenesis, inflammatory response, and stimulate parasympathetic nerves. | [24,25] |
| Sphingosine-1-phospahate (S1P) | Cholangio carcinoma, heart, liver | TCA | Promotes cholangiocarcinoma growth, lipid metabolism, angiogenesis, and cardiac cellular signaling. | [26,27,28,29] |
| Large conductance voltage- and Ca2+-activated potassium (K+) (BK) channels | Liver and intestinal tract | LCA | Improves vascular muscle cells vasodilation. | [30,31] |
| Model | Suggested UDCA Mechanism of Action | Concentration of UDCA Used | References |
|---|---|---|---|
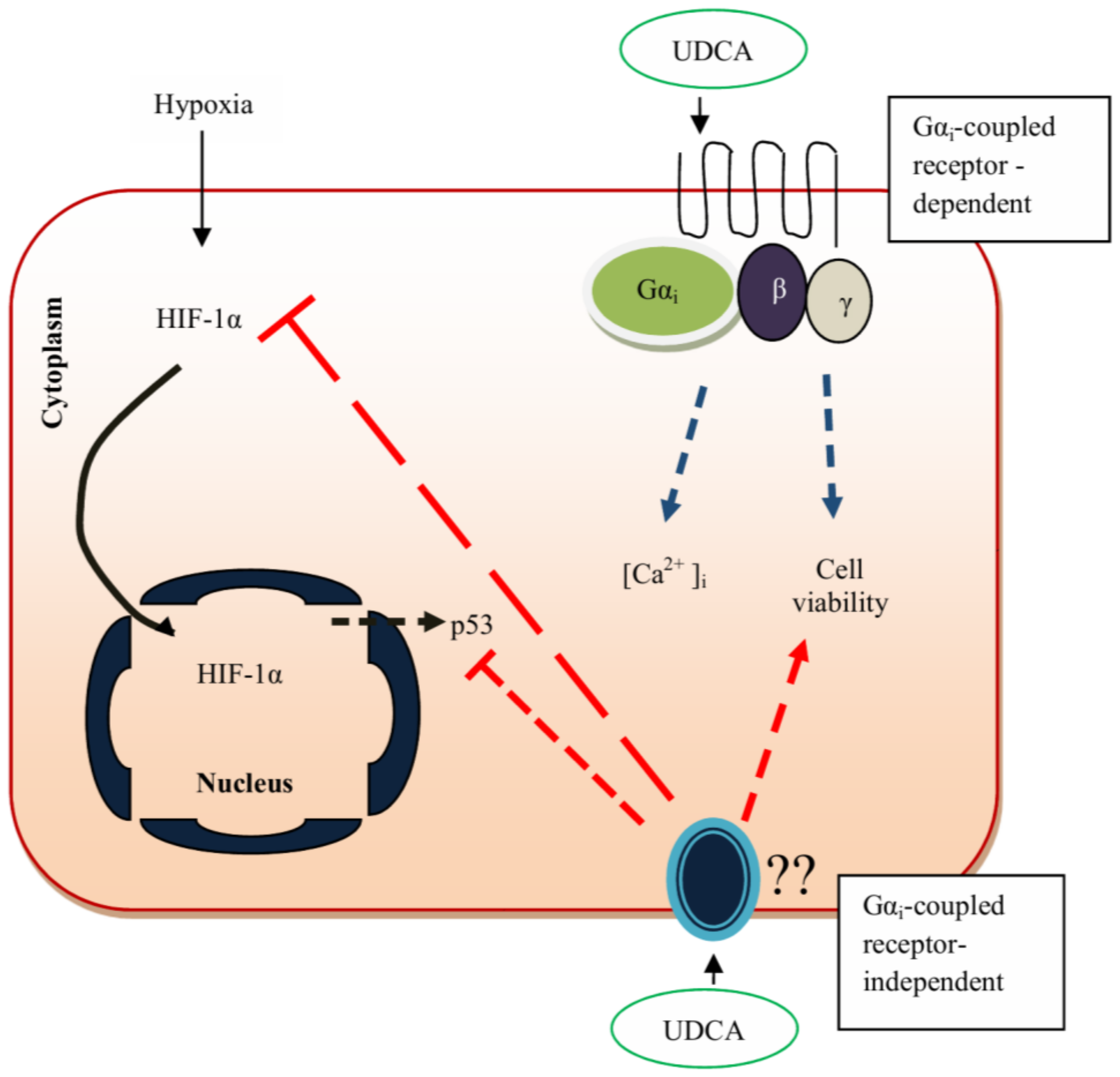
| In vitro rat model of the fetal heart | UDCA induces cAMP release without any effects on contraction rate, which is mediated through TGR5. | 100 µM | [63] |
| In vitro rat model of the cholestatic fetal heart | UDCA activates KATP channels and improves intracellular calcium level. | 100 µM | [91] |
| In vitro rat model of ischemia–reperfusion | UDCA reduces LDH release and enhances the recovery of cardiac contractile function during reperfusion. | 80–160 µM | [101] |
| In vitro and in vivo rat models of ischemia–reperfusion | UDCA inhibits the opening of MPTP and Bcl-2 via PI3K/Akt pathway. | 40 mg/kg | [102] |
| In vivo rat model of metabolic syndrome | UDCA reduces uric acid level and improves insulin resistance of fructose-induced metabolic syndrome rat. | 150 mg/kg | [103] |
| In vitro rat model of hypoxic cells | UDCA inhibits HIF-1α expression, upregulates ERK 1/2, and Akt while downregulating caspase-9 and reactive oxygen species (ROS) generation in cobalt chloride (CoCl2)-induced hypoxic CMs. | 100 µM | [93,104,105] |
| In vivo mouse model of diabetic atherosclerosis | UDCA exerts antiatherogenic activity through reduction of endoplasmic reticulum stress, receptor for advanced glycation end product (RAGE) signaling, and proinflammatory responses of ROS and Nf-κB. | 100 µM | [106] |
| Patients with coronary heart disease | UDCA improves endothelium- and NO-independent vasodilatation that maintains the arterial flow in patients with heart failure. | 13–19 mg/kg | [107] |
| Patients with chronic heart failure | UDCA improves liver function and lowers the level of γ-glutamyl transferase, aspartate transaminase, and soluble TNF-α receptor 1. | 1280 µM | [108] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanafi, N.I.; Mohamed, A.S.; Sheikh Abdul Kadir, S.H.; Othman, M.H.D. Overview of Bile Acids Signaling and Perspective on the Signal of Ursodeoxycholic Acid, the Most Hydrophilic Bile Acid, in the Heart. Biomolecules 2018, 8, 159. https://doi.org/10.3390/biom8040159
Hanafi NI, Mohamed AS, Sheikh Abdul Kadir SH, Othman MHD. Overview of Bile Acids Signaling and Perspective on the Signal of Ursodeoxycholic Acid, the Most Hydrophilic Bile Acid, in the Heart. Biomolecules. 2018; 8(4):159. https://doi.org/10.3390/biom8040159
Chicago/Turabian StyleHanafi, Noorul Izzati, Anis Syamimi Mohamed, Siti Hamimah Sheikh Abdul Kadir, and Mohd Hafiz Dzarfan Othman. 2018. "Overview of Bile Acids Signaling and Perspective on the Signal of Ursodeoxycholic Acid, the Most Hydrophilic Bile Acid, in the Heart" Biomolecules 8, no. 4: 159. https://doi.org/10.3390/biom8040159