Administration of Enalapril Started Late in Life Attenuates Hypertrophy and Oxidative Stress Burden, Increases Mitochondrial Mass, and Modulates Mitochondrial Quality Control Signaling in the Rat Heart

 ,
,  ,
, 
 ,
, 


Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Western Immunoblotting
2.3. Quantification of Mitochondrial DNA Content
2.4. Relative Quantification of Mitochondrial DNA4834 Deletion
2.5. Measurement of Citrate Synthase Activity
2.6. Detection of Mitochondrial DNA Oxidative Damage
2.7. Analysis of TFAM Binding to mtDNA by Mitochondrial Immunoprecipitation
2.8. Determination of Protein Carbonylation
2.9. Statistical Analysis
3. Results
3.1. Morphological and Physiological Characteristics of Experimental Animals
3.2. Oxidative Stress and Mitochondrial DNA Lesions
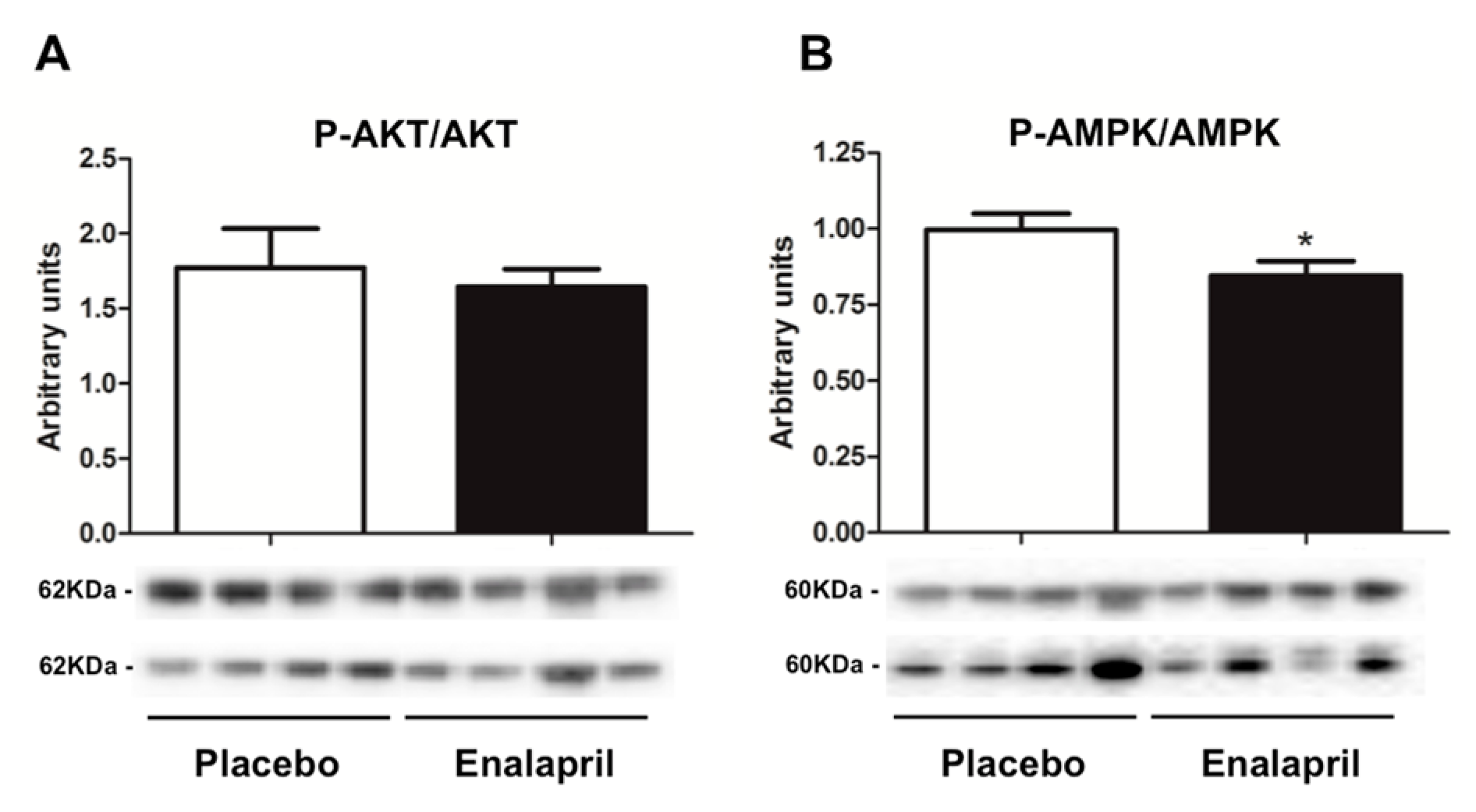
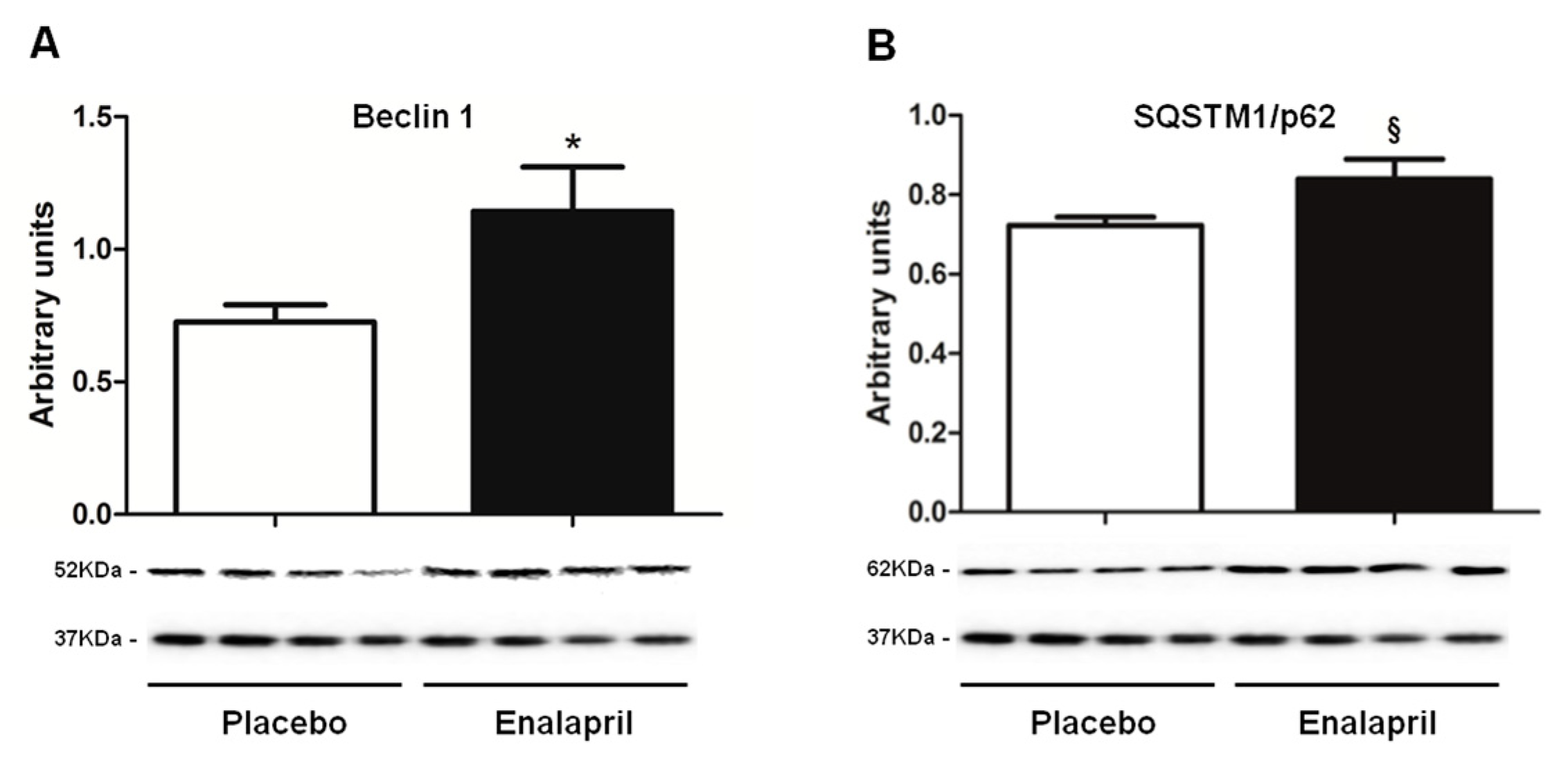
3.3. Protein Expression of Selected Mediators of Energy Metabolism, Mitochondrial Biogenesis, Dynamics and Autophagy
3.4. Analysis of Mitochondrial Transcription Factor A Binding to Specific Regions of Mitochondrial DNA
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wohlgemuth, S.E.; Calvani, R.; Marzetti, E. The interplay between autophagy and mitochondrial dysfunction in oxidative stress-induced cardiac aging and pathology. J. Mol. Cell. Cardiol. 2014, 71, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Hepple, R.T. Impact of aging on mitochondrial function in cardiac and skeletal muscle. Free Radic. Biol. Med. 2016, 98, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Fischer, F.; Hamann, A.; Osiewacz, H.D. Mitochondrial quality control: An integrated network of pathways. Trends Biochem. Sci. 2012, 37, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Judge, S.; Jang, Y.M.; Smith, A.; Hagen, T.; Leeuwenburgh, C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: Implications for the mitochondrial theory of aging. FASEB J. 2005, 19, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Heath, S.-H.; Hagen, T.M. Two subpopulations of mitochondria in the aging rat heart display heterogenous levels of oxidative stress. Free Radic. Biol. Med. 2003, 35, 1064–1072. [Google Scholar] [CrossRef]
- Chiao, Y.A.; Rabinovitch, P.S. The aging heart. Cold Spring Harb. Perspect. Med. 2015, 5, a025148. [Google Scholar] [CrossRef] [PubMed]
- Muftuoglu, M.; Mori, M.P.; de Souza-Pinto, N.C. Formation and repair of oxidative damage in the mitochondrial DNA. Mitochondrion 2014, 17, 164–181. [Google Scholar] [CrossRef] [PubMed]
- Prithivirajsingh, S.; Story, M.D.; Bergh, S.A.; Geara, F.B.; Ang, K.K.; Ismail, S.M.; Stevens, C.W.; Buchholz, T.A.; Brock, W.A. Accumulation of the common mitochondrial DNA deletion induced by ionizing radiation. FEBS Lett. 2004, 571, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Liu, V.W.; Zhang, C.; Nagley, P. Mutations in mitochondrial DNA accumulate differentially in three different human tissues during ageing. Nucleic Acids Res. 1998, 26, 1268–1275. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, S.A.; Hanke, T.; Erasmi, A.W.; Bechtel, M.J.F.; Scharfschwerdt, M.; Meissner, C.; Sievers, H.H.; Gosslau, A. Mitochondrial DNA deletions and the aging heart. Exp. Gerontol. 2006, 41, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Møller, P.; Løhr, M.; Folkmann, J.K.; Mikkelsen, L.; Loft, S. Aging and oxidatively damaged nuclear DNA in animal organs. Free Radic. Biol. Med. 2010, 48, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Steenken, S.; Jovanovic, S.V. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J. Am. Chem. Soc. 1997, 119, 617–618. [Google Scholar] [CrossRef]
- Herrero, A.; Barja, G. 8-oxo-deoxyguanosine levels in heart and brain mitochondrial and nuclear DNA of two mammals and three birds in relation to their different rates of aging. Aging 1999, 11, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Kino, K.; Sugiyama, H. Possible cause of G-C–>C-G transversion mutation by guanine oxidation product, imidazolone. Chem. Biol. 2001, 8, 369–378. [Google Scholar] [CrossRef]
- Ozawa, T. Mechanism of somatic mitochondrial DNA mutations associated with age and diseases. Biochim. Biophys. Acta 1995, 1271, 177–189. [Google Scholar] [CrossRef]
- Fisher, R.P.; Clayton, D.A. Purification and characterization of human mitochondrial transcription factor 1. Mol. Cell. Biol. 1988, 8, 3496–3509. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Maynard, S.; Bayne, A.-C.V.; Sykora, P.; Tian, J.; de Souza-Pinto, N.C.; Croteau, D.L.; Bohr, V.A. The mitochondrial transcription factor A functions in mitochondrial base excision repair. DNA Repair 2010, 9, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Chimienti, G.; Picca, A.; Sirago, G.; Fracasso, F.; Calvani, R.; Bernabei, R.; Russo, F.; Carter, C.S.; Leeuwenburgh, C.; Pesce, V.; et al. Increased TFAM binding to mtDNA damage hot spots is associated with mtDNA loss in aged rat heart. Free Radic. Biol. Med. 2018, 124, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.S.; Giovannini, S.; Seo, D.-O.; Dupree, J.; Morgan, D.; Chung, H.Y.; Lees, H.; Daniels, M.; Hubbard, G.B.; Lee, S.; et al. Differential effects of enalapril and losartan on body composition and indices of muscle quality in aged male Fischer 344 × Brown Norway rats. Age 2011, 33, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Marzetti, E.; Calvani, R.; DuPree, J.; Lees, H.A.; Giovannini, S.; Seo, D.-O.; Buford, T.W.; Sweet, K.; Morgan, D.; Strehler, K.Y.E.; et al. Late-life enalapril administration induces nitric oxide-dependent and independent metabolic adaptations in the rat skeletal muscle. Age 2013, 35, 1061–1075. [Google Scholar] [CrossRef] [PubMed]
- Piotrkowski, B.; Koch, O.R.; De Cavanagh, E.M.V.; Fraga, C.G. Cardiac mitochondrial function and tissue remodelling are improved by a non-antihypertensive dose of enalapril in spontaneously hypertensive rats. Free Radic. Res. 2009, 43, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.S. (University of Alabama at Birmingham, Birmingham, AL, USA). Pilot study for enalapril dose-finding. Unpublished word. 2006. [Google Scholar]
- Nicassio, L.; Fracasso, F.; Sirago, G.; Musicco, C.; Picca, A.; Marzetti, E.; Calvani, R.; Cantatore, P.; Gadaleta, M.N.; Pesce, V. Dietary supplementation with acetyl-l-carnitine counteracts age-related alterations of mitochondrial biogenesis, dynamics and antioxidant defenses in brain of old rats. Exp. Gerontol. 2017, 98, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Fracasso, F.; Pesce, V.; Cantatore, P.; Joseph, A.-M.; Leeuwenburgh, C.; Gadaleta, M.N.; Lezza, A.M.S. Age- and calorie restriction-related changes in rat brain mitochondrial DNA and TFAM binding. Age 2013, 35, 1607–1620. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Srere, P.A. [1] Citrate synthase: [EC 4.1.3.7. Citrate oxaloacetate-lyase (CoA-acetylating)]. Methods Enzymol. 1969, 13, 3–11. [Google Scholar] [CrossRef]
- Pastukh, V.; Ruchko, M.; Gorodnya, O.; Wilson, G.L.; Gillespie, M.N. Sequence-specific oxidative base modifications in hypoxia-inducible genes. Free Radic. Biol. Med. 2007, 43, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Pastukh, V.M.; Gorodnya, O.M.; Gillespie, M.N.; Ruchko, M.V. Regulation of mitochondrial genome replication by hypoxia: The role of DNA oxidation in D-loop region. Free Radic. Biol. Med. 2016, 96, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Pesce, V.; Fracasso, F.; Joseph, A.-M.; Leeuwenburgh, C.; Lezza, A.M.S. A comparison among the tissue-specific effects of aging and calorie restriction on TFAM amount and TFAM-binding activity to mtDNA in rat. Biochim. Biophys. Acta 2014, 1840, 2184–2191. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Kauppila, T.E.S.; Motori, E.; Li, X.; Atanassov, I.; Folz-Donahue, K.; Bonekamp, N.A.; Albarran-Gutierrez, S.; Stewart, J.B.; Larsson, N.-G. Increased total mtDNA copy number cures male infertility despite unaltered mtDNA mutation load. Cell Metab. 2017, 26, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.J.; Reeve, A.K.; Samuels, D.C.; Chinnery, P.F.; Blackwood, J.K.; Taylor, R.W.; Wanrooij, S.; Spelbrink, J.N.; Lightowlers, R.N.; Turnbull, D.M. What causes mitochondrial DNA deletions in human cells? Nat. Genet. 2008, 40, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Moraes, C.T. Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum. Mol. Genet. 2009, 18, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, K.; Kanki, T.; Fukuoh, A.; Kurisaki, H.; Aoki, Y.; Ikeuchi, M.; Kim, S.H.; Hamasaki, N.; Kang, D. The C-terminal tail of mitochondrial transcription factor a markedly strengthens its general binding to DNA. J. Biochem. 2007, 141, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Russell, O.M.; Fruh, I.; Rai, P.K.; Marcellin, D.; Doll, T.; Reeve, A.; Germain, M.; Bastien, J.; Rygiel, K.A.; Cerino, R.; et al. Preferential amplification of a human mitochondrial DNA deletion in vitro and in vivo. Sci. Rep. 2018, 8, 1799. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Lezza, A.M.S. Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions. Useful insights from aging and calorie restriction studies. Mitochondrion 2015, 25, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [PubMed]
- Mungai, P.T.; Waypa, G.B.; Jairaman, A.; Prakriya, M.; Dokic, D.; Ball, M.K.; Schumacker, P.T. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol. Cell. Biol. 2011, 31, 3531–3545. [Google Scholar] [CrossRef] [PubMed]
- Lahair, M.M.; Howe, C.J.; Rodriguez–Mora, O.; McCubrey, J.A.; Franklin, R.A. Molecular pathways leading to oxidative stress-induced phosphorylation of Akt. Antioxid. Redox Signal. 2006, 8, 1749–1756. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Xu, X.; Hu, X.; Fassett, J.; Zhu, G.; Tao, Y.; Li, J.; Huang, Y.; Zhang, P.; Zhao, B.; et al. PGC-1 α regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid. Redox Signal. 2010, 13, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta 2008, 1777, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Nakada, K.; Hayashi, J.-I. Mitochondrial dynamics and aging: Mitochondrial interaction preventing individuals from expression of respiratory deficiency caused by mutant mtDNA. Biochim. Biophys. Acta 2006, 1763, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Y.; Dorn, G.W. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 2011, 109, 1327–1331. [Google Scholar] [CrossRef] [PubMed]
- Givvimani, S.; Pushpakumar, S.B.; Metreveli, N.; Veeranki, S.; Kundu, S.; Tyagi, S.C. Role of mitochondrial fission and fusion in cardiomyocyte contractility. Int. J. Cardiol. 2015, 187, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Eisner, V.; Cupo, R.R.; Gao, E.; Csordás, G.; Slovinsky, W.S.; Paillard, M.; Cheng, L.; Ibetti, J.; Chen, S.R.W.; Chuprun, J.K.; et al. Mitochondrial fusion dynamics is robust in the heart and depends on calcium oscillations and contractile activity. Proc. Natl. Acad. Sci. USA 2017, 114, E859–E868. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Hall, A.R.; Dongworth, R.K.; Kalkhoran, S.; Pyakurel, A.; Scorrano, L.; Hausenloy, D.J. Akt protects the heart against ischaemia-reperfusion injury by modulating mitochondrial morphology. Thromb. Haemost. 2015, 113, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Manufacturer and Catalogue Number | Type | Species | Dilution | Detected Band MW (kDa) |
|---|---|---|---|---|---|
| Anti AKT | Cell Signaling (Berverly, MA, USA) | Polyclonal | Rabbit | 1:20,000 | 60 |
| (sc-9272) | |||||
| Anti AMPK | Cell Signaling | Polyclonal | Rabbit | 1:5000 | 62 |
| (sc-2532) | |||||
| Anti Beclin 1 | Millipore (Burlington, MA, USA) | Polyclonal | Rabbit | 1:300,000 | 52 |
| (AB15417) | |||||
| Anti Drp1 | Abnova (Taiwan, China) | Monoclonal | Mouse | 1:10,000 | 80 |
| (H00010059-M01) | |||||
| Anti FoxO3a | Cell Signaling | Polyclonal | Rabbit | 1:10,000 | 97 |
| (sc-9467) | |||||
| Anti MFN2 | Abnova (Taiwan, China) | Monoclonal | Mouse | 1:10,000 | 80 |
| (H00009927-M03) | |||||
| Anti MnSOD | Assay Designs (Farmingdale, NY, USA) | Polyclonal | Rabbit | 1:40,000 | 26 |
| (SOD-110) | |||||
| Anti P-AKT | Cell Signaling | Polyclonal | Rabbit | 1:5000 | 60 |
| (sc-9271) | |||||
| Anti P-AMPK | Cell Signaling | Polyclonal | Rabbit | 1:5000 | 62 |
| (sc-2531) | |||||
| Anti P-FoxO3a | Cell Signaling | Polyclonal | Rabbit | 1:5000 | 97 |
| (sc-9465) | |||||
| Anti PGC-1α | Santa Cruz Biotechnology (Dallas, TX, USA) | Polyclonal | Goat | 1:10,000 | 91 |
| (sc-5816) | |||||
| Anti PrxIII | Ab Frontier (Seoul, South Korea) | Polyclonal | Rabbit | 1:160,000 | 26 |
| (LF-PA0030) | |||||
| Anti Prx-SO3 | Ab Frontier (Seoul, South Korea) | Polyclonal | Rabbit | 1:30,000 | 26 |
| (LF-PA0004) | |||||
| Anti SQSTM1/p62 | Sigma-Aldrich (St. Louis, MO, USA) | Monoclonal | Rabbit | 1:20,000 | 62 |
| (P0066) | |||||
| Anti TFAM | Santa Cruz Biotechnology (Dallas, TX, USA) | Polyclonal | Goat | 1:30,000 | 25 |
| (sc-19050) | |||||
| Anti GAPDH | Cell Signaling | Monoclonal | Rabbit | 1:100,000 | 37 |
| (sc-2118) |
| Primer Set | Forward Primer | Reverse Primer | (nps) | (nps) |
|---|---|---|---|---|
| β-actin | 5′CCCAGCCATGTACGTAGCCA3′ | 5′CGTCTCCGGAGTCCATCAC3′ | 2181–2200 | 2266–2248 |
| D-loop | 5′CACCCCCTACACCTGAAACTT3′ | 5′TTTGTGTCGGGAAATTTTACCAAT3′ | 16,092–16,112 | 16,250–16,227 |
| mtDNA | 5′GGTTCTTACTTCAGGGCCATCA3′ | 5′TGATTAGACCCGTTACCATCGA3′ | 15,785–15,806 | 15,868–15,847 |
| mtDNA4834 | 5′AAGGACGAACCTGAGCCCTAATA3′ | 5′CGAAGTAGATGATGCGTATACTGTA3′ | 8109–8131 | 13,020–12,996 |
| ND1 | 5′AACGCCCTAACATCAATTGTATTCC3′ | 5′TGGTCATATCGAAAACGGGGG3′ | 3442–3466 | 3590–3570 |
| OriL | 5′CAGCTAAATACCCTACTTACTGG3′ | 5′GCCCCCTTTTTACCAAAAAGCC3′ | 5120–5142 | 5270–5249 |
| Placebo (n = 8) | Enalapril (n = 8) | p-Value | |
|---|---|---|---|
| Body weight, g | 577.3 ± 12.9 | 551.0 ± 16.3 | 0.2228 |
| Heart weight (absolute), g | 1.53 ± 0.03 | 1.37 ± 0.05 | 0.0359 |
| Heart weight by body weight | 2.92 ± 0.06 | 2.63 ± 0.10 | 0.0359 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Picca, A.; Sirago, G.; Pesce, V.; Lezza, A.M.S.; Calvani, R.; Bossola, M.; Villani, E.R.; Landi, F.; Leeuwenburgh, C.; Bernabei, R.; et al. Administration of Enalapril Started Late in Life Attenuates Hypertrophy and Oxidative Stress Burden, Increases Mitochondrial Mass, and Modulates Mitochondrial Quality Control Signaling in the Rat Heart. Biomolecules 2018, 8, 177. https://doi.org/10.3390/biom8040177
Picca A, Sirago G, Pesce V, Lezza AMS, Calvani R, Bossola M, Villani ER, Landi F, Leeuwenburgh C, Bernabei R, et al. Administration of Enalapril Started Late in Life Attenuates Hypertrophy and Oxidative Stress Burden, Increases Mitochondrial Mass, and Modulates Mitochondrial Quality Control Signaling in the Rat Heart. Biomolecules. 2018; 8(4):177. https://doi.org/10.3390/biom8040177
Chicago/Turabian StylePicca, Anna, Giuseppe Sirago, Vito Pesce, Angela Maria Serena Lezza, Riccardo Calvani, Maurizio Bossola, Emanuele Rocco Villani, Francesco Landi, Christiaan Leeuwenburgh, Roberto Bernabei, and et al. 2018. "Administration of Enalapril Started Late in Life Attenuates Hypertrophy and Oxidative Stress Burden, Increases Mitochondrial Mass, and Modulates Mitochondrial Quality Control Signaling in the Rat Heart" Biomolecules 8, no. 4: 177. https://doi.org/10.3390/biom8040177
APA StylePicca, A., Sirago, G., Pesce, V., Lezza, A. M. S., Calvani, R., Bossola, M., Villani, E. R., Landi, F., Leeuwenburgh, C., Bernabei, R., Carter, C. S., & Marzetti, E. (2018). Administration of Enalapril Started Late in Life Attenuates Hypertrophy and Oxidative Stress Burden, Increases Mitochondrial Mass, and Modulates Mitochondrial Quality Control Signaling in the Rat Heart. Biomolecules, 8(4), 177. https://doi.org/10.3390/biom8040177