Mesoporous Silica Nanoparticles Trigger Liver and Kidney Injury and Fibrosis Via Altering TLR4/NF-?B, JAK2/STAT3 and Nrf2/HO-1 Signaling in Rats

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis and Characterization of MSNs
2.2. Animals and Treatments
2.3. Collection and Preparation of Samples
2.4. Assay of Liver and Kidney Function Markers and Pro-Inflammatory Cytokines
2.5. Assay of Oxidative Stress Markers and Antioxidants
2.6. Gene Expression
2.7. Western Blotting
2.8. Histological Examination
2.9. Statistical Analysis
3. Results
3.1. Characterization of MSNs
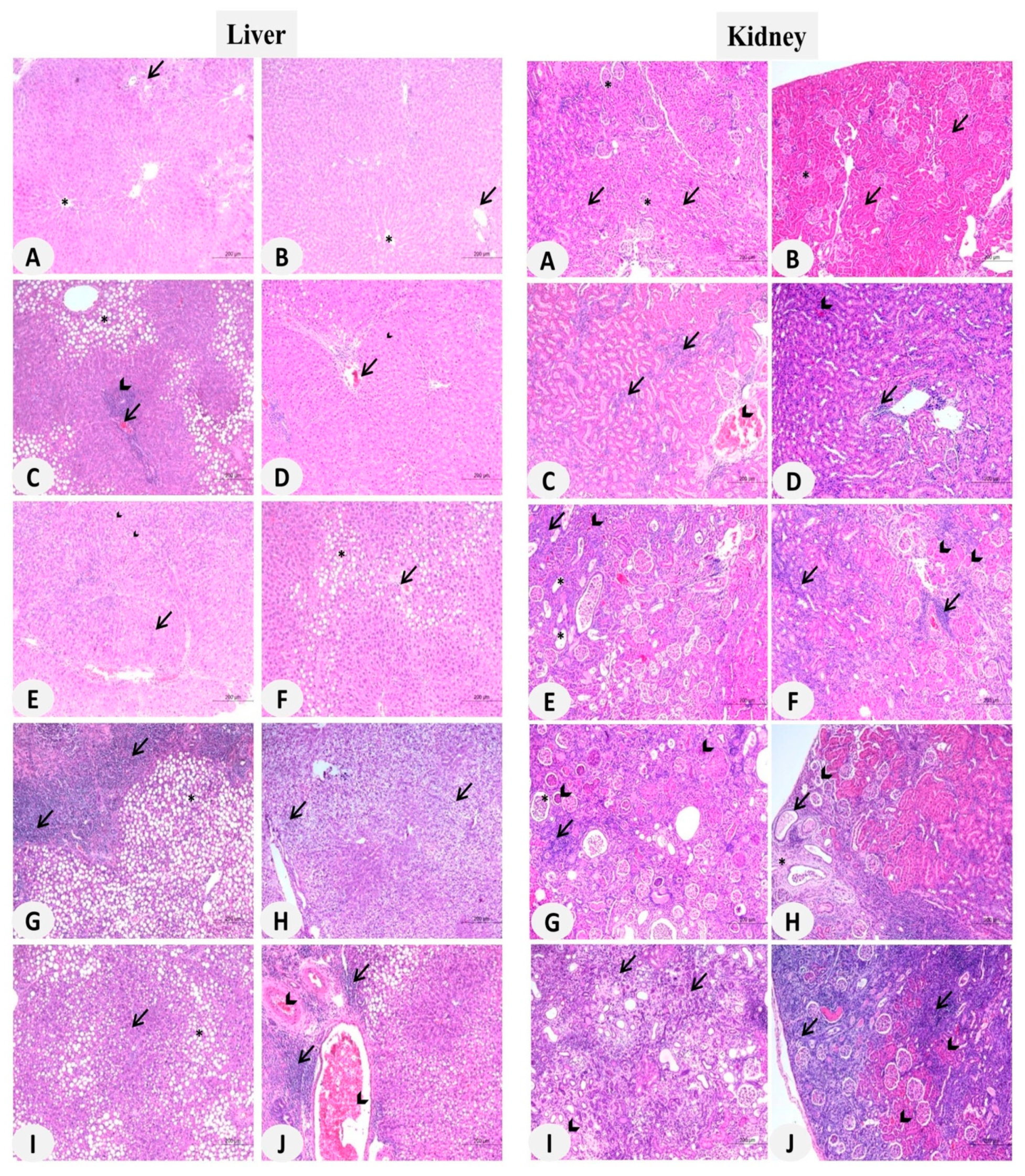
3.2. MSNs Induce Hepatic and Nephrotoxicity in Rats
3.3. MSNs Provoke ROS Production and Oxidative Stress in the Liver and Kidney of Rats
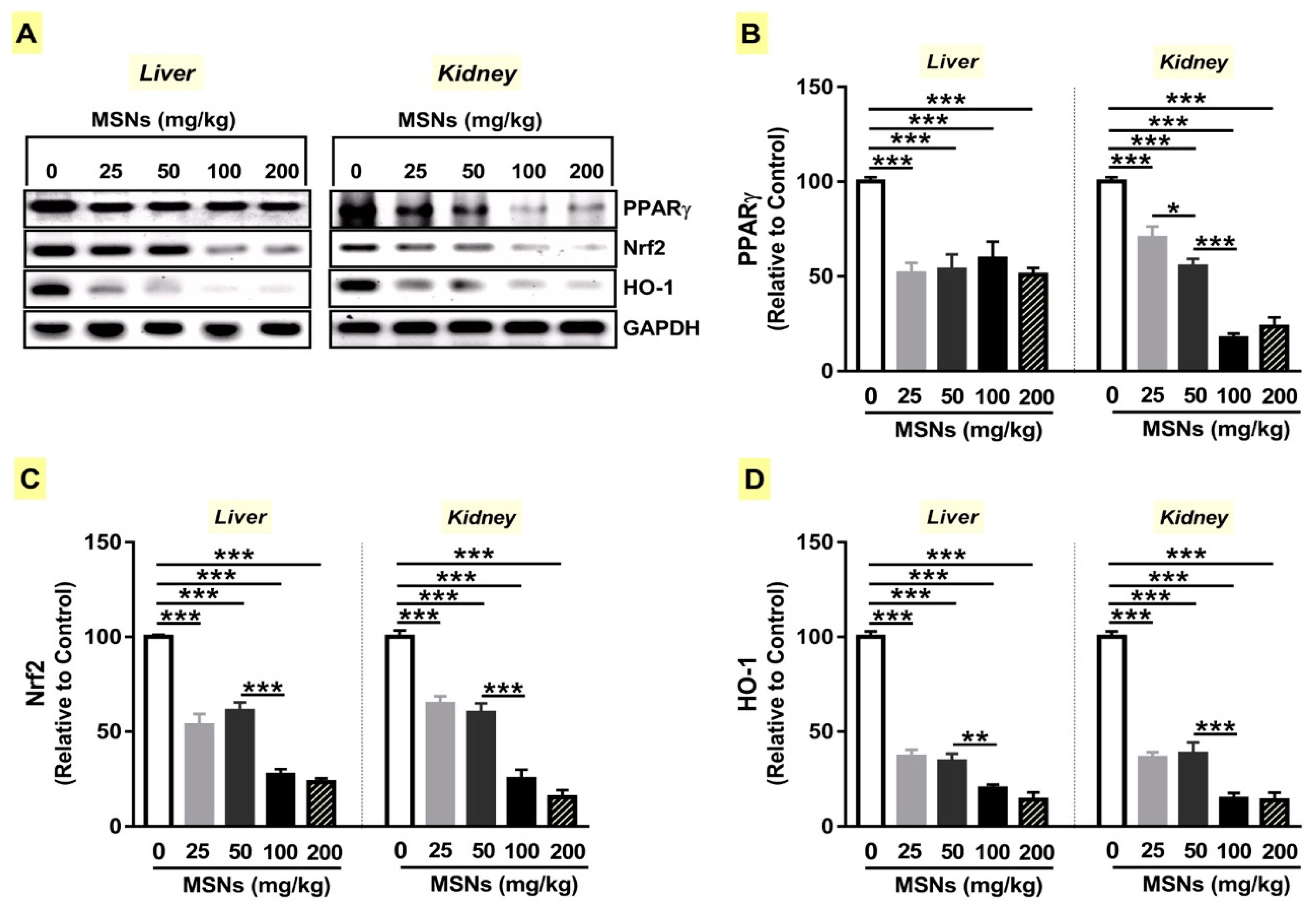
3.4. MSNs Suppress PPARγ Expression and Nrf2/HO-1 Signaling in Liver and Kidney of Rats
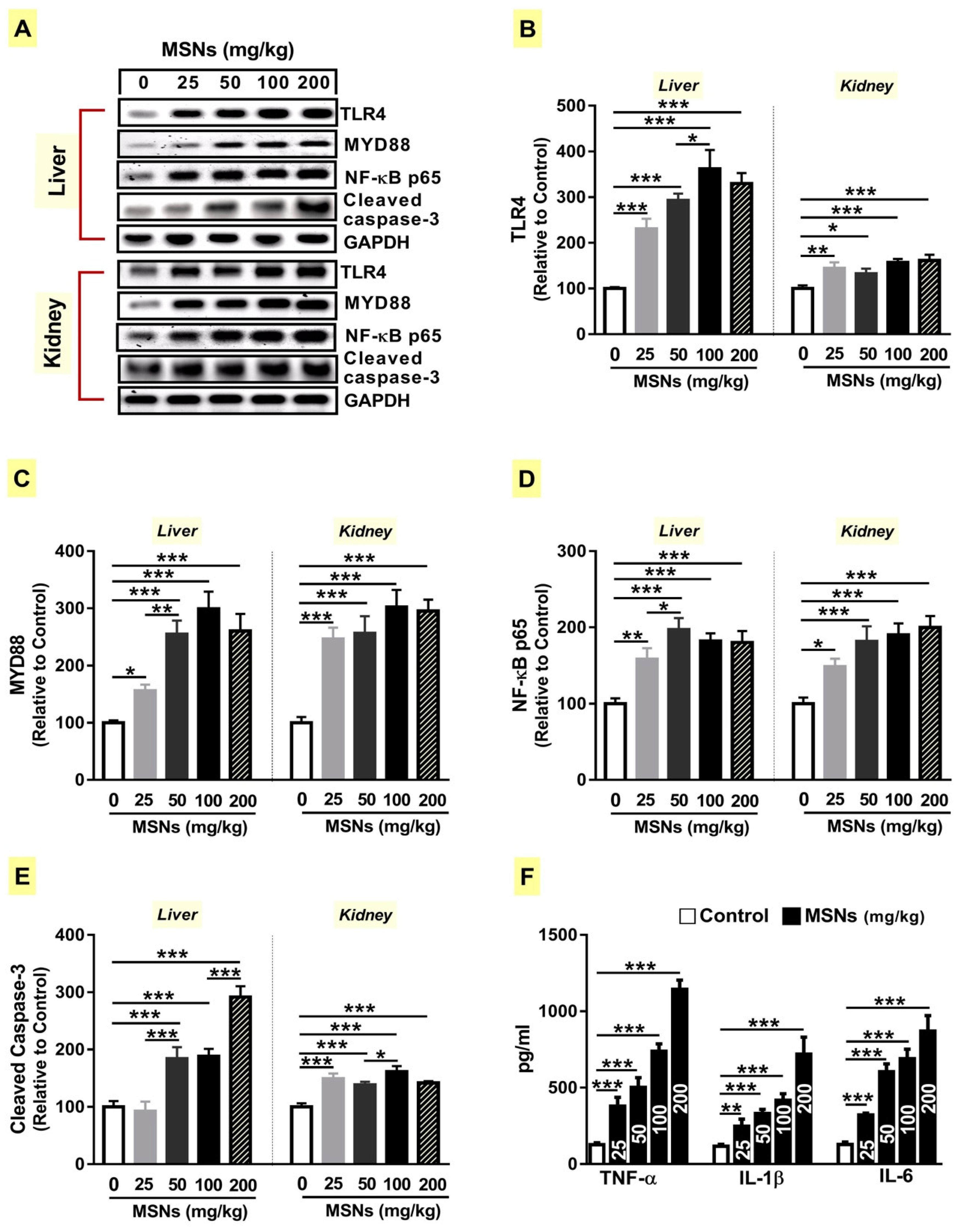
3.5. MSNs Activate TLR4/MyD88/NF-κB Signaling and Apoptosis in the Liver and Kidney of Rats
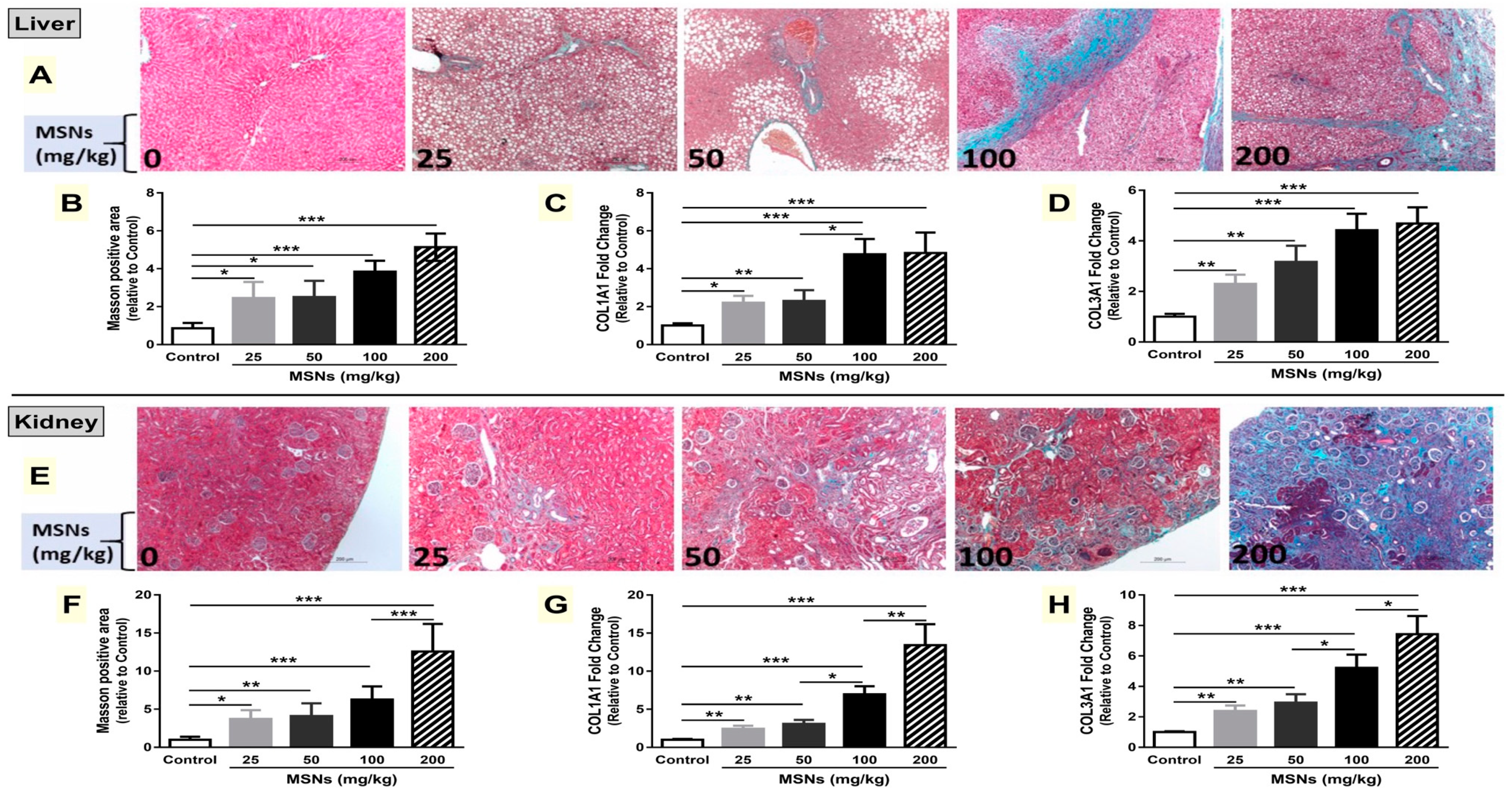
3.6. MSNs Induce Liver and Kidney Fibrosis in Rats
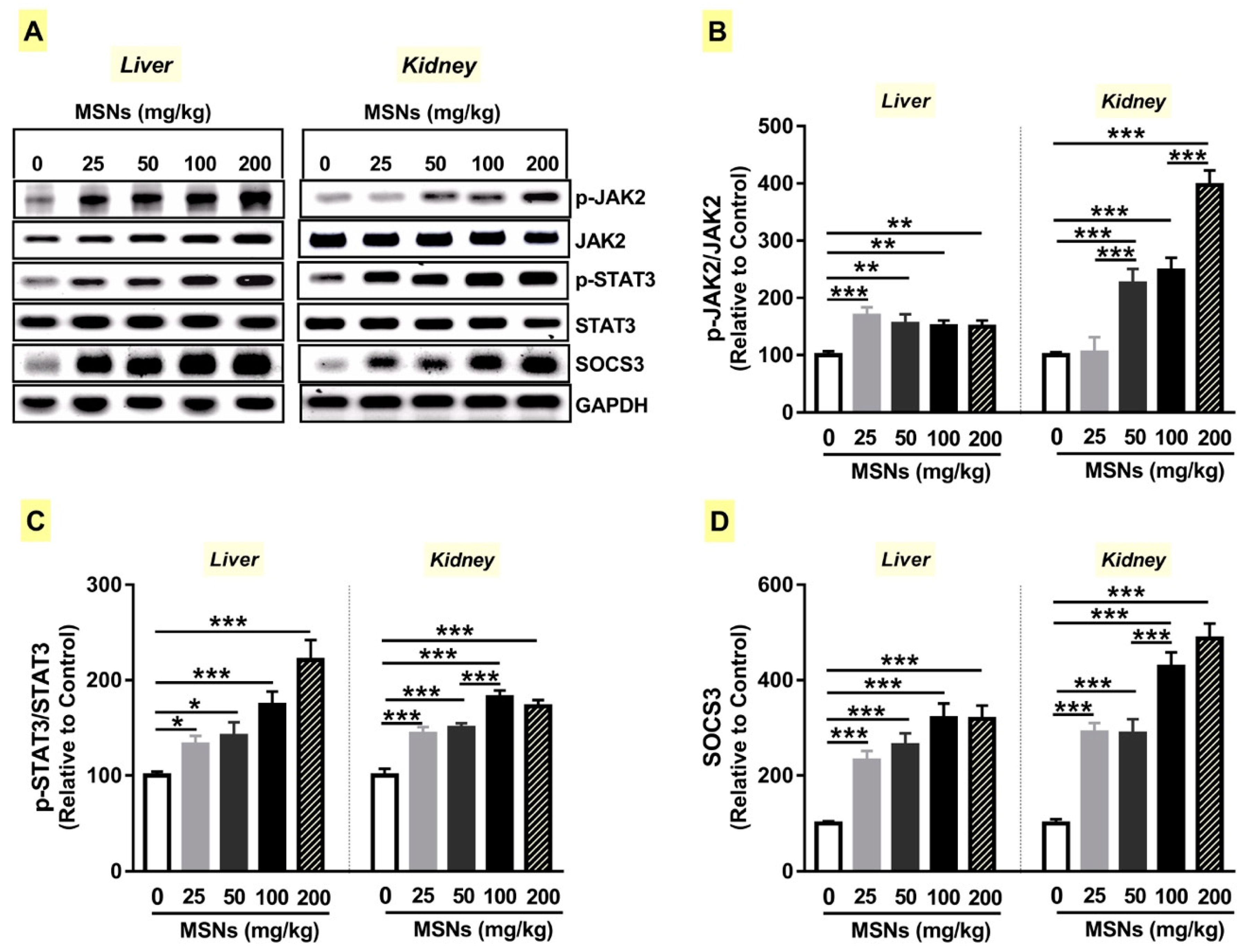
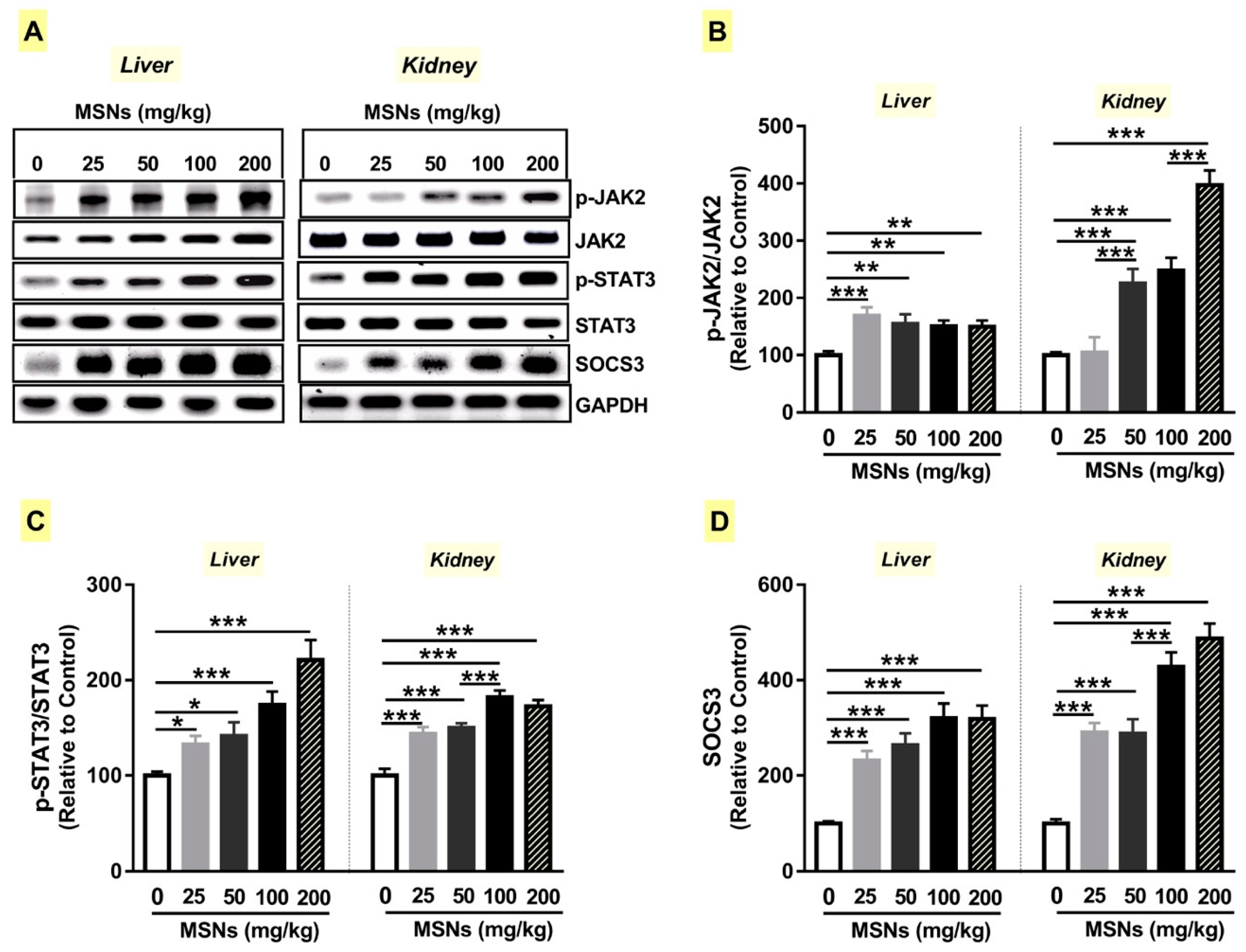
3.7. MSNs Activate JAk2/STAT3 Signaling in the Liver and Kidney of Rats
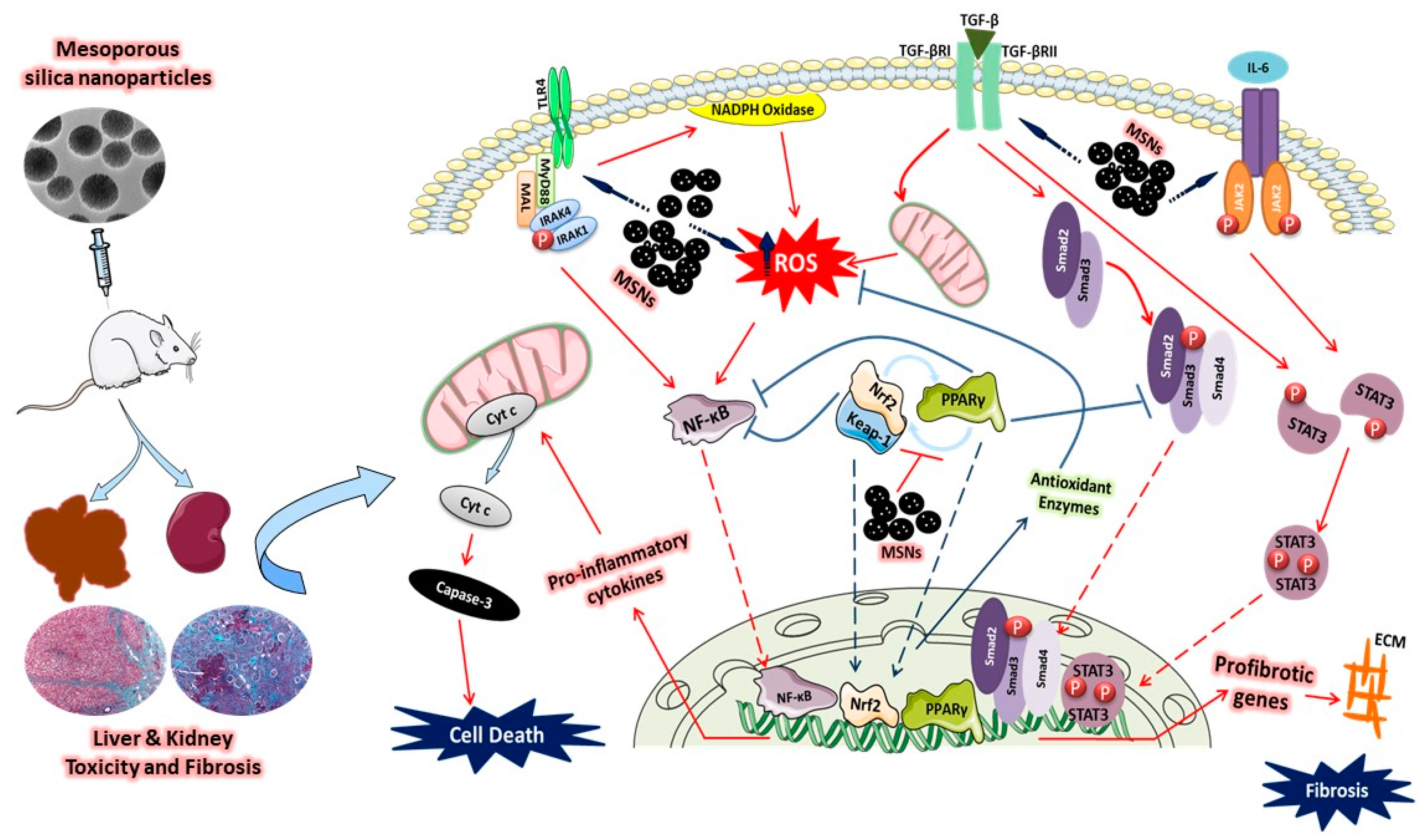
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhou, Y.; Quan, G.; Wu, Q.; Zhang, X.; Niu, B.; Wu, B.; Huang, Y.; Pan, X.; Wu, C. Mesoporous silica nanoparticles for drug and gene delivery. Acta Pharm. Sin. B 2018, 8, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Brinch, A.; Hansen, S.F.; Hartmann, N.B.; Baun, A. Eu regulation of nanobiocides: Challenges in implementing the biocidal product regulation (BPR). Nanomaterials 2016, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Murugadoss, S.; Lison, D.; Godderis, L.; Van Den Brule, S.; Mast, J.; Brassinne, F.; Sebaihi, N.; Hoet, P.H. Toxicology of silica nanoparticles: An update. Arch. Toxicol. 2017, 91, 2967–3010. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, S.; Krystek, P.; Peters, R.J.; Lankveld, D.P.; Bokkers, B.G.; van Hoeven-Arentzen, P.H.; Bouwmeester, H.; Oomen, A.G. Presence and risks of nanosilica in food products. Nanotoxicology 2011, 5, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Younes, M.; Aggett, P.; Aguilar, F.; Crebelli, R.; Dusemund, B.; Filipič, M.; Frutos, M.J.; Galtier, P.; Galtier, P.; Gott, D.; et al. Re-evaluation of silicon dioxide (E 551) as a food additive. EFSA J. 2018, 16, e05088. [Google Scholar]
- Croissant, J.G.; Fatieiev, Y.; Khashab, N.M. Degradability and clearance of silicon, organosilica, silsesquioxane, silica mixed oxide, and mesoporous silica nanoparticles. Adv. Mater. 2017, 29, 1604634. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Patel, K.D.; Leong, K.W.; Kim, H.W. Progress in nanotheranostics based on mesoporous silica nanomaterial platforms. ACS Appl. Mater. Interfaces 2017, 9, 10309–10337. [Google Scholar] [CrossRef] [PubMed]
- Trewyn, B.G.; Giri, S.; Slowing, I.I.; Lin, V.S.Y. Mesoporous silica nanoparticle based controlled release, drug delivery, and biosensor systems. Chem. Commun. 2007, 31, 3236–3245. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, H.L.; Jeong, H.J.; Lim, S.T.; Sohn, M.H.; Kim, D.W. Mesoporous silica nanoparticle pretargeting for pet imaging based on a rapid bioorthogonal reaction in a living body. Angew. Chem. Int. Ed. 2013, 52, 10549–10552. [Google Scholar] [CrossRef]
- Jafari, S.; Derakhshankhah, H.; Alaei, L.; Fattahi, A.; Varnamkhasti, B.S.; Saboury, A.A. Mesoporous silica nanoparticles for therapeutic/diagnostic applications. Biomed. Pharmacother. 2019, 109, 1100–1111. [Google Scholar] [CrossRef]
- Rascol, E.; Pisani, C.; Dorandeu, C.; Nyalosaso, J.L.; Charnay, C.; Daurat, M.; Da Silva, A.; Devoisselle, J.-M.; Gaillard, J.-C.; Armengaud, J.; et al. Biosafety of mesoporous silica nanoparticles. Biomimetics 2018, 3, 22. [Google Scholar] [CrossRef]
- Chen, L.; Liu, J.; Zhang, Y.; Zhang, G.; Kang, Y.; Chen, A.; Feng, X.; Shao, L. The toxicity of silica nanoparticles to the immune system. Nanomedicine 2018, 13, 1939–1962. [Google Scholar] [CrossRef] [Green Version]
- Leung, C.C.; Yu, I.T.; Chen, W. Silicosis. Lancet 2012, 379, 2008–2018. [Google Scholar] [CrossRef]
- Hozayen, W.G.; Mahmoud, A.M.; Desouky, E.M.; El-Nahass, E.-S.; Soliman, H.A.; Farghali, A.A. Cardiac and pulmonary toxicity of mesoporous silica nanoparticles is associated with excessive ros production and redox imbalance in wistar rats. Biomed. Pharmacother. 2019, 109, 2527–2538. [Google Scholar] [CrossRef]
- Yu, Y.; Duan, J.; Li, Y.; Li, Y.; Jing, L.; Yang, M.; Wang, J.; Sun, Z. Silica nanoparticles induce liver fibrosis via TGF-β(1)/Smad3 pathway in icr mice. Int. J. Nanomed. 2017, 12, 6045–6057. [Google Scholar] [CrossRef]
- Waters, K.M.; Masiello, L.M.; Zangar, R.C.; Tarasevich, B.J.; Karin, N.J.; Quesenberry, R.D.; Bandyopadhyay, S.; Teeguarden, J.G.; Pounds, J.G.; Thrall, B.D. Macrophage responses to silica nanoparticles are highly conserved across particle sizes. Toxicol. Sci. Off. J. Soc. Toxicol. 2009, 107, 553–569. [Google Scholar] [CrossRef]
- Chen, X.; Zhouhua, W.; Jie, Z.; Xinlu, F.; Jinqiang, L.; Yuwen, Q.; Zhiying, H. Renal interstitial fibrosis induced by high-dose mesoporous silica nanoparticles via the NF-κB signaling pathway. Int. J. Nanomed. 2015, 10, 1–22. [Google Scholar] [CrossRef]
- Ahmed, S.M.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Winkler, H.C.; Kornprobst, J.; Wick, P.; von Moos, L.M.; Trantakis, I.; Schraner, E.M.; Bathke, B.; Hochrein, H.; Suter, M.; Naegeli, H. MyD88-dependent pro-interleukin-1β induction in dendritic cells exposed to food-grade synthetic amorphous silica. Part. Fibre Toxicol. 2017, 14, 21. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Ogata, H.; Chinen, T.; Yoshida, T.; Kinjyo, I.; Takaesu, G.; Shiraishi, H.; Iida, M.; Kobayashi, T.; Yoshimura, A. Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-β1 production. Oncogene 2006, 25, 2520–2530. [Google Scholar] [CrossRef] [PubMed]
- Ricote, M.; Huang, J.T.; Welch, J.S.; Glass, C.K. The peroxisome proliferator-activated receptor (PPARγ) as a regulator of monocyte/macrophage function. J. Leukoc. Biol. 1999, 66, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.C.; Hsu, S.L.; Wu, C.L.; Liu, W.C.; Hsueh, C.M. Peroxisome proliferator-activated receptor gamma (PPARγ) plays a critical role in the development of tgfbeta resistance of H460 cell. Cell. Signal. 2011, 23, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Melichian, D.S.; Chang, E.; Warner-Blankenship, M.; Ghosh, A.K.; Varga, J. Rosiglitazone abrogates bleomycin-induced scleroderma and blocks profibrotic responses through peroxisome proliferator-activated receptor-gamma. Am. J. Pathol. 2009, 174, 519–533. [Google Scholar] [CrossRef] [PubMed]
- AbouAitah, K.; Swiderska-Sroda, A.; Farghali, A.A.; Wojnarowicz, J.; Stefanek, A.; Gierlotka, S.; Opalinska, A.; Allayeh, A.K.; Ciach, T.; Lojkowski, W. Folic acid-conjugated mesoporous silica particles as nanocarriers of natural prodrugs for cancer targeting and antioxidant action. Oncotarget 2018, 9, 26466–26490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmoud, A.M.; Wilkinson, F.L.; Jones, A.M.; Wilkinson, J.A.; Romero, M.; Duarte, J.; Alexander, M.Y. A novel role for small molecule glycomimetics in the protection against lipid-induced endothelial dysfunction: Involvement of akt/enos and Nrf2/are signaling. Biochim. Biophys. Acta 2017, 1861, 3311–3322. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2011, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Okuno, Y.; Matsuda, M.; Kobayashi, H.; Morita, K.; Suzuki, E.; Fukuhara, A.; Komuro, R.; Shimabukuro, M.; Shimomura, I. Adipose expression of catalase is regulated via a novel remote PPARγ-responsive region. Biochem. Biophys. Res. Commun. 2008, 366, 698–704. [Google Scholar] [CrossRef]
- Linkov, I.; Satterstrom, F.K.; Corey, L.M. Nanotoxicology and nanomedicine: Making hard decisions. Nanomed. Nanotechnol. Biol. Med. 2008, 4, 167–171. [Google Scholar] [CrossRef]
- Kumar, R.; Roy, I.; Ohulchanskky, T.Y.; Vathy, L.A.; Bergey, E.J.; Sajjad, M.; Prasad, P.N. In vivo biodistribution and clearance studies using multimodal organically modified silica nanoparticles. ACS Nano 2010, 4, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Li, Y.; Wang, W.; Jin, M.; Du, Z.; Li, Y.; Duan, J.; Yu, Y.; Sun, Z. Acute toxicity of amorphous silica nanoparticles in intravenously exposed ICR mice. PLoS ONE 2013, 8, e61346. [Google Scholar] [CrossRef] [PubMed]
- Nishimori, H.; Kondoh, M.; Isoda, K.; Tsunoda, S.; Tsutsumi, Y.; Yagi, K. Silica nanoparticles as hepatotoxicants. Eur. J. Pharm. Biopharm. 2009, 72, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, T.; Fu, C.; Tan, L.; Meng, X.; Liu, H. Biodistribution, excretion, and toxicity of mesoporous silica nanoparticles after oral administration depend on their shape. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1915–1924. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Li, Y.; Liu, X.; Jin, M.; Zhang, L.; Du, Z.; Guo, C.; Huang, P.; Sun, Z. Cytotoxicity and mitochondrial damage caused by silica nanoparticles. Toxicol. In Vitro 2011, 25, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Zuo, D.; Duan, Z.; Jia, Y.; Chu, T.; He, Q.; Yuan, J.; Dai, W.; Li, Z.; Xing, L.; Wu, Y. Amphipathic silica nanoparticles induce cytotoxicity through oxidative stress mediated and p53 dependent apoptosis pathway in human liver cell line HL-7702 and rat liver cell line BRL-3a. Colloids Surf. B Biointerfaces 2016, 145, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Napierska, D.; Thomassen, L.C.; Lison, D.; Martens, J.A.; Hoet, P.H. The nanosilica hazard: Another variable entity. Part. Fibre Toxicol. 2010, 7, 39. [Google Scholar] [CrossRef]
- Xue, Y.; Chen, Q.; Ding, T.; Sun, J. Sio(2) nanoparticle-induced impairment of mitochondrial energy metabolism in hepatocytes directly and through a kupffer cell-mediated pathway in vitro. Int. J. Nanomed. 2014, 9, 2891–2903. [Google Scholar]
- Fubini, B.; Hubbard, A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic. Biol. Med. 2003, 34, 1507–1516. [Google Scholar] [CrossRef]
- Kojima, S.; Negishi, Y.; Tsukimoto, M.; Takenouchi, T.; Kitani, H.; Takeda, K. Purinergic signaling via p2x7 receptor mediates IL-1β production in kupffer cells exposed to silica nanoparticle. Toxicology 2014, 321, 13–20. [Google Scholar] [CrossRef]
- Satta, S.; Mahmoud, A.M.; Wilkinson, F.L.; Yvonne Alexander, M.; White, S.J. The role of NrfF2 in cardiovascular function and disease. Oxidative Med. Cell. Longev. 2017, 2017, 9237263. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I. An Nrf2/small maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.M.; Germoush, M.O.; Al-Anazi, K.M.; Mahmoud, A.H.; Farah, M.A.; Allam, A.A. Commiphora molmol protects against methotrexate-induced nephrotoxicity by up-regulating Nrf2/ARE/HO-1 signaling. Biomed. Pharmacother. 2018, 106, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.M.; Hussein, O.E.; Hozayen, W.G.; Abd El-Twab, S.M. Methotrexate hepatotoxicity is associated with oxidative stress, and down-regulation of PPARγ and Nrf2: Protective effect of 18beta-glycyrrhetinic acid. Chem. Biol. Interact. 2017, 270, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.M.; Zaki, A.R.; Hassan, M.E.; Mostafa-Hedeab, G. Commiphora molmol resin attenuates diethylnitrosamine/phenobarbital-induced hepatocarcinogenesis by modulating oxidative stress, inflammation, angiogenesis and Nrf2/ARE/HO-1 signaling. Chem. Biol. Interact. 2017, 270, 41–50. [Google Scholar] [CrossRef] [PubMed]
- ALHaithloul, H.A.S.; Alotaibi, M.F.; Bin-Jumah, M.; Elgebaly, H.; Mahmoud, A.M. Olea europaea leaf extract up-regulates Nrf2/ARE/HO-1 signaling and attenuates cyclophosphamide-induced oxidative stress, inflammation and apoptosis in rat kidney. Biomed. Pharmacother. 2019, 111, 676–685. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Hussein, O.E.; Abd El-Twab, S.M.; Hozayen, W.G. Ferulic acid protects against methotrexate nephrotoxicity via activation of Nrf2/ARE/HO-1 signaling and PPARγ, and suppression of NF-κB/NLRP3 inflammasome axis. Food Funct. 2019, 10, 4593–4607. [Google Scholar] [CrossRef]
- Aladaileh, S.H.; Abukhalil, M.H.; Saghir, S.A.M.; Hanieh, H.; Alfwuaires, M.A.; Almaiman, A.A.; Bin-Jumah, M.; Mahmoud, A.M. Galangin activates Nrf2 signaling and attenuates oxidative damage, inflammation, and apoptosis in a rat model of cyclophosphamide-induced hepatotoxicity. Biomolecules 2019, 9, 346. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Monestier, M.; Esmon, N.L.; Esmon, C.T. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J. Immunol. 2011, 187, 2626–2631. [Google Scholar] [CrossRef]
- Smith, K.D. Toll-like receptors in kidney disease. Curr. Opin. Nephrol. Hypertens. 2009, 18, 189–196. [Google Scholar] [CrossRef]
- Yu, M.; Li, H.; Liu, Q.; Liu, F.; Tang, L.; Li, C.; Yuan, Y.; Zhan, Y.; Xu, W.; Li, W.; et al. Nuclear factor p65 interacts with keap1 to repress the Nrf2-are pathway. Cell. Signal. 2011, 23, 883–892. [Google Scholar] [CrossRef]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Van der Zande, M.; Vandebriel, R.J.; Groot, M.J.; Kramer, E.; Herrera Rivera, Z.E.; Rasmussen, K.; Ossenkoppele, J.S.; Tromp, P.; Gremmer, E.R.; Peters, R.J.B.; et al. Sub-chronic toxicity study in rats orally exposed to nanostructured silica. Part. Fibre Toxicol. 2014, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; ten Dijke, P. TGF-β in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Hozayen, W.G.; Hasan, I.H.; Shaban, E.; Bin-Jumah, M. Umbelliferone ameliorates CCL4-induced liver fibrosis in rats by upregulating PPARγ and attenuating oxidative stress, inflammation, and TGF-β1/Smad3 signaling. Inflammation 2019, 42, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, I.; Xu, S.; Denton, C.P.; Abraham, D.J.; Ponticos, M. Stat3 controls COL1A2 enhancer activation cooperatively with junb, regulates type i collagen synthesis posttranscriptionally, and is essential for lung myofibroblast differentiation. Mol. Biol. Cell 2018, 29, 84–95. [Google Scholar] [CrossRef]
- Li, C.; Iness, A.; Yoon, J.; Grider, J.R.; Murthy, K.S.; Kellum, J.M.; Kuemmerle, J.F. Noncanonical Stat3 activation regulates excess TGF-β1 and collagen i expression in muscle of stricturing crohn’s disease. J. Immunol. 2015, 194, 3422–3431. [Google Scholar] [CrossRef]
- Seo, H.Y.; Jeon, J.H.; Jung, Y.A.; Jung, G.S.; Lee, E.J.; Choi, Y.K.; Park, K.G.; Choe, M.S.; Jang, B.K.; Kim, M.K.; et al. Fyn deficiency attenuates renal fibrosis by inhibition of phospho-Stat3. Kidney Int. 2016, 90, 1285–1297. [Google Scholar] [CrossRef]
- Matsui, F.; Babitz, S.A.; Rhee, A.; Hile, K.L.; Zhang, H.; Meldrum, K.K. Mesenchymal stem cells protect against obstruction-induced renal fibrosis by decreasing Stat3 activation and Stat3-dependent MMP-9 production. Am. J. Physiol. Ren. Physiol. 2017, 312, F25–F32. [Google Scholar] [CrossRef]
- Leask, A.; Abraham, D.J. TGF-β signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Sumova, B.; Mallano, T.; Chen, C.W.; Distler, A.; Bergmann, C.; Ludolph, I.; Horch, R.E.; Gelse, K.; Ramming, A.; et al. Activation of Stat3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat. Commun. 2017, 8, 1130. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Cho, S.J.; Kang, M.J.; Chapoval, S.P.; Lee, P.J.; Noble, P.W.; Yehualaeshet, T.; Lu, B.; Flavell, R.A.; Milbrandt, J.; et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor β1-induced pulmonary fibrosis. J. Exp. Med. 2004, 200, 377–389. [Google Scholar] [CrossRef]
- Takehara, T.; Tatsumi, T.; Suzuki, T.; Rucker, E.B., 3rd; Hennighausen, L.; Jinushi, M.; Miyagi, T.; Kanazawa, Y.; Hayashi, N. Hepatocyte-specific disruption of Bcl-xL leads to continuous hepatocyte apoptosis and liver fibrotic responses. Gastroenterology 2004, 127, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Tao, Q.; Sun, M.; Wu, J.Z.; Yang, W.; Jian, P.; Peng, J.; Hu, Y.; Liu, C.; Liu, P. Kupffer cells are associated with apoptosis, inflammation and fibrotic effects in hepatic fibrosis in rats. Lab. Investig. 2010, 90, 1805–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Chan, C.-C.; Kwon, O.-S.; Liu, S.; McGhee, J.; Stimpson, S.A.; Chen, L.Z.; Harrington, W.W.; Symonds, W.T.; Rockey, D.C. Regulation of peroxisome proliferator-activated receptor-γ in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G902–G911. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Chen, Y.; Ding, G.; Xu, Y.; Bai, M.; Zhang, Y.; Jia, Z.; Huang, S.; Zhang, A. Renal tubular epithelium-targeted peroxisome proliferator-activated receptor-γ maintains the epithelial phenotype and antagonizes renal fibrogenesis. Oncotarget 2016, 7, 64690–64701. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer Sequence (5′-3′) | Reverse Primer Sequence (5′-3′) |
|---|---|---|
| COL1A1 | GTACATCAGCCCAAACCCCA | CAGGATCGGAACCTTCGCTT |
| COL3A1 | AGGGCAGGGAACAACTGATG | GGTCCCACATTGCACAAAGC |
| β-actin | AGGAGTACGATGAGTCCGGC | CGCAGCTCAGTAACAGTCCG |
| Control | MSNs | ||||
|---|---|---|---|---|---|
| 25 mg/kg | 50 mg/kg | 100 mg/kg | 200 mg/kg | ||
| ALT (U/L) | 21.63 ± 1.71 | 82.86 ± 7.45 *** | 109.71 ± 3.19 *** | 111.36 ± 3.79 *** | 137.07 ± 4.69 *** |
| AST (U/L) | 47.27 ± 2.34 | 153.06 ± 5.09 *** | 157.20 ± 13.65 *** | 175.19 ± 9.87 *** | 179.00 ± 4.39 *** |
| ALP (U/L) | 166.50 ± 7.97 | 305.31 ± 10.74 *** | 313.09 ± 8.23 *** | 589.14 ± 27.16 *** | 735.93 ± 25.09 *** |
| Bilirubin (mg/dl) | 0.52 ± 0.07 | 1.86 ± 0.17 * | 1.83 ± 0.26 * | 1.82 ± 0.21 * | 2.95 ± 0.42 *** |
| Albumin (g/dl) | 3.37 ± 0.14 | 2.78 ± 0.15 | 2.69 ± 0.21 * | 2.52 ± 0.17 ** | 2.14 ± 0.11 *** |
| Creatinine (mg/dl) | 0.59 ± 0.02 | 2.34 ± 0.10 *** | 2.57 ± 0.13 *** | 2.53 ± 0.11 *** | 3.19 ± 0.24 *** |
| Urea (mg/dl) | 27.51 ± 1.72 | 55.87 ± 4.65 ** | 60.07 ± 2.86 ** | 58.46 ± 3.06 ** | 81.85 ± 5.99 *** |
| Control | MSNs (mg/kg) | ||||
|---|---|---|---|---|---|
| 25 | 50 | 100 | 200 | ||
| Liver | |||||
| Degenerative changes | - | ++ | ++ | +++ | +++ |
| Necrosis | - | ++ | ++ | +++ | +++ |
| Leukocyte infiltration | - | - | - | - | - |
| Hepatic parenchyma | - | - | - | - | - |
| Portal area | - | + | + | ++ | ++ |
| Congestion | - | + | ++ | ++ | ++ |
| Granulomatous reactions | - | - | - | + | + |
| Fatty changes | - | + | + | + | + |
| Kupffer cell activation | - | + | ++ | ++ | +++ |
| Kidney | |||||
| Tubular degeneration | - | + | ++ | ++ | +++ |
| Tubular necrosis | - | - | ++ | ++ | +++ |
| Glomerulonephritis | - | + | + | ++ | +++ |
| Glomerular atrophy | - | + | - | + | +++ |
| Glomerular hypercellularity | - | + | ++ | ++ | ++ |
| Bowman’s capsule dilation | - | - | - | - | + |
| Leukocyte infiltration | - | ++ | +++ | +++ | +++ |
| Chronic nephritis | - | + | ++ | ++ | +++ |
| Others | - | - | Cast | Cast, edema, tubular dilation, cystic dilation | Cystic dilation, cast |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmoud, A.M.; Desouky, E.M.; Hozayen, W.G.; Bin-Jumah, M.; El-Nahass, E.-S.; Soliman, H.A.; Farghali, A.A. Mesoporous Silica Nanoparticles Trigger Liver and Kidney Injury and Fibrosis Via Altering TLR4/NF-?B, JAK2/STAT3 and Nrf2/HO-1 Signaling in Rats. Biomolecules 2019, 9, 528. https://doi.org/10.3390/biom9100528
Mahmoud AM, Desouky EM, Hozayen WG, Bin-Jumah M, El-Nahass E-S, Soliman HA, Farghali AA. Mesoporous Silica Nanoparticles Trigger Liver and Kidney Injury and Fibrosis Via Altering TLR4/NF-?B, JAK2/STAT3 and Nrf2/HO-1 Signaling in Rats. Biomolecules. 2019; 9(10):528. https://doi.org/10.3390/biom9100528
Chicago/Turabian StyleMahmoud, Ayman M., Ekram M. Desouky, Walaa G. Hozayen, May Bin-Jumah, El-Shaymaa El-Nahass, Hanan A. Soliman, and Ahmed A. Farghali. 2019. "Mesoporous Silica Nanoparticles Trigger Liver and Kidney Injury and Fibrosis Via Altering TLR4/NF-?B, JAK2/STAT3 and Nrf2/HO-1 Signaling in Rats" Biomolecules 9, no. 10: 528. https://doi.org/10.3390/biom9100528
APA StyleMahmoud, A. M., Desouky, E. M., Hozayen, W. G., Bin-Jumah, M., El-Nahass, E.-S., Soliman, H. A., & Farghali, A. A. (2019). Mesoporous Silica Nanoparticles Trigger Liver and Kidney Injury and Fibrosis Via Altering TLR4/NF-?B, JAK2/STAT3 and Nrf2/HO-1 Signaling in Rats. Biomolecules, 9(10), 528. https://doi.org/10.3390/biom9100528