Attenuation in Nicotinic Acetylcholine Receptor α9 and α10 Subunit Double Knock-Out Mice of Experimental Autoimmune Encephalomyelitis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. EAE Induction
2.3. In Vivo Central Nervous System Bioluminescence
2.4. Histological Analyses
2.5. Statistical Analyses
3. Results
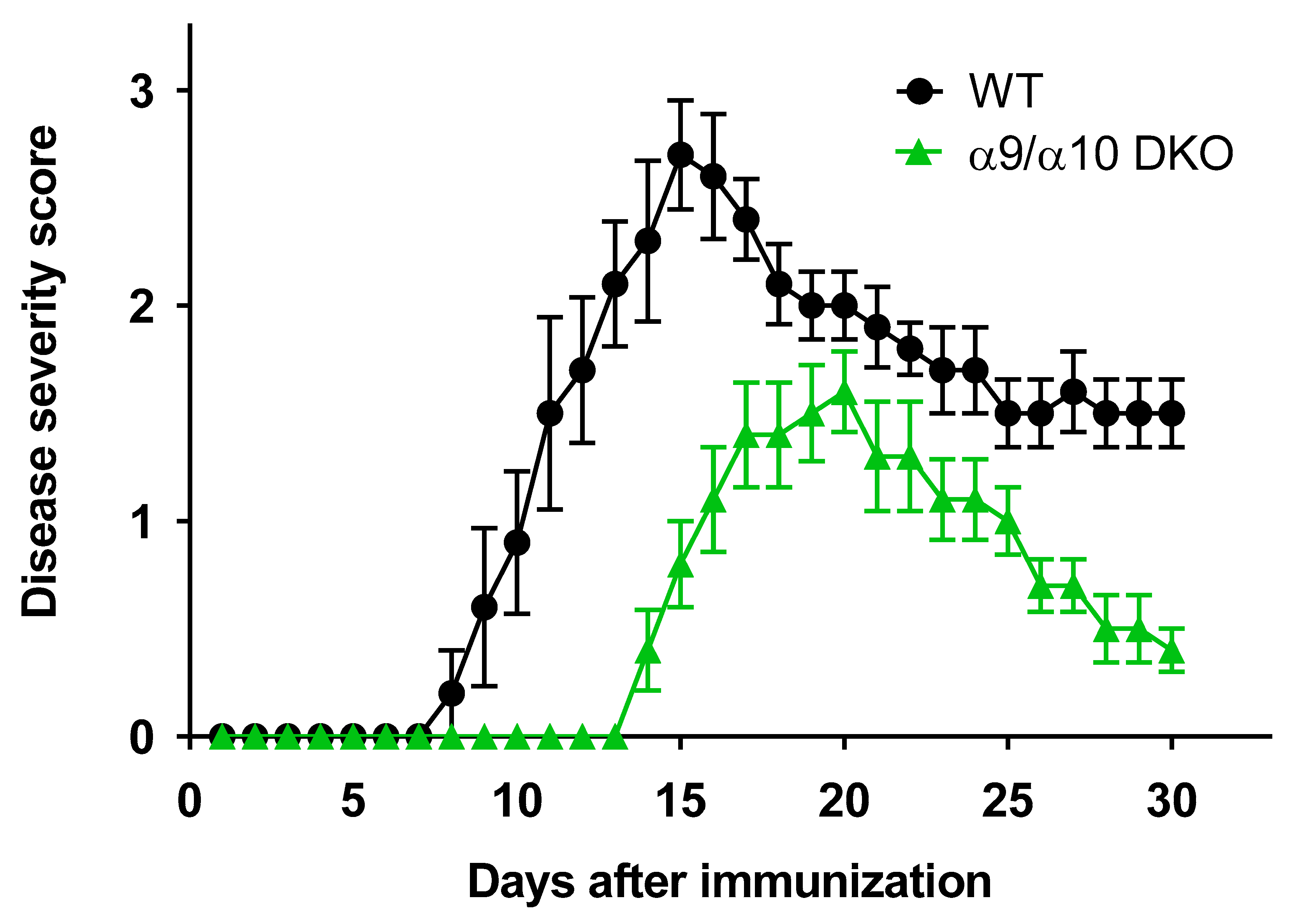
3.1. Attenuated EAE Disease Severity in α9/α10 DKO Mice
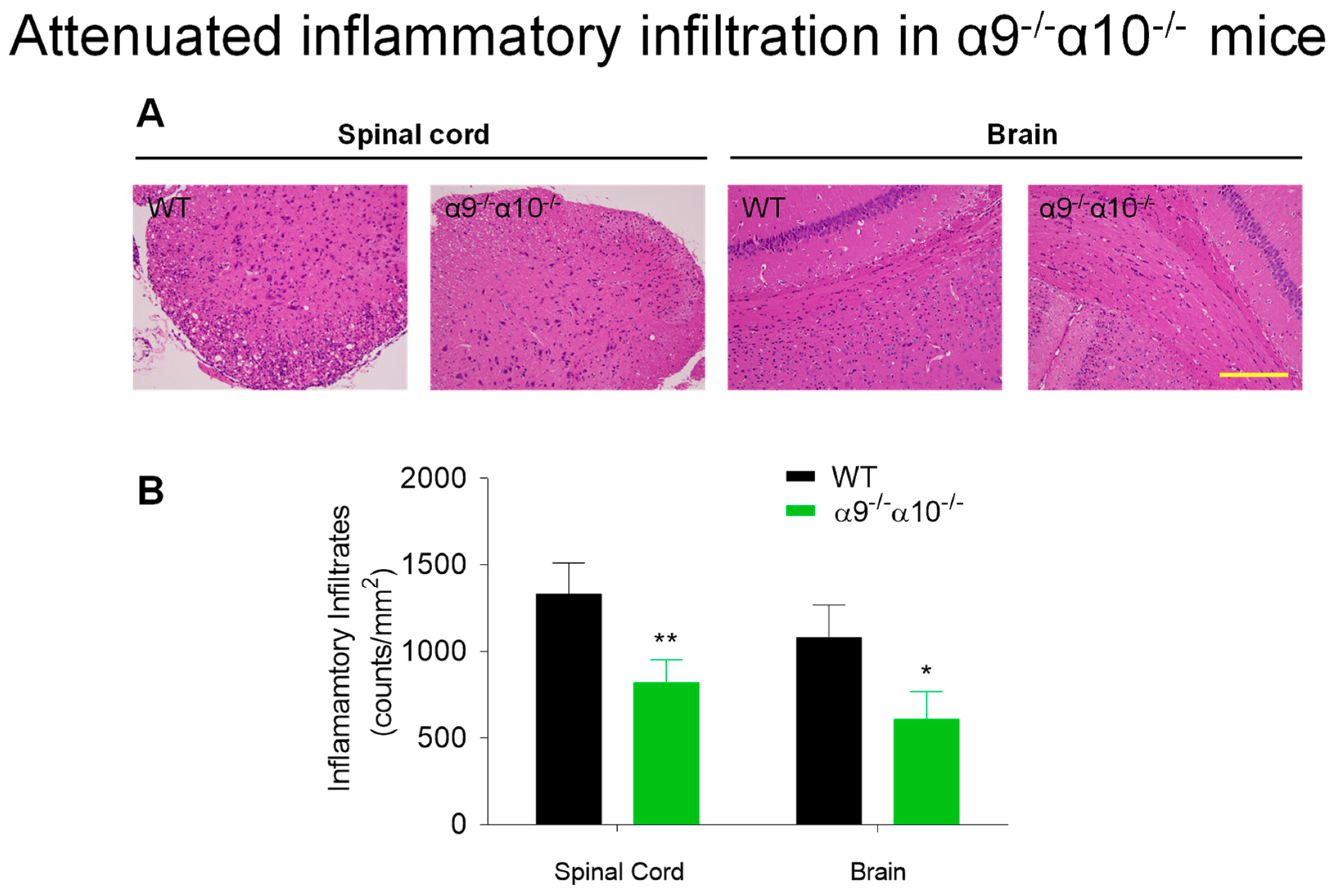
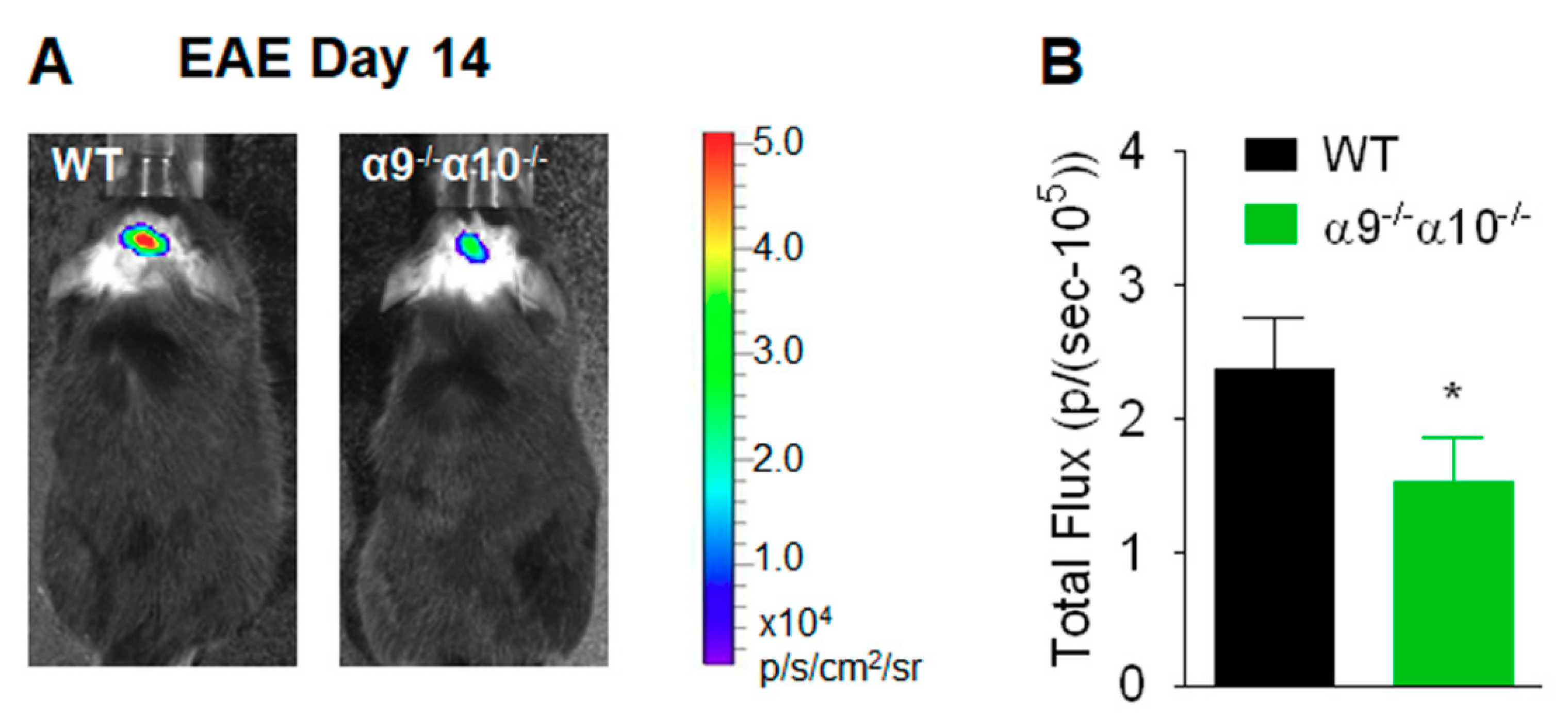
3.2. Diminished Inflammatory Indices in EAE α9/α10 DKO Mice
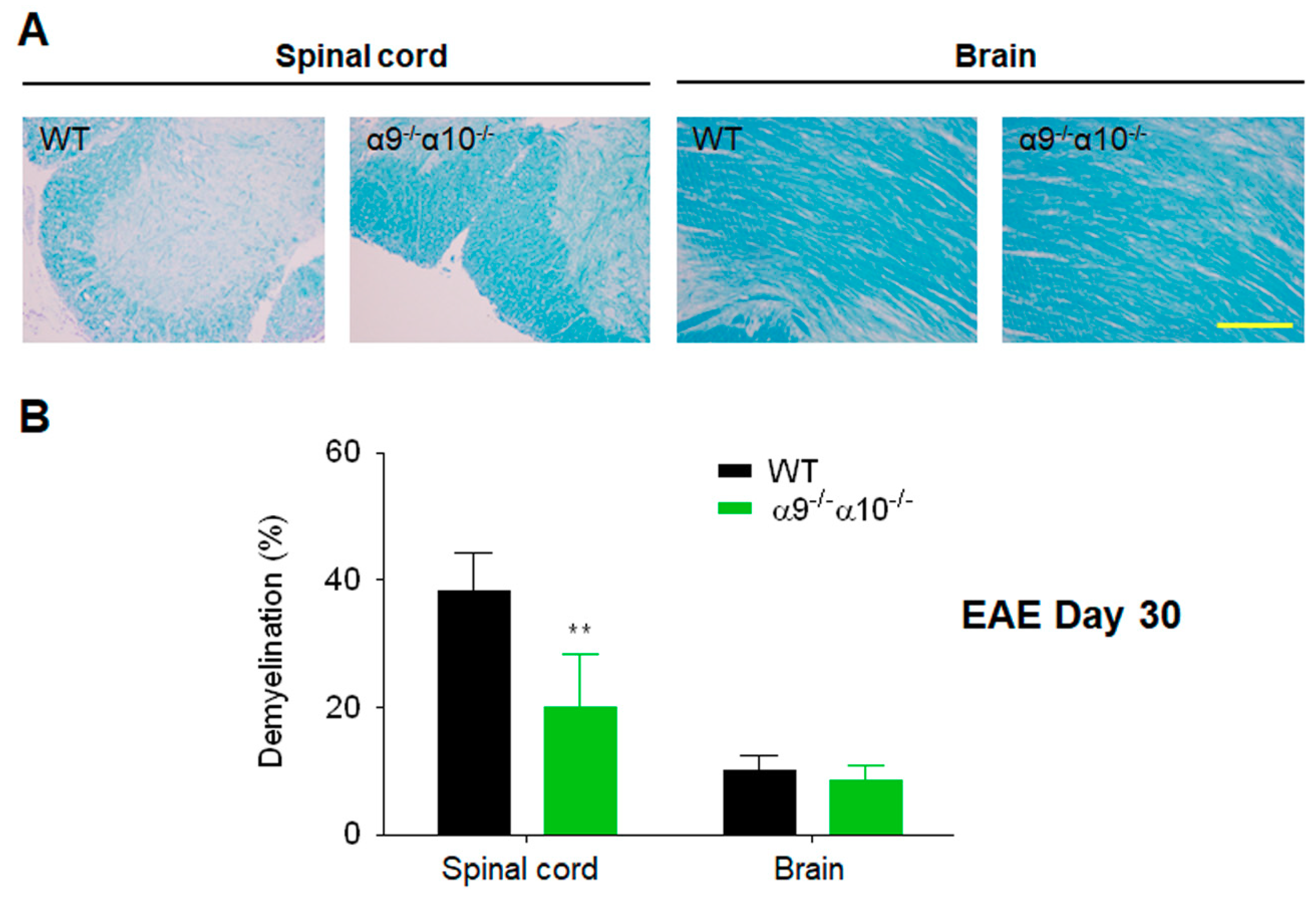
3.3. Demyelination is Reduced in EAE α9/α10 DKO Mice
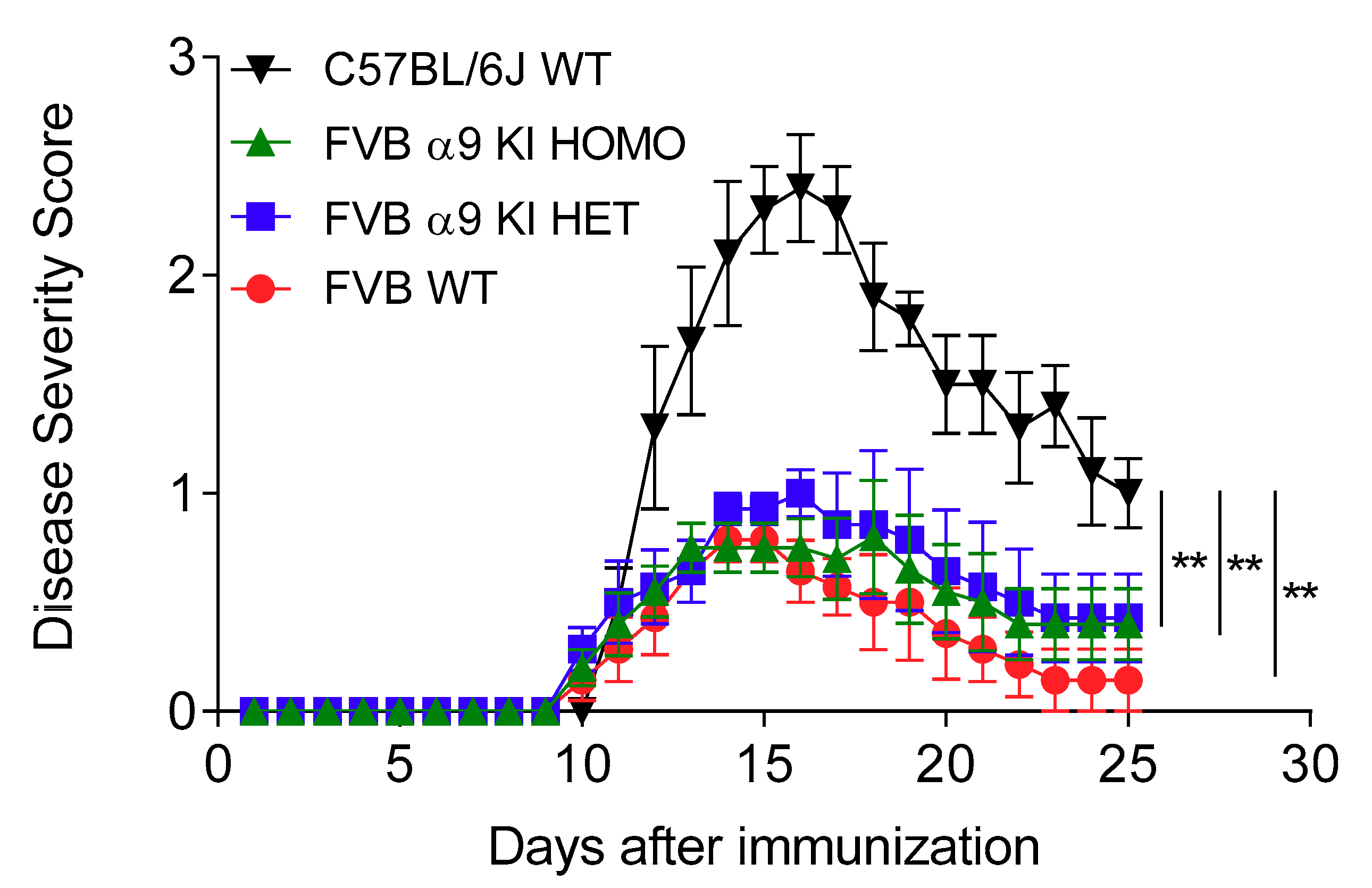
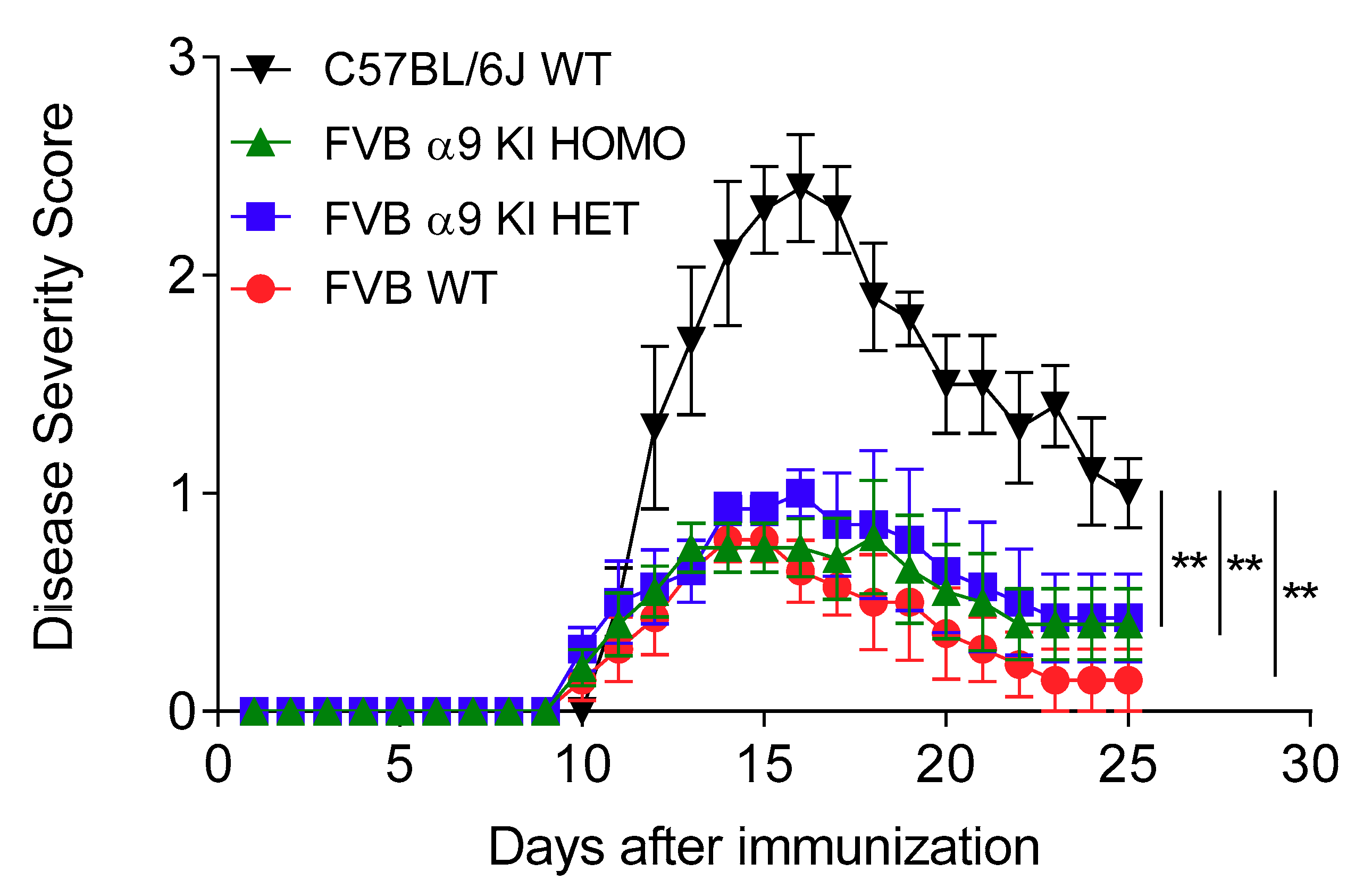
3.4. Effects of a nAChR α9 Subunit Gain-of-Function Mutation
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jensen, A.A.; Frolund, B.; Lijefors, T.; Krogsgaard-Larsen, P. Neuronal nicotinic acetylcholine receptors: Structural revelations, target identifications, and therapeutic inspirations. J. Med. Chem. 2005, 48, 4705–4745. [Google Scholar] [CrossRef] [PubMed]
- Lukas, R.J.; Bencherif, M. Recent developments in nicotinic acetylcholine receptor biology. In Biological and Biophysical Aspects of Ligand-Gated Ion Channel Receptor Superfamilies; Arias, H.R., Ed.; Research Signpost: Kerala, India, 2006; pp. 27–59. [Google Scholar]
- Taly, A.; Corringer, P.-J.; Guedin, D.; Lestage, P.; Changeux, J.-P. Nicotinic receptors: Allosteric transitions and therapeutic targets in the nervous system. Nat. Rev. Drug Discov. 2009, 8, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Lukas, R.J.; Changeux, J.P.; Le Novere, N.; Albuquerque, E.X.; Balfour, D.J.K.; Berg, D.K.; Bertrand, D.; Chiappinelli, V.A.; Clarke, P.B.S.; Collins, A.C.; et al. International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol. Rev. 1999, 51, 397–401. [Google Scholar] [PubMed]
- Grando, S.A.; Kawashima, K.; Kirkpatrick, C.J.; Meurs, H.; Wessler, I. The non-neuronal cholinergic system: Basic science, therapeutic implications and new perspectives. Life Sci. 2012, 91, 969–972. [Google Scholar] [CrossRef]
- Wessler, I.; Kirkpatrick, C.J. Acetylcholine beyond neurons: The non-neuronal cholinergic system in humans. Br. J. Pharmacol. 2008, 154, 1558–1571. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, K.; Fujii, T. Expression of non-neuronal acetylcholine in lymphocytes and its contribution to the regulation of immune function. Front. Biosci. 2004, 9, 2063–2085. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, K.; Yoshikawa, K.; Fujii, Y.X.; Moriwaki, Y.; Misawa, H. Expression and function of genes encoding cholinergic components in murine immune cells. Life Sci. 2007, 80, 2314–2319. [Google Scholar] [CrossRef]
- Kuo, Y.-P.; Lucero, L.; Michaels, J.; DeLuca, D.; Lukas, R.J. Differential expression of nicotinic acetylcholine receptor subunits in fetal and neonatal mouse thymus. J. Neuroimmun. 2002, 130, 140–154. [Google Scholar] [CrossRef]
- Sato, K.Z.; Fujii, T.; Watanabe, Y.; Yamada, S.; Ando, T.; Kazuko, F.; Kawashima, K. Diversity of mRNA expression for muscarinic acetylcholine receptor subtypes and neuronal nicotinic acetylcholine receptor subunits in human mononuclear leukocytes and leukemic cell lines. Neurosci. Lett. 1999, 266, 17–20. [Google Scholar] [CrossRef]
- Reardon, C.; Duncan, G.S.; Brüstle, A.; Brenner, D.; Tusche, M.W.; Olofsson, P.S.; Rosas-Ballina, M.; Tracey, K.J.; Mak, T.W. Lymphocyte-derived ACh regulates local innate but not adaptive immunity. Proc. Natl. Acad. Sci. 2013, 110, 1410–1415. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, K.; Klein, T.W.; Friedman, H.; Yamamoto, Y. Involvement of nicotinic acetylcholine receptors in suppression of antimicrobial activity and cytokine responses of alveolar macrophages to Legionella pneumophila infection by nicotine. J. Immunol. 2001, 167, 6518–6524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middlebrook, A.J.; Michaels, J.; Lukas, R.J.; DeLuca, D. Effects of Nicotine Exposure on Developing Murine and Human Thymocytes in Fetal Thumus Organ Culture; FASEB Conference on Neuroimmunology: Copper Mountain, CO, USA, 2000. [Google Scholar]
- Fujii, T.; Takada-Takatori, Y.; Kawashima, K. Basic and clinical aspects of non-neuronal acetylcholine: Expression of an independent, non-neuronal cholinergic system in lymphocytes and its clinical significance in immunotherapy. J. Pharmacol. Sci. 2008, 106, 186–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, J.; Simard, A.R.; Turner, G.H.; Wu, J.; Whiteaker, P.; Lukas, R.J.; Shi, F.D. Attenuation of CNS inflammatory responses by nicotine involves alpha7 and non-alpha7 nicotinic receptors. Exp. Neurol. 2011, 227, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middlebrook, A.J.; Martina, C.; Chang, Y.; Lukas, R.J.; DeLuca, D. Murine fetal thymus organ cultures treated with nicotine exhibit a reduction in mature thymocytes and expanded populations of immature thymocytes. J. Immunol. 2002, 169, 2915–2924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simard, A.R.; Gan, Y.; St-Pierre, S.; Kousari, A.; Patel, V.; Whiteaker, P.; Morley, B.J.; Lukas, R.; Shi, F.-D. Differential modulation of experimental autoimmune encephalomyelitis by a9*- and β2*-nicotinic acetylcholine receptors. Immunol. Cell Biol. 2013, 91, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.; Diamond, B. Autoimmune diseases. N Engl. J. Med. 2001, 345, 340–350. [Google Scholar] [CrossRef]
- Vigano, S.; Perreau, M.; Pantaleo, G.; Harari, A. Positive and negative regulation of cellular immune responses in physiologic conditions and diseases. Clin. Dev. Immunol. 2012, 2012, 485781. [Google Scholar] [CrossRef]
- Nikoopour, E.; Schwartz, J.A.; Singh, B. Therapeutic benefits of regulating inflammation in autoimmunity. Inflamm. Allergy Drug Targets 2008, 7, 203–210. [Google Scholar] [CrossRef]
- Weiner, H.L.; Selkoe, D.J. Inflammation and therapeutic vaccination in CNS diseases. Nature 2002, 420, 879–884. [Google Scholar] [CrossRef]
- Wekerle, H. The viral triggering of autoimmune disease. Nat. Med. 1998, 4, 770–771. [Google Scholar] [CrossRef]
- Pierson, E.; Simmons, S.B.; Castelli, L.; Goverman, J.M. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol. Rev. 2012, 248, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Laveti, D.; Kumar, M.; Hemalatha, R.; Sistla, R.; Naidu, V.G.; Talla, V.; Verma, V.; Kaur, N.; Nagpal, R. Anti-inflammatory treatments for chronic diseases: A review. Inflamm. Allergy Drug Targets 2013, 12, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Czirr, E.; Wyss-Coray, T. The immunology of neurodegeneration. J. Clin. Investig. 2012, 122, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Wraith, D.C.; Nicholson, L.B. The adaptive immune system in diseases of the central nervous system. J. Clin. Investig. 2012, 122, 1172–1179. [Google Scholar] [CrossRef]
- Steinman, L. Multiple sclerosis: A coordinated immunological attack against myelin in the central nervous system. Cell 1996, 85, 299–302. [Google Scholar] [CrossRef] [Green Version]
- Sardi, F.; Fassina, L.; Venturini, L.; Inguscio, M.; Guerriero, F.; Rolfo, E.; Ricevuti, G. Alzheimer’s disease, autoimmunity and inflammation. The good, the bad and the ugly. Autoimmun. Rev. 2011, 11, 149–153. [Google Scholar] [CrossRef]
- Nizri, E.; Irony-Tur-Sinai, M.; Lory, O.; Orr-Urtreger, A.; Lavi, E.; Brenner, T. Activation of the cholinergic anti-inflammatory system by nicotine attenuates neuroinflammation via suppression of Th1 and Th17 responses. J. Immunol. 2009, 183, 6681–6688. [Google Scholar] [CrossRef] [Green Version]
- Shi, F.D.; Piao, W.H.; Kuo, Y.P.; Campagnolo, D.I.; Vollmer, T.L.; Lukas, R.J. Nicotinic attenuation of central nervous system inflammation and autoimmunity. J. Immunol. 2009, 182, 1730–1739. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Whiteaker, P.; Morley, B.J.; Shi, F.-D.; Lukas, R.J. Distinctive roles for α7*- and α9*-nicotinic acetylcholine receptors in inflammatory and autoimmune responses in the murine experimental autoimmune encephalomyelitis model of multiple sclerosis. Front. Cell. Neurosci. 2017, 11, 287. [Google Scholar] [CrossRef]
- Zoli, M.; Pucci, S.; Vilella, A.; Gotti, C. Neuronal and extraneuronal nicotinic acetylcholine receptors. Curr. Neuropharmacol. 2018, 16, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Morley, B.J.; Whiteaker, P.; Elgoyhen, A.B. Commentary: Nicotinic acetylcholine receptor α9 and α10 subunits are expressed in the brain of mice. Front. Cell. Neurosci. 2018, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Razani-Boroujerdi, S.; Boyd, R.T.; Dávila-García, M.I.; Nandi, J.S.; Mishra, N.C.; Singh, S.P.; Pena-Philippides, J.C.; Langley, R.; Sopori, M.L. T cells express alpha7-nicotinic acetylcholine receptor subunits that require a functional TCR and leukocyte-specific protein tyrosine kinase for nicotine-induced Ca2+ response. J. Immunol. 2007, 179, 2889–2898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecker, A.; Küllmar, M.; Wilker, S.; Richter, K.; Zakrzewicz, A.; Atanasova, S.; Mathes, V.; Timm, T.; Lerner, S.; Klein, J.; et al. Phosphocholine-modified macromolecules and canonical nicotinic agonists inhibit ATP-induced IL-1β release. J. Immunol. 2015, 195, 2325–2334. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Mathes, V.; Fronius, M.; Althaus, M.; Hecker, A.; Krasteva-Christ, G.; Padberg, W.; Hone, A.J.; McIntosh, J.M.; Zakrzewicz, A.; et al. Phosphocholine-an agonist of metabotropic but not of ionotropic functions of a9-containing nicotinic acetylcholine receptors. Sci. Rep. 2016, 6, 28660. [Google Scholar] [CrossRef]
- Zakrzewicz, A.; Richter, K.; Agné, A.; Wilker, S.; Siebers, K.; Fink, B.; Krasteva-Christ, G.; Althaus, M.; Padberg, W.; Hone, A.J.; et al. Canonical and novel non-canonical cholinergic agonists inhibit ATP-induced release of monocytic interleukin-1b via different combinations of nicotinic acetylcholine receptor subunits a7, a9 and a10. Front. Cell. Neurosci. 2017, 11, 189. [Google Scholar] [CrossRef] [Green Version]
- Dunckley, T.; Lukas, R.J. Nicotine modulates the expression of a diverse set of genes in the neuronal SH-SY5Y cell line. J. Biol. Chem. 2003, 14, 14. [Google Scholar] [CrossRef] [Green Version]
- Dunckley, T.; Lukas, R.J. Nicotinic modulation of gene expression in SH-SY5Y neuroblastoma cells. Brain Res. 2006, 1116, 39–49. [Google Scholar] [CrossRef]
- Nakayama, H.; Numakawa, T.; Ikeuchi, T.; Hatanaka, H. Nicotine-induced phosphorylation of extracellular signal-regulated protein kinase and CREB in PC12h cells. J. Neurochem. 2001, 79, 489–498. [Google Scholar] [CrossRef] [Green Version]
- King, J.R.; Nordman, J.C.; Bridges, S.P.; Lin, M.K.; Kabbani, N. Identification and characterization of a G protein-binding cluster in α7 nicotinic acetylcholine receptors. J. Biol. Chem. 2015, 290, 20060–20070. [Google Scholar] [CrossRef] [Green Version]
- Koval, L.; Lykhmus, O.; Zhmak, M.; Khruschov, A.; Tsetlin, V.; Magrini, E.; Viola, A.; Chernyavsky, A.; Qian, J.; Grando, S.; et al. Differential involvement of α4β2, α7 and α9α10 nicotinic acetylcholine receptors in B lymphocyte activation in vitro. Int. J. Biochem. Cell. Biol. 2011, 43, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.S.; Ferris, R.L.; Matthews, T.; Hiel, H.; Lopez-Albaitero, A.; Lustig, L.R. Characterization of the human nicotinic acetylcholine receptor subunit alpha (alpha) 9 (CHRNA9) and alpha (alpha) 10 (CHRNAIO) in lymphocytes. Life Sci. 2004, 76, 263–280. [Google Scholar] [CrossRef] [PubMed]
- Morley, B.J.; Dolan, D.F.; Ohlemiller, K.; Simmons, D.D. Generation and characterization of α9 and α10 nicotinic acetylcholine receptor subunit knockout mice on a C57Bl/6J background. Front. Neurosci. 2017, 11, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morley, B.J.; Lysakowski, A.; Vijayakumar, S.; Menapace, D.; Jones, T.A. Nicotinic acetylcholine receptors regulate vestibular afferent gain and activation timing. J. Comp. Neurol. 2017, 525, 1216–1233. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.L.; Souza, F.G.O.; Bruce, K.S.; Strang, C.E.; Morley, B.J.; Keyser, K.T. Acetylcholine receptors in the retinas of the alpha7 nicotinic acetylcholine receptor knockout mouse. Mol. Vis. 2014, 20, 1328–1356. [Google Scholar] [PubMed]
- Taranda, J.; Maison, S.F.; Ballestero, J.A.; Katz, E.; Savino, J.; Vetter, D.E.; Boulter, J.; Liberman, M.C.; Fuchs, P.A.; Elgoyhen, A.B. A point mutation in the hair cell nicotinic cholinergic receptor prolongs cochlear inhibition and enhances noise protection. PLoS Biol. 2009, 7, e18. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Liu, R.; Piao, W.; Zhou, Q.; Vollmer, T.L.; Campagnolo, D.I.; Xiang, R.; La Cava, A.; Van Kaer, L.; Shi, F.D. Central nervous system (CNS)-resident natural killer cells suppress Th17 responses and CNS autoimmune pathology. J. Exp. Med. 2010, 207, 1907–1921. [Google Scholar] [CrossRef]
- Bai, X.F.; Li, O.; Zhou, Q.; Zhang, H.; Joshi, P.S.; Zheng, X.; Liu, Y.; Wang, Y.; Zheng, P.; Liu, Y. CD24 controls expansion and persistence of autoreactive T cells in the central nervous system during experimental autoimmune encephalomyelitis. J. Exp. Med. 2004, 200, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.D.; Karpus, W.J. Experimental Autoimmune Encephalomyelitis in the Mouse; Current Protocols in Immunology; Wiley: Hoboken, NJ, USA, 2007; Chapter 15: Unit 15 11. [Google Scholar]
- Puchacz, E.; Buisson, B.; Bertrand, D.; Lukas, R.J. Functional expression of nicotinic acetylcholine receptors containing rat a7 subunits in human SH-SY5Y neuroblastoma cells. FEBS Lett. 1994, 354, 155–159. [Google Scholar] [CrossRef] [Green Version]
- Plazas, P.V.; De Rosa, M.J.; Gomez-Casati, M.E.; Verbitsky, M.; Weisstaub, N.; Katz, E.; Bouzat, C.; Elgoyhen, A.B. Key roles of hydrophobic rings of TM2 in gating of the alpha9alpha10 nicotinic cholinergic receptor. Br. J. Pharmacol. 2005, 145, 963–974. [Google Scholar] [CrossRef]
- Palace, J. Making the diagnosis of multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2001, 71, ii3–ii8. [Google Scholar] [PubMed]
- Shytle, R.D.; Silver, A.A.; Lukas, R.J.; Newman, M.B.; Sheehan, D.V.; Sanberg, P.R. Nicotinic acetylcholine receptors as targets for antidepressants. Mol. Psychiatry 2002, 7, 525–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, T.; Mashimo, M.; Moriwaki, Y.; Misawa, H.; Ono, S.; Horiguchi, K.; Kawashima, K. Expression and function of the cholinergic system in immune cells. Front. Immunol. 2017, 8, 1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabley, J.G.; Pacher, P.; Southan, G.J.; Salzman, A.L.; Szabo, C. Nicotine reduces the incidence of type I diabetes in mice. J. Pharmacol. Exp. Ther. 2002, 300, 876–881. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liao, H.; Ochani, M.; Justiniani, M.; Lin, X.; Yang, L.; Al-Abed, Y.; Metz, C.; Miller, E.J.; Tracey, K.J.; et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat. Med. 2004, 10, 1216–1221. [Google Scholar] [CrossRef]
- Emre, M.; de Decker, C. Effects of cigarette smoking on motor functions in patients with multiple sclerosis. Arch. Neurol. 1992, 49, 1243–1247. [Google Scholar] [CrossRef]
- Handel, A.E.; Williamson, A.J.; Disanto, G.; Dobson, R.; Giovannoni, G.; Ramagopalan, S.V. Smoking and multiple sclerosis: An updated meta-analysis. PLoS ONE 2011, 6, e16149. [Google Scholar] [CrossRef]
- Salzer, J.; Hallmans, G.; Nystrom, M.; Stenlund, H.; Wadell, G.; Sundstrom, P. Smoking as a risk factor for multiple sclerosis. Mult. Scler. 2013, 19, 1022–1027. [Google Scholar] [CrossRef] [Green Version]
- Hedström, A.; Hillert, J.; Olsson, T.; Alfredsson, L. Nicotine might have a protective effect in the etiology of multiple sclerosis. Mult. Scler. J. 2013, 19, 1009–1013. [Google Scholar] [CrossRef]
- Hedstrom, A.K.; Baarnhielm, M.; Olsson, T.; Alfredsson, L. Tobacco smoking, but not Swedish snuff use, increases the risk of multiple sclerosis. Neurology 2009, 73, 696–701. [Google Scholar] [CrossRef]
- Ulloa, L. The vagus nerve and the nicotinic anti-inflammatory pathway. Nat. Rev. Drug Discov. 2005, 4, 673–684. [Google Scholar] [CrossRef]
- Cloëz-Tayarani, I.; Changeux, J.P. Nicotine and serotonin in immune regulation and inflammatory processes: A perspective. J. Leukoc. Biol. 2007, 81, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J. Reflex control of immunity. Nat. Rev. Immunol. 2009, 9, 418–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippini, P.; Cesario, A.; Fini, M.; Locatelli, F.; Rutella, S. The Yin and Yang of non-neuronal α7-nicotinic receptors in inflammation and autoimmunity. Curr. Drug Targets 2012, 13, 644–655. [Google Scholar] [CrossRef]
- Piao, W.H.; Campagnolo, D.; Dayao, C.; Lukas, R.J.; Wu, J.; Shi, F.D. Nicotine and inflammatory neurological disorders. Acta Pharmacol. Sin. 2009, 30, 715–722. [Google Scholar] [CrossRef] [Green Version]
- Hao, J.; Shi, F.-D.; Abdelwahab, M.; Shi, S.X.; Simard, A.; Whiteaker, P.; Lukas, R.J.; Zhou, Q. Nicotinic receptor β2 determines NK cell-dependent metastasis in a murine model of metastatic lung cancer. PLoS ONE 2013, 8, e57495. [Google Scholar] [CrossRef]
- Ellison, M.; Feng, Z.P.; Park, A.J.; Zhang, X.; Olivera, B.M.; McIntosh, J.M.; Norton, R.S. Alpha-RgIA, a novel conotoxin that blocks the alpha9alpha10 nAChR: Structure and identification of key receptor-binding residues. J. Mol. Biol. 2008, 377, 1216–1227. [Google Scholar] [CrossRef] [Green Version]
- Ellison, M.; Haberlandt, C.; Gomez-Casati, M.E.; Watkins, M.; Elgoyhen, A.B.; McIntosh, J.M.; Olivera, B.M. Alpha-RgIA: A novel conotoxin that specifically and potently blocks the alpha9alpha10 nAChR. Biochemistry 2006, 45, 1511–1517. [Google Scholar] [CrossRef]
- Yu, R.; Kompella, S.N.; Adams, D.J.; Craik, D.J.; Kaas, Q. Determination of the α-conotoxin Vc1.1 binding site on the α9α10 nicotinic acetylcholine receptor. J. Med. Chem. 2013, 56, 3557–3567. [Google Scholar] [CrossRef]
- Romero, H.K.; Christensen, S.B.; Mannelli, L.D.; Gajewiak, J.; Ramachandra, R.; Elmslie, K.S.; Vetter, D.E.; Ghelardini, C.; Iadonato, S.P.; Mercado, J.L.; et al. Inhibition of α9α10 nicotinic acetylcholine receptors prevents chemotherapy-induced neuropathic pain. Proc. Natl. Acad. Sci. 2017, 114, E1825–E1832. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, J.M.; Absalom, N.; Chebib, M.; Elgoyhen, A.B.; Vincler, M. Alpha9 nicotinic acetylcholine receptors and the treatment of pain. Biochem. Pharmacol. 2009, 78, 693–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C57BL/6J WT | α9/α10 DKO | p (WT vs. DKO) | |
|---|---|---|---|
| Mean EAE score (day 15) | 2.7 ± 0.3 | 0.8 ± 0.2 | 0.0004 |
| Peak disease severity | 2.7 ± 0.3 | 1.6 ± 0.2 | 0.006 |
| Cumulative disease score | 39.3 ± 2.6 | 16.8 ± 2.3 | 0.0002 |
| Incidence | 5/5 | 5/5 | - |
| C57BL/6J WT | FBV WT | FBV α9 KI HET | FBV α9 KI HOMO | |
|---|---|---|---|---|
| Mean EAE score (day 15) | 2.3 ± 0.2 | 0.8 ± 0.1 | 0.9 ± 0.1 | 0.8 ± 0.1 |
| p versus C57BL/6J WT | 0.00002 | 0.00002 | 0.00001 | |
| Peak disease severity | 2.4 ± 0.2 | 0.9 ± 0.1 | 1.0 ± 0.1 | 0.8 ± 0.3 |
| p versus C57BL/6J WT | 0.0005 | 0.005 | 0.001 | |
| Cumulative disease score | 24.1 ± 1.2 | 6.6 ± 2.0 | 10.4 ± 2.9 | 9.0 ± 1.9 |
| p versus C57BL/6J WT | 0.00007 | 0.004 | 0.002 | |
| Incidence | 5/5 | 7/7 | 10/10 | 5/5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Li, M.; Whiteaker, P.; Shi, F.-D.; Morley, B.J.; Lukas, R.J. Attenuation in Nicotinic Acetylcholine Receptor α9 and α10 Subunit Double Knock-Out Mice of Experimental Autoimmune Encephalomyelitis. Biomolecules 2019, 9, 827. https://doi.org/10.3390/biom9120827
Liu Q, Li M, Whiteaker P, Shi F-D, Morley BJ, Lukas RJ. Attenuation in Nicotinic Acetylcholine Receptor α9 and α10 Subunit Double Knock-Out Mice of Experimental Autoimmune Encephalomyelitis. Biomolecules. 2019; 9(12):827. https://doi.org/10.3390/biom9120827
Chicago/Turabian StyleLiu, Qiang, Minshu Li, Paul Whiteaker, Fu-Dong Shi, Barbara J. Morley, and Ronald J. Lukas. 2019. "Attenuation in Nicotinic Acetylcholine Receptor α9 and α10 Subunit Double Knock-Out Mice of Experimental Autoimmune Encephalomyelitis" Biomolecules 9, no. 12: 827. https://doi.org/10.3390/biom9120827