Differential Effects of Yeast NADH Dehydrogenase (Ndi1) Expression on Mitochondrial Function and Inclusion Formation in a Cell Culture Model of Sporadic Parkinson’s Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of PD Cybrids and Cell Culture
2.2. Viral Transfection
2.3. Immunocytochemistry
2.4. Immunoblots
2.5. Quantitative Real-Time PCR (qRT-PCR)
2.6. Cellular Respiration (OXPHOS)
2.7. Aggregated Protein and CLB Measurements
2.8. Mitochondrial Movement
3. Results
3.1. Ndi1 Expression in PD61 Cybrid and SH-SY5Y Cell Lines
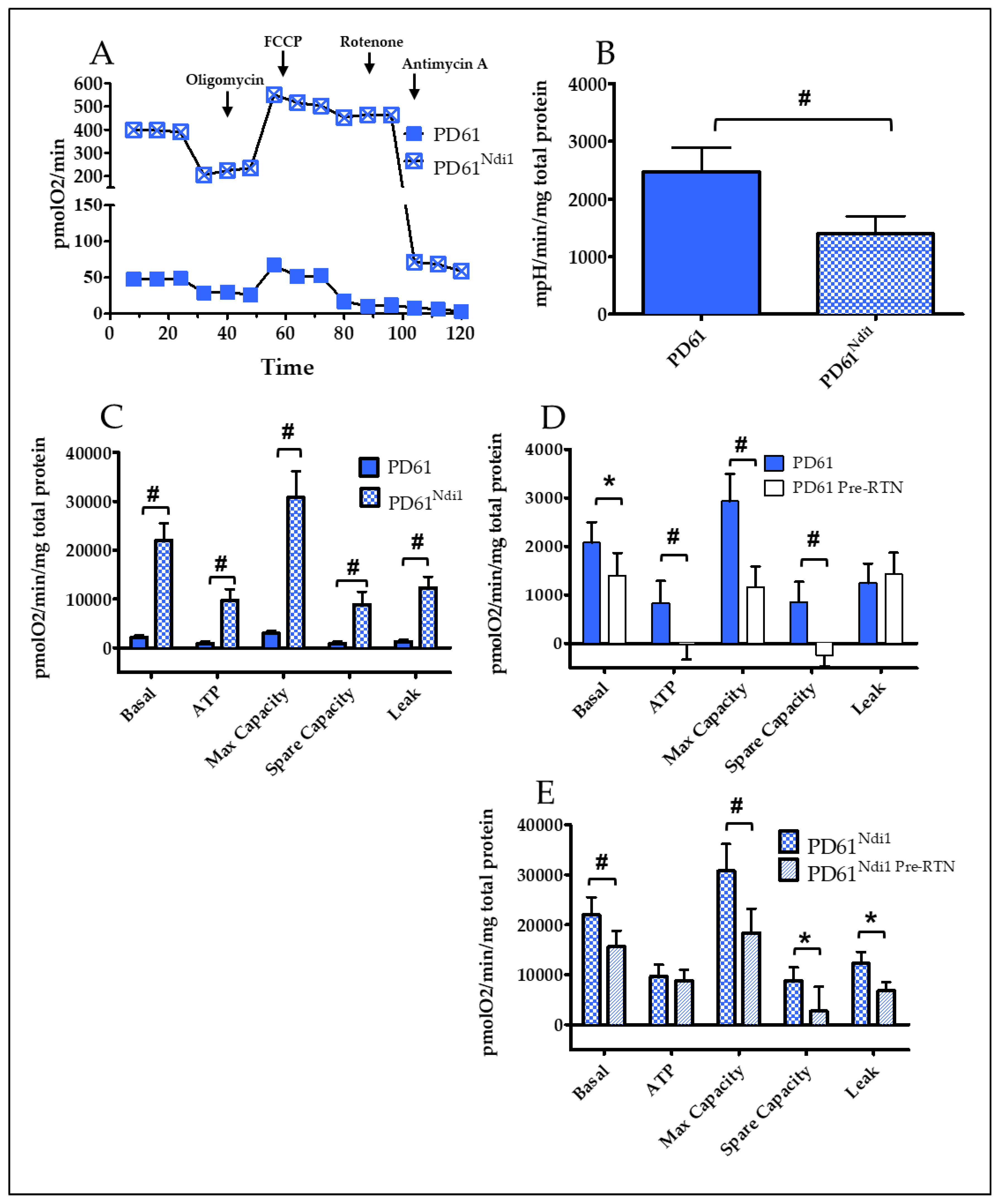
3.2. Effects of Ndi1 Expression on Oxygen Consumption Rates (OCR) and Extracellular Acidification Rates (ECAR)
3.3. PD61/PD61Ndi1
3.4. SH-SY5Y/SH-SY5YNdi1
3.5. Effects of Ndi1 Expression on Mitochondrial Movement
3.6. Effects of Ndi1 Expression on Endogenous Mitochondrial Gene Expression
3.7. Effects of Ndi1 Expression on Mitochondrial Biogenesis
3.8. Effects of Ndi1 Expression on ETC Assembly
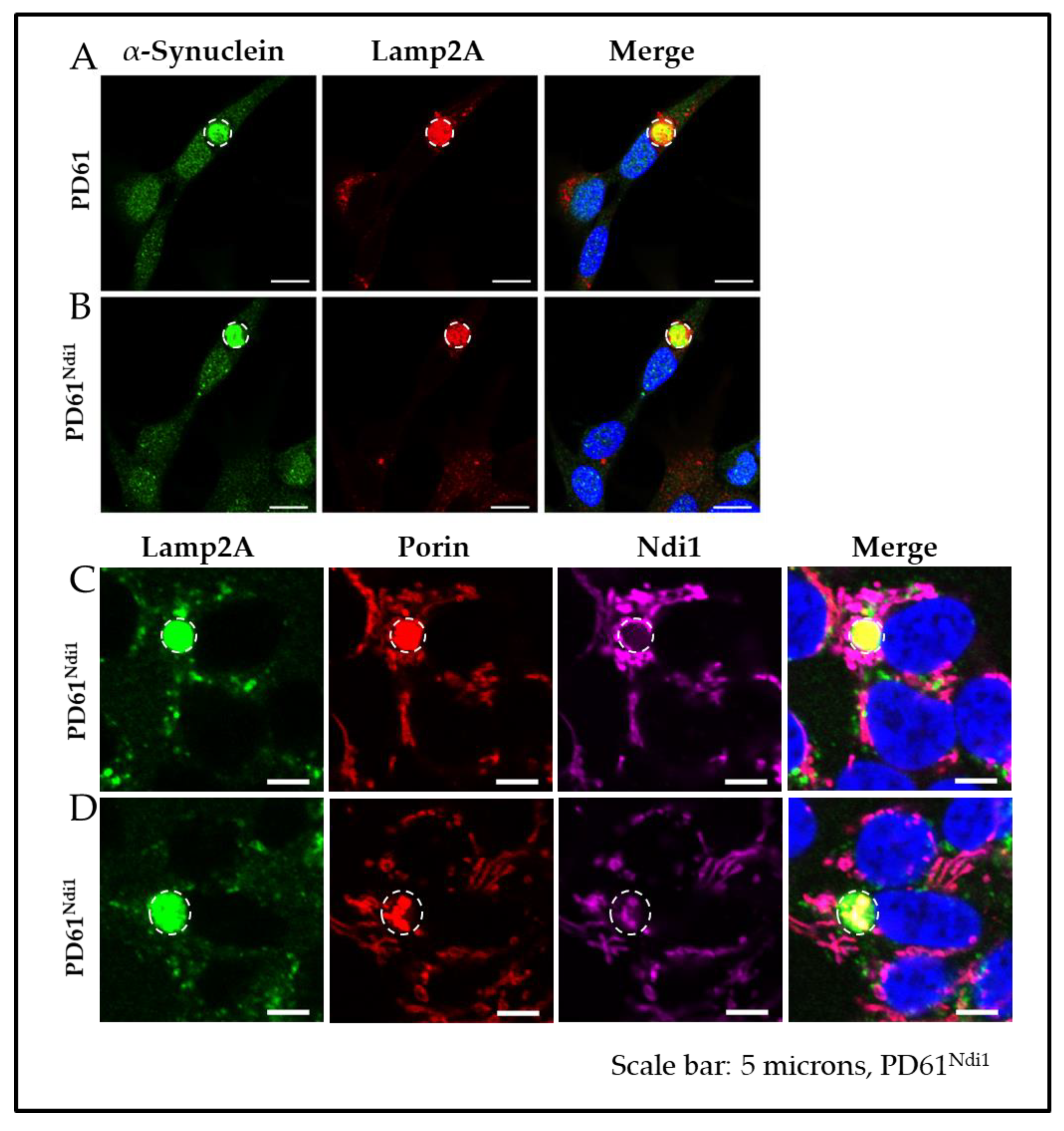
3.9. Effects on Ndi1 Expression on αSYN
3.10. Effects of Ndi1 Expression on Levels of Protein Aggregates and CLB
4. Discussion
4.1. Cellular Consequences of Ndi1 Expression in PD61
4.2. Cellular Consequences of Ndi1 Expression in SH-SY5Y
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Greenland, J.C.; Williams-Gray, C.H.; Barker, R.A. The clinical heterogeneity of Parkinson’s disease and its therapeutic implications. Eur. J. Neurosci. 2018. [Google Scholar] [CrossRef]
- Cova, I.; Priori, A. Diagnostic biomarkers for Parkinson’s disease at a glance: Where are we? J. Neural Transm. (Vienna) 2018, 125, 1417–1432. [Google Scholar] [CrossRef] [PubMed]
- Titova, N.; Chaudhuri, K.R. Nonmotor Parkinson’s and future directions. Int. Rev. Neurobiol. 2017, 134, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W. Parkinson’s disease and parkinsonism: Neuropathology. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Johnson, M.E.; Stecher, B.; Labrie, V.; Brundin, L.; Brundin, P. Triggers, Facilitators, and aggravators: Redefining Parkinson’s disease pathogenesis. Trends Neurosci. 2019, 42, 4–13. [Google Scholar] [CrossRef]
- Larsen, S.B.; Hanss, Z.; Kruger, R. The genetic architecture of mitochondrial dysfunction in Parkinson’s disease. Cell Tissue Res. 2018, 373, 21–37. [Google Scholar] [CrossRef]
- Ammal Kaidery, N.; Thomas, B. Current perspective of mitochondrial biology in Parkinson’s disease. Neurochem. Int. 2018, 117, 91–113. [Google Scholar] [CrossRef]
- Dolle, C.; Flones, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.B.; Haugarvoll, K.; et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 2016, 7, 13548. [Google Scholar] [CrossRef]
- Van der Bliek, A.M.; Sedensky, M.M.; Morgan, P.G. Cell Biology of the Mitochondrion. Genetics 2017, 207, 843–871. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; De Rasmo, D. Complex I deficiencies in neurological disorders. Trends Mol. Med. 2013, 19, 61–69. [Google Scholar] [CrossRef]
- Sanchez-Caballero, L.; Guerrero-Castillo, S.; Nijtmans, L. Unraveling the complexity of mitochondrial complex I assembly: A dynamic process. Biochim. Biophys. Acta 2016, 1857, 980–990. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Parks, J.K.; Miller, S.W.; Tuttle, J.B.; Trimmer, P.A.; Sheehan, J.P.; Bennett, J.P., Jr.; Davis, R.E.; Parker, W.D., Jr. Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann. Neurol. 1996, 40, 663–671. [Google Scholar] [CrossRef]
- Haas, R.H.; Nasirian, F.; Nakano, K.; Ward, D.; Pay, M.; Hill, R.; Shults, C.W. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson’s disease. Ann. Neurol. 1995, 37, 714–722. [Google Scholar] [CrossRef]
- Arthur, C.R.; Morton, S.L.; Dunham, L.D.; Keeney, P.M.; Bennett, J.P., Jr. Parkinson’s disease brain mitochondria have impaired respirasome assembly, age-related increases in distribution of oxidative damage to mtDNA and no differences in heteroplasmic mtDNA mutation abundance. Mol. Neurodegener. 2009, 4, 37. [Google Scholar] [CrossRef]
- Winklhofer, K.F.; Haass, C. Mitochondrial dysfunction in Parkinson’s disease. Biochim. Biophys. Acta 2010, 1802, 29–44. [Google Scholar] [CrossRef]
- Cannon, J.R.; Greenamyre, J.T. Neurotoxic in vivo models of Parkinson’s disease recent advances. Prog. Brain Res. 2010, 184, 17–33. [Google Scholar] [CrossRef]
- Sherer, T.B.; Richardson, J.R.; Testa, C.M.; Seo, B.B.; Panov, A.V.; Yagi, T.; Matsuno-Yagi, A.; Miller, G.W.; Greenamyre, J.T. Mechanism of toxicity of pesticides acting at complex I: Relevance to environmental etiologies of Parkinson’s disease. J. Neurochem. 2007, 100, 1469–1479. [Google Scholar] [CrossRef]
- Heller, A.; Brockhoff, G.; Goepferich, A. Targeting drugs to mitochondria. Eur. J. Pharm. Biopharm. 2012, 82, 1–18. [Google Scholar] [CrossRef]
- Pienaar, I.S.; Chinnery, P.F. Existing and emerging mitochondrial-targeting therapies for altering Parkinson’s disease severity and progression. Pharmacol. Ther. 2013, 137, 1–21. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Beal, M.F. Mitochondria targeted therapeutic approaches in Parkinson’s and Huntington’s diseases. Mol. Cell. Neurosci. 2012, 55, 101–114. [Google Scholar] [CrossRef]
- Mo, J.J.; Liu, L.Y.; Peng, W.B.; Rao, J.; Liu, Z.; Cui, L.L. The effectiveness of creatine treatment for Parkinson’s disease: An updated meta-analysis of randomized controlled trials. BMC Neurol. 2017, 17, 105. [Google Scholar] [CrossRef]
- Beal, M.F.; Oakes, D.; Shoulson, I.; Henchcliffe, C.; Galpern, W.R.; Haas, R.; Juncos, J.L.; Nutt, J.G.; Voss, T.S.; Ravina, B.; et al. A Randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol. 2014, 71, 543–552. [Google Scholar] [CrossRef]
- Smith, R.A.; Murphy, M.P. Mitochondria-targeted antioxidants as therapies. Discov. Med. 2011, 11, 106–114. [Google Scholar]
- Navarro, A.; Boveris, A. Brain mitochondrial dysfunction in aging, neurodegeneration, and Parkinson’s disease. Front. Aging Neurosci. 2010, 2. [Google Scholar] [CrossRef]
- Schapira, A.H.; Patel, S. Targeting mitochondria for neuroprotection in Parkinson disease. JAMA Neurol. 2014, 16, 965–973. [Google Scholar] [CrossRef]
- Barber-Singh, J.; Seo, B.B.; Matsuno-Yagi, A.; Yagi, T. Protective role of rAAV-NDI1, serotype 5, in an acute MPTP mouse Parkinson’s model. Parkinson’s Dis. 2010, 2011, 438370. [Google Scholar] [CrossRef]
- Chang, J.C.; Hoel, F.; Liu, K.H.; Wei, Y.H.; Cheng, F.C.; Kuo, S.J.; Tronstad, K.J.; Liu, C.S. Peptide-mediated delivery of donor mitochondria improves mitochondrial function and cell viability in human cybrid cells with the MELAS A3243G mutation. Sci. Rep. 2017, 7, 10710. [Google Scholar] [CrossRef]
- Marres, C.A.; de Vries, S.; Grivell, L.A. Isolation and inactivation of the nuclear gene encoding the rotenone-insensitive internal NADH: Ubiquinone oxidoreductase of mitochondria from Saccharomyces cerevisiae. Eur. J. Biochem. FEBS 1991, 195, 857–862. [Google Scholar] [CrossRef]
- De Vries, S.; Grivell, L.A. Purification and characterization of a rotenone-insensitive NADH:Q6 oxidoreductase from mitochondria of Saccharomyces cerevisiae. Eur. J. Biochem. FEBS 1988, 176, 377–384. [Google Scholar] [CrossRef]
- Park, J.S.; Li, Y.F.; Bai, Y. Yeast NDI1 improves oxidative phosphorylation capacity and increases protection against oxidative stress and cell death in cells carrying a Leber’s hereditary optic neuropathy mutation. Biochim. Biophys. Acta 2007, 1772, 533–542. [Google Scholar] [CrossRef]
- Seo, B.B.; Matsuno-Yagi, A.; Yagi, T. Modulation of oxidative phosphorylation of human kidney 293 cells by transfection with the internal rotenone-insensitive NADH-quinone oxidoreductase (NDI1) gene of Saccharomyces cerevisiae. Biochim. Biophys. Acta 1999, 1412, 56–65. [Google Scholar] [CrossRef]
- Bai, Y.; Hajek, P.; Chomyn, A.; Chan, E.; Seo, B.B.; Matsuno-Yagi, A.; Yagi, T.; Attardi, G. Lack of complex I activity in human cells carrying a mutation in mtDNA-encoded ND4 subunit is corrected by the Saccharomyces cerevisiae NADH-quinone oxidoreductase (NDI1) gene. J. Biol. Chem. 2001, 276, 38808–38813. [Google Scholar] [CrossRef]
- Seo, B.B.; Wang, J.; Flotte, T.R.; Yagi, T.; Matsuno-Yagi, A. Use of the NADH-quinone oxidoreductase (NDI1) gene of Saccharomyces cerevisiae as a possible cure for complex I defects in human cells. J. Biol. Chem. 2000, 275, 37774–37778. [Google Scholar] [CrossRef]
- Seo, B.B.; Kitajima-Ihara, T.; Chan, E.K.; Scheffler, I.E.; Matsuno-Yagi, A.; Yagi, T. Molecular remedy of complex I defects: Rotenone-insensitive internal NADH-quinone oxidoreductase of Saccharomyces cerevisiae mitochondria restores the NADH oxidase activity of complex I-deficient mammalian cells. Proc. Natl. Acad. Sci. USA 1998, 95, 9167–9171. [Google Scholar] [CrossRef]
- Marella, M.; Seo, B.B.; Flotte, T.R.; Matsuno-Yagi, A.; Yagi, T. No immune responses by the expression of the yeast Ndi1 protein in rats. PLoS ONE 2011, 6, e25910. [Google Scholar] [CrossRef]
- Cannino, G.; El-Khoury, R.; Pirinen, M.; Hutz, B.; Rustin, P.; Jacobs, H.T.; Dufour, E. Glucose modulates respiratory complex I activity in response to acute mitochondrial dysfunction. J. Biol. Chem. 2012, 287, 38729–38740. [Google Scholar] [CrossRef]
- Marella, M.; Seo, B.B.; Matsuno-Yagi, A.; Yagi, T. Mechanism of cell death caused by complex I defects in a rat dopaminergic cell line. J. Biol. Chem. 2007, 282, 24146–24156. [Google Scholar] [CrossRef]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev. Cell 2017, 40, 583–594.e6. [Google Scholar] [CrossRef]
- Yagi, T.; Seo, B.B.; Nakamaru-Ogiso, E.; Marella, M.; Barber-Singh, J.; Yamashita, T.; Kao, M.C.; Matsuno-Yagi, A. Can a single subunit yeast NADH dehydrogenase (Ndi1) remedy diseases caused by respiratory complex I defects? Rejuvenation Res. 2006, 9, 191–197. [Google Scholar] [CrossRef]
- Chadderton, N.; Palfi, A.; Millington-Ward, S.; Gobbo, O.; Overlack, N.; Carrigan, M.; O’Reilly, M.; Campbell, M.; Ehrhardt, C.; Wolfrum, U.; et al. Intravitreal delivery of AAV-NDI1 provides functional benefit in a murine model of Leber hereditary optic neuropathy. Eur. J. Hum. Genet. 2013, 21, 62–68. [Google Scholar] [CrossRef]
- Cho, J.; Hur, J.H.; Graniel, J.; Benzer, S.; Walker, D.W. Expression of yeast NDI1 rescues a Drosophila complex I assembly defect. PLoS ONE 2012, 7, e50644. [Google Scholar] [CrossRef]
- Marella, M.; Seo, B.B.; Thomas, B.B.; Matsuno-Yagi, A.; Yagi, T. Successful amelioration of mitochondrial optic neuropathy using the yeast NDI1 gene in a rat animal model. PLoS ONE 2010, 5, e11472. [Google Scholar] [CrossRef]
- Sanz, A.; Soikkeli, M.; Portero-Otin, M.; Wilson, A.; Kemppainen, E.; McIlroy, G.; Ellila, S.; Kemppainen, K.K.; Tuomela, T.; Lakanmaa, M.; et al. Expression of the yeast NADH dehydrogenase Ndi1 in Drosophila confers increased lifespan independently of dietary restriction. Proc. Natl. Aca. Sci. USA 2010, 107, 9105–9110. [Google Scholar] [CrossRef]
- Barber-Singh, J.; Seo, B.B.; Nakamaru-Ogiso, E.; Lau, Y.S.; Matsuno-Yagi, A.; Yagi, T. Neuroprotective effect of long-term NDI1 gene expression in a chronic mouse model of Parkinson disorder. Rejuvenation Res. 2009, 12, 259–267. [Google Scholar] [CrossRef]
- Albert, K.; Voutilainen, M.H.; Domanskyi, A.; Airavaara, M. AAV vector-mediated gene delivery to substantia nigra dopamine neurons: Implications for gene therapy and disease models. Genes (Basel) 2017, 8, E63. [Google Scholar] [CrossRef]
- Richardson, J.R.; Caudle, W.M.; Guillot, T.S.; Watson, J.L.; Nakamaru-Ogiso, E.; Seo, B.B.; Sherer, T.B.; Greenamyre, J.T.; Yagi, T.; Matsuno-Yagi, A.; et al. Obligatory role for complex I inhibition in the dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicol. Sci. 2007, 95, 196–204. [Google Scholar] [CrossRef]
- Seo, B.B.; Marella, M.; Yagi, T.; Matsuno-Yagi, A. The single subunit NADH dehydrogenase reduces generation of reactive oxygen species from complex I. FEBS Lett. 2006, 580, 6105–6108. [Google Scholar] [CrossRef]
- Sherer, T.B.; Betarbet, R.; Testa, C.M.; Seo, B.B.; Richardson, J.R.; Kim, J.H.; Miller, G.W.; Yagi, T.; Matsuno-Yagi, A.; Greenamyre, J.T. Mechanism of toxicity in rotenone models of Parkinson’s disease. J. Neurosci. 2003, 23, 10756–10764. [Google Scholar] [CrossRef]
- Vilain, S.; Esposito, G.; Haddad, D.; Schaap, O.; Dobreva, M.P.; Vos, M.; Van Meensel, S.; Morais, V.A.; De Strooper, B.; Verstreken, P. The yeast complex I equivalent NADH dehydrogenase rescues pink1 mutants. PLoS Genet. 2012, 8, e1002456. [Google Scholar] [CrossRef]
- Trimmer, P.A.; Borland, M.K.; Keeney, P.M.; Bennett, J.P., Jr.; Parker, W.D., Jr. Parkinson’s disease transgenic mitochondrial cybrids generate Lewy inclusion bodies. J. Neurochem. 2004, 88, 800–812. [Google Scholar] [CrossRef]
- Stellos, K.; Panagiota, V.; Kogel, A.; Leyhe, T.; Gawaz, M.; Laske, C. Predictive value of platelet activation for the rate of cognitive decline in Alzheimer’s disease patients. J. Cereb. Blood Flow Metab. 2010, 30, 1817–1820. [Google Scholar] [CrossRef]
- Kocer, A.; Yaman, A.; Niftaliyev, E.; Duruyen, H.; Eryilmaz, M.; Kocer, E. Assessment of platelet indices in patients with neurodegenerative diseases: Mean platelet volume was increased in patients with Parkinson’s disease. Curr. Gerontol. Geriatr. Res. 2013, 2013, 986254. [Google Scholar] [CrossRef]
- Silva, D.F.; Selfridge, J.E.; Lu, J.; E, L.; Roy, N.; Hutfles, L.; Burns, J.M.; Michaelis, E.K.; Yan, S.; Cardoso, S.M.; et al. Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines. Hum. Mol. Genet. 2013, 22, 3931–3946. [Google Scholar] [CrossRef]
- Llobet, L.; Gomez-Duran, A.; Iceta, R.; Iglesias, E.; Montoya, J.; Martin-Martinez, J.; Ara, J.R.; Ruiz-Pesini, E. Stressed cybrids model demyelinated axons in multiple sclerosis. Metab. Brain Dis. 2013, 28, 639–645. [Google Scholar] [CrossRef]
- Trimmer, P.A.; Bennett, J.P., Jr. The cybrid model of sporadic Parkinson’s disease. Exp. Neurol. 2009, 218, 320–325. [Google Scholar] [CrossRef]
- Yu, Q.; Du, F.; Douglas, J.T.; Yu, H.; Yan, S.S.; Yan, S.F. Mitochondrial dysfunction triggers synaptic deficits via activation of p38 MAP kinase signaling in differentiated Alzheimer’s disease trans-mitochondrial cybrid cells. J. Alzheimers Dis. 2017, 59, 223–239. [Google Scholar] [CrossRef]
- Yu, Q.; Fang, D.; Swerdlow, R.H.; Yu, H.; Chen, J.X.; Yan, S.S. Antioxidants rescue mitochondrial transport in differentiated Alzheimer’s disease trans-mitochondrial cybrid cells. J. Alzheimers Dis. 2016, 54, 679–690. [Google Scholar] [CrossRef]
- Pignataro, D.; Francia, S.; Zanetta, F.; Brenna, G.; Brandini, S.; Olivieri, A.; Torroni, A.; Biamonti, G.; Montecucco, A. A missense MT-ND5 mutation in differentiated Parkinson disease cytoplasmic hybrid induces ROS-dependent DNA damage esponse amplified by DROSHA. Sci. Rep. 2017, 7, 9528. [Google Scholar] [CrossRef]
- Keeney, P.M.; Dunham, L.D.; Quigley, C.K.; Morton, S.L.; Bergquist, K.E.; Bennett, J.P., Jr. Cybrid models of Parkinson’s disease show variable mitochondrial biogenesis and genotype-respiration relationships. Exp. Neurol. 2009, 220, 374–382. [Google Scholar] [CrossRef]
- Cronin-Furman, E.N.; Borland, M.K.; Bergquist, K.E.; Bennett, J.P., Jr.; Trimmer, P.A. Mitochondrial quality, dynamics and functional capacity in Parkinson’s disease cybrid cell lines selected for Lewy body expression. Mol. Neurodegener. 2013, 8, 6. [Google Scholar] [CrossRef]
- Dranka, B.P.; Hill, B.G.; Darley-Usmar, V.M. Mitochondrial reserve capacity in endothelial cells: The impact of nitric oxide and reactive oxygen species. Free Radic. Biol. Med. 2010, 48, 905–914. [Google Scholar] [CrossRef]
- Borland, M.K.; Trimmer, P.A.; Rubinstein, J.D.; Keeney, P.M.; Mohanakumar, K.; Liu, L.; Bennett, J.P., Jr. Chronic, low-dose rotenone reproduces Lewy neurites found in early stages of Parkinson’s disease, reduces mitochondrial movement and slowly kills differentiated SH-SY5Y neural cells. Mol. Neurodegener. 2008, 3, 21. [Google Scholar] [CrossRef]
- Trimmer, P.A.; Schwartz, K.M.; Borland, M.K.; De Taboada, L.; Streeter, J.; Oron, U. Reduced axonal transport in Parkinson’s disease cybrid neurites is restored by light therapy. Mol. Neurodegener. 2009, 4, 26. [Google Scholar] [CrossRef]
- Gerencser, A.A.; Neilson, A.; Choi, S.W.; Edman, U.; Yadava, N.; Oh, R.J.; Ferrick, D.A.; Nicholls, D.G.; Brand, M.D. Quantitative microplate-based respirometry with correction for oxygen diffusion. Anal. Chem. 2009, 81, 6868–6878. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; E, L.; Aires, D.; Lu, J. Glycolysis-respiration relationships in a neuroblastoma cell line. Biochim. Biophys. Acta 2013, 1830, 2891–2898. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Nuclear control of respiratory gene expression in mammalian cells. J. Cell. Biochem. 2006, 97, 673–683. [Google Scholar] [CrossRef]
- Diaz, F.; Moraes, C.T. Mitochondrial biogenesis and turnover. Cell Calcium 2008, 44, 24–35. [Google Scholar] [CrossRef]
- Gaspari, M.; Larsson, N.G.; Gustafsson, C.M. The transcription machinery in mammalian mitochondria. Biochim. Biophys. Acta 2004, 1659, 148–152. [Google Scholar] [CrossRef]
- Capaldi, R.A.; Murray, J.; Byrne, L.; Janes, M.S.; Marusich, M.F. Immunological approaches to the characterization and diagnosis of mitochondrial disease. Mitochondrion 2004, 4, 417–426. [Google Scholar] [CrossRef]
- Beyer, K. Alpha-synuclein structure, posttranslational modification and alternative splicing as aggregation enhancers. Acta Neuropathol. 2006, 112, 237–251. [Google Scholar] [CrossRef]
- Ischiropoulos, H. Oxidative modifications of alpha-synuclein. Ann. N. Y. Acad. Sci. 2003, 991, 93–100. [Google Scholar] [CrossRef]
- Camilleri, A.; Zarb, C.; Caruana, M.; Ostermeier, U.; Ghio, S.; Hogen, T.; Schmidt, F.; Giese, A.; Vassallo, N. Mitochondrial membrane permeabilisation by amyloid aggregates and protection by polyphenols. Biochim. Biophys. Acta 2013, 1828, 2532–2543. [Google Scholar] [CrossRef]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T., Jr. Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry 2000, 39, 2552–2563. [Google Scholar] [CrossRef]
- Lendel, C.; Bertoncini, C.W.; Cremades, N.; Waudby, C.A.; Vendruscolo, M.; Dobson, C.M.; Schenk, D.; Christodoulou, J.; Toth, G. On the mechanism of nonspecific inhibitors of protein aggregation: Dissecting the interactions of alpha-synuclein with Congo red and lacmoid. Biochemistry 2009, 48, 8322–8334. [Google Scholar] [CrossRef]
- Marella, M.; Seo, B.B.; Yagi, T.; Matsuno-Yagi, A. Parkinson’s disease and mitochondrial complex I: A perspective on the Ndi1 therapy. J. Bioenerget. Biomembr. 2009, 41, 493–497. [Google Scholar] [CrossRef]
- Marella, M.; Seo, B.B.; Nakamaru-Ogiso, E.; Greenamyre, J.T.; Matsuno-Yagi, A.; Yagi, T. Protection by the NDI1 gene against neurodegeneration in a rotenone rat model of Parkinson’s disease. PLoS ONE 2008, 3, e1433. [Google Scholar] [CrossRef]
- DeCorby, A.; Gaskova, D.; Sayles, L.C.; Lemire, B.D. Expression of Ndi1p, an alternative NADH:ubiquinone oxidoreductase, increases mitochondrial membrane potential in a C. elegans model of mitochondrial disease. Biochim. Biophys. Acta 2007, 1767, 1157–1163. [Google Scholar] [CrossRef]
- Rustin, P.; Jacobs, H.T. Respiratory chain alternative enzymes as tools to better understand and counteract respiratory chain deficiencies in human cells and animals. Physiol. Plant. 2009, 137, 362–370. [Google Scholar] [CrossRef]
- Borland, M.K.; Mohanakumar, K.P.; Rubinstein, J.D.; Keeney, P.M.; Xie, J.; Capaldi, R.; Dunham, L.D.; Trimmer, P.A.; Bennett, J.P., Jr. Relationships among molecular genetic and respiratory properties of Parkinson’s disease cybrid cells show similarities to Parkinson’s brain tissues. Biochim. Biophys. Acta 2009, 1792, 68–74. [Google Scholar] [CrossRef]
- Desler, C.; Hansen, T.L.; Frederiksen, J.B.; Marcker, M.L.; Singh, K.K.; Juel Rasmussen, L. Is There a link between mitochondrial reserve respiratory capacity and aging? J. Aging Res. 2012, 2012, 192503. [Google Scholar] [CrossRef]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef]
- Belarbi, K.; Cuvelier, E.; Destee, A.; Gressier, B.; Chartier-Harlin, M.C. NADPH oxidases in Parkinson’s disease: A systematic review. Mol. Neurodegener. 2017, 12, 84. [Google Scholar] [CrossRef]
- Austin, S.; St-Pierre, J. PGC1alpha and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125, 4963–4971. [Google Scholar] [CrossRef]
- Choi, S.W.; Gerencser, A.A.; Nicholls, D.G. Bioenergetic analysis of isolated cerebrocortical nerve terminals on a microgram scale: Spare respiratory capacity and stochastic mitochondrial failure. J. Neurochem. 2009, 109, 1179–1191. [Google Scholar] [CrossRef]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg effect: 80 years on. Biochem. Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondrial medicine and the neurodegenerative mitochondriopathies. Pharmaceuticals (Basel) 2009, 2, 150–167. [Google Scholar] [CrossRef]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef]
- Zurita Rendon, O.; Shoubridge, E.A. Early complex I assembly defects result in rapid turnover of the NDI1 subunit. Hum. Mol. Genet. 2012, 21, 3815–3824. [Google Scholar] [CrossRef]
- Picard, M.; Shirihai, O.S.; Gentil, B.J.; Burelle, Y. Mitochondrial morphology transitions and functions: Implications for retrograde signaling? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R393–R406. [Google Scholar] [CrossRef]
- Austin, S.; Klimcakova, E.; St-Pierre, J. Impact of PCG-1α on the topology and rate of superoxide production by the mitochondrial electron transport chain. Free Radic. Biol. Med. 2011, 51, 2243–2248. [Google Scholar] [CrossRef]
- Srivastava, S.; Diaz, F.; Iommarini, L.; Aure, K.; Lombes, A.; Moraes, C.T. PGC-1alpha/beta induced expression partially compensates for respiratory chain defects in cells from patients with mitochondrial disorders. Hum. Mol. Genet. 2009, 18, 1805–1812. [Google Scholar] [CrossRef]
- Andreux, P.A.; Houtkooper, R.H.; Auwerx, J. Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug Discov. 2013, 12, 465–483. [Google Scholar] [CrossRef]
- Kuusisto, E.; Parkkinen, L.; Alafuzoff, I. Morphogenesis of Lewy bodies: Dissimilar incorporation of alpha-synuclein, ubiquitin, and p62. J. Neuropathol. Exp. Neurol. 2003, 62, 1241–1253. [Google Scholar] [CrossRef]
- Nashine, S.; Cohen, P.; Chwa, M.; Lu, S.; Nesburn, A.B.; Kuppermann, B.D.; Kenney, M.C. Humanin G (HNG) protects age-related macular degeneration (AMD) transmitochondrial ARPE-19 cybrids from mitochondrial and cellular damage. Cell Death Dis. 2017, 8, e2951. [Google Scholar] [CrossRef]
- Seo, B.B.; Nakamaru-Ogiso, E.; Flotte, T.R.; Yagi, T.; Matsuno-Yagi, A. A single-subunit NADH-quinone oxidoreductase renders resistance to mammalian nerve cells against complex I inhibition. Mol. Ther. J. Am. Soc. Gene Ther. 2002, 6, 336–341. [Google Scholar] [CrossRef]
- Schneider, L.; Giordano, S.; Zelickson, B.R.; M, S.J.; G, A.B.; Ouyang, X.; Fineberg, N.; Darley-Usmar, V.M.; Zhang, J. Differentiation of SH-SY5Y cells to a neuronal phenotype changes cellular bioenergetics and the response to oxidative stress. Free Radic. Biol. Med. 2011, 51, 2007–2017. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cronin-Furman, E.N.; Barber-Singh, J.; Bergquist, K.E.; Yagi, T.; Trimmer, P.A. Differential Effects of Yeast NADH Dehydrogenase (Ndi1) Expression on Mitochondrial Function and Inclusion Formation in a Cell Culture Model of Sporadic Parkinson’s Disease. Biomolecules 2019, 9, 119. https://doi.org/10.3390/biom9040119
Cronin-Furman EN, Barber-Singh J, Bergquist KE, Yagi T, Trimmer PA. Differential Effects of Yeast NADH Dehydrogenase (Ndi1) Expression on Mitochondrial Function and Inclusion Formation in a Cell Culture Model of Sporadic Parkinson’s Disease. Biomolecules. 2019; 9(4):119. https://doi.org/10.3390/biom9040119
Chicago/Turabian StyleCronin-Furman, Emily N., Jennifer Barber-Singh, Kristen E. Bergquist, Takao Yagi, and Patricia A. Trimmer. 2019. "Differential Effects of Yeast NADH Dehydrogenase (Ndi1) Expression on Mitochondrial Function and Inclusion Formation in a Cell Culture Model of Sporadic Parkinson’s Disease" Biomolecules 9, no. 4: 119. https://doi.org/10.3390/biom9040119