
Genetic Structure and Geographical Differentiation of Traditional Rice (Oryza sativa L.) from Northern Vietnam

 ,
,
Abstract
:1. Introduction
2. Results
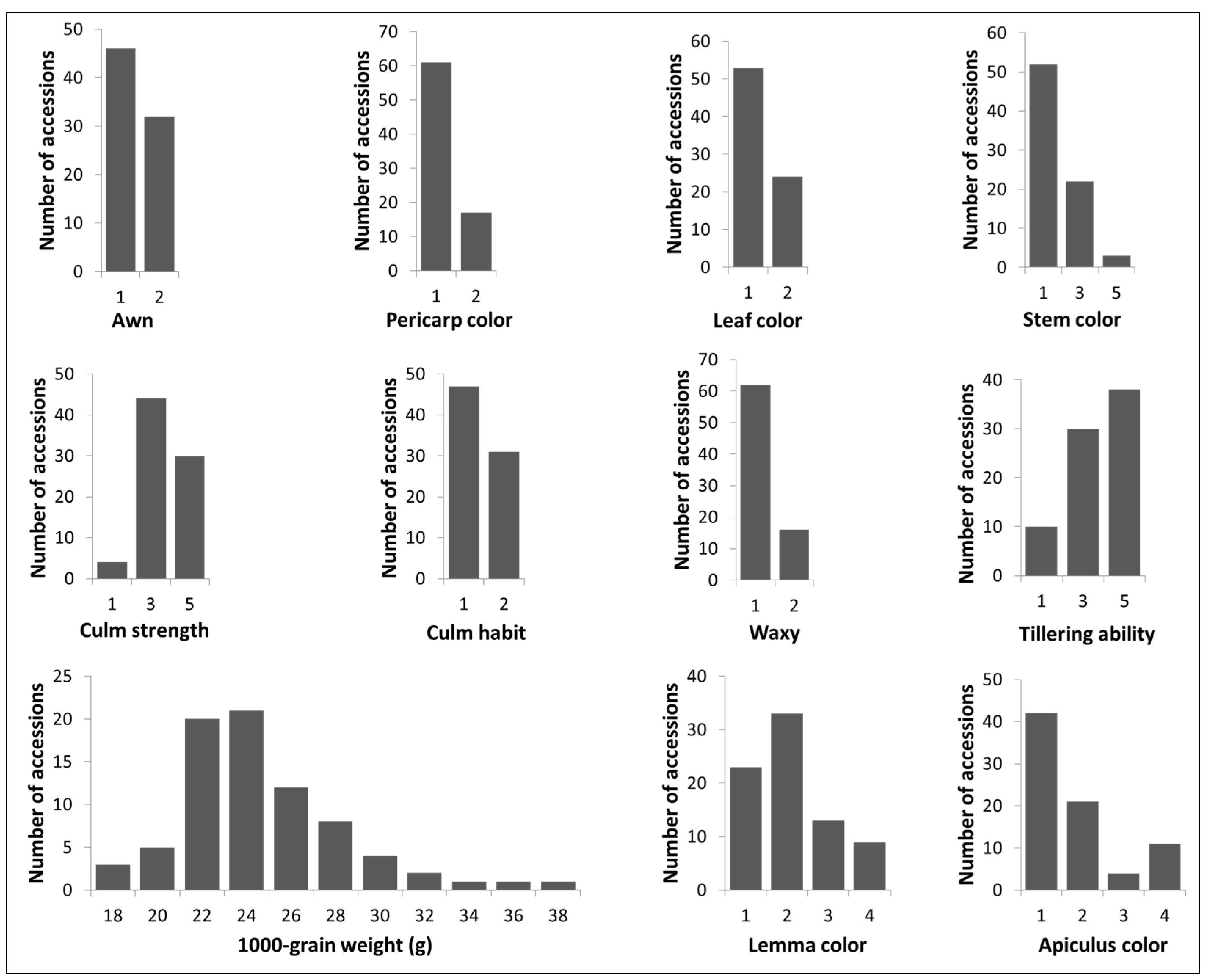
2.1. Assessment of Phenotype
2.2. Marker Polymorphisms
2.3. Allele Specificity
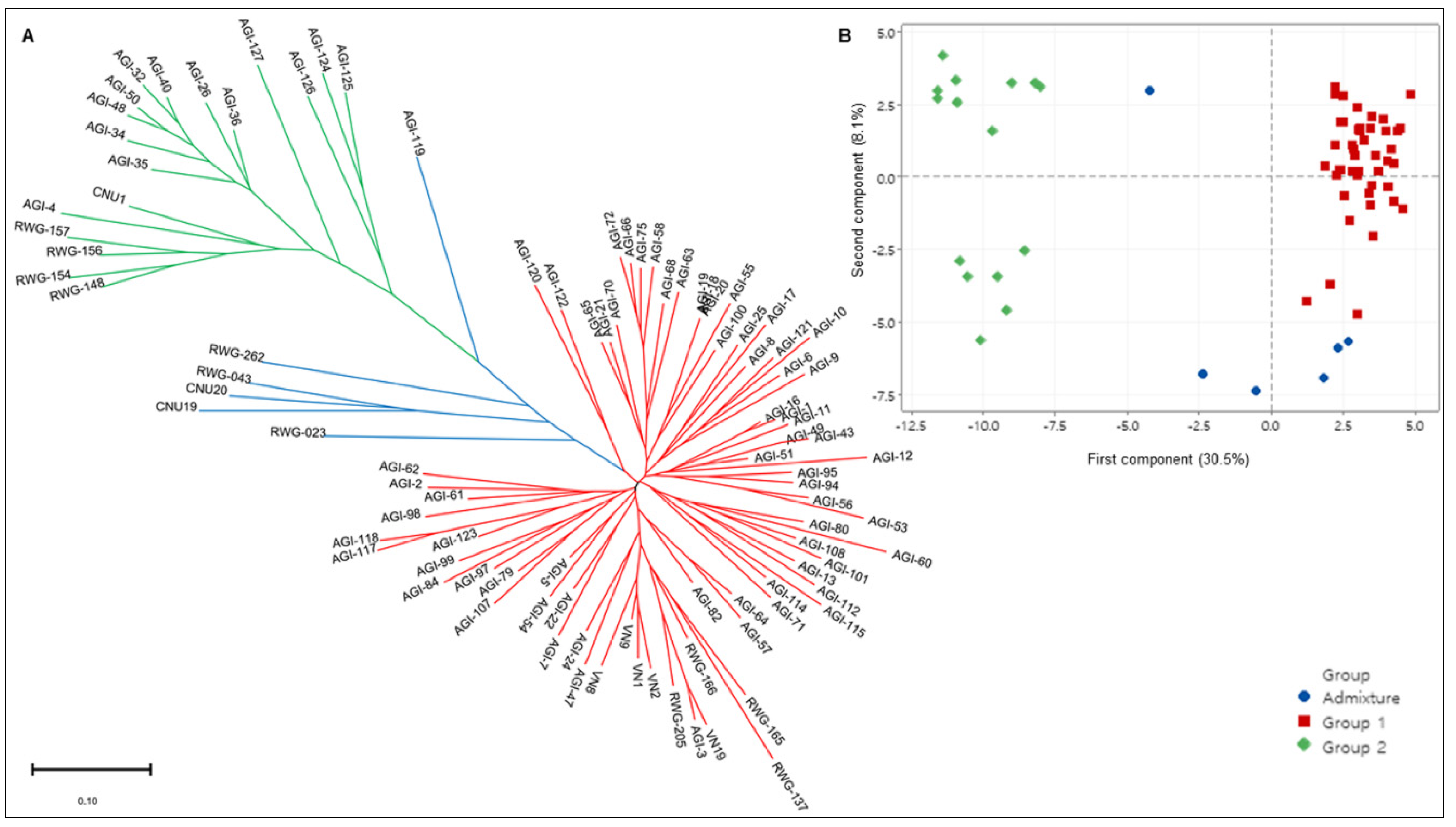
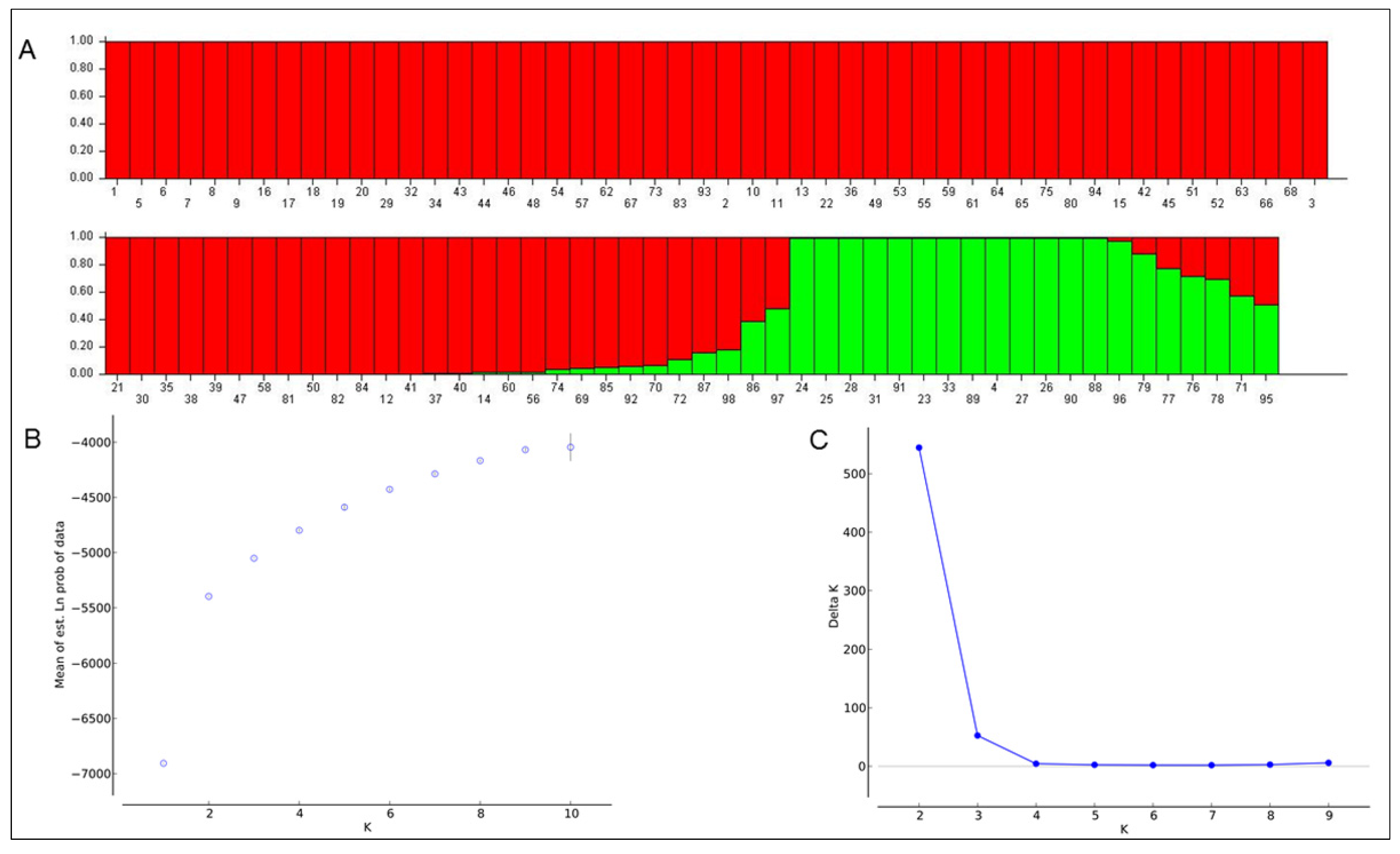
2.4. Population Structure Analysis
2.5. Distinguishing Indica/Japonica Subspecies
2.6. Genetic Differentiation between Subgroups/Regions
2.7. Genetic Diversity within Subgroups and Geographic Distribution of Genetic Diversity
2.8. Contribution of Population Structure to Phenotypic Variation
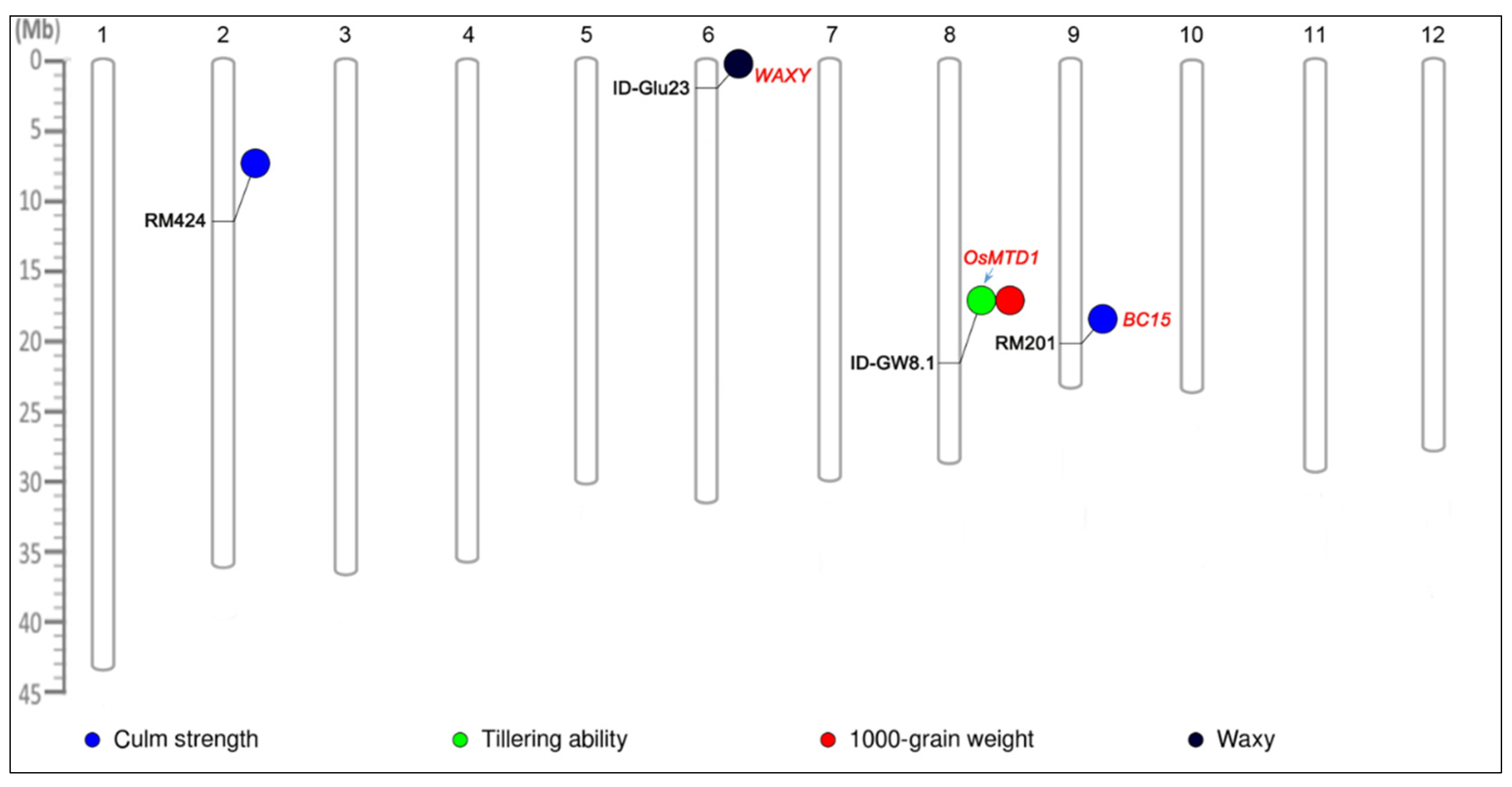
2.9. Association Analysis within AGI Collection
2.10. Performance of Traits Relevant to Different Alleles of Significant Loci
3. Discussion
3.1. Genetic Diversity
3.2. Population Structure and Factors Affecting Genetic Differentiation
3.3. Distinguishing Indica and Japonica Subspecies
3.4. Marker-Trait Association Analysis
3.5. Rare Allele
4. Materials and Methods
4.1. Plant Materials
4.2. Phenotypic Screening and Statistical Analysis
4.3. SSR and InDel Markers Analysis
4.4. KASP (Kompetitive Allele-Specific PCR) Analysis
4.5. Distinguishing Indica-Japonica Subspecies
4.6. Genetic Diversity and Population Structure Analysis
4.7. Marker-Trait Associations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molina, J.; Sikora, M.; Garud, N.; Flowers, J.M.; Rubinstein, S.; Reynolds, A.; Huang, P.; Jackson, S.; Schaal, B.A.; Bustamante, C.D.; et al. Molecular evidence for a single evolutionary origin of domesticated rice. Proc. Natl. Acad. Sci. USA 2011, 108, 8351–8356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Londo, J.P.; Chiang, Y.C.; Hung, K.H.; Chiang, T.Y.; Schaal, B.A. Phylogeography of Asian wild rice, Oryza rufipogon, reveals multiple independent domestications of cultivated rice, Oryza sativa. Proc. Natl. Acad. Sci. USA 2006, 103, 9578–9583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nachimuthu, V.V.; Muthurajan, R.; Duraialaguraja, S.; Sivakami, R.; Pandian, B.A.; Ponniah, G.; Gunasekaran, K.; Swaminathan, M.; K, K.S.; Sabariappan, R. Analysis of Population Structure and Genetic Diversity in Rice Germplasm Using SSR Markers: An Initiative Towards Association Mapping of Agronomic Traits in Oryza sativa. Rice 2015, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Cui, D.; Tang, C.; Lu, H.; Li, J.; Ma, X.; A, X.; Han, B.; Yang, Y.; Dong, C.; Zhang, F.; et al. Genetic differentiation and restricted gene flow in rice landraces from Yunnan, China: Effects of isolation-by-distance and isolation-by-environment. Rice 2021, 14, 54. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.B.; Weale, M.E. Population genomics: Linkage disequilibrium holds the key. Curr. Biol. 2001, 11, R576–R579. [Google Scholar] [CrossRef] [Green Version]
- Buckler, E.S.t.; Thornsberry, J.M. Plant molecular diversity and applications to genomics. Curr. Opin. Plant Biol. 2002, 5, 107–111. [Google Scholar] [CrossRef]
- Chang, T.-T. The origin, evolution, cultivation, dissemination, and diversification of Asian and African rices. Euphytica 1976, 25, 425–441. [Google Scholar] [CrossRef]
- Higham, C.; Lu, T.L.D. The origins and dispersal of rice cultivation. Antiquity 1998, 72, 867–877. [Google Scholar] [CrossRef]
- Phung, N.T.; Mai, C.D.; Mournet, P.; Frouin, J.; Droc, G.; Ta, N.K.; Jouannic, S.; Le, L.T.; Do, V.N.; Gantet, P.; et al. Characterization of a panel of Vietnamese rice varieties using DArT and SNP markers for association mapping purposes. BMC Plant Biol. 2014, 14, 371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, G.T.; Van Dinh, L.; Nguyen, T.T.; Ta, N.K.; Gathignol, F.; Mai, C.D.; Jouannic, S.; Tran, K.D.; Khuat, T.H.; Do, V.N.; et al. Genome-wide Association Study of a Panel of Vietnamese Rice Landraces Reveals New QTLs for Tolerance to Water Deficit During the Vegetative Phase. Rice 2019, 12, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, T.D.; Gathignol, F.; Vu, H.T.; Nguyen, K.L.; Tran, L.H.; Vu, H.T.T.; Dinh, T.X.; Lazennec, F.; Pham, X.H.; Very, A.A.; et al. Genome-Wide Association Mapping of Salinity Tolerance at the Seedling Stage in a Panel of Vietnamese Landraces Reveals New Valuable QTLs for Salinity Stress Tolerance Breeding in Rice. Plants 2021, 10, 1088. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.; Santos, B.; Khanh, T.D.; Trung, K.H.; Duong, T.D.; Doai, N.T.P.; Khoa, N.T.; Ha, D.T.T.; Diep, N.T.; Dung, K.T.; et al. Resequencing of 672 Native Rice Accessions to Explore Genetic Diversity and Trait Associations in Vietnam. Rice 2021, 14, 52. [Google Scholar] [CrossRef] [PubMed]
- Pusadee, T.; Jamjod, S.; Chiang, Y.C.; Rerkasem, B.; Schaal, B.A. Genetic structure and isolation by distance in a landrace of Thai rice. Proc. Natl. Acad. Sci. USA 2009, 106, 13880–13885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goudarzi, F.; Hemami, M.R.; Rancilhac, L.; Malekian, M.; Fakheran, S.; Elmer, K.R.; Steinfartz, S. Geographic separation and genetic differentiation of populations are not coupled with niche differentiation in threatened Kaiser’s spotted newt (Neurergus kaiseri). Sci. Rep. 2019, 9, 6239. [Google Scholar] [CrossRef] [Green Version]
- Gregorius, H.R. The relationship between the concepts of genetic diversity and differentiation. Theor. Appl. Genet. 1987, 74, 397–401. [Google Scholar] [CrossRef]
- Xiong, H.; Shi, A.; Mou, B.; Qin, J.; Motes, D.; Lu, W.; Ma, J.; Weng, Y.; Yang, W.; Wu, D. Genetic Diversity and Population Structure of Cowpea (Vigna unguiculata L. Walp). PLoS ONE 2016, 11, e0160941. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Chung, J.-W.; Ma, K.-H.; Kim, T.-S.; Kim, S.-M.; Shin, D.-I.; Kim, C.-H.; Koo, H.-M.; Park, Y. Analysis of Genetic Diversity and Population Structure of Rice Cultivars from Korea, China and Japan using SSR Markers. Genes Genom. 2009, 31, 283–292. [Google Scholar] [CrossRef]
- Wright, S. Evolution and the Genetics of Populations. A Treatise in Four Volumes; Variability within and among Natural Populations; University of Chicago Press: Chicago, IL, USA, 1978; Volume 4. [Google Scholar]
- Oka, H.-I.; Morishima, H. Phylogenetic differentiation of cultivated rice, XXIII. Potentiality of wild progenitors to evolve the Indica and Japonica types of rice cultivars. Euphytica 1982, 31, 41–50. [Google Scholar] [CrossRef]
- Chen, W.-B.; Sato, Y.-I.; Nakamura, I.; Nakai, H. Indica-japonica differentiation in Chinese rice landraces. Euphytica 1993, 74, 195–201. [Google Scholar] [CrossRef]
- Oka, H.-I. Origin of Cultivated Rice; Japan Scientific Societies Press: Tokyo, Japan, 1988. [Google Scholar]
- Muto, C.; Ishikawa, R.; Olsen, K.M.; Kawano, K.; Bounphanousay, C.; Matoh, T.; Sato, Y.I. Genetic diversity of the wx flanking region in rice landraces in northern Laos. Breed. Sci. 2016, 66, 580–590. [Google Scholar] [CrossRef] [Green Version]
- Taddesse, L.; Fukuta, Y.; Ishikawa, R. Genetic study of diversity and blast resistance in Ethiopian rice cultivars adapted to different ecosystems. Breed. Sci. 2020, 70, 303–312. [Google Scholar] [CrossRef]
- Wanchana, S.; Toojinda, T.; Tragoonrung, S.; Vanavichit, A. Duplicated coding sequence in the waxy allele of tropical glutinous rice (Oryza sativa L.). Plant Sci. 2003, 165, 1193–1199. [Google Scholar] [CrossRef]
- Liu, Q.; Shen, G.; Peng, K.; Huang, Z.; Tong, J.; Kabir, M.H.; Wang, J.; Zhang, J.; Qin, G.; Xiao, L. The alteration in the architecture of a T-DNA insertion rice mutant osmtd1 is caused by up-regulation of MicroRNA156f. J. Integr. Plant Biol. 2015, 57, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Zhang, B.; Dai, Y.; Zhang, L.; Shang-Guan, K.; Peng, Y.; Zhou, Y.; Zhu, Z. Brittle culm15 encodes a membrane-associated chitinase-like protein required for cellulose biosynthesis in rice. Plant Physiol. 2012, 159, 1440–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Valdés, M.H.; Burgueño, J.; Singh, S.; Martínez, O.; Sansaloni, C.P. An informational view of accession rarity and allele specificity in germplasm banks for management and conservation. PLoS ONE 2018, 13, e0193346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, T.; Wu, J.; Itoh, T.; Numa, H.; Antonio, B.; Sasaki, T. The Nipponbare genome and the next-generation of rice genomics research in Japan. Rice 2016, 9, 33. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Li, S.; Ren, H. OsFH15, a class I formin, interacts with microfilaments and microtubules to regulate grain size via affecting cell expansion in rice. Sci. Rep. 2017, 7, 6538. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Jin, F.; Song, M.H.; Suh, J.P.; Hwang, H.G.; Kim, Y.G.; McCouch, S.R.; Ahn, S.N. Fine mapping of a yield-enhancing QTL cluster associated with transgressive variation in an Oryza sativa x O. rufipogon cross. Theor. Appl. Genet. 2008, 116, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Sun, Y.; Li, J.; Su, L.; Zheng, X.; Wang, X.; Li, K.; Yang, Q.; Qiao, W. Identify QTLs for grain size and weight in common wild rice using chromosome segment substitution lines across six environments. Breed. Sci. 2017, 67, 472–482. [Google Scholar] [CrossRef] [Green Version]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research--an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef] [Green Version]
- Luong, N.; Jeon, Y.-A.; Shim, K.-C.; Kim, S.; Lee, H.-S.; Adeva, C.; Do, V.A.; Ahn, S.-N. Characterization of the Spikelet Number per Panicle QTL qSPP7 Using a Nearly Isogenic Line Derived from an Interspecific Cross in Rice. Plant Breed. Biotechnol. 2019, 7, 245–256. [Google Scholar] [CrossRef]
- Cheon, K.-S.; Baek, J.; Cho, Y.-I.; Jeong, Y.-M.; Lee, Y.-Y.; Oh, J.; Won, Y.; Kang, D.-Y.; Oh, H.; Lim, S.; et al. Single Nucleotide Polymorphism (SNP) Discovery and Kompetitive Allele-Specific PCR (KASP) Marker Development with Korean Japonica Rice Varieties. Plant Breed. Biotechnol. 2018, 6, 391–403. [Google Scholar] [CrossRef]
- Yang, G.; Chen, S.; Chen, L.; Sun, K.; Huang, C.; Zhou, D.; Huang, Y.; Wang, J.; Liu, Y.; Wang, H.; et al. Development of a core SNP arrays based on the KASP method for molecular breeding of rice. Rice 2019, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Kanno, A.; Watanabe, N.; Nakamura, I.; Hirai, A. Variations in chloroplast DNA from rice (Oryza sativa): Differences between deletions mediated by short direct-repeat sequences within a single species. Theor. Appl. Genet. 1993, 86, 579–584. [Google Scholar] [CrossRef]
- Tang, J.; Xia, H.; Cao, M.; Zhang, X.; Zeng, W.; Hu, S.; Tong, W.; Wang, J.; Wang, J.; Yu, J.; et al. A comparison of rice chloroplast genomes. Plant Physiol. 2004, 135, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Sano, R.; Morishima, H. Indica-Japonica differentiation of rice cultivars viewed from variations in key characters and isozymes, with special reference to landraces from the Himalayan hilly areas. Theor. Appl. Genet. 1992, 84, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [Green Version]
- Li, F.P.; Lee, Y.S.; Kwon, S.W.; Li, G.; Park, Y.J. Analysis of genetic diversity and trait correlations among Korean landrace rice (Oryza sativa L.). Genet. Mol. Res. 2014, 13, 6316–6331. [Google Scholar] [CrossRef]
- Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Bhattarai, U.; Subudhi, P.K. Genetic Diversity, Population Structure, and Marker-Trait Association for Drought Tolerance in US Rice Germplasm. Plants 2019, 8, 530. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.Z.; Khalequzzaman, M.; Bashar, M.K.; Ivy, N.A.; Mian, M.A.K.; Pittendrigh, B.R.; Haque, M.M.; Ali, M.P. Variability Assessment of Aromatic Rice Germplasm by Pheno-Genomic traits and Population Structure Analysis. Sci. Rep. 2018, 8, 9911. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Earl, D.A.; vonHoldt, B.M. Structure harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Assoumane, A.; Zoubeirou, A.M.; Rodier-Goud, M.; Favreau, B.; Bezançon, G.; Verhaegen, D. Highlighting the occurrence of tetraploidy in Acacia senegal (L.) Willd. and genetic variation patterns in its natural range revealed by DNA microsatellite markers. Tree Genet. Genomes 2013, 9, 93–106. [Google Scholar] [CrossRef]
- Guo, Y.; Chen, S.; Li, Z.; Cowling, W.A. Center of Origin and Centers of Diversity in an Ancient Crop, Brassica rapa (Turnip Rape). J. Hered. 2014, 105, 555–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | df 1 | SS | MS | Est. Var. | Percent |
|---|---|---|---|---|---|
| Among subgroups | 1 | 1.350 | 1.350 | 0.040 | 17% |
| Within subgroup | 90 | 17.852 | 0.198 | 0.198 | 83% |
| Total | 91 | 19.202 | 0.238 | 100% | |
| Pairwise population PhiPT values 2 | |||||
| Group 1 | Group 2 | ||||
| Group 1 | 0.000 | 0.001 | |||
| Group 2 | 0.167 | 0.000 | |||
| Source | df 1 | SS | MS | Est. Var. | Percent | Overall PhiPT | Probability |
|---|---|---|---|---|---|---|---|
| Among regions | 5 | 3.242 | 0.648 | 0.029 | 13% | 0.130 | 0.002 |
| Within region | 92 | 17.774 | 0.193 | 0.193 | 87% | ||
| Total | 97 | 21.016 | 0.222 | 100% | |||
| Pairwise population PhiPT values | |||||||
| NW 2 | NE | RRD | NCC | S | Inter | ||
| NW | 0.000 | ||||||
| NE | 0.188 | 0.000 | |||||
| RRD | 0.067 | 0.194 | 0.000 | ||||
| NCC | 0.052 | 0.116 | 0.040 | 0.000 | |||
| S | 0.125 | 0.155 | 0.200 | 0.097 | 0.000 | ||
| Inter | 0.190 | 0.104 | 0.220 | 0.135 | 0.078 | 0.000 | |
| Groups | Statistics 1 | |||||
|---|---|---|---|---|---|---|
| MAF | SS | Na | GD | He | PIC | |
| Full set | 0.681 | 98 | 3.333 | 0.432 | 0.011 | 0.382 |
| Group 1 (indica) | 0.777 | 74 | 2.978 | 0.306 A | 0.011 | 0.273 A |
| Group 2 (japonica) | 0.758 | 18 | 2.356 | 0.324 A | 0.004 | 0.276 A |
| Admixture | - | 6 | - | - | - | - |
| AGI collection | 0.700 | 79 | 3.156 | 0.402 | 0.012 | 0.356 |
| Group 1 (indica) | 0.776 | 65 | 2.867 | 0.299 A | 0.012 | 0.268 A |
| Group 2 (japonica) | 0.776 | 13 | 2.178 | 0.294 A | 0.005 | 0.252 A |
| Admixture | - | 1 | - | - | - | - |
| Six regions | ||||||
| Northeast | 0.718 | 14 | 2.533 | 0.376 A | 0.003 | 0.327 A |
| Northwest | 0.677 | 17 | 2.733 | 0.424 A | 0.016 | 0.366 A |
| Red River Delta | 0.725 | 26 | 2.867 | 0.367 A | 0.016 | 0.326 A |
| North-central coast | 0.698 | 21 | 2.867 | 0.400 A | 0.012 | 0.351 A |
| Southern | 0.880 | 5 | 1.467 | 0.166 B | 0.004 | 0.136 B |
| International | 0.615 | 15 | 2.800 | 0.473 A | 0.005 | 0.406 A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luong, N.H.; Linh, L.-H.; Shim, K.-C.; Adeva, C.; Lee, H.-S.; Ahn, S.-N. Genetic Structure and Geographical Differentiation of Traditional Rice (Oryza sativa L.) from Northern Vietnam. Plants 2021, 10, 2094. https://doi.org/10.3390/plants10102094
Luong NH, Linh L-H, Shim K-C, Adeva C, Lee H-S, Ahn S-N. Genetic Structure and Geographical Differentiation of Traditional Rice (Oryza sativa L.) from Northern Vietnam. Plants. 2021; 10(10):2094. https://doi.org/10.3390/plants10102094
Chicago/Turabian StyleLuong, Ngoc Ha, Le-Hung Linh, Kyu-Chan Shim, Cheryl Adeva, Hyun-Sook Lee, and Sang-Nag Ahn. 2021. "Genetic Structure and Geographical Differentiation of Traditional Rice (Oryza sativa L.) from Northern Vietnam" Plants 10, no. 10: 2094. https://doi.org/10.3390/plants10102094