
Cytotoxic Phenylpropanoid Derivatives and Alkaloids from the Flowers of Pancratium maritimum L.




1
Department of Natural Products, Faculty of Pharmacy, King Abdulaziz University, P.O. Box 80260, Jeddah 21589, Saudi Arabia
2
Natural Products Unit, King Fahd Medical Research Center, King Abdulaziz University, P.O. Box 80260, Jeddah 21589, Saudi Arabia
3
Department of Medical Laboratory Sciences, Faculty of Applied Medical Sciences, King Abdulaziz University, P.O. Box 80260, Jeddah 21589, Saudi Arabia
4
Department of Pharmacy Practice, Faculty of Pharmacy, King Abdulaziz University, P.O. Box 80260, Jeddah 21589, Saudi Arabia
*
Authors to whom correspondence should be addressed.
Plants 2022, 11(4), 476; https://doi.org/10.3390/plants11040476
Submission received: 8 January 2022
/
Revised: 4 February 2022
/
Accepted: 5 February 2022
/
Published: 9 February 2022
(This article belongs to the Collection Bioactive Compounds in Plants)
Abstract
:Regarding our growing interest in identifying biologically active leads from Amaryllidaceous plants, the flowers of Pancratium maritimum L. (Amaryllidaceae) were investigated. Purification of the cytotoxic fractions of the alcoholic extract of the flowers gave a new glycoside, 3-[4-(β-D-glucopyranosyloxy)phenyl]-2-(Z)-propenoic acid methyl ester (1), together with the previously reported compounds 3-methoxy-4-(β-D-glucopyranosyloxy)benzoic acid methyl ester (2), 3-(4-methoxyphenyl)propan-1-ol-1-O-β-D-glucopyranoside (3), (E)-3-(4-hydroxyphenyl)acrylic acid methyl ester (4), caffeic acid (5), dihydrocaffeic acid methyl ester (6), and pancratistatin (7). Interestingly, compounds 1 and 2 are phenolic-O-glycosides, while the glucose moiety in 3 is attached to the propanol side chain. This is the first report about the existence of 1–6 in the genus Pancratium. Further, glycosides 1–3 from the Amaryllidaceae family are reported on here for the first time. The structures of 1–7 were determined by analyses of their 1D (1H and 13C) and 2D (COSY, HMQC, HMBC) NMR spectra, and by high-resolution mass spectral measurements. Pancratistatin displayed potent and selective growth inhibitory effects against MDA-MB-231, HeLa, and HCT 116 cells with an IC50 value down to 0.058 µM, while it possessed lower selectivity towards the normal human dermal fibroblasts with IC50 of 6.6 µM.
1. Introduction
The Amaryllidaceae family consist of about 85 genera and 1100 species; they are well known for their ornamental values and biologically-active alkaloids [1]. Plants of the genus Pancratium are bulbous monocotyledons, which grow on sand banks and sandy coastal environments [2]. Although Pancratium includes about 15 species, distributed throughout the Mediterranean, Africa, and Asia, only three species of this genus, P. maximum, P. sickenbergeri, and P. tortuosum, are indigenous to Saudi Arabia [3]. In Egypt, there are four species belonging to the genus Pancratium, namely P. arabicum, P. maritimum L., P. sickenbergeri, and P. tortuosum [4]. The extracts of the bulbs and flowers of P. maritimum possess analgesic, antifungal, and anticancer activities. Furthermore, these extracts possess purgative, hypotensive, emetic, and anti-inflammatory effects [5].
Cancer is a worldwide health concern and accounts for 8 million deaths worldwide each year, with almost 600,000 deaths in the United States only [6]. Plant-derived alkaloids, such as vinblastine, vincristine, and paclitaxel, are valuable and important antitumor drugs [7].
Chemists and pharmacologists worldwide have substantially considered members of the Amaryllidaceae family, due to their complex and diverse alkaloids and biological and pharmacological properties. Amaryllidaceous alkaloids display antimalarial [8,9,10], analgesic [11], acetylcholinesterase [12], and antifungal [13] activities. After the discovery of pancratistatin [14], with its narciclasine-backbone and its significant antitumor effects [15], representative alkaloids of this class have been evaluated as potential cytotoxic agents [16]. Later on, the anticancer potential of the semisynthetic derivatives of narciclasine [17], lycorine [18], and crinine [19] were investigated.
On the contrary, the non-alkaloidal constituents of Amaryllidaceae plants have been overlooked by researchers. However, since the identification of chromones, chromones, acetophenones, and their glycosides from Pancratium biflorum [20,21,22], several studies were carried out to explore the non-alkaloidal constituents of the genus Pancratium [23,24,25], resulting in identification of different chromones and acetophenones and their glycosides, flavonoids, chalcones, and others [20,21,22,23,24,25].
As a continuation of our work in exploring the secondary metabolites of Amaryllidaceous plants [26,27,28,29,30], the cytotoxic fractions of the alcoholic extracts of the fresh flowers of P. maritimum L. (Sea Daffodil) were investigated. One new glycoside (1), together with the previously reported compounds 2–7, were purified from the active fractions of the flower extracts. In this work, we describe the purification, structural assignments, and the cytotoxic effects of the compounds.
2. Results and Discussion
2.1. Structure of Compound 1
Compound 1 (Figure 1) possesses the molecular formula C16H20O8, as shown by a pseudomolecular ion peak at m/z 363.1055 [M + Na]+ in the (+)-HRESIMS (Figure S1), suggesting seven degrees of unsaturation. The structure of 1 was assigned from interpretation of its 1D (Figures S2–S5) and 2D NMR spectra (Figures S6–S8). The 1H and 13C NMR data of 1 (Table 1) showed signals for 1,4-disubstituted benzene moiety linked to Z-propenoic acid methyl ester and O-β-glucopyranoside units. The 1H and 13C NMR signals at δH/C 130.4 (qC, C-1), 7.68 (d, J = 8.7 Hz)/133.0 (2 × CH, H-2,6/C-2,6), 7.08 (d, J = 8.7 Hz)/117.1 (2 × CH, H-3,5/C-3,5), 159.9 (qC, C-4) supported the assignment of the 1,4-substituted benzene moiety.
The propenoic acid methyl ester moiety was assigned from the 1H and 13C NMR resonances at δH/C 6.93 (d, J = 12.7 Hz)/144.3 (CH, H-7/C-7), 5.87 (d, J = 12.7 Hz)/117.6 (H-8/C-8), 168.6 (qC, C-9) and 3.72 (s)/51.8 (CH3, H3-10/C-10). The Z configuration at Δ7,8 was assigned from the coupling constants of 12.7 Hz between the olefinic protons H-7 and H-8. In addition, HMBC from H-7 and H-8 to C-9 and from H3-10 to C-9 supported the assignment.
The 1H/13C resonances at δH/C 4.98 (d, J = 7.2 Hz)/102.0 (CH, H-1′/C-1′), 3.48 (m)/74.9 (CH, H-2′/C-2′), 3.49 (m)/78.0 (CH, H-3′/C-3′), 3.41 (m)/71.4 (CH, H-4′/C-4′), 3.49 (m)/78.3 (CH, H-5′/C-5′) and 3.91 (t, J = 11.9 Hz), 3.72 (m)/62.5 (CH2, H2-6′/C-6′) proposed the presence of a glucopyranoside unit. The continuous spin–spin COSY couplings from the anomeric proton H-1′ (5.04 ppm) to H2-6′ (3.91 and 3.71 ppm) supported the assignment of the glucopyranoside moiety. The coupling constant of the anomeric proton (H-1′, d, J = 7.2 Hz) supported the β configuration of the glucopyranoside unit (Table 1).
The attachment of the Z-propenoic acid methyl ester to the benzene ring at C-1 was supported by HMBC correlations from H-7 and H-8 to C-1, and from H-2,6 to C-7. Similarly, the placement of the β-D-glucopyranoside moiety at C-4 of the benzene moiety was supported by HMBC cross-peaks from H-3,5 to C-1′ and from H-1′ to C-4. Thus, 1 was assigned as 4-(β-D-glucopyranosyloxy)phenyl]-2-(Z)-propenoic acid methyl ester, and it is reported here as a new natural product.
2.2. Structure of Compound 2
Compound 2 (Figure 1) showed the molecular formula C15H20O9 as obtained from the pseudomolecular ion peak at m/z 367.1003 [M + Na]+ in the (+)-HRESIMS (Figure S9), suggesting six degrees of unsaturation. The structure of 2 was assigned from interpretation of its 1D (Figures S10–S13) and 2D NMR spectra (Figures S14–S16). The 1H and 13C NMR data of 2 (Table 2) showed resonances for 3,4-disubstituted benzoic acid methyl ester. The 1H and 13C NMR resonances at δH/C 125.5 (qC, C-1), 7.62 (d, J = 1.8 Hz)/114.1 (CH, H-2/C-2), 150.5 (qC, C-3), 152.2 (qC, C-4), 7.23 (d, J = 8.5 Hz)/116.5 (CH, H-5/C-5), 7.65 (dd, J = 8.5 and 1.8 Hz)/ 124.6 (CH, H-6/C-6), 168.3 (qC, C-7) and 3.89 (s)/52.6 (CH3, H3-8/C-8) proposed the existence of 3,4-disubstituted benzoic acid methyl ester. The COSY correlation between H-6 and H-2 and between H-6 and H-5 supported the existence of an ABX system and the assignment of these protons (Figure 2). In addition, the protonated carbons were assigned from HSQC correlations (Table 2).
The substituents at C-3 and C-4 were assigned as a methoxyl (δH/C 3.92 (s)/56.2) group and an O-β-glucopyranoside unit, respectively. The presence of O-β-glucopyranoside moiety was established from the 1H and 13C NMR resonances at δH/C 5.04 (d, J = 7.7 Hz)/102.0 (CH, H-1′/C-1′), 3.55 (dd, J = 9.1 and 7.7 Hz)/74.8 (CH, H-2′/C-2′), 3.48 (m)/77.9 (CH, H-3′/C-3′), 3.42 (t, J = 9.1 Hz)/71.3 (CH, H-4′/C-4′), 3.50 (t, J = 9.1 Hz)/78.4 (CH, H-5′/C-5′) and 3.82 (m), 3.71 (dd, J = 11.0 and 4.5 Hz)/62.5 (CH2, H2-6′/C-6′). The continuous spin-spin COSY couplings from the anomeric proton H-1′ (5.04 ppm) to H2-6′ (3.91 and 3.71 ppm) supported the assignment of the glucopyranoside moiety. The coupling constant of the anomeric proton (H-1′, d, J = 7.7) supported the β configuration of the glucopyranoside unit (Table 2 and Figure 2).
The placement of the OCH3 group at C-3 was supported by HMBC correlations from H-5 to C-3 and from H3-9 to C-3, while the HMBC from H-1′ to C-4 and from H-5 to C-1′ secured the placement of the glucopyranoside moiety at C-4. The 1H and 13C NMR data of 2 are similar to those of 3-methoxy-4-(β-D-glucopyranosyloxy)benzoic acid methyl ester [31], which was previously reported from the Solanaceous plant Lycium schweinfurthii (family: Solanaceae) [31]. Thus, compound 2 was assigned as 3-methoxy-4-(β-D-glucopyranosyloxy)benzoic acid methyl ester. This is the first report about the existence of compound 2 in the Amaryllidaceae family.
2.3. Structure of Compound 3
Compound 3 (Figure 1) displayed molecular formula C16H24O7 as supported by the pseudomolecular ion peak at m/z 351.1419 [M + Na]+ in the (+)-HRESIMS (Figure S17), requiring five degrees of unsaturation. The structure of 3 was assigned from interpretation of its 1D (Figures S18–S21) and 2D NMR spectra (Figures S22–S24). The 1H and 13C NMR spectra of 3 displayed signals for 3-(4-methoxyphenyl)propan-1-ol linked to a β-O-glucopyranoside unit at the OH of the propanol side chain. The 1H and 13C NMR resonances at δH/C 159.4 (qC, C-1), 6.83 (d, J = 8.5 Hz)/114.8 (CH, H-2,6/C-2,6), 7.13 (d, J = 8.5 Hz)/130.5 (CH, H-3,5/C-3,5), 135.4 (qC, C-4), 2.67 (t, J = 7.6 Hz)/32.3 (CH2, C-7), 1.90 (m)/32.9 (CH2, C-8), 3.92 (dt, J = 9.6, 6.5) 3.54 (dt, J = 9.6, 6.5 Hz)/70.1 (CH2, C-9), and 3.77 (s)/55.7 (CH3, H3-10/C-10) were assigned as 3-(4-methoxyphenyl)propan-1-ol moiety (Table 3).
The β-glucopyranoside moiety was assigned from 1H and 13C NMR signals at 4.25 (d, J = 7.8 Hz)/104.5 (CH, H-1′/C-1′), 3.21 (dd, J = 9.0 and 7.8 Hz)/75.2 (CH, H-2′/C-2′), 3.37 (t, J = 9.0 Hz)/78.2 (CH, H-3′/C-3′), 3.31 (t, J = 9.0 Hz)/71.7 (CH, H-4′/C-4′), 3.27 (m)/77.9 (CH, H-5′/C-5′) and 3.88 (dd, J = 11.9 and 2.2 Hz), 3.69 (dd, J = 11.9 and 6.1 Hz)/62.8 (CH2, H2-6′/C-6′) (Table 3). The chemical shift of the anomeric proton (H-1′) at δH 4.25 and its coupling constant of 7.8 Hz supported the β configuration of the glucopyranoside moiety.
The COSY experiment displayed three coupling systems, including the coupling between H-2,6 and H-3,5 in the aromatic moiety, the vicinal couplings in the aliphatic side chain from H2-7 to H2-8 and from H2-8 to H2-9, and the contiguous coupling system within the glucopyranoside moiety from H-1′ to H2-6′ (Figure 2). The placement of the OCH3 group at C-1 was assigned from HMBC of H3-10 to C-1 (Table 3 and Figure 2). Similarly, the attachment of the glucose moiety at the terminal OH of the propanol moiety was assigned from HMBC correlations from H2-9 to C-1′ and from H-1′ to C-9. The NMR data of 3 are similar to those of 3-(4′-methoxyphenyl)-propanol 1-O-β-glucopyranoside, which was previously isolated from the plant Mediasia macrophylla (family: Apiaceae) [32]. Thus, 3 was assigned 3-(4-methoxyphenyl)propan-1-ol-1-O-β-D-glucopyranoside. This is the first report about the occurrence of compound 3 in the Amaryllidaceae family.
2.4. Structure of Compound 4
Compound 4 (Figure 1) displayed molecular C10H10O3 as obtained from (+)-HRESIMS (Figure S25). Its 1H NMR spectrum (Figure S26) revealed resonances for 1,4-disubstituted benzene ring (Table 4) at δH 7.48 (2H, d, J = 8.5 Hz, H-2,6) and 6.82 (2H, d, J = 8.5 Hz, H-3,5). In addition, signals at δH 7.67 (1H, d, J = 15.9 Hz, H-7), 6.37 (1H, d, J = 15.9 Hz, H-8), and 3.73 (3H, s, H3-10) suggested the presence of acrylic acid methyl ester. Furthermore, the 13C NMR spectrum of 4 (Figure S27) displayed signals for six methines at δC 131.2 (C-2,6), 116.9 (C-3,C-5), 146.5 (C-7), and 115.9 (C-8), one methoxyl at δC 52.9 (C-10), and three quaternary carbons at δC 127.4 (C-1), 161.2 (C-4) and 169.0 (C-9). These 1H and 13C NMR data are similar to those of (E)-3-(4-hydroxyphenyl)acrylic acid methyl ester [33], which was previously isolated from the mangrove-derived actinobacterium Saccharomonospora oceani VJDS-3 [33]. Thus, 4 was assigned as (E)-3-(4-hydroxyphenyl)acrylic acid methyl ester. This is the first report about the existence of this compound in the genus Pancratium.
2.5. Structure of Compound 5
Compound 5 (Figure 1) displayed molecular formula C9H8O4 as supported from (+)-HRESIMS (Figure S28). Its 1H NMR spectrum (Figure S29) revealed resonances for five protons in the olefinic/aromatic region (Table 4). The 1H signals are counted for an ABX system (H-2, H-5 and H-6) at δH 7.05 (d, J = 1.5 Hz, H-2), 6.79 (d, J = 8.0 Hz, H-5) and 6.94 (dd, J = 8.0 and 1.5 Hz, H-6) and signals for an E-configured double bond (H-7 and H-8) at δH 7.54 (d, J = 15.8 Hz, H-7) and 6.24 (d, J = 15.8 Hz, H-8). Furthermore, the 13C NMR spectrum of 5 (Figure S30) showed resonances for nine carbons including five methines at δC 115.1, 116.5, 122.8, 146.9, and 116.0 are assigned for C-2, C-5, C-6, C-7, and C-8, respectively, and four quaternary carbons at 127.9 (C-1), 146.8 (C-3), 149.4 (C-4), and 171.1 (C-9) (Table 4). These 1H and 13C NMR data are similar to those reported for caffeic acid (3,4-dihydroxycinnamic acid) [34], which was isolated before from Hippeastrum vittatum (family: Amaryllidaceae) [34]. Thus, 5 was assigned as caffeic acid (3,4-dihydroxycinnamic acid). This is the first occurrence of this compound in the genus Pancratium.
2.6. Structure of Compound 6
Compound 6 (Figure 1) displayed molecular C10H12O4 as supported from (+)-HRESIMS (Figure S31). Its 1H NMR spectrum (Figure S32) revealed resonances for an ABX system (H-2, H-5 and H-6) at δH 6.63 (d, J = 1.9 Hz, H-2), 6.68 (d, J = 8.0 Hz, H-5) and 6.51 (dd, J = 8.0 and 1.9 Hz, H-6) and signals for an ethylene moiety (H2-7 and H2-8) at δH 2.77 (t, J = 7.6 Hz, H2-7) and 2.57 (t, J = 7.6 Hz, H2-8) and one methoxyl at δH 3.65 (s, 3H, H3-10). Furthermore, the 13C NMR spectrum of 6 (Figure S33) showed resonances for nine carbons including three methines at δC 116.5, 116.4, 120.5 assigned for C-2, C-5, C-6, respectively, two methylenes at 31.1 (C-7) and 37.1 (C-8), one methoxyl at δC 52.1 (C-10), and four quaternary carbons at 133.6 (C-1), 146.3 (C-3), 144.7 (C-4), and 175.4 (C-10) (Table 4). These 1H and 13C NMR data are similar to those reported for dihydrocaffeic acid methyl ester, which was isolated from Hippeastrum vittatum (family: Amaryllidaceae) [34]. Thus, 6 was assigned as dihydrocaffeic acid methyl ester. This is the first report about the existence of this compound in the genus Pancratium.
2.7. Structure of Compound 7
Compound 7 (Figure 1) possesses molecular formula C14H15NO8 as supported from the pseudo-molecular ion peak at m/z 348.0689 [M + Na]+ in the (+)-HRESIMS (Figure S34). The 1H and 13C NMR data (Figures S35 and S36) of 7 are similar to those reported for (+)-pancratistatin [14,35,36], which was isolated previously from Pancratium littorale (family: Amaryllidaceae). Accordingly, 7 was assigned as pancratistatin.
2.8. Antiproliferation Activities of the Compounds
Compounds 1–7 were evaluated for their antiproliferative and growth inhibition activities against MDA-MB-231, HeLa, and HCT 116 cell lines (Table 5). Pancratistatin displayed a potent growth inhibitory activity towards these cell lines with IC50 values 0.14, 0.058, and 0.10 μM, respectively. On the other hand, compounds 1–3 and 5 were inactive at the level of 10 μM against these cells. Due to the high potency of compound 7, a concentration of 10 µM was set as a cutoff value in this assay.
To evaluate the selectivity of pancratistatin against cancer cell lines, it was evaluated against the normal human dermal fibroblasts (NHDF). Pancratistatin displayed less selectivity towards NHDF with IC50 of 6.6 µM, suggesting a higher selectivity (66-fold) towards the tested cancer cell lines (Table 5 and Figure 3).
It is well known that pancratistatin possess a wide-range selectivity towards various cancer cell lines [14,37,38] with IC50 ranging from 0.043 to 0.098 µM [37,38]. Furthermore, pancratistatin induce apoptosis through targeting mitochondria in cancer cells [39,40]. Moreover, it causes flipping of phosphatidyl-serine, activation of caspase-3, generation of reactive oxygen species (ROS), and loss of the mitochondrial membrane leading to apoptosis [41].
However, it is worth mentioning that this is the first report, to the best of our knowledge, about the evaluation of pancratistatin against MDA-MB-231, HeLa, HCT 116, and NHDF cell lines.
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations of the compounds were measured on a JASCO DIP-370 digital polarimeter at 25 °C at the sodium D-line (589 nm). The IR spectra were recorded on a Shimadzu Infrared-400 spectrophotometer (Shimadzu, Kyoto, Japan). The 1D and 2D NMR spectra (chemical shifts in ppm, coupling constants in Hz) were recorded on Bruker Avance DRX 600 MHz (600 MHz for 1H and 150 MHz for 13C NMR) (Bruker, Rheinstetten, Germany) and Bruker Ascend 850 MHz (850 MHz for 1H and 213 MHz for 13C NMR) (Bruker BioSpin, Billerica, MA, USA) spectrometers. Positive ion HRESIMS data were obtained with a Micromass Q-Tof equipped with leucine enkephalin lock spray, using m/z 556.2771 [M + H]+ as a reference mass. Sephadex LH-20 (0.25–0.1 mm, Pharmacia) was used for column chromatography. Precoated silica gel 60 F-254 plates (Merck) were used for TLC. HPLC purifications were performed on Shim-Pack, PREP-ODS (H) (20 × 250 mm), Cosmosil ARII C18 (20 × 250 mm), and Atlantis Prep OBD T3 Column (10 × 250 mm, 5 µm).
3.2. Botanical Materials
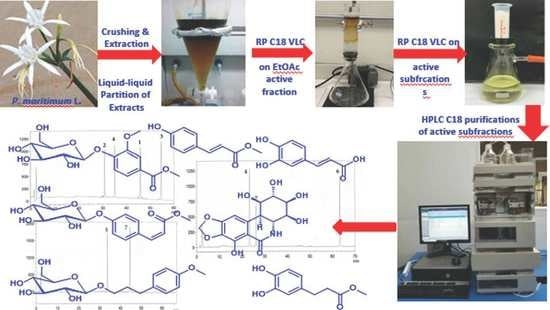
The fresh entire flowers of P. maritimum L. with most of their stalks (Figure 4) were collected from the coastal sandy beaches of the Egyptian Governorate Maṭrūḥ, Egypt. The plant materials were kindly identified at the Department of Botany, Faculty of Science, Suez Canal University, Ismailia, Egypt. A voucher specimen was kept at the Department of Pharmacognosy, Faculty of Pharmacy, Suez Canal University under code number DYPM-1.
3.3. Purification of Compounds 1–7
The fresh flowers with their stalks (1.8 kg) were crushed into small pieces and soaked directly in 70% ethanol (1.5 L) (Scheme 1). After receiving the materials in the laboratory, the mixture was sonicated for an additional 6 h at room temperature and the alcoholic solution was filtered. This process was repeated thrice and the combined hydro-alcoholic extracts were evaporated under reduced pressure. The resulting suspension was dissolved in 300 mL H2O and successively extracted with n-hexane (3 × 200 mL), CH2Cl2 (3 × 200 mL), and ethyl acetate (3 × 200 mL). Finally, 500 mL of H2O were added to the remaining mother aqueous layer before gentle shaking with n-BuOH (3 × 250 mL), since n-butanol has some solubility in water. The inactive fractions (n-hexane, CH2Cl2, and n-BuOH) were kept for any future investigations. The cytotoxic ethyl acetate residue (690 mg) was chromatographed on RP C18 silica column (30 × 4 cm) using H2O-MeOH gradients to give 10 fractions (Fractions 1-10). The cytotoxic fraction (Fraction 1, 330 mg), which was eluted with 80% H2O (IC50 = 4.4 μg/mL against HeLa cells), was repartitioned on Sep-Pak ODS Cartridge (10 g, Waters) using H2O-MeOH gradients to afford seven fractions (Fractions A-G) (Scheme 1). The cytotoxic fraction C (27 mg) (IC50 = 1.95 μg/mL against HeLa cells), which was eluted with 20% MeOH, was purified on HPLC column (Shim-Pack, PREP-ODS (H), 20 × 250 mm) using 25% CH3CN at 3.8 mL/min to give compounds 5 (tR = 34.8 min, 3.1 mg) and 7 (tR = 44.6 min, 2.3 mg) (Figure S37). Similarly, the cytotoxic fraction D (19 mg) (IC50 = 2.12 μg/mL against HeLa cells), which was eluted with 30% MeOH, was purified on RP C18 HPLC column (Cosmosil ARII, 20 × 250 mm) using 30% MeOH at 5.5 mL/min to give compounds 4 (tR = 23.3 min, 7.4 mg) and 7 (tR = 26.1 min, 2.4 mg) and 6 (tR = 63.3 min, 1.9 mg) (Figure S38). Finally, the less cytotoxic fraction E (29 mg) (IC50 = 13.2 μg/mL against HeLa cells), which was eluted with 40% MeOH, was purified on RP C18 HPLC column (Atlantis Prep OBD T3 Column, 10 × 250 mm, 5 µm) using 35% CH3CN at 2 mL/min to give compounds 2 (tR = 31.3 min, 3.3 mg), 4 (tR = 35.1 min, 3.7 mg), 1 (tR = 46.0 min, 3.8 mg), 3 (tR = 55.1 min, 4.1 mg) (Figure S39 and Scheme 1).
3.4. Spectroscopic Data of Compounds 1–7
3.4.1. 3-[4-(β-D-glucopyranosyloxy)phenyl]-2-(Z)-propenoic Acid Methyl Ester (1)
Yellow powder; [α]D—31.5° (MeOH, c = 0.1); IR νmax (film) 3345, 1665, 1600, 1035 cm−1; NMR data: see Table 2; (+)-HRESIMS m/z 363.1055 (calcd for C16H20O8Na [M + Na]+, 363.1050).
3.4.2. 3-Methoxy-4-(β-D-glucopyranosyloxy)benzoic Acid Methyl Ester (2)
White powder; [α]D—23.9° (MeOH, c = 0.1); IR νmax (film) 3350, 1700, 1035 cm−1; NMR data: see Table 1; (+)-HRESIMS m/z 367.1003 (calcd for C15H20O9Na [M + Na]+, 367.0999).
3.4.3. 3-(4-Methoxyphenyl)propan-1-ol-1-O-β-D-glucopyranoside (3)
Yellowish powder; [α]D—27.5° (MeOH, c = 0.1); IR νmax (film) 3455, 1647, 1595, 627 cm−1; NMR data: see Table 3; (+)-HRESIMS m/z 351.1419 (calcd for C16H24O7Na [M + Na]+, 351.1414).
3.4.4. (E)-3-(4-Hydroxyphenyl)acrylic Acid Methyl Ester (4)
Amorphous powder; IR νmax (film) 2938, 1687, 1599 cm−1; NMR data: see Table 4; HRESIMS m/z 179.0709 (calcd for C10H11O3 [M + H]+, 179.0607).
3.4.5. Caffeic Acid (5)
Off-white solid; IR νmax (film) 3421, 1665, 1518, 1025, 627 cm−1; NMR data: see Table S4; (+)-HRESIMS m/z 181.0501 (calcd for C9H9O4 [M + H]+, 181.0495).
3.4.6. Dihydrocaffeic Acid Methyl Ester (6)
Off-white powder; IR νmax (film) 3385, 1647, 630 cm−1; NMR data: see Table 4; (+)-HRESIMS m/z 197.0815 (calcd for C10H13O4 [M + H]+, 197.0808).
3.4.7. Pancratistatin (7)
White powder; [α]D + 48.5° (DMSO, c = 0.1); UV (MeOH) λmax: 280, 237 nm. IR (film) νmax: 3419, 1672, 1629, 1468, 1358, 1083, 1047 cm−1; NMR data: see Figures S28 and S29. (+)-HRESIMS m/z 348.0693 (calcd for C14H15NO8Na [M + Na]+, 348.0689).
3.5. Antiproliferative and Growth Inhibition Effects of Compounds 1–7
3.5.1. Culture of Cell Lines
HCT116 (Colorectal carcinoma, ATCC CCL-247) and HeLa (human cervical carcinoma, ATCC CCL-2) cells were cultured in RPMI 1640 medium with 10% FBS, and 1% penicillin–streptomycin, while MDA-MB-231 cells (triple-negative breast cancer, ATCC HTB-26) were cultured in DMEM medium with 1% penicillin–streptomycin and 10% FBS.
The normal human dermal fibroblasts (NHDF) cells were cultured in fibroblast basal medium complemented with penicillin–streptomycin (1% v/v), insulin (5 µg/mL), heat-inactivated fetal bovine serum (FBS) (2% v/v), and basic fibroblast growth factor (1 ng/mL).
3.5.2. Evaluation of the Antiproliferative Activity
The evaluation of the antiproliferative effects compounds 1–7 were performed using MTT assay, as reported earlier [42,43,44]. In short, the cells were incubated at 37 °C overnight in 5% CO2/air. After that, the compounds were added at the top row of a 96-well microtiter plate; a descendant serial dilution (1:4) of the concentration was performed followed by incubation of the cells with the compounds for 72 h. Using the Cell Titer 96 AQueous non-radioactive cell proliferation protocol, the cell viability was estimated at 490 nm on a Molecular Devices Emax microplate reader. The IC50 values of the compounds (expressed in μM) were evaluated using the program SoftMax Pro. A concentration of 10 µM was set as a cutoff value in this assay.
4. Conclusions
Partition of the cytotoxic fractions of the alcoholic extract of the fresh flowers of P. maritimum L. afforded a potent cytotoxic alkaloid, pancratistatin (7), along with three glycosides (1–3) and three phenylpropanoid derivatives (4–6). Their structures were determined by analyses of their NMR and HRESIMS spectral data. We should note that compound 1 is reported here as a new natural product, while compounds 2 and 3 are reported here for the first time from the Amaryllidaceae family. On the contrary, 4–6 are reported here for the first time from the genus Pancratium.
Pancratistatin displayed highly potent activity against the HCT116, HeLa, and MDA-MB-231 cell lines down to 0.058 µM. Furthermore, pancratistatin showed less selectivity towards the normal human dermal fibroblasts (NHDF) with a IC50 value of 6.6 µM compared to the average IC50 value of 0.099 µM against the cancerous cell lines HCT116, HeLa, and MDA-MB-231, suggesting a higher selectivity (66-fold) towards these cancerous cells.
On the other hand, compounds 1–3 and 5 were inactive against these cell lines at a concentration of 10 µM. The results clearly indicate that pancratistatin is a powerful lead with potent antiproliferative and growth inhibitory activities against different cancer cell lines.
The results of the current study highlight the importance of medicinal plants as a great source of bioactive compounds. In the long-term, the results of this study and similar studies will have broader and long-term impacts, including opening new channels of cooperation with the private sector in the areas of biotechnology and drug discovery, and exploring indigenous plants for their chemical diversity and biomedical importance.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/plants11040476/s1, Figures S1–S39: (+)-HRESIMS, 1H NMR, 13C NMR, DEPT, COSY, HSQC, and HMBC spectra of compounds 1–7, and HPLC chromatograms for purification of 1–7.
Author Contributions
Conceptualization, D.T.A.Y. and L.A.S.; methodology, L.A.S., A.E.A. and D.T.A.Y.; formal analysis, D.T.A.Y.; L.A.S. and A.E.A.; investigation, L.A.S., D.T.A.Y. and A.E.A.; resources, D.T.A.Y.; data curation, L.A.S. and D.T.A.Y.; writing—original draft preparation, L.A.S. and D.T.A.Y.; writing—review and editing, D.T.A.Y., L.A.S. and A.E.A.; supervision, D.T.A.Y.; project administration, D.T.A.Y.; funding acquisition, D.T.A.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Institutional Fund Projects under grant no. IFPIP: 188-166-1442.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article or Supplementary Material.
Acknowledgments
This research work was funded by Institutional Fund Projects under grant no. (IFPIP: 188-166-1442). Therefore, the authors gratefully acknowledge technical and financial support from the Ministry of Education and King Abdulaziz University, DSR, Jeddah, Saudi Arabia.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jin, Z. Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep. 2016, 33, 1318–1343. [Google Scholar] [CrossRef] [PubMed]
- Willis, J.C. Amaryllidaceae. In A Dictionary of the Flowering Plants & Ferns, 8th ed.; Shaw, A.H.K., Ed.; Cambridge University Press: Cambridge, UK, 1998; p. 847. [Google Scholar]
- Collenette, S. (Ed.) Wildflowers of Saudi Arabia; National Commission for Wildlife Conservation and Development (NCWCD): Riyadh, Saudi Arabia, 1999; pp. 39–40. [Google Scholar]
- Täckholm, V. (Ed.) Students’ Flora of Egypt, 2nd ed.; Cairo University: Cairo, Egypt, 1974; p. 657. [Google Scholar]
- El-Hadidy, A.; Abd El-Ghani, M.; Amer, W.; Hassan, R. Morphological and molecular differentiation between Egyptian species of Pancratium L. (Amaryllidaceae). Acta Biol. Cracov. Bot. 2012, 54, 53–64. [Google Scholar] [CrossRef]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer Statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cragg, G.M.; Newman, D.J. Plants as source of anticancer agents. J. Ethnopharmacol. 2005, 100, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cedrón, J.; Gutiérrez, L.; Flores, N.; Ravelo, A.; Estévez-Braun, A. Synthesis and antimalarial activity of new haemanthamine-type derivatives. Bioorg. Med. Chem. 2012, 20, 5464–5472. [Google Scholar] [CrossRef]
- Cedrón, J.; Gutiérrez, L.; Flores, N.; Ravelo, A.; Estévez-Braun, A. Preparation and antimalarial activity of semisynthetic lycorenine derivative. Eur. J. Med. Chem. 2013, 63, 722–730. [Google Scholar] [CrossRef]
- Cedrón, J.; Gutiérrez, L.; Flores, N.; Ravelo, A.; Estévez-Braun, A. Synthesis and antiplasmodial activity of lycorine derivatives. Bioorg. Med. Chem. 2010, 18, 4694–4701. [Google Scholar] [CrossRef]
- Citoglu, G.; Acikara, O.; Yilmaz, B.; Özbek, H. Evaluation of analgesic, anti-inflammatory and hepatoprotective effects of lycorine from Sternbergia fisheriana (Herbert) Rupr. Fitoterapia 2012, 83, 81–87. [Google Scholar] [CrossRef]
- McNulty, J.; Nair, J.; Little, J.; Brennan, D.; Bastida, J. Structure-activity studies on acetylcholinesterase inhibition in the lycorine series of Amaryllidaceae alkaloids. Bioorg. Med. Chem. Lett. 2010, 20, 5290–5294. [Google Scholar] [CrossRef]
- Evidente, A.; Andolfi, A.; Abou-Donia, A.H.; Touema, S.M.; Hammoda, H.M.; Shawky, E.; Motta, A. (−)-Amarbellisine, a lycorine-type alkaloid from Amaryllis belladonna L. growing in Egypt. Phytochemistry 2004, 65, 2113–2118. [Google Scholar] [CrossRef]
- Pettit, G.R.; Gaddamidi, V.; Herald, D.L.; Singh, S.B.; Cragg, G.M.; Schmidt, J.M.; Boettner, F.E.; Williams, M.; Sagawa, Y. Antineoplastic agents, 120. Pancratium littorale. J. Nat. Prod. 1986, 49, 995–1002. [Google Scholar] [CrossRef]
- Kornienko, A.; Evidente, A. Chemistry, biology, and medicinal potential of narciclasine and its congeners. Chem. Rev. 2008, 108, 1982–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cedrón, J.; Del Arco-Aguilar, M.; Estévez-Braun, A.; Ravelo, A. Chemistry and biology of Pancratium alkaloids. Alkaloids Chem. Biol. 2010, 68, 1–37. [Google Scholar]
- Ingrassia, L.; Lefranc, F.; Dewelle, J.; Pottier, L.; Mathieu, V.; Spiegl-Kreinecker, S.; Sauvage, S.; El Yazidi, M.; Dehoux, M.; Berger, W.; et al. Structure-activity relationship analysis of novel derivatives of narciclasine (an Amaryllidaceae isocarbostyril derivative) as potential anticancer agent. J. Med. Chem. 2009, 52, 1100–1114. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yuan, H.; Zhang, X.; Li, Y.; Shang, L.; Yin, Z. Novel lycorine derivatives as anticancer agents: Synthesis and in vitro biological evaluation. Molecules 2014, 19, 2469–2480. [Google Scholar] [CrossRef] [PubMed]
- Evidente, A.; Kireev, A.; Jenkins, A.; Romero, A.; Steelant, W.; Van Slambrouck, S.; Kornienko, A. Biological evaluation of structurally diverse Amaryllidaceae alkaloids and their synthetic derivatives: Discovery of novel leads for anticancer drug design. Planta Med. 2009, 75, 501–507. [Google Scholar] [CrossRef] [Green Version]
- Ghosal, S.; Singh, S.P.; Bhagat, M.P.; Kumar, Y. Three chromones from bulbs of Pancratium biflorum. Phytochemistry 1980, 21, 2943–2946. [Google Scholar] [CrossRef]
- Ghosal, S.; Kumar, Y.; Singh, S.P.; Ahad, K. Biflorin, a chromone-C-glucoside from Pancratium biflorum. Phytochemistry 1983, 22, 2591–2593. [Google Scholar] [CrossRef]
- Ghosal, S.; Mittal, P.; Kumar, Y.; Singh, S.K. Free and glucosyloxy acetophenones from Pancratium biflorum. Phytochemistry 1989, 28, 3193–3196. [Google Scholar] [CrossRef]
- Youssef, D.T.A.; Ramadan, M.A.; Khalifa, A.A. Acetophenones, a chalcone, a chromone and flavonoids from Pancratium maritimum. Phytochemistry 1998, 49, 2579–2583. [Google Scholar] [CrossRef]
- Ali, A.A.; Makboul, M.A.; Attia, A.A.; Ali, D.T. Chromones and flavans from Pancratium maritimum. Phytochemistry 1990, 29, 625–627. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Mohamed, G.A.; Shaala, L.A.; Youssef, D.T.A. Non-alkaloidal compounds from the bulbs of the Egyptian plant Pancratium maritimum. Z. Naturforsch. C. 2014, 69, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youssef, D.T.A. Alkaloids of the flowers of Hippeastrum vittatum. J. Nat. Prod. 2001, 64, 839–841. [Google Scholar] [CrossRef]
- Youssef, D.T.A.; Khalifa, A.A. Cytotoxic quaternary alkaloids from the flowers of Narcissus tazetta. Pharmazie 2001, 56, 818–822. [Google Scholar] [PubMed]
- Youssef, D.T.A. Further alkaloids from the flowers of Pancratium maritimum. Pharmazie 1999, 54, 535–537. [Google Scholar]
- Youssef, D.T.A.; Frahm, A.W. Alkaloids of the flowers of Pancratium maritimum. Planta Med. 1998, 64, 669–670. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Mohamed, G.A.; Shaala, L.A.; Youssef, D.T.A.; El Sayed, K.A. New alkaloids from Pancratium maritimum. Planta Med. 2013, 79, 1480–1484. [Google Scholar] [CrossRef] [Green Version]
- Elbermawi, A.; Halim, A.F.; Mansour, E.S.; Ahmad, K.F.; Ashour, A.; Amen, Y.; Shimizu, K. A new glucoside with a potent α-glucosidase inhibitory activity from Lycium schweinfurthii. Nat. Prod. Res. 2021, 35, 976–983. [Google Scholar] [CrossRef]
- Kurimoto, S.; Okasaka, M.; Kashiwada, Y.; Kodzhimatov, O.K.; Takaishi, Y. Four new glucosides from the aerial parts of Mediasia macrophylla. J. Nat. Med. 2011, 65, 180–185. [Google Scholar] [CrossRef]
- Indupalli, M.; Muvva, V.; Mangamuri, U.; Munaganti, R.K.; Naragani, K. Bioactive compounds from mangrove derived rare actinobacterium Saccharomonospora oceani VJDS-3. 3 Biotech 2018, 8, 103. [Google Scholar] [CrossRef]
- Youssef, D.T.A. Cytotoxic phenolics from the flowers of Hippeastrum vittatum. Bull. Pharm. Sci. Assiut 2005, 28, 143–148. [Google Scholar] [CrossRef]
- Pettit, G.R.; Gaddamidi, V.; Cragg, G.M.; Herald, D.L.; Sagawa, Y. Isolation and structure of pancratistatin. J. Chem. Soc. Chem. Commun. 1984, 24, 1693–1694. [Google Scholar] [CrossRef]
- Pettit, G.R.; Melody, N.; Herald, D.L. Antineoplastic agents. 450. Synthesis of (+)-pancratistatin from (+)-narciclasine as relay. J. Org. Chem. 2001, 66, 2583–2587. [Google Scholar] [PubMed]
- Ingrassia, L.; Lefranc, F.; Mathieu, V.; Darro, F.; Kiss, R. Amaryllidaceae isocarbostyril alkaloids and their derivatives as promising antitumor agents. Trans. Oncol. 2008, 1, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.J.; van Staden, J. Cytotoxicity studies of lycorine alkaloids of the Amaryllidaceae. Nat. Prod. Commun. 2014, 9, 1193–1210. [Google Scholar] [CrossRef]
- McLachlan, A.; Kekre, N.; McNulty, J.; Pandey, S. Pancratistatin: A natural anti-cancer compound that targets mitochondria specifically in cancer cells to induce apoptosis. Apoptosis 2005, 10, 619–630. [Google Scholar] [CrossRef]
- Griffin, C.; McNulty, J.; Pandey, S. Pancratistatin induces apoptosis and autophagy in metastatic prostate cancer cells. Int. J. Oncol. 2011, 38, 1549–1556. [Google Scholar]
- Kekre, N.; Griffin, C.; McNulty, J.; Pandey, S. Pancratistatin causes early activation of caspase-3 and the flipping of phosphatidyl serine followed by rapid apoptosis specifically in human lymphoma cells. Cancer Chemother. Pharmacol. 2005, 56, 29–38. [Google Scholar] [CrossRef]
- Shaala, L.A.; Youssef, D.T.A. Cytotoxic psammaplysin analogues from the Verongid Red Sea sponge Aplysinella species. Biomolecules 2019, 9, 841. [Google Scholar] [CrossRef] [Green Version]
- Youssef, D.T.A.; Mooberry, S.L. Hurghadolide A and swinholide I, potent actin-microfilament disrupters from the Red Sea sponge Theonella swinhoei. J. Nat. Prod. 2006, 69, 154–157. [Google Scholar] [CrossRef]
- Shaala, L.A.; Youssef, D.T.A.; Hemimycalins, C.-E. Cytotoxic and antimicrobial alkaloids with hydantoin and 2-iminoimidazolidin-4-one backbones from the Red Sea marine sponge Hemimycale sp. Mar. Drugs 2021, 19, 691. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structures of compounds 1–7.

Figure 2.
Key COSY and HMBC correlations of 1–3.

Figure 3.
Comparison of the growth inhibitory effects of pancratistatin (7) against cancer cell lines versus NHDF.
Figure 3.
Comparison of the growth inhibitory effects of pancratistatin (7) against cancer cell lines versus NHDF.

Figure 4.
Photograph of the flowers of Pancratium maritimum L.

Scheme 1.
Fractionation of the extract the flowers of P. maritimum L. and purification of 1–7.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
NMR data of 1 (850 MHz for 1H and 213 for 13C, CD3OD).
| Position | δC, Type | δH (Mult., J in Hz) | HMBC |
|---|---|---|---|
| 1 | 130.4, C | ||
| 2 | 133.0, CH | 7.68 (d, 8.7) | C-4, C-7 |
| 3 | 117.1, CH | 7.08 (d, 8.7) | C-1 |
| 4 | 159.9, C | ||
| 5 | 117.1, CH | 7.08 (d, 8.7) | C-1 |
| 6 | 133.0, CH | 7.68 (d, 8.7) | C-4, C-7 |
| 7 | 144.3, CH | 6.93 (d, 12.7) | C-2, C-6, C-9 |
| 8 | 117.6, CH | 5.87 (d, 12.7) | C-1, C-9 |
| 9 | 168.6, C | ||
| 10 | 51.8, CH3 | 3.72 (s) | C-9 |
| 1′ | 102.0, CH | 4.98 (d, 7.2) | C-4 |
| 2′ | 74.9, CH | 3.48 (m) | C-1′ |
| 3′ | 78.0, CH | 3.49 (m) | C-1′ |
| 4′ | 71.4, CH | 3.41 (m) | C-6′ |
| 5′ | 78.3, CH | 3.49 (m) | C-1′, C-3′ |
| 6′ | 62.5, CH2 | 3.91 (t, 11.9), 3.72 (m) | C-4′ |
Table 2.
NMR data of 2 (850 MHz for 1H and 213 for 13C, CD3OD).
| Position | δC, Type | δH (Mult., J in Hz) | HMBC |
|---|---|---|---|
| 1 | 125.5, C | ||
| 2 | 114.1, CH | 7.62 (d, 1.8) | C-3, C-6, C-7 |
| 3 | 150.5, C | ||
| 4 | 152.2, C | ||
| 5 | 116.5, CH | 7.23 (d, 8.5) | C-1, C-3, C-4 |
| 6 | 124.6, CH | 7.65 (dd, 8.5, 1.8) | C-1, C-4, C-7 |
| 7 | 168.3, C | ||
| 8 | 52.6, CH3 | 3.89 (s) | C-7 |
| 9 | 56.7, CH3 | 3.92 (s) | C-3, C-1′ |
| 1′ | 102.0, CH | 5.04 (d, 7.7) | C-4 |
| 2′ | 74.8, CH | 3.55 (dd, 9.1, 7.7) | C-3′ |
| 3′ | 77.9, CH | 3.48 (m) | C-1′ |
| 4′ | 71.3, CH | 3.42 (t, 9.1) | C-3′, C-5′ |
| 5′ | 78.4, CH | 3.50 (t, 9.1) | C-1′ |
| 6′ | 62.5, CH2 | 3.82 (m), 3.71 (dd, 11.0, 4.5) | C-5′ |
Table 3.
NMR data of 3 (850 MHz for 1H and 213 for 13C, CD3OD).
| Position | δC, Type | δH (Mult., J in Hz) | HMBC |
|---|---|---|---|
| 1 | 159.4, C | ||
| 2 | 114.8, CH | 6.83 (d, 8.5) | C-1, C-4 |
| 3 | 130.5, CH | 7.13 (d, 8.5) | C-1, C-7 |
| 4 | 135.4, C | ||
| 5 | 130.5, CH | 7.13 (d, 8.5) | C-1, C-7 |
| 6 | 114.8, CH | 6.83 (d, 8.5) | C-1 |
| 7 | 32.3, CH2 | 2.67 (t, 7.6) | C-4, C-8, C-9 |
| 8 | 32.9, CH2 | 1.90 (m) | C4, C-7, C-9 |
| 9 | 70.1, CH2 | 3.92 (dt, 9.6, 6.5), 3.54 (dt, 9.6, 6.5) | C-7, C-8, C-1′ |
| 10 | 55.7, CH3 | 3.77 (s) | C-1 |
| 1′ | 104.5, CH | 4.25 (d, 7.8) | C-9, C-3′ |
| 2′ | 75.2, CH | 3.21 (dd, 9.0, 7.8) | C-1′, C-3′ |
| 3′ | 78.2, CH | 3.37 (t, 9.0) | C-1′ |
| 4′ | 71.7, CH | 3.31 (t, 9.0) | C-3′, C-5′, C-6′ |
| 5′ | 77.9, CH | 3.27 (m) | C-3′, C-6′ |
| 6′ | 62.8, CH2 | 3.88 (dd, 11.9, 2.2), 3.69 (dd, 11.9, 6.1) |
Table 4.
NMR data of 4–6 (850 MHz for 1H and 213 for 13C, CD3OD).
| Position | δC, Type | δH (Mult., J in Hz) | δC, Type | δH (Mult., J in Hz) | δC, Type a | δH (Mult., J in Hz) a |
|---|---|---|---|---|---|---|
| 1 | 127.4, C | 127.9, C | 133.6, C | |||
| 2 | 131.2, CH | 7.48 (d, 8.5) | 115.1, CH | 7.05 (d, 1.5) | 116.5, CH | 6.63 (d, 1.9) |
| 3 | 116.9, CH | 6.82 (d, 8.5) | 146.8, C | 146.3, C | ||
| 4 | 161.2, C | 149.4, C | 144.7, C | |||
| 5 | 116.9, CH | 6.82 (d, 8.5) | 116.5, CH | 6.79 (d, 8.0) | 116.4, CH | 6.68 (d, 8.0) |
| 6 | 131.2, CH | 7.48 (d, 8.5) | 122.8, CH | 6.94 (dd, 8.0, 1.5) | 120.5, CH | 6.51 (dd, 8.0, 1.9) |
| 7 | 146.5, CH | 7.67 (d, 15.9) | 146.9, CH | 7.54 (d, 15.8) | 31.1, CH2 | 2.77 (t, 7.6) |
| 8 | 115.9, CH | 6.37 (d, 15.9) | 116.0, CH | 6.24 (d, 15.8) | 37.1, CH2 | 2.57 (t, 7.6) |
| 9 | 169.0, C | 171.1, C | 175.4, C | |||
| 10 | 52.9, CH3 | 3.73 (s) | 52.1, CH3 | 3.65 (s) |
a Data acquired at 600 MHZ for 1H and 150 MHz for 13C.
Table 5.
Antiproliferative and growth inhibition effects of 1–7.
| Compound | IC50 (μM) (Mean + SEM) a | |||
|---|---|---|---|---|
| MDA-MB-231 | HeLa | HCT 116 | NHDF | |
| 1 | ≥10.0 | ≥10.0 | ≥10.0 | NT |
| 2 | ≥10.0 | ≥10.0 | ≥10.0 | NT |
| 3 | ≥10.0 | ≥10.0 | ≥10.0 | NT |
| 4 | NT | NT | NT | NT |
| 5 | ≥10.0 | ≥10.0 | ≥10.0 | NT |
| 6 | NT | NT | NT | NT |
| 7 | 0.14 ± 0.002 | 0.058 ± 0.001 | 0.10 ± 0.005 | 6.6 ± 0.034 |
| 5-FU b | 13.0 ± 0.30 | 12.3 ± 0.25 | 4.6 ± 0.23 | NT |
(a) The results are the mean of three independent experiments; (b) 5-Fluorouracil, a positive drug; NHDF: normal human dermal fibroblasts.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Youssef, D.T.A.; Shaala, L.A.; Altyar, A.E. Cytotoxic Phenylpropanoid Derivatives and Alkaloids from the Flowers of Pancratium maritimum L. Plants 2022, 11, 476. https://doi.org/10.3390/plants11040476
AMA Style
Youssef DTA, Shaala LA, Altyar AE. Cytotoxic Phenylpropanoid Derivatives and Alkaloids from the Flowers of Pancratium maritimum L. Plants. 2022; 11(4):476. https://doi.org/10.3390/plants11040476
Chicago/Turabian StyleYoussef, Diaa T. A., Lamiaa A. Shaala, and Ahmed E. Altyar. 2022. "Cytotoxic Phenylpropanoid Derivatives and Alkaloids from the Flowers of Pancratium maritimum L." Plants 11, no. 4: 476. https://doi.org/10.3390/plants11040476
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.