
Two Arabidopsis Splicing Factors, U2AF65a and U2AF65b, Differentially Control Flowering Time by Modulating the Expression or Alternative Splicing of a Subset of FLC Upstream Regulators

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
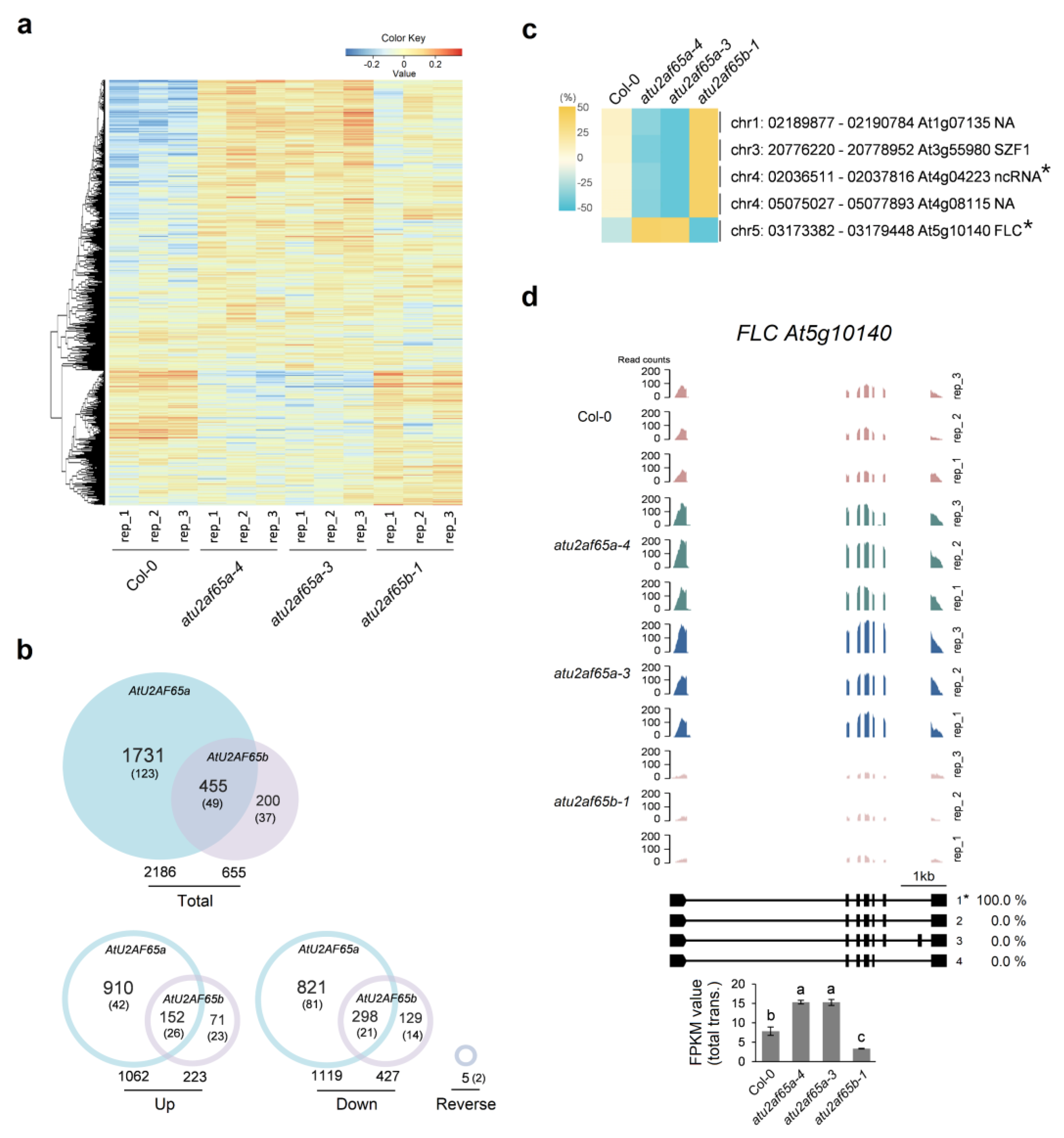
2.1. RNA Sequencing Analysis in the Shoot Apex-Enriched Samples of atu2af65a and atu2af65b Mutants
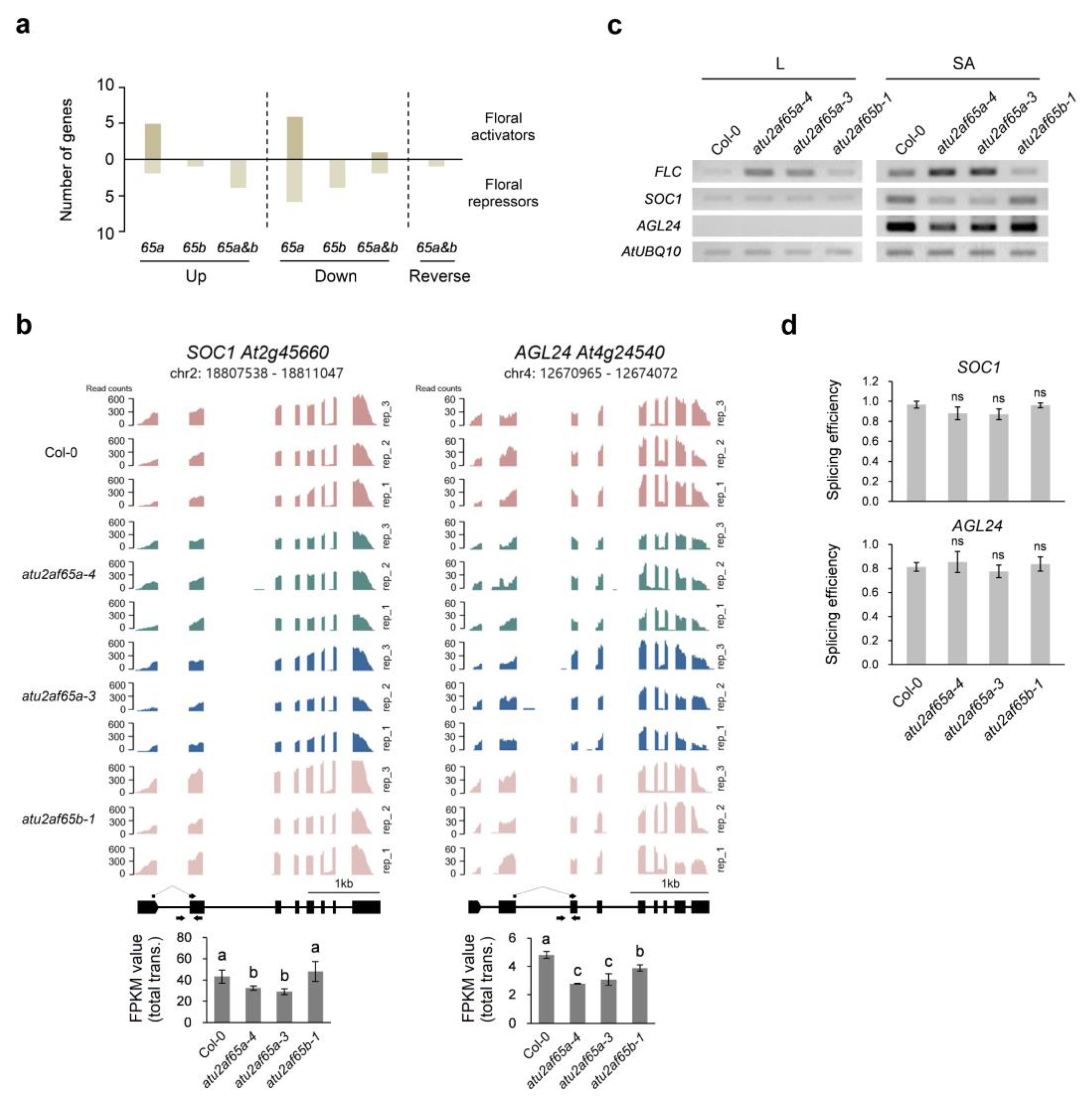
2.2. Downregulation of SOC1 and AGL24 Expression in the Shoot Apices of the atu2af65a Mutants Is Due to Upregulation of FLC Expression
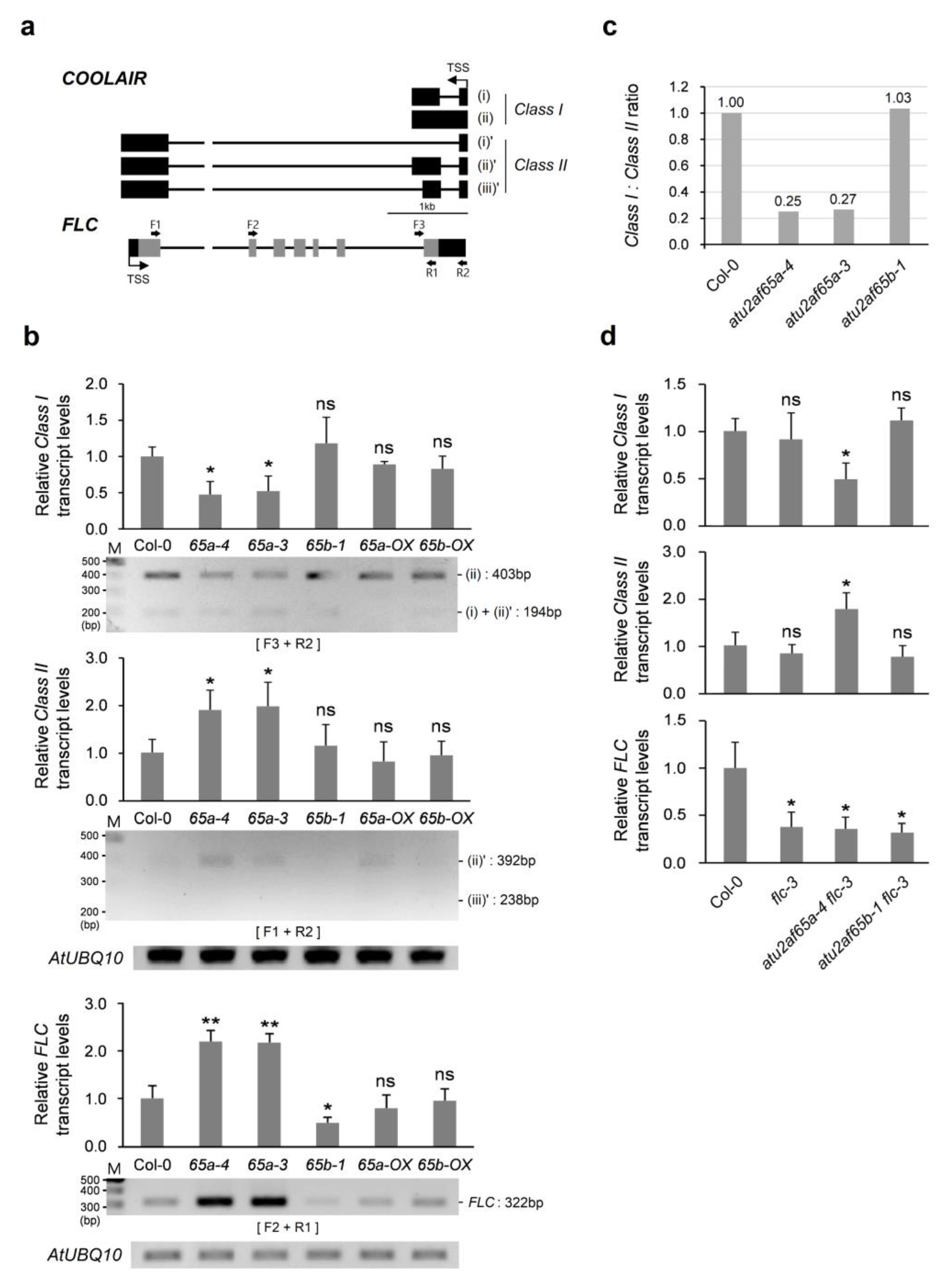
2.3. COOLAIR Is Involved in Misregulation of FLC Expression in the Shoot Apices of the atu2af65a Mutants
2.4. EDM2 and PP2A-b’ɤ Genes Are Involved in Change in FLC Expression in the atu2af65a or atu2af65b Mutants
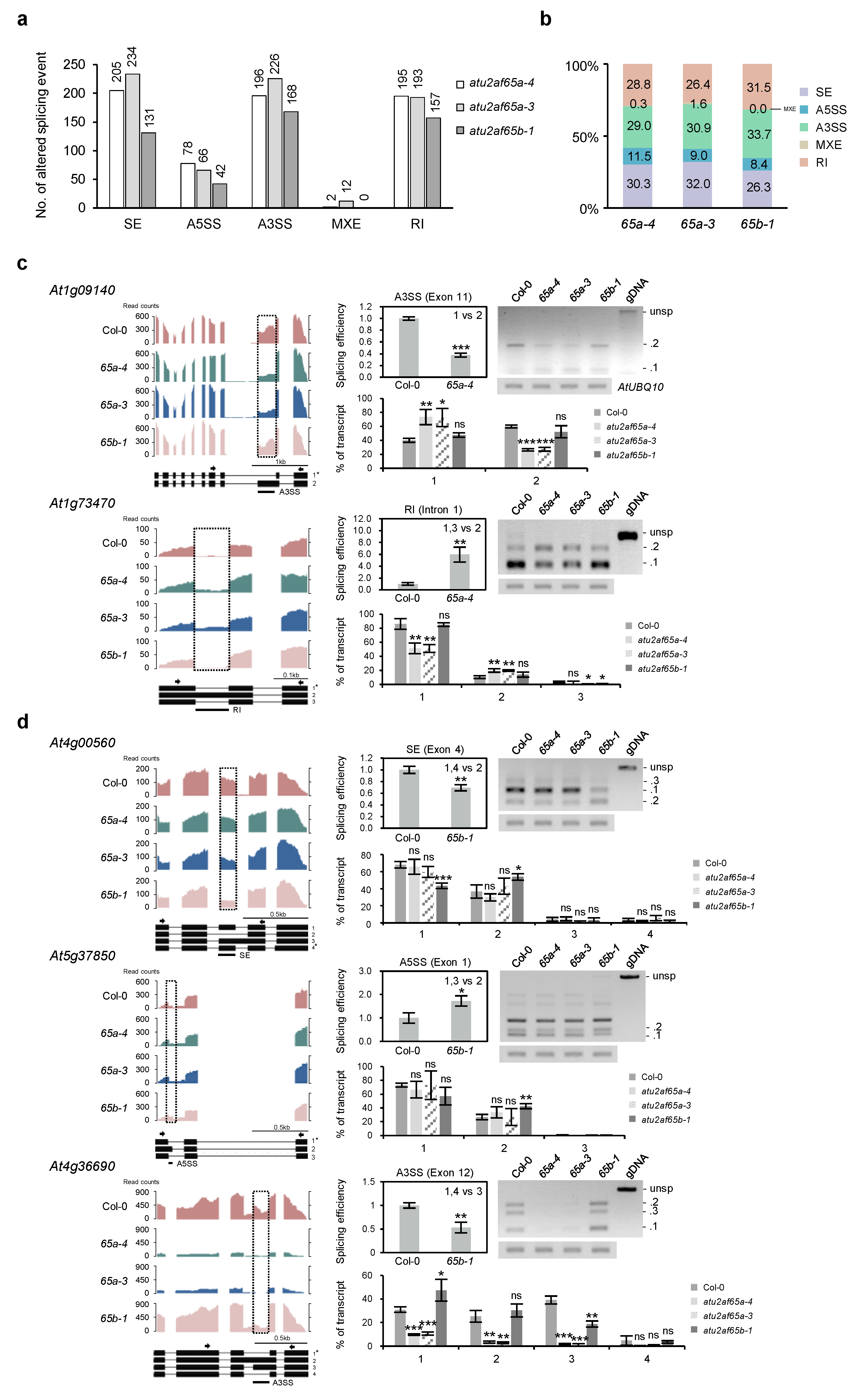
2.5. Global-Scale Changes in Splicing Events in the atu2af65a and atu2af65b Mutants
2.6. Alternative Splicing of AtU2AF65a Pre-mRNA could Be Altered Similarly by Mutations in AtU2AF65b and AtSF1 Genes as Well as Lower Ambient Growth Temperature
3. Discussion
4. Materials and Methods
4.1. Plant Material and Growth Conditions
4.2. RNA Extraction, RT-PCR and Real-Time Quantitative PCR
4.3. Preparation of Shoot Apices and RNA-seq
4.4. Nuclear/Cytoplasmic Fractionation and Isolation of Nuclear Extracts
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barton, M.K.; Poething, R.S. Formation of the shoot apical meristem in Arabidopsis thaliana: An analysis of development in the wild type and in the shoot meristemless mutant. Development 1993, 119, 823–831. [Google Scholar] [CrossRef]
- Aida, M.; Ishida, T.; Tasaka, M. Shoot apical meristem and cotyledon formation during Arabidopsis embryogenesis: Interaction among the cup–shaped cotyledon and shoot meristemless genes. Development 1999, 126, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Long, J.A.; Barton, M.K. The development of apical embryonic pattern in Arabidopsis. Development 1998, 125, 3027–3035. [Google Scholar] [CrossRef] [PubMed]
- Simpson, G.G.; Gendall, A.R.; Dean, C. When to switch to flowering. Annu. Rev. Cell Dev. Biol. 1999, 99, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Boss, P.K.; Bastow, R.M.; Mylne, J.S.; Dean, C. Multiple pathways in the decision to flower: Enabling, promoting, and resetting. Plant Cell 2004, 16 (Suppl. S1), S18–S31. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Xu, Y.; Tan, E.L.; Kumar, P.P. Agamous–like 24, a dosage–dependent mediator of the flowering signals. Porc. Natl. Acad. Sci. USA 2002, 99, 16336–16341. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Chen, H.; Er, H.L.; Soo, H.M.; Kumar, P.P.; Han, J.H.; Liou, Y.C.; Yu, H. Direct interaction of AGL24 and SOC1 integrates flowering signals in Arabidopsis. Development 2008, 135, 1481–1491. [Google Scholar] [CrossRef] [Green Version]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Kadam, U.S.; Schulz, B.; Irudayaraj, J. Detection and quantification of alternative splice sites in Arabidopsis genes AtDCL2 and AtPTB2 with highly sensitive surface enhanced Raman spectroscopy (SERS) and gold nanoprobes. FEBS Lett. 2014, 588, 1637–1643. [Google Scholar] [CrossRef] [Green Version]
- Kadam, U.S.; Schulz, B.; Irudayaraj, J.M.K. Multiplex single–cell quantification of rare RNA transcripts from protoplasts in a model plant system. Plant J. 2017, 90, 1187–1195. [Google Scholar] [CrossRef] [Green Version]
- Martín, G.; Márquez, Y.; Mantica, F.; Duque, P.; Irimia, M. Alternative splicing landscapes in Arabidopsis thaliana across tissues and stress conditions highlight major functional differences with animals. Genome Biol. 2021, 22, 35. [Google Scholar] [CrossRef]
- Park, H.-Y.; Lee, H.T.; Lee, J.H.; Kim, J.-K. Arabidopsis U2AF65 Regulates Flowering Time and the Growth of Pollen Tubes. Front. Plant Sci. 2019, 10, 569. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.H.; Park, H.-Y.; Lee, K.C.; Thu, M.P.; Kim, S.-K.; Suh, M.C.; Kang, H.; Kim, J.-K. A homolog of splicing factor SF1 is essential for development and is involved in the alternative splicing of pre–mRNA in Arabidopsis thaliana. Plant J. 2014, 78, 591–603. [Google Scholar] [CrossRef]
- Wang, B.B.; Brendel, V. Molecular characterization and phylogeny of U2AF35 homologs in plants. Plant Physiol. 2006, 140, 624–636. [Google Scholar] [CrossRef] [Green Version]
- Zuo, P.; Maniatis, T. The splicing factor U2AF35 mediates critical protein–protein interactions in constitutive and enhancer–dependent splicing. Genes Dev. 1996, 10, 1356–1368. [Google Scholar] [CrossRef] [Green Version]
- Tholen, J.; Razew, M.; Weis, F.; Galej, W.P. Structural basis of branch site recognition by the human spliceosome. Science 2022, 375, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Peled–Zehavi, H.; Berglund, J.A.; Rosbash, M.; Frankel, A.D. Recognition of RNA branch point sequences by the KH domain of splicing factor 1 (mammalian branch point binding protein) in a splicing factor complex. Mol. Cell. Biol. 2001, 21, 5232–5241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koncz, C.; Dejong, F.; Villacorta, N.; Szakonyi, D.; Koncz, Z. The spliceosome–activating complex: Molecular mechanisms underlying the function of a pleiotropic regulator. Front. Plant Sci. 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhage, L.; Severing, E.I.; Bucher, J.; Lammers, M.; Busscher–Lange, J.; Bonnema, G.; Rodenburg, N.; Proveniers, M.C.; Angenent, G.C.; Immink, R.G. Splicing–related genes are alternatively spliced upon changes in ambient temperatures in plants. PLoS ONE 2017, 12, e0172950. [Google Scholar] [CrossRef] [Green Version]
- Xiong, F.; Ren, J.J.; Yu, Q.; Wang, Y.Y.; Lu, C.C.; Kong, L.J.; Otegui, M.S.; Wang, X.L. AtU2AF65b functions in abscisic acid mediated flowering via regulating the precursor messenger RNA splicing of ABI5 and FLC in Arabidopsis. New Phytol. 2019, 223, 277–292. [Google Scholar] [CrossRef]
- Zhang, X.N.; Mount, S.M. Two alternatively spliced isoforms of the Arabidopsis SR45 protein have distinct roles during normal plant development. Plant Physiol. 2009, 150, 1450–1458. [Google Scholar] [CrossRef] [Green Version]
- Michaels, S.D.; Amasino, R.M. Flowering locus cencodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell 1999, 5, 949–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA–Seq data. Porc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Jiang, H.; Xu, Y.; Li, H.; Wu, X.; Xie, Q.; Li, C. The CCCH–type zinc finger proteins AtSZF1 and AtSZF2 regulate salt stress responses in Arabidopsis. Plant Cell Physiol. 2007, 48, 1148–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, F.; Elowitz, M.B. Constitutive splicing and economies of scale in gene expression. Nat. Struct. Mol. Biol. 2019, 26, 424–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, D.; Mao, F.; Tian, Y.; Lin, X.; Gu, L.; Gu, H.; Qu, L.J.; Wu, Y.; Wu, Z. The Features and Regulation of Co–transcriptional Splicing in Arabidopsis. Mol. Plant 2020, 13, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.D.; Lin, Y.; Ren, Q.P.; Wang, Y.Y.; Xiong, F.; Wang, X.L. RNA Splicing of FLC Modulates the Transition to Flowering. Front. Plant Sci. 2019, 10, 1625. [Google Scholar] [CrossRef]
- Csorba, T.; Questa, J.I.; Sun, Q.; Dean, C. Antisense coolair mediates the coordinated switching of chromatin states at FLC during vernalization. Porc. Natl. Acad. Sci. USA 2014, 111, 16160–16165. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Marquardt, S.; Lister, C.; Swiezewski, S.; Dean, C. Targeted 3’ processing of antisense transcripts triggers Arabidopsis FLC chromatin silencing. Science 2010, 327, 94–97. [Google Scholar] [CrossRef]
- Marquardt, S.; Raitskin, O.; Wu, Z.; Liu, F.; Sun, Q.; Dean, C. Functional consequences of splicing of the antisense transcript COOLAIR on FLC transcription. Mol. Cell 2014, 54, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Hornyik, C.; Terzi, L.C.; Simpson, G.G. The spen family protein FPA controls alternative cleavage and polyadenylation of RNA. Dev. Cell 2010, 18, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazin, J.; Romero, N.; Rigo, R.; Charon, C.; Blein, T.; Ariel, F.; Crespi, M. Nuclear Speckle RNA Binding Proteins Remodel Alternative Splicing and the Non–coding Arabidopsis Transcriptome to Regulate a Cross–Talk Between Auxin and Immune Responses. Front. Plant Sci. 2018, 9, 1209. [Google Scholar] [CrossRef] [PubMed]
- Kalyna, M.; Simpson, C.G.; Syed, N.H.; Lewandowska, D.; Marquez, Y.; Kusenda, B.; Marshall, J.; Fuller, J.; Cardle, L.; McNicol, J.; et al. Alternative splicing and nonsense–mediated decay modulate expression of important regulatory genes in Arabidopsis. Nucleic Acids Res. 2012, 40, 2454–2469. [Google Scholar] [CrossRef] [Green Version]
- Heidari, B.; Nemie–Feyissa, D.; Kangasjarvi, S.; Lillo, C. Antagonistic Regulation of Flowering Time through Distinct Regulatory Subunits of Protein Phosphatase 2A. PLoS ONE 2013, 8, e67987. [Google Scholar] [CrossRef]
- Tsuchiya, T.; Eulgem, T. The Arabidopsis defense component EDM2 affects the floral transition in an FLC–dependent manner. Plant J. 2010, 62, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Kardailsky, I.; Shukla, V.K.; Ahn, J.H.; Dagenais, N.; Christensen, S.K.; Nguyen, J.T.; Chory, J.; Harrison, M.J.; Weigel, D. Activation tagging of the floral inducer FT. Science 1999, 286, 1962–1965. [Google Scholar] [CrossRef] [Green Version]
- Swiezewski, S.; Liu, F.; Magusin, A.; Dean, C. Cold–induced silencing by long antisense transcripts of an Arabidopsis Polycomb target. Nature 2009, 462, 799–802. [Google Scholar] [CrossRef]
- Vagner, S.; Ruegsegger, U.; Gunderson, S.I.; Keller, W.; Mattaj, I.W. Position–dependent inhibition of the cleavage step of pre–mRNA 3’–end processing by U1 snRNP. RNA 2000, 6, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Millevoi, S.; Loulergue, C.; Dettwiler, S.; Karaa, S.Z.; Keller, W.; Antoniou, M.; Vagner, S. An interaction between U2AF 65 and CF I(m) links the splicing and 3’ end processing machineries. EMBO J. 2006, 25, 4854–4864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macknight, R.; Bancroft, I.; Page, T.; Lister, C.; Schmidt, R.; Love, K.; Westphal, L.; Murphy, G.; Sherson, S.; Cobbett, C.; et al. FCA, a gene controlling flowering time in Arabidopsis, encodes a protein containing RNA–binding domains. Cell 1997, 89, 737–745. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Cho, Y.S.; Yoon, H.S.; Suh, M.C.; Moon, J.; Lee, I.; Weigel, D.; Yun, C.H.; Kim, J.K. Conservation and divergence of FCA function between Arabidopsis and rice. Plant Mol. Biol. 2005, 58, 823–838. [Google Scholar] [CrossRef]
- Tian, Y.; Zheng, H.; Zhang, F.; Wang, S.; Ji, X.; Xu, C.; He, Y.; Ding, Y. PRC2 recruitment and H3K27me3 deposition at FLC require FCA binding of coolair. Sci. Adv. 2019, 5, 7246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drexler, H.L.; Choquet, K.; Merens, H.E.; Tang, P.S.; Simpson, J.T.; Churchman, L.S. Revealing nascent RNA processing dynamics with nano–COP. Nat. Protoc. 2021, 16, 1343–1375. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Long, Y.; Zhang, H.; Li, Z.; Liu, Z.; Zhao, Y.; Lu, D.; Jin, X.; Deng, X.; Xia, R.; et al. Post–transcriptional splicing of nascent RNA contributes to widespread intron retention in plants. Nat. Plants 2020, 6, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Dent, C.; Liang, H.; Lv, J.; Shang, G.; Liu, Y.; Feng, F.; Wang, F.; Pang, J.; Li, X.; et al. CRY2 interacts with CIS1 to regulate thermosensory flowering via FLM alternative splicing. Nat. Commun. 2022, 13, 7045. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene–set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Moro, B.; Kisielow, M.; Borrero, V.B.; Bouet, A.; Brosnan, C.A.; Bologna, N.G. Nuclear RNA purification by flow cytometry to study nuclear processes in plants. STAR Protoc. 2021, 2, 100320. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.T.; Park, H.-Y.; Lee, K.C.; Lee, J.H.; Kim, J.-K. Two Arabidopsis Splicing Factors, U2AF65a and U2AF65b, Differentially Control Flowering Time by Modulating the Expression or Alternative Splicing of a Subset of FLC Upstream Regulators. Plants 2023, 12, 1655. https://doi.org/10.3390/plants12081655
Lee HT, Park H-Y, Lee KC, Lee JH, Kim J-K. Two Arabidopsis Splicing Factors, U2AF65a and U2AF65b, Differentially Control Flowering Time by Modulating the Expression or Alternative Splicing of a Subset of FLC Upstream Regulators. Plants. 2023; 12(8):1655. https://doi.org/10.3390/plants12081655
Chicago/Turabian StyleLee, Hee Tae, Hyo-Young Park, Keh Chien Lee, Jeong Hwan Lee, and Jeong-Kook Kim. 2023. "Two Arabidopsis Splicing Factors, U2AF65a and U2AF65b, Differentially Control Flowering Time by Modulating the Expression or Alternative Splicing of a Subset of FLC Upstream Regulators" Plants 12, no. 8: 1655. https://doi.org/10.3390/plants12081655