

Integrated Transcriptomic and Proteomic Characterization of a Chromosome Segment Substitution Line Reveals the Regulatory Mechanism Controlling the Seed Weight in Soybean
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
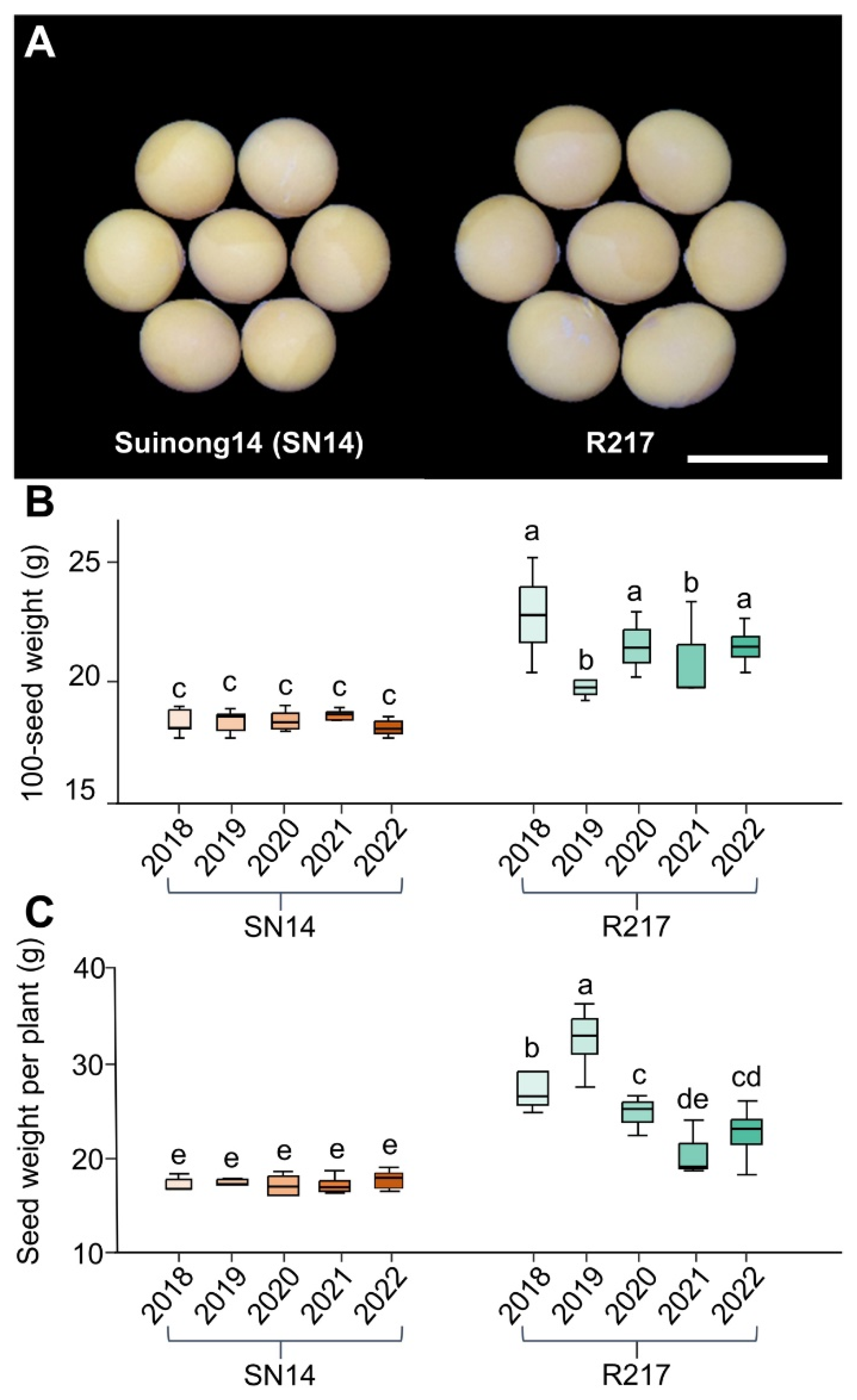
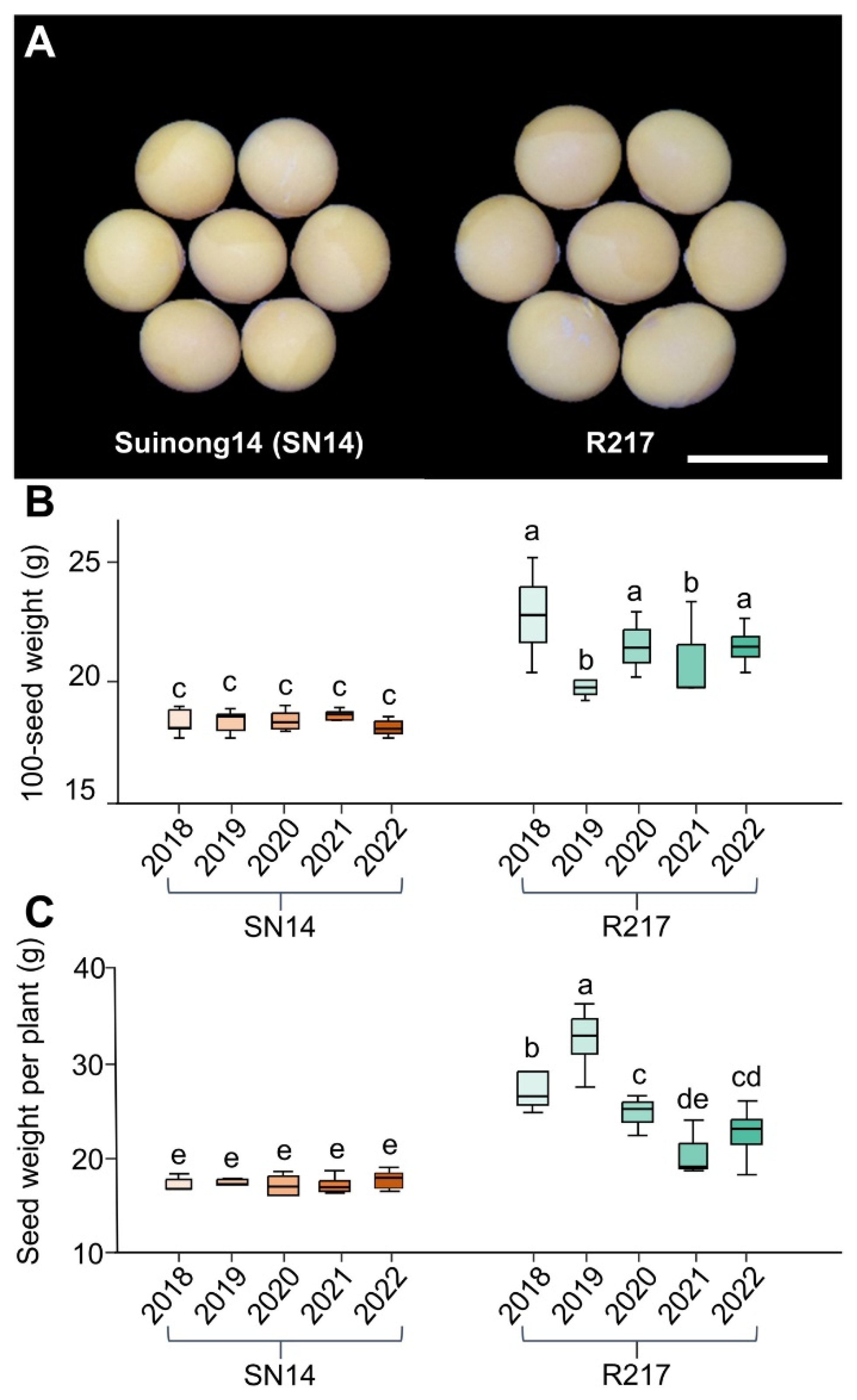
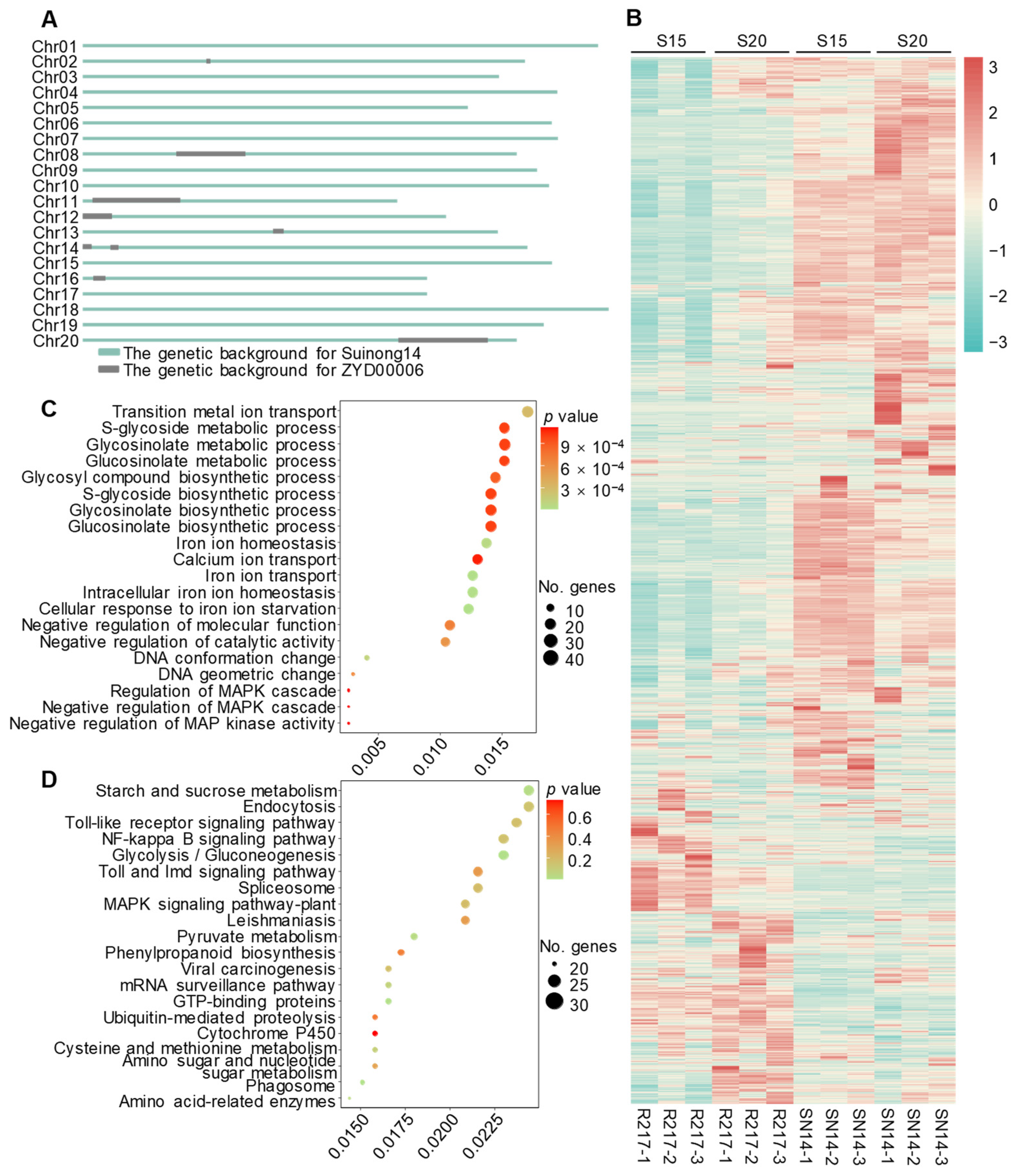
2.1. Phenotypic Variation of the Large Seed CSSL R217
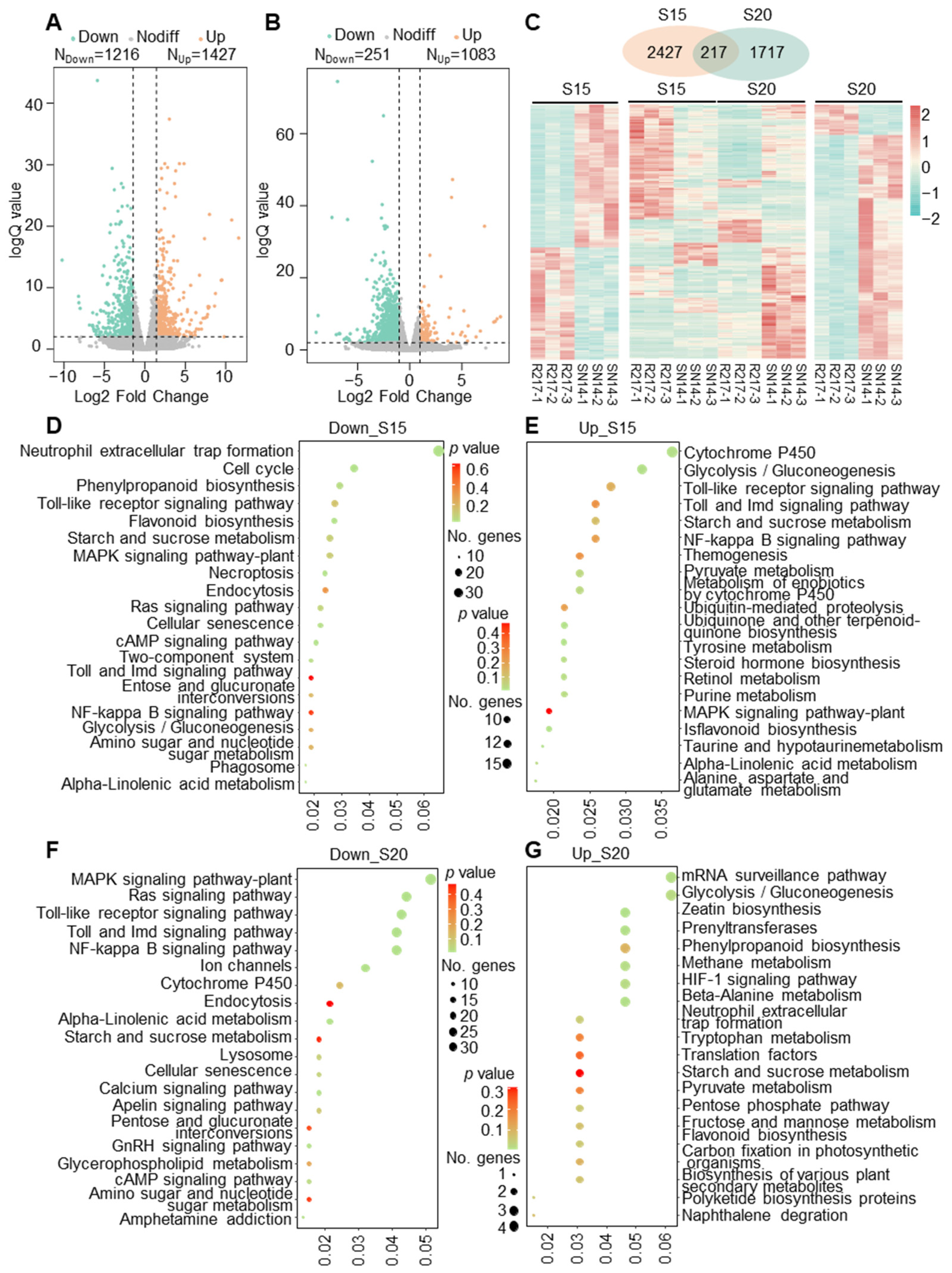
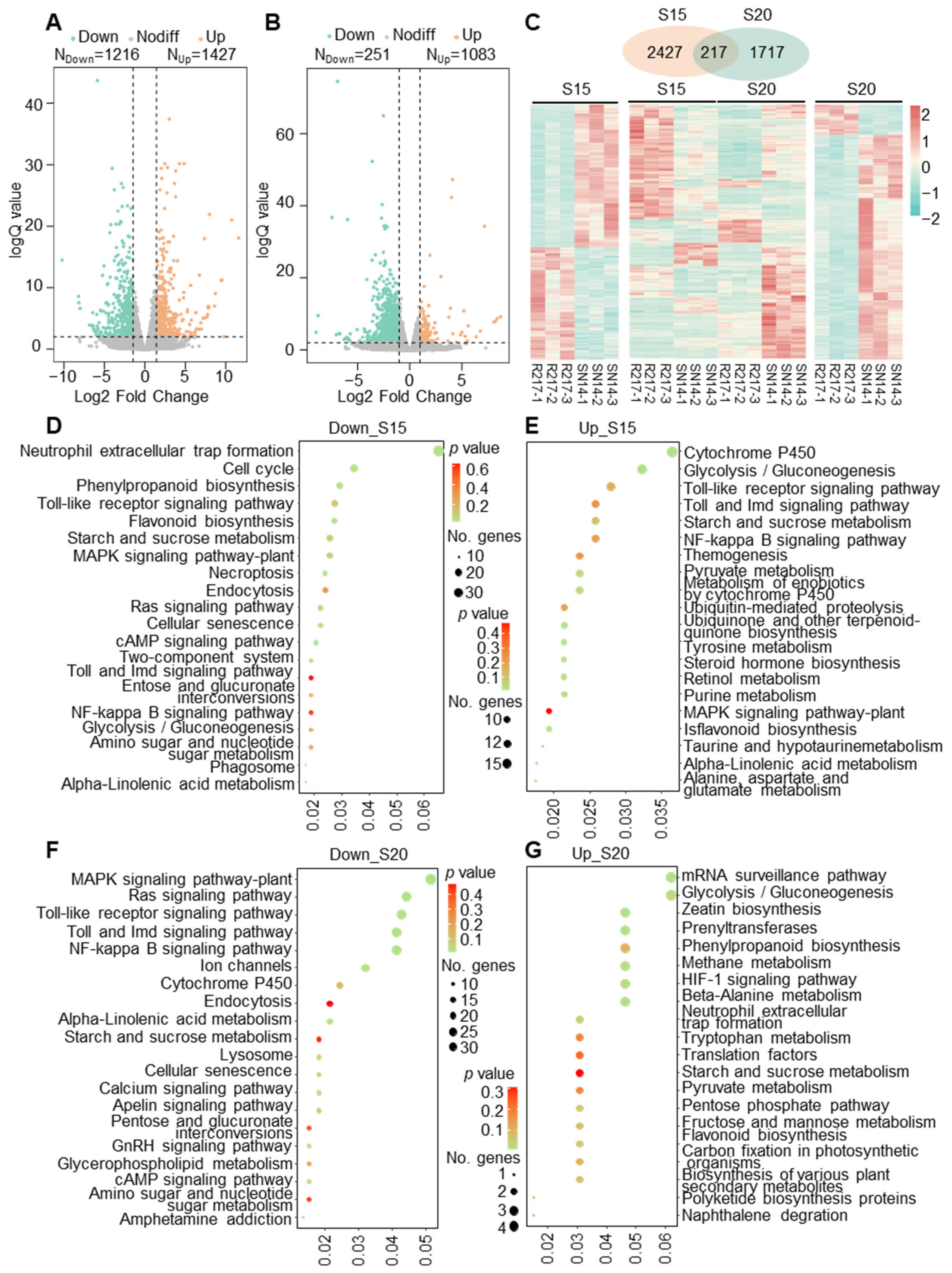
2.2. Transcriptomic Comparison between R217 and SN14
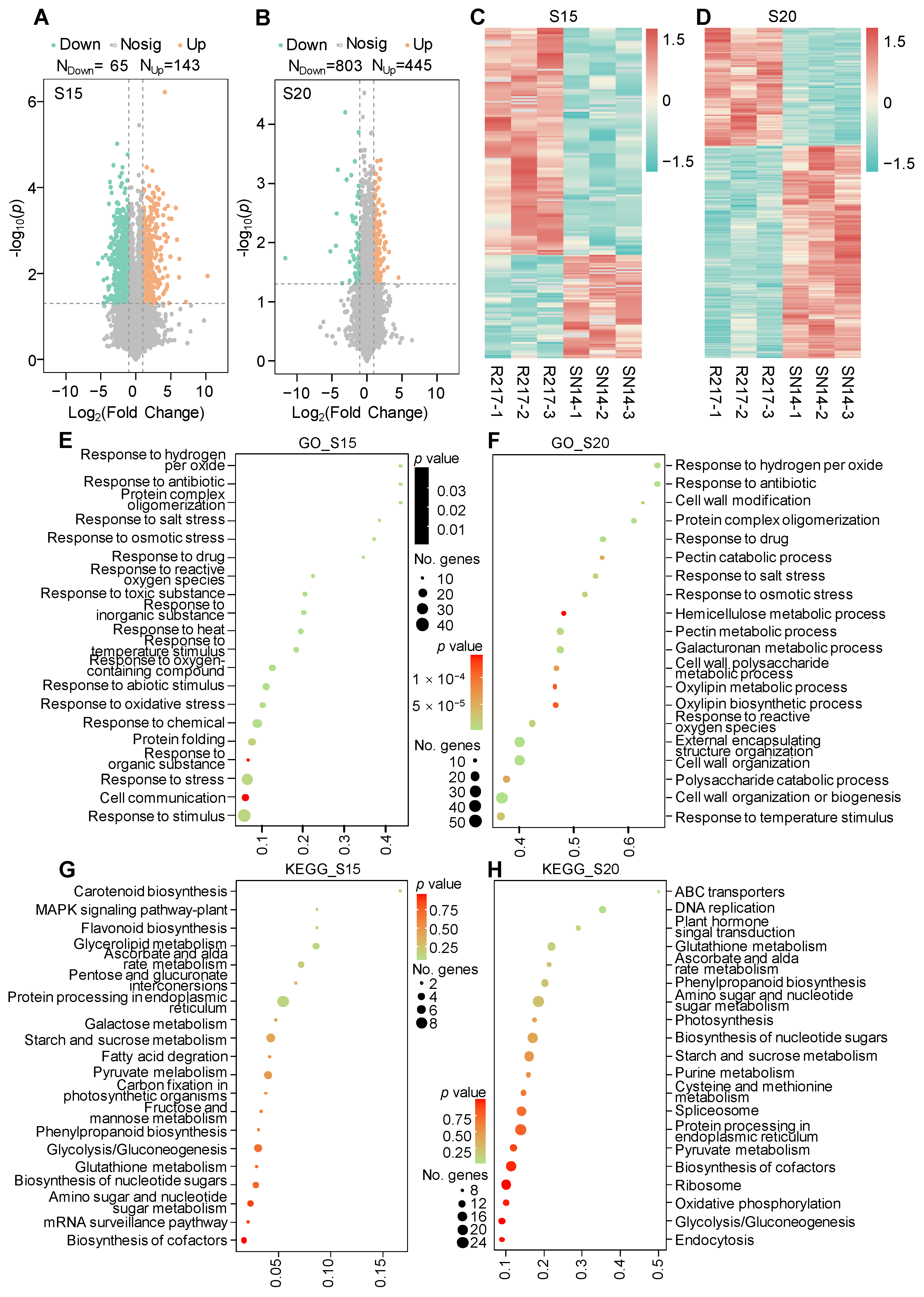
2.3. Proteomic Analysis of R217 and SN14
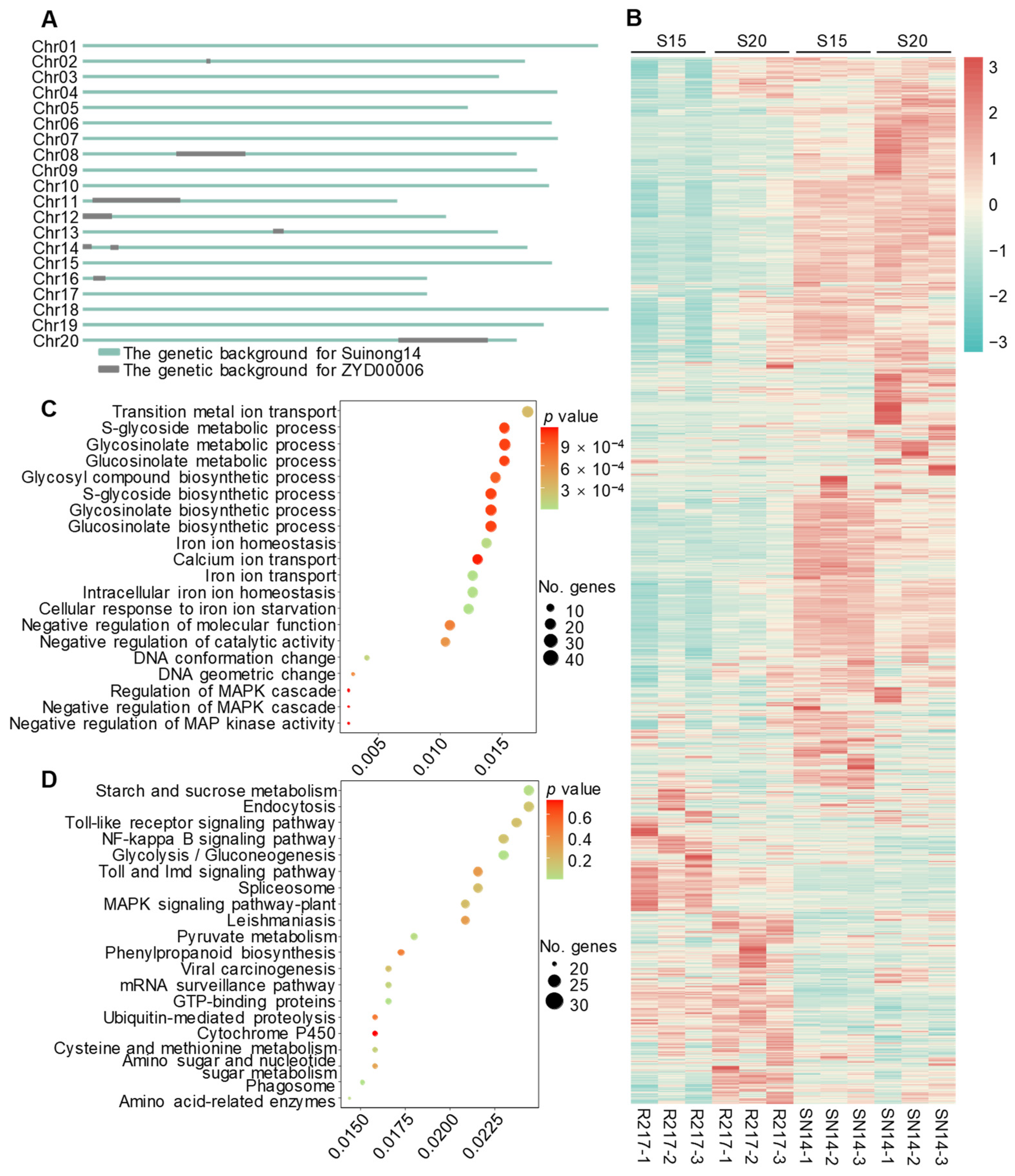
2.4. Analysis of Genes on the Substituted Chromosomal Segments of R217
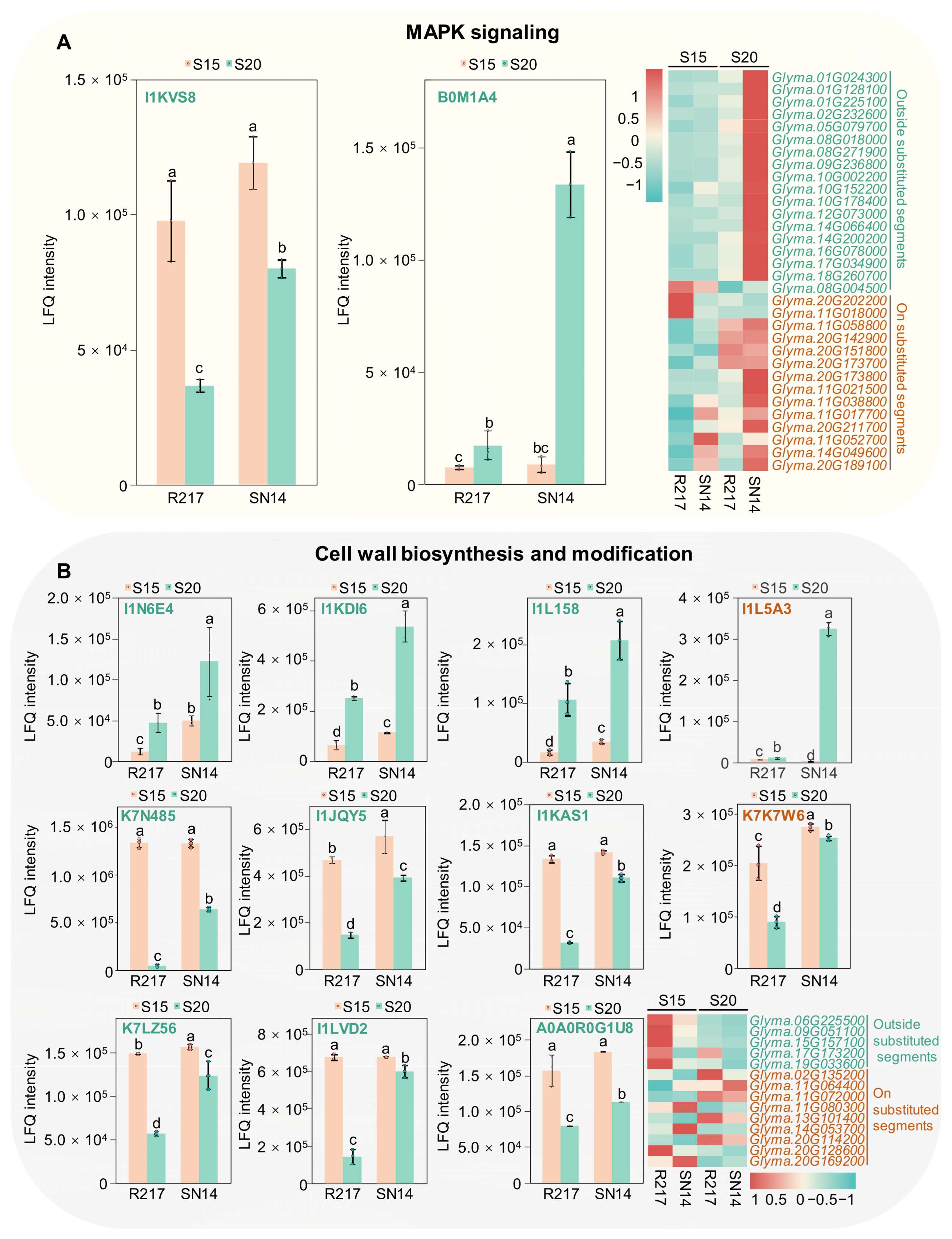
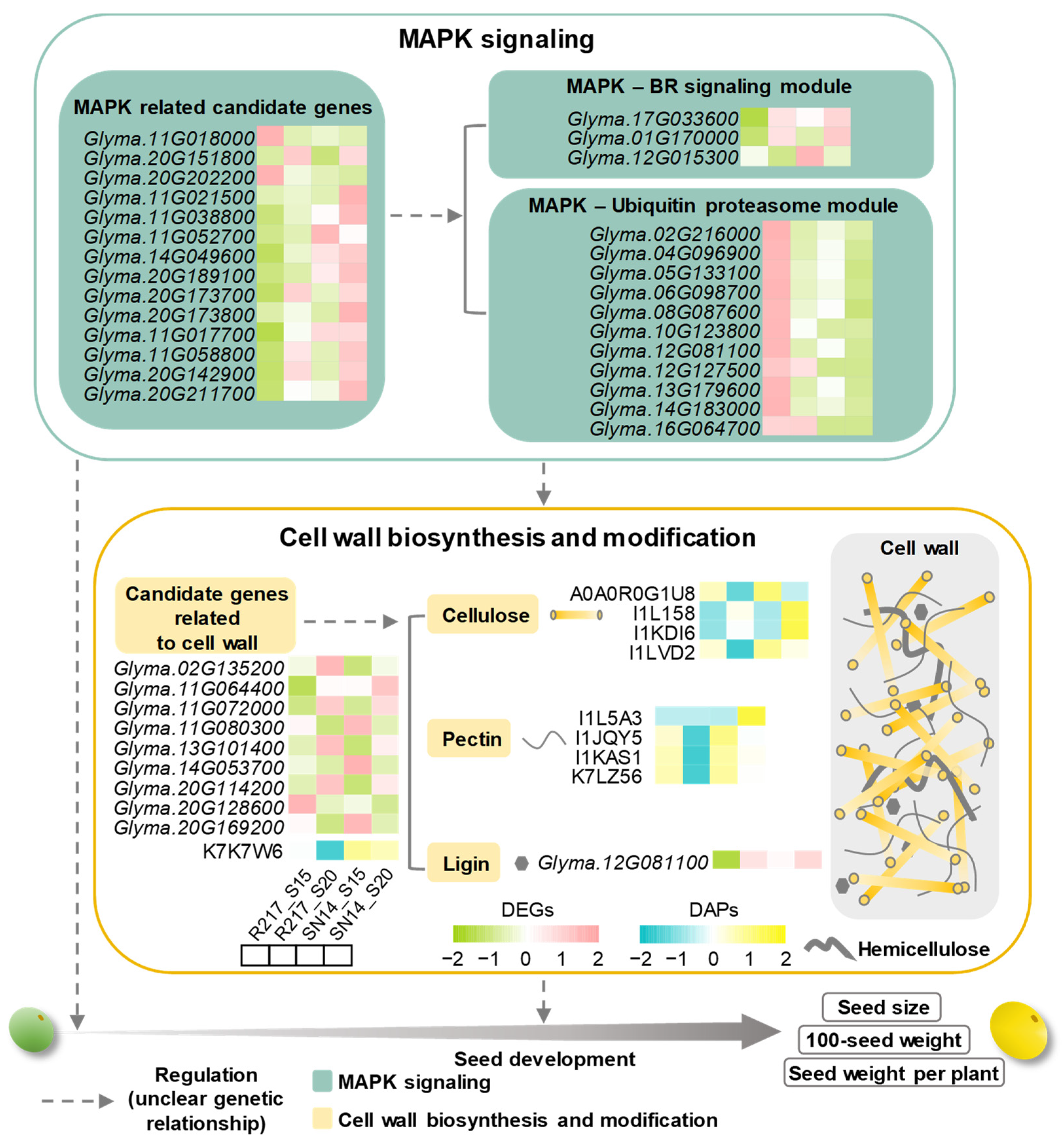
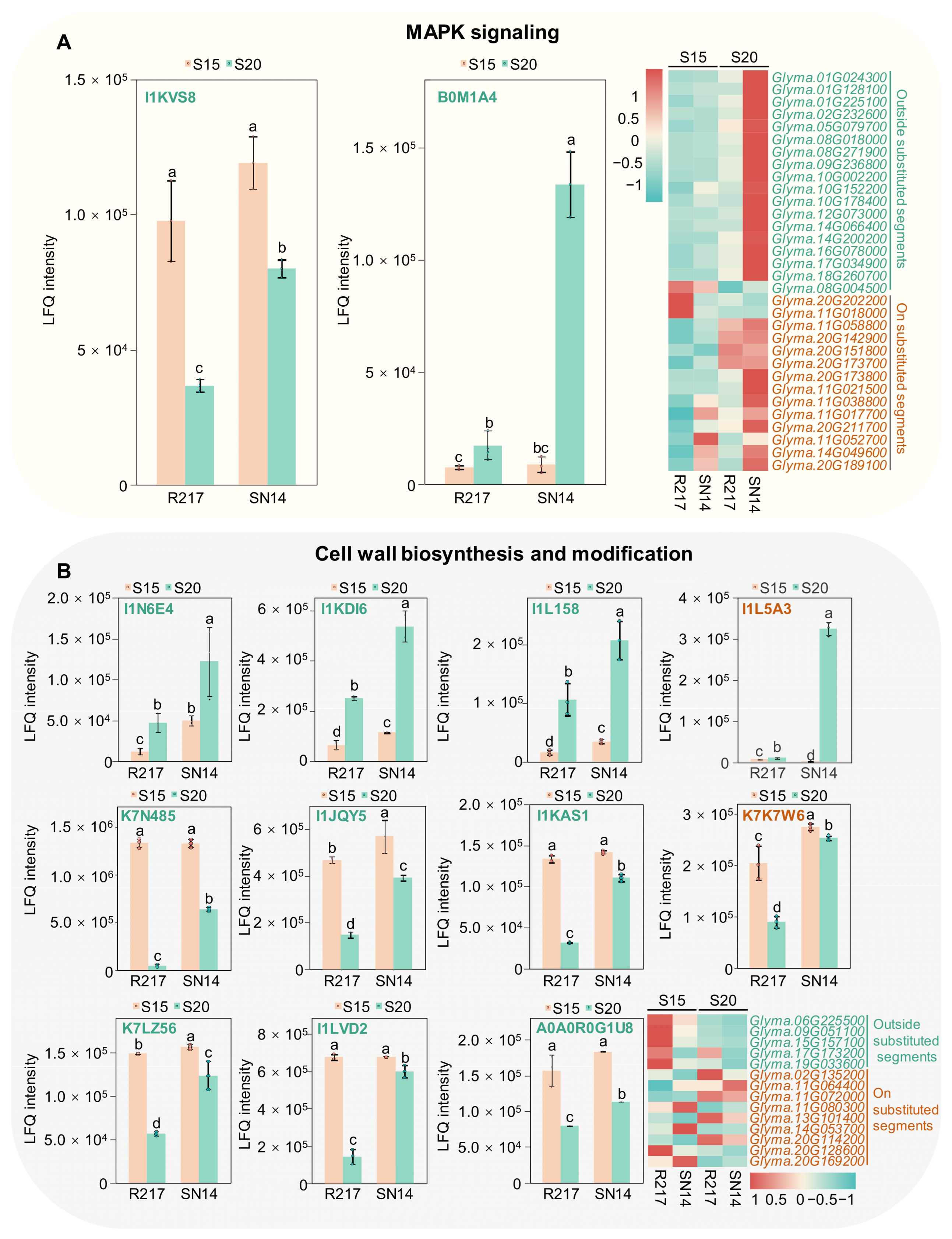
2.5. Potential Molecular Genetic Mechanism of Seed Weight
3. Discussion
3.1. CSSLs Facilitate Identifying Seed Size-Regulating Genes in Soybean
3.2. Integrated Transcriptomic and Proteomic Analyses Reveal the Seed Weight-Related Regulatory Pathways
3.3. Potential Regulatory Mechanisms Involved in Seed Size Control
4. Materials and Methods
4.1. Plant Materials
4.2. Seed Weight and Size Evaluation
4.3. RNA-Seq Analysis
4.4. Protein Extraction and Digestion
4.5. LC-MS/MS Analysis
4.6. Proteomic Data Processing
4.7. Bioinformatics Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Karikari, B.; Wang, Z.; Zhou, Y.; Yan, W.; Feng, J.; Zhao, T. Identification of quantitative trait nucleotides and candidate genes for soybean seed weight by multiple models of genome-wide association study. BMC Plant Biol. 2020, 20, 404. [Google Scholar] [CrossRef]
- Nguyen, C.X.; Paddock, K.J.; Zhang, Z.Y.; Stacey, M.G. GmKIX8-1 regulates organ size in soybean and is the causative gene for the major seed weight QTL qSw17-1. New Phytol. 2021, 229, 920–934. [Google Scholar] [CrossRef]
- Lu, X.; Xiong, Q.; Cheng, T.; Li, Q.T.; Liu, X.L.; Bi, Y.D.; Li, W.; Zhang, W.K.; Ma, B.; Lai, Y.C.; et al. A PP2C-1 allele underlying a quantitative trait locus enhances soybean 100-seed weight. Mol. Plant 2017, 10, 670–684. [Google Scholar] [CrossRef]
- Duan, Z.B.; Zhang, M.; Zhang, Z.F.; Liang, S.; Fan, L.; Yang, X.; Yuan, Y.Q.; Pan, Y.; Zhou, G.A.; Liu, S.L.; et al. Natural allelic variation of GmST05 controlling seed size and quality in soybean. Plant Biotechnol. J. 2022, 20, 1807–1818. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.H.; Ma, R.R.; Huang, W.X.; Hou, J.J.; Fang, C.; Wang, L.S.; Yuan, Z.H.; Sun, Q.; Dong, X.H.; et al. Identification of ST1 reveals a selection involving hitchhiking of seed morphology and oil content during soybean domestication. Plant Biotechnol. J. 2022, 20, 1110–1121. [Google Scholar] [CrossRef]
- Cai, Z.; Xian, P.; Cheng, Y.; Zhong, Y.; Yang, Y.; Zhou, Q.; Lian, T.; Ma, Q.; Nian, H.; Ge, L. MOTHER-OF-FT-AND-TFL1 regulates the seed oil and protein content in soybean. New Phytol. 2023, 239, 905–919. [Google Scholar] [CrossRef]
- Wei, S.M.; Yong, B.; Jiang, H.W.; Wang, Y.; Li, B.B.; Yang, C.; Zhu, W.W.; An, Z.H.; Chen, Q.S.; He, C.Y. A loss-of-function mutant allele of a glycosyl hydrolase gene has been co-opted for seed weight control during soybean domestication. J. Integr. Plant Biol. 2023, 65, 2469–2689. [Google Scholar] [CrossRef]
- Wang, S.D.; Liu, S.L.; Wang, J.; Yokosho, K.; Zhou, B.; Yu, Y.C.; Liu, Z.; Frommer, W.B.; Ma, J.F.; Chen, L.Q.; et al. Simultaneous changes in seed size, oil content and protein content driven by selection of SWEET homologues during soybean domestication. Natl. Sci. Rev. 2020, 7, 1776–1786. [Google Scholar] [CrossRef]
- Zhu, W.W.; Yang, C.; Yong, B.; Wang, Y.; Li, B.B.; Gu, Y.Z.; Wei, S.M.; An, Z.H.; Sun, W.K.; Qiu, L.J.; et al. An enhancing effect attributed to a nonsynonymous mutation in SOYBEAN SEED SIZE 1, a SPINDLY-like gene, is exploited in soybean domestication and improvement. New Phytol. 2022, 236, 1375–1392. [Google Scholar] [CrossRef]
- Gu, Y.Z.; Li, W.; Jiang, H.W.; Wang, Y.; Gao, H.H.; Liu, M.; Chen, Q.S.; Lai, Y.C.; He, C.Y. Differential expression of a WRKY gene between wild and cultivated soybeans correlates to seed size. J. Exp. Bot. 2017, 68, 2717–2729. [Google Scholar] [CrossRef]
- Du, D.; Gao, X.; Geng, J.; Li, Q.; Li, L.; Lv, Q.; Li, X. Identification of key proteins and networks related to grain development in wheat (Triticum aestivum L.) by Comparative Transcription and Proteomic Analysis of Allelic Variants in TaGW2-6A. Front. Plant Sci. 2016, 7, 922. [Google Scholar] [CrossRef]
- Gao, H.; Wang, Y.; Li, W.; Gu, Y.; Lai, Y.; Bi, Y.; He, C. Transcriptomic comparison reveals genetic variation potentially underlying seed developmental evolution of soybeans. J Exp. Bot. 2018, 69, 5089–5104. [Google Scholar] [CrossRef]
- Ma, C.; Zhou, J.; Chen, G.; Bian, Y.; Lv, D.; Li, X.; Wang, Z.; Yan, Y. iTRAQ-based quantitative proteome and phosphoprotein characterization reveals the central metabolism changes involved in wheat grain development. BMC Genom. 2014, 15, 1029. [Google Scholar] [CrossRef]
- Yu, T.; Li, G.; Dong, S.; Liu, P.; Zhang, J.; Zhao, B. Proteomic analysis of maize grain development using iTRAQ reveals temporal programs of diverse metabolic processes. BMC Plant Biol. 2016, 16, 241. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, Y.; Wang, Q.; Du, C.; Xiong, W.; Li, X.; Zhu, S.; Li, Y. iTRAQ-based proteomics analysis and network integration for kernel tissue development in maize. Int. J. Mol. Sci. 2017, 18, 1840. [Google Scholar] [CrossRef]
- Zeng, D.; Cui, J.; Yin, Y.; Zhang, M.; Shan, S.; Liu, M.Y.; Cheng, D.; Lu, W.; Sun, Y. Proteomic analysis in different development stages on SP0 generation of rice seeds after space flight. Life Sci. Space Res. 2020, 26, 34–45. [Google Scholar] [CrossRef]
- Qing, D.; Zhou, Y.; Pan, Y.; Yang, X.; Li, J.; Zhou, W.; Liang, H.; Chen, W.; Chen, L.; Lu, C.; et al. TMT-based quantitative proteomic analysis of indica rice cultivars reveals that novel components of the signaling pathways might play a role in grain length regulation. J. Proteom. 2023, 270, 104745. [Google Scholar] [CrossRef]
- Han, X.; Li, J.; Zhao, Y.; Zhang, Z.; Jiang, H.; Wang, J.; Feng, X.; Zhang, Y.; Du, Z.; Wu, X.; et al. Integrated transcriptomic and proteomic characterization of a chromosome segment substitution line reveals a new regulatory network controlling the seed storage profile of soybean. Food Energy Secur. 2022, 11, e381. [Google Scholar] [CrossRef]
- Jin, X.; Zhai, H.; Wang, P.; Zhang, X.; Wu, X.; Zhang, H.; Chen, S.; Huang, Z. Physiological and omics analysis of maize inbred lines during late grain development. Genes Genom. 2022, 44, 993–1006. [Google Scholar] [CrossRef]
- Li, N.; Xu, R.; Li, Y.H. Molecular networks of seed size control in plants. Annu. Rev. Plant Biol. 2019, 70, 435–463. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Liu, X.; Pai, Q.; Wang, Y.; Wu, X. Molecular network for regulation of seed size in plants. Int. J. Mol. Sci. 2023, 24, 10666. [Google Scholar] [CrossRef]
- Duan, P.; Rao, Y.; Zeng, D.; Yang, Y.; Xu, R.; Zhang, B.; Dong, G.; Qian, Q.; Li, Y. SMALL GRAIN 1, which encodes a mitogen-activated protein kinase kinase 4, influences grain size in rice. Plant J. 2014, 77, 547–557. [Google Scholar] [CrossRef]
- Liu, S.; Hua, L.; Dong, S.; Chen, H.; Zhu, X.; Jiang, J.; Zhang, F.; Li, Y.; Fang, X.; Chen, F. OsMAPK6, a mitogen-activated protein kinase, influences rice grain size and biomass production. Plant J. 2015, 84, 672–681. [Google Scholar] [CrossRef]
- Xu, R.; Duan, P.; Yu, H.; Zhou, Z.; Zhang, B.; Wang, R.; Li, J.; Zhang, G.; Zhuang, S.; Lyu, J.; et al. Control of grain size and weight by the OsMKKK10-OsMKK4-OsMAPK6 Signaling pathway in rice. Mol. Plant 2018, 11, 860–873. [Google Scholar] [CrossRef]
- Xu, R.; Yu, H.; Wang, J.; Duan, P.; Zhang, B.; Li, J.; Li, Y.; Xu, J.; Lyu, J.; Li, N.; et al. A mitogen-activated protein kinase phosphatase influences grain size and weight in rice. Plant J. 2018, 95, 937–946. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, H.; Su, J.; Wang, H.; Zhu, Q.; Liu, Y.; Xu, J.; Lukowitz, W.; Zhang, S. Maternal control of embryogenesis by MPK6 and its upstream MKK4/MKK5 in Arabidopsis. Plant J. 2017, 92, 1005–1019. [Google Scholar] [CrossRef]
- Guo, T.; Chen, K.; Dong, N.Q.; Shi, C.L.; Ye, W.W.; Gao, J.P.; Shan, J.X.; Lin, H.X. GRAIN SIZE AND NUMBER1 negatively regulates the OsMKKK10-OsMKK4-OsMPK6 cascade to coordinate the trade-off between grain number per panicle and grain size in rice. Plant Cell 2018, 30, 871–888. [Google Scholar] [CrossRef]
- Wu, X.; Cai, X.; Zhang, B.; Wu, S.; Wang, R.; Li, N.; Li, Y.; Sun, Y.; Tang, W. ERECTA regulates seed size independently of its intracellular domain via MAPK-DA1-UBP15 signaling. Plant Cell 2022, 34, 3773–3789. [Google Scholar] [CrossRef]
- Xiao, W.; Hu, S.; Zou, X.; Cai, R.; Liao, R.; Lin, X.; Yao, R.; Guo, X. Lectin receptor-like kinase LecRK-VIII.2 is a missing link in MAPK signaling-mediated yield control. Plant Physiol. 2021, 187, 303–320. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Li, X.; Zhou, W.; Ren, Y.; Wang, Z.; Liu, Z.; Tang, J.; Tong, H.; Fang, J.; Bu, Q. Transcription factor OsWRKY53 positively regulates brassinosteroid signaling and plant architecture. Plant Physiol. 2017, 175, 1337–1349. [Google Scholar] [CrossRef]
- Liu, Z.; Mei, E.; Tian, X.; He, M.; Tang, J.; Xu, M.; Liu, J.; Song, L.; Li, X.; Wang, Z.; et al. OsMKKK70 regulates grain size and leaf angle in rice through the OsMKK4-OsMAPK6-OsWRKY53 signaling pathway. J. Integr. Plant Biol. 2021, 63, 2043–2057. [Google Scholar] [CrossRef]
- Baez, L.A.; Tichá, T.; Hamann, T. Cell wall integrity regulation across plant species. Plant Mol. Biol. 2022, 109, 483–504. [Google Scholar] [CrossRef]
- Gigli-Bisceglia, N.; Engelsdorf, T.; Hamann, T. Plant cell wall integrity maintenance in model plants and crop species-relevant cell wall components and underlying guiding principles. Cell Mol. Life Sci. 2020, 77, 2049–2077. [Google Scholar] [CrossRef] [PubMed]
- Iwai, H. Virtual issue: Cell wall functions in plant growth and environmental responses. J. Plant Res. 2021, 134, 1155–1158. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, M.; Viana, V.E.; Banfi, C.; Cassina, V.; Corti, R.; Herrera-Ubaldo, H.; Babolin, N.; Guazzotti, A.; Kiegle, E.; Gregis, V.; et al. Cell wall modifications by α-XYLOSIDASE1 are required for control of seed and fruit size in Arabidopsis. J. Exp. Bot. 2022, 73, 1499–1515. [Google Scholar] [CrossRef]
- Li, B.; Liu, H.; Zhang, Y.; Kang, T.; Zhang, L.; Tong, J.; Xiao, L.; Zhang, H. Constitutive expression of cell wall invertase genes increases grain yield and starch content in maize. Plant Biotechnol. J. 2013, 11, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wilkinson, L.G.; Aubert, M.K.; Houston, K.; Shirley, N.J.; Tucker, M.R. Ovule cell wall composition is a maternal determinant of grain size in barley. New Phytol. 2023, 237, 2136–2147. [Google Scholar] [CrossRef]
- Mira, J.P.; Arenas-M, A.; Calderini, D.F.; Canales, J. Integrated transcriptome analysis identified key expansin genes associated with wheat cell wall, grain weight and yield. Plants 2023, 12, 2868. [Google Scholar] [CrossRef]
- Jin, Y.; Ni, D.A.; Ruan, Y.L. Posttranslational elevation of cell wall invertase activity by silencing its inhibitor in tomato delays leaf senescence and increases seed weight and fruit hexose level. Plant Cell 2009, 21, 2072–2089. [Google Scholar] [CrossRef]
- Ishimaru, K.; Hirotsu, N.; Madoka, Y.; Murakami, N.; Hara, N.; Onodera, H.; Kashiwagi, T.; Ujiie, K.; Shimizu, B.; Onishi, A.; et al. Loss of function of the IAA-glucose hydrolase gene TGW6 enhances rice grain weight and increases yield. Nat. Genet. 2013, 45, 707–711. [Google Scholar] [CrossRef]
- Sestili, F.; Pagliarello, R.; Zega, A.; Saletti, R.; Pucci, A.; Botticella, E.; Masci, S.; Tundo, S.; Moscetti, I.; Foti, S.; et al. Enhancing grain size in durum wheat using RNAi to knockdown GW2 genes. Theor. Appl. Genet. 2019, 132, 419–429. [Google Scholar] [CrossRef]
- Wang, S.; Yang, H.; Mei, J.; Liu, X.; Wen, Z.; Zhang, L.; Xu, Z.; Zhang, B.; Zhou, Y. Rice homeobox protein KNAT7 integrates the pathways regulating cell expansion and wall stiffness. Plant Physiol. 2019, 181, 669–682. [Google Scholar] [CrossRef]
- Dong, N.Q.; Sun, Y.; Guo, T.; Shi, C.L.; Zhang, Y.M.; Kan, Y.; Xiang, Y.H.; Zhang, H.; Yang, Y.B.; Li, Y.C.; et al. UDP-glucosyltransferase regulates grain size and abiotic stress tolerance associated with metabolic flux redirection in rice. Nat. Commun. 2020, 11, 2629. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.J.; Zhang, H.P.; Cao, J.J.; Zhu, X.F.; Wang, S.X.; Jiang, H.; Wu, Z.Y.; Lu, J.; Chang, C.; Sun, G.L.; et al. Characterization of an IAA-glucose hydrolase gene TaTGW6 associated with grain weight in common wheat (Triticum aestivum L.). Mol. Breed. 2016, 36, 25. [Google Scholar] [CrossRef]
- Terao, A.; Hyodo, H.; Satoh, S.; Iwai, H. Changes in the distribution of cell wall polysaccharides in early fruit pericarp and ovule, from fruit set to early fruit development, in tomato (Solanum lycopersicum). J. Plant Res. 2013, 126, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Sun, X.; Liu, B.; Kong, F.; Pan, X.; Zhang, H. A polygalacturonase gene PG031 regulates seed coat permeability with a pleiotropic effect on seed weight in soybean. Theor. Appl. Genet. 2022, 135, 1603–1618. [Google Scholar] [CrossRef]
- Eshed, Y.; Zamir, D. A genomic library of Lycopersicon pennellii in L. esculentum: A tool for fine mapping of genes. Euphytica 1994, 79, 175–179. [Google Scholar] [CrossRef]
- Pillen, K.; Zacharias, A.; Léon, J. Advanced backcross QTL analysis in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 107, 340–352. [Google Scholar] [CrossRef]
- Shen, G.J.; Xing, Y.Z. Two novel QTLs for heading date are identified using a set of chromosome segment substitution lines in rice (Oryza sativa L.). J. Genet. Genom. 2014, 41, 659–662. [Google Scholar] [CrossRef]
- Zhu, D.; Li, X.M.; Wang, Z.M.; You, C.Y.; Nie, X.H.; Sun, J.; Zhang, X.L.; Zhang, D.W.; Lin, Z.X. Genetic dissection of an allotetraploid interspecific CSSLs guides interspecific genetics and breeding in cotton. BMC Genom. 2020, 21, 431. [Google Scholar] [CrossRef]
- Xin, D.W.; Qi, Z.M.; Jiang, H.W.; Hu, Z.B.; Zhu, R.S.; Hu, J.H.; Han, H.Y.; Hu, G.H.; Liu, C.Y.; Chen, Q.S. QTL Location and epistatic effect analysis of 100-seed weight using wild soybean (Glycine soja Sieb. & Zucc.) chromosome segment substitution lines. PLoS ONE 2016, 11, e0149380. [Google Scholar]
- Zheng, H.Y.; Hou, L.L.; Xie, J.G.; Cao, F.B.; Wei, R.; Yang, M.L.; Qi, Z.M.; Zhu, R.S.; Zhang, Z.G.; Xin, D.W.; et al. Construction of chromosome segment substitution lines and inheritance of seed-pod characteristics in wild soybean. Front. Plant Sci. 2022, 13, 869455. [Google Scholar] [CrossRef]
- Clemente, T.E.; Cahoon, E.B. Soybean oil: Genetic approaches for modification of functionality and total content. Plant Physiol. 2009, 151, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.M.; Li, W.T.; Zeng, R.Z.; Zhang, Z.M.; Zhang, G.Q. Identification of QTLs on substituted segments in single segment substitution lines of rice. J. Genet. Genom. 2004, 31, 1395–1400. [Google Scholar]
- Thomson, M. Substitution mapping of dth1.1, a flowering-time quantitative trait locus (QTL) associated with transgressive variation in rice, reveals multiple sub-QTL. Genetics 2006, 172, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Eshed, Y.; Zamir, D. An introgression line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated QTL. Genetics 1995, 141, 1147–1162. [Google Scholar] [CrossRef]
- Yang, H.Y.; Wang, W.B.; He, Q.Y.; Xiang, S.H.; Gai, J.Y. Identifying a wild allele conferring small seed size, high protein content and low oil content using chromosome segment substitution lines in soybean. Theor. Appl. Genet. 2019, 132, 2793–2807. [Google Scholar] [CrossRef]
- Deshmukh, R.; Sonah, H.; Patil, G.; Chen, W.; Prince, S.; Mutava, R.; Vuong, T.; Valliyodan, B.; Nguyen, H.T. Integrating omic approaches for abiotic stress tolerance in soybean. Front. Plant Sci. 2014, 5, 244. [Google Scholar] [CrossRef]
- Peng, Z.; He, S.; Gong, W.; Xu, F.; Pan, Z.; Jia, Y.; Geng, X.; Du, X. Integration of proteomic and transcriptomic profiles reveals multiple levels of genetic regulation of salt tolerance in cotton. BMC Plant Biol. 2018, 18, 128. [Google Scholar] [CrossRef] [PubMed]
- Shah, T.; Xu, J.; Zou, X.; Cheng, Y.; Nasir, M.; Zhang, X. Omics approaches for engineering wheat production under abiotic stresses. Int. J. Mol. Sci. 2018, 19, 2390. [Google Scholar] [CrossRef]
- Xu, Y.; Zhao, Y.; Wang, X.; Ma, Y.; Li, P.; Yang, Z.; Zhang, X.; Xu, C.; Xu, S. Incorporation of parental phenotypic data into multi-omic models improves prediction of yield-related traits in hybrid rice. Plant Biotechnol. J. 2021, 19, 261–272. [Google Scholar] [CrossRef]
- Zhou, Q.; Fu, Z.; Liu, H.; Wang, J.; Guo, Z.; Zhang, X.; Tian, R.; Liu, Y.; Qu, J.; Li, W.; et al. Mining novel kernel size-related genes by pQTL mapping and multi-omics integrative analysis in developing maize kernels. Plant Biotechnol. J. 2021, 19, 1489–1491. [Google Scholar] [CrossRef]
- Liu, Y.; Yi, C.; Liu, Q.; Wang, C.; Wang, W.; Han, F.; Hu, X. Multi-omics profiling identifies candidate genes controlling seed size in peanut. Plants 2022, 11, 3276. [Google Scholar] [CrossRef]
- Wang, Z.; Tao, S.; Liu, S.; Jia, M.; Cui, D.; Sun, G.; Deng, Z.; Wang, F.; Kong, X.; Fu, M.; et al. A multi-omics approach for rapid identification of large genomic lesions at the wheat dense spike (wds) locus. Front. Plant Sci. 2022, 13, 850302. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Jiao, F.; Wang, Q.; Zhang, E.; Song, X.; Pei, Y.; Li, J.; Zhao, M.; Guo, X. Multiomics comparative analysis of the maize large grain mutant tc19 identified pathways related to kernel development. BMC Genom. 2023, 24, 537. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, Y.; Zhang, L.; Yang, Y.; Wu, Q.; Hu, G.; Wang, W.; Li, J.; Huang, Z. Integrated transcriptomic and proteomic analysis of a cytoplasmic male sterility line and associated maintainer line in soybean. Front. Plant Sci. 2023, 14, 1098125. [Google Scholar] [CrossRef]
- Li, J.; Chen, L.; Zhi, X.; Wang, J.; Lu, Y.; Tian, Z.; Wu, M.; Shan, Y.; Chen, H.; Liao, W.; et al. Integrated transcriptome and proteome analysis reveals molecular responses of soybean anther under high-temperature stress. Front. Plant Sci. 2023, 14, 1187922. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, S. Mitogen-activated protein kinase cascades in plant signaling. J. Integr. Plant Biol. 2022, 64, 301–341. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, S. Mitogen-activated protein kinase cascades in signaling plant growth and development. Trends Plant Sci. 2015, 20, 56–64. [Google Scholar] [CrossRef]
- Guo, T.; Lu, Z.Q.; Shan, J.X.; Ye, W.W.; Dong, N.Q.; Lin, H.X. ERECTA1 acts upstream of the OsMKKK10-OsMKK4-OsMPK6 cascade to control spikelet number by regulating cytokinin metabolism in rice. Plant Cell 2020, 32, 2763–2779. [Google Scholar] [CrossRef] [PubMed]
- Mao, G.; Meng, X.; Liu, Y.; Zheng, Z.; Chen, Z.; Zhang, S. Phosphorylation of a WRKY transcription factor by two pathogen-responsive MAPKs drives phytoalexin biosynthesis in Arabidopsis. Plant Cell 2011, 23, 1639–1653. [Google Scholar] [CrossRef]
- Du, L.; Li, N.; Chen, L.; Xu, Y.; Li, Y.; Zhang, Y.; Li, C.; Li, Y. The ubiquitin receptor DA1 regulates seed and organ size by modulating the stability of the ubiquitin-specific protease UBP15/SOD2 in Arabidopsis. Plant Cell 2014, 26, 665–677. [Google Scholar] [CrossRef]
- Dong, H.; Dumenil, J.; Lu, F.H.; Na, L.; Vanhaeren, H.; Naumann, C.; Klecker, M.; Prior, R.; Smith, C.; McKenzie, N.; et al. Ubiquitylation activates a peptidase that promotes cleavage and destabilization of its activating E3 ligases and diverse growth regulatory proteins to limit cell proliferation in Arabidopsis. Genes Dev. 2017, 31, 197–208. [Google Scholar] [CrossRef]
- Kumar, M.; Campbell, L.; Turner, S. Secondary cell walls: Biosynthesis and manipulation. J. Exp. Bot. 2016, 67, 515–531. [Google Scholar] [CrossRef]
- Zhong, R.; Ye, Z.H. Regulation of cell wall biosynthesis. Curr. Opin. Plant Biol. 2007, 10, 564–752. [Google Scholar] [CrossRef]
- Lin, F.; Manisseri, C.; Fagerström, A.; Peck, M.L.; Vega-Sánchez, M.E.; Williams, B.; Chiniquy, D.M.; Saha, P.; Pattathil, S.; Conlin, B.; et al. Cell wall composition and candidate biosynthesis gene expression during rice development. Plant Cell Physiol. 2016, 57, 2058–2075. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Yu, H.X.; Ye, W.W.; Shan, J.X.; Dong, N.Q.; Guo, T.; Kan, Y.; Xiang, Y.H.; Zhang, H.; Yang, Y.B.; et al. A rice QTL GS3.1 regulates grain size through metabolic-flux distribution between flavonoid and lignin metabolons without affecting stress tolerance. Commun. Biol. 2021, 4, 1171. [Google Scholar] [CrossRef] [PubMed]
- Kohlberger, M.; Thalhamer, T.; Weiss, R.; Tenhaken, R. Arabidopsis MAP-Kinase 3 phosphorylates UDP-glucose dehydrogenase: A key enzyme providing UDP-sugar for cell wall biosynthesis. Plant Mol. Biol. Rep. 2018, 36, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Y. MPK4 negatively regulates the l-arabinose synthesis of cell wall in Arabidopsis. Biochem. Biophys. Res. Commun. 2022, 613, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote) omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Wu, T.Z.; Hu, E.Q.; Xu, S.B.; Chen, M.J.; Guo, P.F.; Dai, Z.Z.; Feng, T.Z.; Zhou, L.; Tang, W.L.; Zhan, L.; et al. ClusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Galensa, K. ggplot2: Elegant graphics for data analysis (2nd ed.). Comput. Rev. 2017, 58, 457–458. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, S.; Yu, Z.; Du, F.; Cao, F.; Yang, M.; Liu, C.; Qi, Z.; Chen, Q.; Zou, J.; Wang, J. Integrated Transcriptomic and Proteomic Characterization of a Chromosome Segment Substitution Line Reveals the Regulatory Mechanism Controlling the Seed Weight in Soybean. Plants 2024, 13, 908. https://doi.org/10.3390/plants13060908
Wei S, Yu Z, Du F, Cao F, Yang M, Liu C, Qi Z, Chen Q, Zou J, Wang J. Integrated Transcriptomic and Proteomic Characterization of a Chromosome Segment Substitution Line Reveals the Regulatory Mechanism Controlling the Seed Weight in Soybean. Plants. 2024; 13(6):908. https://doi.org/10.3390/plants13060908
Chicago/Turabian StyleWei, Siming, Zhenhai Yu, Fangfang Du, Fubin Cao, Mingliang Yang, Chunyan Liu, Zhaoming Qi, Qingshan Chen, Jianan Zou, and Jinhui Wang. 2024. "Integrated Transcriptomic and Proteomic Characterization of a Chromosome Segment Substitution Line Reveals the Regulatory Mechanism Controlling the Seed Weight in Soybean" Plants 13, no. 6: 908. https://doi.org/10.3390/plants13060908