Conjunctive Analyses of BSA-Seq and BSR-Seq to Reveal the Molecular Pathway of Leafy Head Formation in Chinese Cabbage

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Bulked-Segregant Analysis
2.1.1. Sequencing Data Analysis of Four DNA Bulks
2.1.2. Association Analysis of BSA-Seq Data
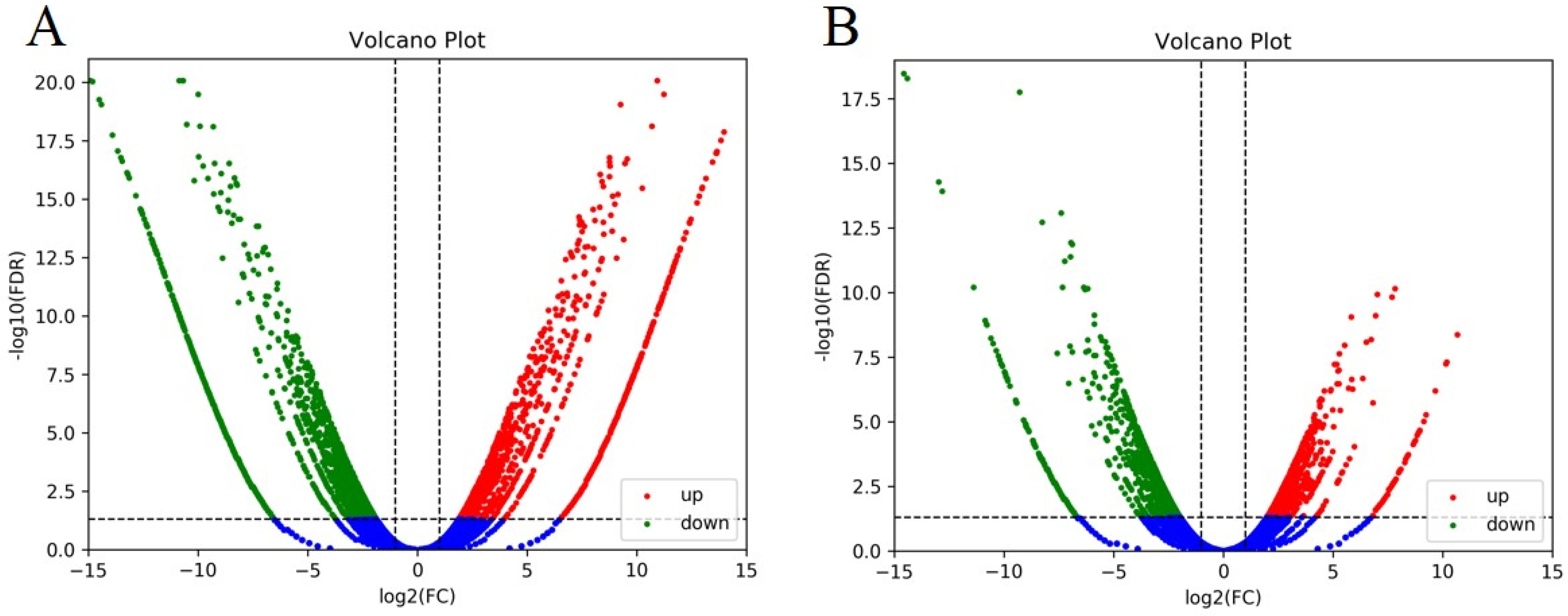
2.2. Bulked Segregant RNA-Seq Analysis
2.2.1. Sequencing Data Analysis of Four cDNA Bulks
2.2.2. Comparison of Gene Expression Files between Two Set Pairs of cDNA Sequencing Bulks
2.2.3. Association Analysis of BSR-Seq Data
2.3. Validation of Quantitative Real Time PCR for Key Genes Associated with Leafy Head Formation in Chinese Cabbage
3. Discussion
4. Materials and Methods
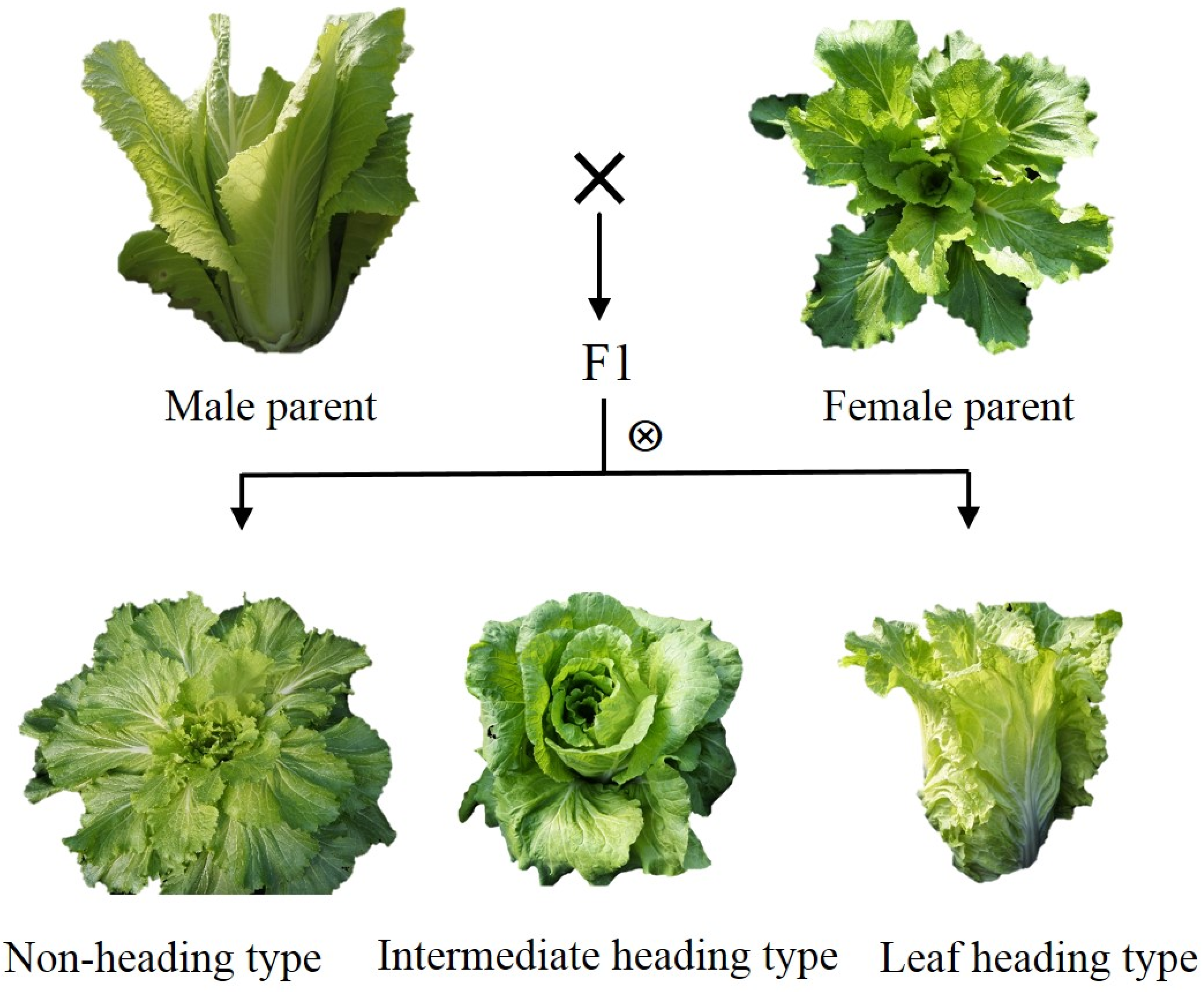
4.1. Plant Materials
4.2. Methods of Bulked-Segregant Analysis
4.2.1. The Construction of Illumina Library of BSA-Seq
4.2.2. Alignment with the Reference Genome and Variant Calling
4.2.3. Mapping of Candidate Genomic Regions by Association Analysis
4.3. Bulked Segregant RNA-Seq Analysis
4.3.1. cDNA Library Preparation of BSR-Seq
4.3.2. Analysis of BSR-Seq Data
4.4. Real-Time Quantitative PCR for Validation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gómez-Campo, C. Biology of Brassica Coenospecies; Elsevier: New York, NY, USA, 1999; Volume 4, pp. 33–52. ISBN 0-444-50278-5. [Google Scholar]
- He, Y.K.; Xue, W.X.; Sun, Y.D.; Yu, X.H.; Liu, P.L. Leafy head formation of the progenies of transgenic plants of Chinese cabbage with exogenous auxin genes. Cell Res. 2000, 10, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Dixon, G.R. Vegetable Brassicas and Related Crucifers; CABI: London, UK, 2007; pp. 1–35. ISBN 978-0-85199-395-9. [Google Scholar]
- Gao, L.W.; Lyu, S.W.; Tang, J.; Zhou, D.Y.; Bonnema, G.; Xiao, D.; Hou, X.L.; Zhang, C.W. Genome-wide analysis of auxin transport genes identifies the hormone responsive patterns associated with leafy head formation in Chinese cabbage. Sci. Rep. 2017, 7, 42229. [Google Scholar] [CrossRef] [PubMed]
- Ito, H. Effect of temperature and photoperiod on head formation of leafy head of Chinese cabbage. J. Hort. Assoc. Jpn. 1957, 26, 154–162. [Google Scholar] [CrossRef]
- Mao, Y.; Wu, F.; Yu, X.; Bai, J.; Zhong, W.; He, Y. MicroRNA319a-targeted Brassica rapa ssp. pekinensis TCP genes modulate head shape in Chinese cabbage by differential cell division arrest in leaf regions. Plant Physiol. 2014, 164, 710–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.; Sun, R.; Hou, X.; Zheng, H.; Zhang, F.; Zhang, Y.; Liu, B.; Liang, J.; Zhuang, M.; Liu, Y.; et al. Subgenome parallel selection is associated with morphotype diversification and convergent crop domestication in Brassica rapa and Brassica oleracea. Nat. Genet. 2016, 48, 1218–1224. [Google Scholar] [CrossRef] [PubMed]
- Gu, A.; Meng, C.; Chen, Y.; Wei, L.; Dong, H.; Lu, Y.; Wang, Y.; Chen, X.; Zhao, J.; Shen, S. Coupling Seq-BSA and RNA-Seq analyses reveal the molecular pathway and genes associated with heading type in Chinese cabbage. Front. Genet. 2017, 8, 176. [Google Scholar] [CrossRef] [Green Version]
- Woodward, A.W.; Bartel, B. Auxin: Regulation, action, and interaction. Ann. Bot. 2005, 95, 707–735. [Google Scholar] [CrossRef] [Green Version]
- Monfared, M.M.; Carles, C.C.; Rossignol, P.; Pires, H.R.; Fletcher, J.C. The ULT1 and ULT2 trxG genes play overlapping roles in Arabidopsis development and gene regulation. Mol. Plant 2013, 6, 1564–1579. [Google Scholar] [CrossRef] [Green Version]
- Tsukaya, H. Mechanism of leaf-shape determination. Annu. Rev. Plant Biol. 2006, 57, 477–496. [Google Scholar] [CrossRef]
- Liu, Z.; Jia, L.; Mao, Y.; He, Y. Classification and quantification of leaf curvature. J. Exp. Bot. 2010, 61, 2757–2767. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wu, F.; Bai, J.; He, Y. BrpSPL9 (Brassica rapa ssp. pekinensis SPL9) controls the earliness of heading time in Chinese cabbage. Plant Biotechnol. J. 2014, 12, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Gao, L.; Liu, W.; Song, L.; Xiao, D.; Liu, T.; Hou, X.; Zhang, C. Transcription coactivator ANGUSTIFOLIA3 (AN3) regulates leafy head formation in Chinese cabbage. Front. Plant Sci. 2019, 10, 520. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Han, M.; Liu, Y.; Lin, X.; Liu, X.; Zhu, H.; He, Y.; Zhang, Q.; Liu, J. Whole-genome resequencing from bulked-segregant analysis reveals gene set based association analyses for the Vibrio anguillarum resistance of turbot (Scophthalmus maximus). Fish Shellfish Immunol. 2019, 88, 76–83. [Google Scholar] [CrossRef]
- Pasam, R.K.; Sharma, R.; Malosetti, M.; Van Eeuwijk, F.A.; Haseneyer, G.; Kilian, B.; Graner, A. Genome-wide association studies for agronomical traits in a world wide spring barley collection. BMC Plant Biol. 2011, 12, 16. [Google Scholar] [CrossRef] [Green Version]
- Burke, J.M.; Burger, J.C.; Chapman, M.A. Crop evolution: From genetics to genomics. Curr. Opin. Genet. Dev. 2007, 17, 525–532. [Google Scholar] [CrossRef]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef]
- Livaja, M.; Wang, Y.; Wieckhorst, S.; Haseneyer, G.; Seidel, M.; Hahn, V.; Knapp, S.J.; Taudien, S.; Schön, C.-C.; Bauer, E. BSTA: A targeted approach combines bulked segregant analysis with next-generation sequencing and de novo transcriptome assembly for SNP discovery in sunflower. BMC Genom. 2013, 14, 628. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Yeh, C.-T.; Tang, H.M.; Nettleton, D.; Schnable, P.S. Gene mapping via bulked segregant RNA-Seq (BSR-Seq). PLoS ONE 2012, 7, e36406. [Google Scholar] [CrossRef] [Green Version]
- Gross, S.D.; Anderson, R.A. Casein kinase I: Spatial organization and positioning of a multifunctional protein kinase family. Cell. Signal. 1998, 10, 699–711. [Google Scholar] [CrossRef]
- De Groot, R.; Den Hertog, J.; Vandenheede, J.; Goris, J.; Sassone-Corsi, P. Multiple and cooperative phosphorylation events regulate the CREM activator function. EMBO J. 1993, 12, 3903–3911. [Google Scholar] [CrossRef] [PubMed]
- Knippschild, U.; Gocht, A.; Wolff, S.; Huber, N.; Löhler, J.; Stöter, M. The casein kinase 1 family: Participation in multiple cellular processes in eukaryotes. Cell. Signal. 2005, 17, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.-T.; Dai, C.; Liu, H.-T.; Xue, H.-W. Arabidopsis casein kinase1 proteins CK1.3 and CK1.4 phosphorylate cryptochrome2 to regulate blue light signaling. Plant Cell 2013, 25, 2618–2632. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Xu, Z.H.; Luo, D.; Xue, H.W. Roles of OsCKI1, a rice casein kinase I, in root development and plant hormone sensitivity. Plant J. 2003, 36, 189–202. [Google Scholar] [CrossRef]
- Zhang, J.-H.; Sun, H.-L.; Zhao, X.-Y.; Liu, X.-M. Arabidopsis casein kinase 1-like 8 enhances NaCl tolerance, early flowering, and the expression of flowering-related genes. J. Plant Interact. 2016, 11, 138–145. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.-T.; Xue, H.-W. Casein kinase 1 Regulates ethylene synthesis by phosphorylating and promoting the turnover of ACS5. Cell Rep. 2014, 9, 1692–1702. [Google Scholar] [CrossRef] [Green Version]
- Polko, J.K.; Van Zanten, M.; Van Rooij, J.A.; Marée, A.F.M.; Voesenek, L.A.C.J.; Peeters, A.J.M.; Pierik, R. Ethylene-induced differential petiole growth in Arabidopsis thaliana involves local microtubule reorientation and cell expansion. New Phytol. 2012, 193, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Fiorani, F.; Bögemann, G.M.; Visser, E.J.W.; Lambers, H.; Voesenek, L.A.C.J. Ethylene emission and responsiveness to applied ethylene vary among Poa species that inherently differ in leaf elongation rates. Plant Physiol. 2002, 129, 1382–1390. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Reid, D.M. The role of endogenous ethylene in the expansion of Helianthus annuus leaves. Can. J. Bot. 1997, 75, 501–508. [Google Scholar] [CrossRef]
- Vogel, J.P.; Woeste, K.E.; Theologis, A.; Kieber, J.J. Recessive and dominant mutations in the ethylene biosynthetic gene ACS5 of Arabidopsis confer cytokinin insensitivity and ethylene overproduction, respectively. Proc. Natl. Acad. Sci. USA 1998, 95, 4766–4771. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.-C.; Shuai, B.; Springer, P.S. The Arabidopsis LATERAL ORGAN BOUNDARIES—Domain gene ASYMMETRIC LEAVES2 functions in the repression of KNOX gene expression and in adaxial-abaxial patterning. Plant Cell 2003, 15, 2241–2252. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Xu, Y.; Dong, A.; Sun, Y.; Pi, L.; Xu, Y.; Huang, H. Novel as1 and as2 defects in leaf adaxial-abaxial polarity reveal the requirement for ASYMMETRIC LEAVES1 and 2 and ERECTA functions in specifying leaf adaxial identity. Development 2003, 130, 4097–4107. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T.; Nukazuka, A.; Tsukaya, H. Leaf adaxial–abaxial polarity specification and lamina outgrowth: Evolution and development. Plant Cell Physiol. 2012, 53, 1180–1194. [Google Scholar] [CrossRef] [PubMed]
- Candela, H.; Johnston, R.; Gerhold, A.; Foster, T.; Hake, S. The milkweed pod1 gene encodes a KANADI protein that is required for abaxial/adaxial patterning in maize leaves. Plant Cell 2008, 20, 2073–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.-J.; Kayum, M.A.; Thamilarasan, S.K.; Nath, U.K.; Park, J.-I.; Chung, M.-Y.; Hur, Y.; Nou, I.-S. Molecular characterisation and expression profiling of calcineurin B-like (CBL) genes in Chinese cabbage under abiotic stresses. Funct. Plant Biol. 2017, 44, 739–750. [Google Scholar] [CrossRef]
- Meena, M.K.; Ghawana, S.; Sardar, A.; Dwivedi, V.; Khandal, H.; Roy, R.; Chattopadhyay, D. Investigation of genes encoding calcineurin B-like protein family in legumes and their expression analyses in chickpea (Cicer arietinum L.). PLoS ONE 2015, 10, e0123640. [Google Scholar] [CrossRef]
- Kolukisaoglu, Ü.; Weinl, S.; Blazevic, D.; Batistic, O.; Kudla, J. Calcium Sensors and Their Interacting Protein Kinases: Genomics of the Arabidopsis and Rice CBL-CIPK Signaling Networks. Plant Physiol. 2004, 134, 43–58. [Google Scholar] [CrossRef] [Green Version]
- Kudla, J.; Xu, Q.; Harter, K.; Gruissem, W.; Luan, S. Genes for calcineurin B-like proteins in Arabidopsis are differentially regulated by stress signals. Proc. Natl. Acad. Sci. USA 1999, 96, 4718–4723. [Google Scholar] [CrossRef] [Green Version]
- Zik, M.; Arazi, T.; Snedden, W.A.; Fromm, H. Two isoforms of glutamate decarboxylase in Arabidopsis are regulated by calcium/calmodulin and differ in organ distribution. Plant Mol. Biol. 1998, 37, 967–975. [Google Scholar] [CrossRef]
- Zhang, F.; Li, L.; Jiao, Z.; Chen, Y.; Liu, H.; Chen, X.; Fu, J.; Wang, G.; Zheng, J. Characterization of the calcineurin B-Like (CBL) gene family in maize and functional analysis of ZmCBL9 under abscisic acid and abiotic stress treatments. Plant Sci. 2016, 253, 118–129. [Google Scholar] [CrossRef]
- Abbasi, F.; Onodera, H.; Toki, S.; Tanaka, H.; Komatsu, S. OsCDPK13, a calcium-dependent protein kinase gene from rice, is induced by cold and gibberellin in rice leaf sheath. Plant Mol. Biol. 2004, 55, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Nelissen, H.; Rymen, B.; Jikumaru, Y.; Demuynck, K.; Van Lijsebettens, M.; Kamiya, Y.; Inzé, D.; Beemster, G.T.S. A local maximum in gibberellin levels regulates maize leaf growth by spatial control of cell division. Curr. Biol. 2012, 22, 1183–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olszewski, N.; Sun, T.P.; Gubler, F. Gibberellin signaling: Biosynthesis, catabolism, and response pathways. Plant Cell 2002, 14, 61–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Tang, D.; Shen, Y.; Qin, B.; Hong, L.; You, A.; Li, M.; Wang, X.; Yu, H.; Gu, M.; et al. Activation of gibberellin 2-oxidase 6 decreases active gibberellin levels and creates a dominant semi-dwarf phenotype in rice (Oryza sativa L.). J. Genet. Genom. 2010, 37, 23–36. [Google Scholar] [CrossRef]
- Magome, H.; Yamaguchi, S.; Hanada, A.; Kamiya, Y.; Oda, K. dwarf and delayed-flowering 1, a novel Arabidopsis mutant deficient in gibberellin biosynthesis because of overexpression of a putative AP2 transcription factor. Plant J. 2004, 37, 720–729. [Google Scholar] [CrossRef]
- Per, T.S.; Khan, M.I.R.; Anjum, N.A.; Masood, A.; Hussain, S.J.; Khan, N.A. Jasmonates in plants under abiotic stresses: Crosstalk with other phytohormones matters. Environ. Exp. Bot. 2018, 145, 104–120. [Google Scholar] [CrossRef]
- Wasternack, C.; Strnad, M. Jasmonate signaling in plant stress responses and development—Active and inactive compounds. New Biotechnol. 2016, 33, 604–613. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, F.; Melotto, M.; Yao, J.; He, S.Y. Jasmonate signaling and manipulation by pathogens and insects. J. Exp. Bot. 2017, 68, 1371–1385. [Google Scholar] [CrossRef]
- Ren, R.; Xu, J.; Zhang, M.; Liu, G.; Yao, X.; Zhu, L.; Hou, Q. Identification and molecular mapping of a gummy stem blight resistance gene in wild watermelon (Citrullus amarus) germplasm PI 189225. Plant Dis. 2019. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bulk | Clean Reads | Date Generated (Gb) | Q30 (%) | Genome Coverage10× (%) | Average Depth (×) | SNP Number | Alignment Efficiency (%) |
|---|---|---|---|---|---|---|---|
| R01 | 37948696 | 11384608800 | 92.89 | 93.43 | 27.5384 | 1693338 | 97 |
| R02 | 36842550 | 11052765000 | 93.55 | 92.44 | 26.8289 | 1663130 | 97.26 |
| R03 | 107061946 | 32118583800 | 93.10 | 97.51 | 74.5499 | 1671014 | 96.71 |
| R04 | 105760141 | 31728042300 | 92.81 | 97.69 | 75.1521 | 1744548 | 97.02 |
| Method | Chromosome | Start | End | Size(M) | Genes |
|---|---|---|---|---|---|
| SNP-index | Scaffold000193 | 62295 | 162295 | 0.1 | 5 |
| small indels-index | A05 | 10696712 | 10796712 | 0.1 | 13 |
| small indels-index | Scaffold000111 | 57736 | 157736 | 0.1 | 12 |
| small indels-index | Scaffold000167 | 153224 | 253224 | 0.1 | 10 |
| Bulk | Clean Reads | Date Generated (Gb) | Q30 (%) | SNP Number | Alignment Efficiency (%) |
|---|---|---|---|---|---|
| T01 | 21748815 | 6524644500 | 92.63 | 252616 | 87.56 |
| T02 | 17800431 | 5340129300 | 92.70 | 187276 | 89.90 |
| T03 | 62663675 | 18799102500 | 91.87 | 285539 | 88.27 |
| T04 | 52154937 | 15646481100 | 92.45 | 292860 | 88.98 |
| Method | Chromosome | Start | End | Size(M) | Genes |
|---|---|---|---|---|---|
| SNP-index | A02 | 18137278 | 21860151 | 3.72 | 401 |
| SNP-index | Scaffold000111 | 134474 | 596970 | 0.46 | 45 |
| SNP-index | Scaffold000167 | 11168 | 12357 | 0.01 | 1 |
| SNP-index | Scaffold000262 | 0 | 45603 | 0.05 | 2 |
| SNP-index | Scaffold000302 | 27795 | 28067 | 0.00 | 1 |
| SNP-index | Scaffold000317 | 0 | 27038 | 0.03 | 6 |
| small indels-index | A02 | 34693040 | 35853039 | 1.31 | 141 |
| small indels-index | Scaffold000111 | 399069 | 499069 | 0.10 | 10 |
| Gene Name | Sense Primer | Anti-Sense Primer |
|---|---|---|
| BrYUCCA5 | GTGTGTTCAGTCCGCCCGTTA | ATTCCATCTCTGACCCGGAAACG |
| BrGH3.5 | TACGAGCTTGTTGTCACCACTT | TGCGTTCTTTACCGCGTTCT |
| BrCBL9 | TCCACGGCTGCCAGAGAGTT | GCCATCGTCAACAACTGAGCTGC |
| BrAct | TTGCTATTCAGGCTGTTCT | CACCATCACCAGAGTCAA |
| BrSAUR46 | CCACTCTTCAAGGCACTGCTGGA | GGACAACGTCGAGGAAGGTGCT |
| BrMYB1 | AGCTCCCGTAAAGCACCATC | TGTAGTTGCGGTTGTGTGGT |
| BrIAA30 | CTCGGACTCAGCTTCGGCTC | CATGATCATTTCTTGATCAGCAGCC |
| BrMYB51 | ACCTCCCCGAGATTCCAGAG | ATCCCGGCTTCTCGTTGTTA |
| BrGH3.5 | GCCGCAAGAATGTGGTGTTG | CATCGAATGGGACGAGGTGT |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Hou, Z.; Gao, L.; Xiao, D.; Hou, X.; Zhang, C.; Yan, J.; Song, L. Conjunctive Analyses of BSA-Seq and BSR-Seq to Reveal the Molecular Pathway of Leafy Head Formation in Chinese Cabbage. Plants 2019, 8, 603. https://doi.org/10.3390/plants8120603
Li R, Hou Z, Gao L, Xiao D, Hou X, Zhang C, Yan J, Song L. Conjunctive Analyses of BSA-Seq and BSR-Seq to Reveal the Molecular Pathway of Leafy Head Formation in Chinese Cabbage. Plants. 2019; 8(12):603. https://doi.org/10.3390/plants8120603
Chicago/Turabian StyleLi, Rui, Zhongle Hou, Liwei Gao, Dong Xiao, Xilin Hou, Changwei Zhang, Jiyong Yan, and Lixiao Song. 2019. "Conjunctive Analyses of BSA-Seq and BSR-Seq to Reveal the Molecular Pathway of Leafy Head Formation in Chinese Cabbage" Plants 8, no. 12: 603. https://doi.org/10.3390/plants8120603