
Shotgun Proteomics as a Powerful Tool for the Study of the Proteomes of Plants, Their Pathogens, and Plant–Pathogen Interactions

 ,
,  , and
, and 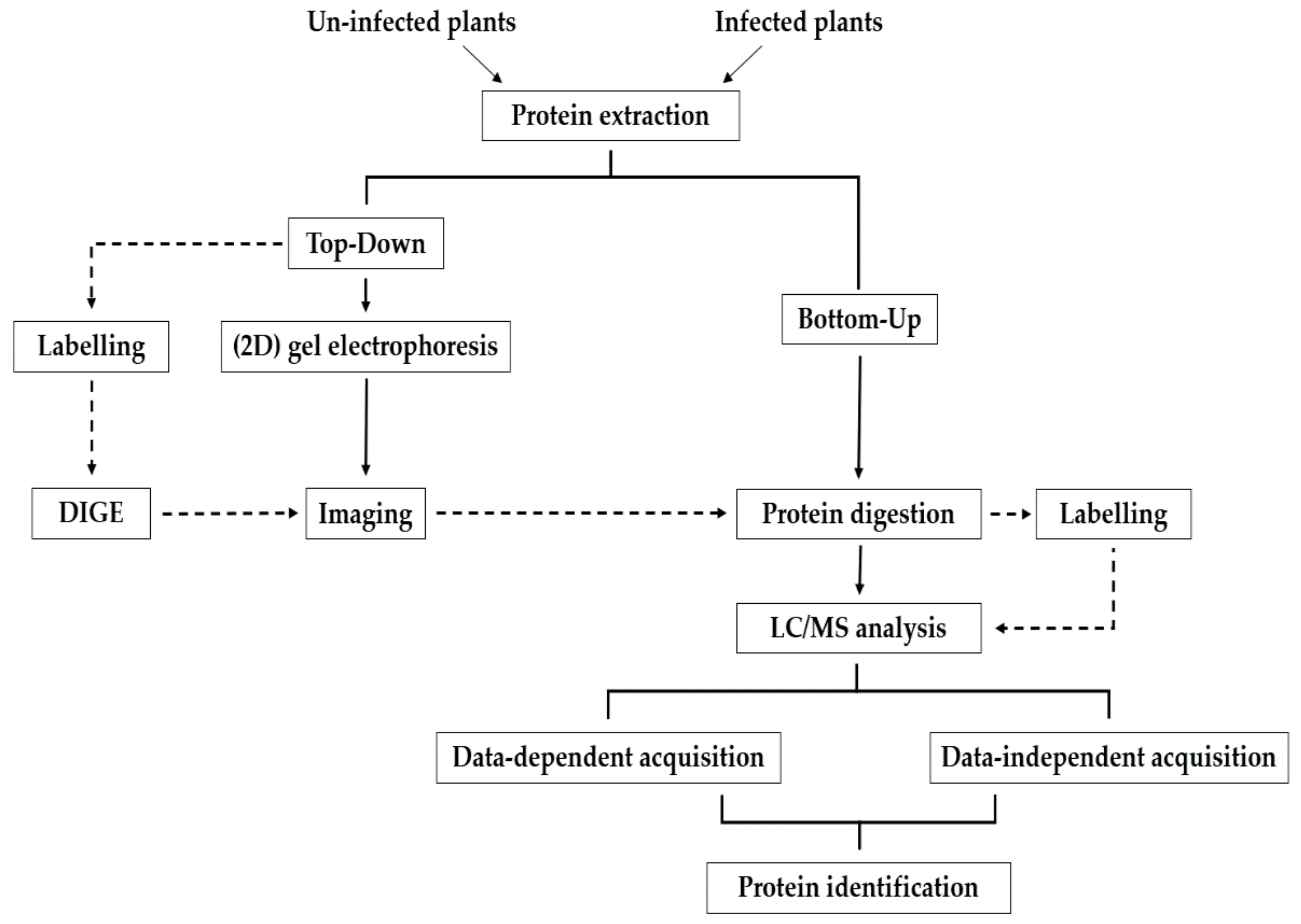{kind=link}
{kind=link}
Abstract
:1. Introduction
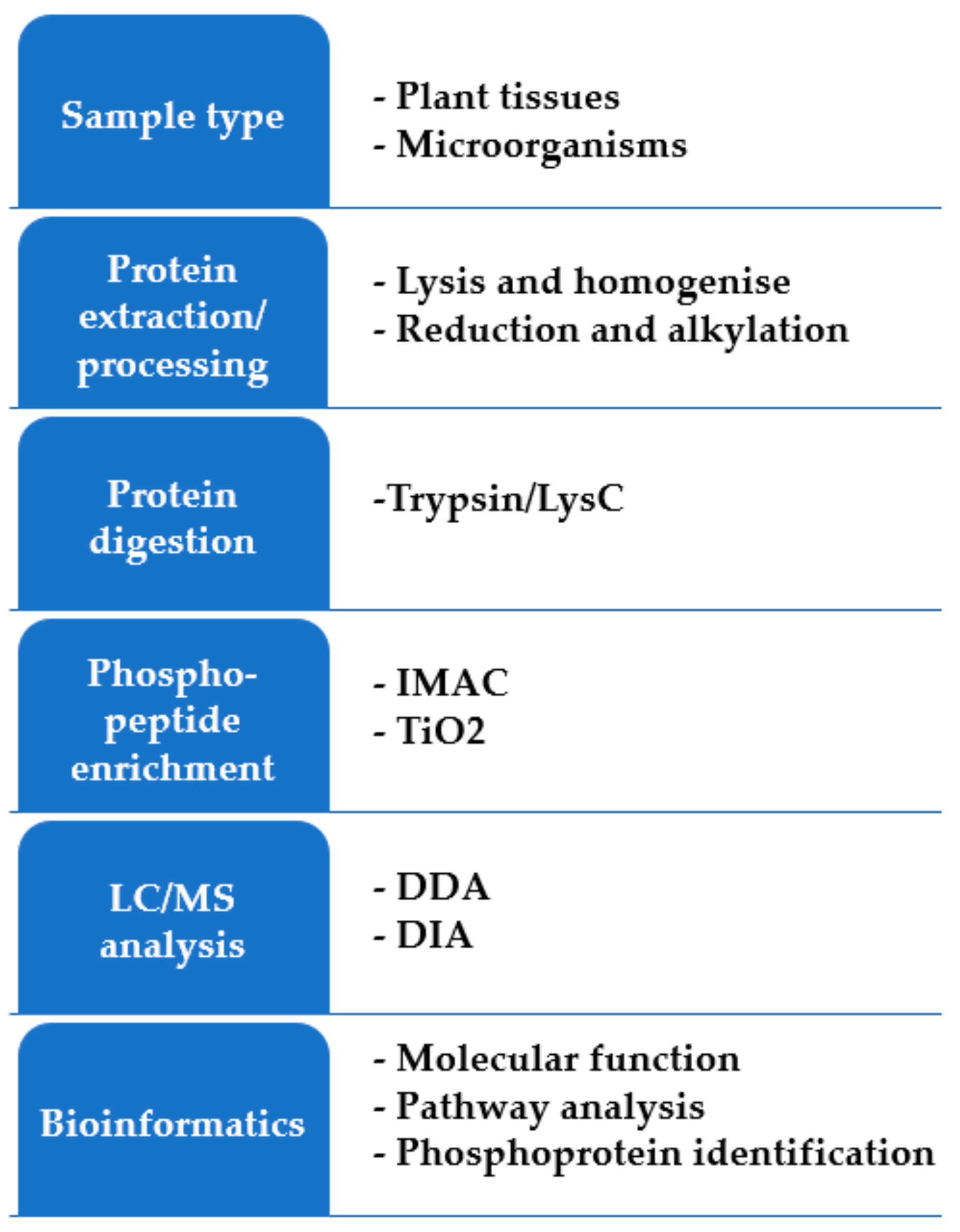
2. Sample Preparation Prior to LC-MS/MS
2.1. Protein Extraction
2.2. Sample Cleanup
3. MS Strategies
4. Post-Translational Modifications
5. Bioinformatics
6. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Boyd, L.A.; Ridout, C.; O’Sullivan, D.M.; Leach, J.E.; Leung, H. Plant-pathogen interactions: Disease resistance in modern agriculture. Trends Genet. 2013, 29, 233–240. [Google Scholar] [CrossRef]
- Gupta, R.; Lee, S.E.; Agrawal, G.K.; Rakwal, R.; Park, S.; Wang, Y.; Kim, S.T. Understanding the plant-pathogen interactions in the context of proteomics-generated apoplastic proteins inventory. Front. Plant Sci. 2015, 6, 352. [Google Scholar] [CrossRef] [Green Version]
- Balotf, S.; Tegg, R.S.; Nichols, D.S.; Wilson, C.R. Spore Germination of the Obligate Biotroph Spongospora subterranea: Transcriptome Analysis Reveals Germination Associated Genes. Front. Microbiol. 2021, 12, 1557. [Google Scholar] [CrossRef]
- Hayden, K.J.; Garbelotto, M.; Knaus, B.J.; Cronn, R.C.; Rai, H.; Wright, J.W. Dual RNA-seq of the plant pathogen Phytophthora ramorum and its tanoak host. Tree Genet. Genom. 2014, 10, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Schneider, D.J.; Collmer, A. Studying plant-pathogen interactions in the genomics era: Beyond molecular Koch’s postulates to systems biology. Annu. Rev. Phytopathol. 2010, 48, 457–479. [Google Scholar] [CrossRef] [Green Version]
- Haddad, N.; Johnson, N.; Kathariou, S.; Métris, A.; Phister, T.; Pielaat, A.; Tassou, C.; Wells-Bennik, M.H.; Zwietering, M.H. Next generation microbiological risk assessment—Potential of omics data for hazard characterisation. Int. J. Food Microbiol. 2018, 287, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Sanchez, J.-C.; Gooley, A.A.; Appel, R.D.; Humphery-Smith, I.; Hochstrasser, D.F.; Williams, K.L. Progress with Proteome Projects: Why all Proteins Expressed by a Genome Should be Identified and How to Do It. Biotechnol. Genet. Eng. Rev. 1996, 13, 19–50. [Google Scholar] [CrossRef] [Green Version]
- Lum, K.K.; Cristea, I.M. Proteomic approaches to uncovering virus–host protein interactions during the progression of viral infection. Expert Rev. Proteom. 2016, 13, 325–340. [Google Scholar] [CrossRef]
- Glinski, M.; Weckwerth, W. The role of mass spectrometry in plant systems biology. Mass Spectrom. Rev. 2006, 25, 173–214. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Weckwerth, W. Mass spectrometry untangles plant membrane protein signaling networks. Trends Plant Sci. 2020, 25, 930–944. [Google Scholar] [CrossRef]
- Meyer, M.D.; Ryck, J.D.; Goormachtig, S.; Van Damme, P. Keeping in Touch with Type-III Secretion System Effectors: Mass Spectrometry-Based Proteomics to Study Effector–Host Protein–Protein Interactions. Int. J. Mol. Sci. 2020, 21, 6891. [Google Scholar] [CrossRef] [PubMed]
- Prathi, N.B.; Palit, P.; Madhu, P.; Ramesh, M.; Laha, G.; Balachandran, S.; Madhav, M.S.; Sundaram, R.; Mangrauthia, S.K. Proteomic and transcriptomic approaches to identify resistance and susceptibility related proteins in contrasting rice genotypes infected with fungal pathogen Rhizoctonia solani. Plant Physiol. Biochem. 2018, 130, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Sinitcyn, P.; Rudolph, J.D.; Cox, J. Computational methods for understanding mass spectrometry—Based shotgun proteomics data. Annu. Rev. Biomed. Data Sci. 2018, 1, 207–234. [Google Scholar] [CrossRef]
- Walther, T.C.; Mann, M. Mass spectrometry—Based proteomics in cell biology. J. Cell Biol. 2010, 190, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Da Veiga Leprevost, F.; Haynes, S.E.; Avtonomov, D.M.; Chang, H.-Y.; Shanmugam, A.K.; Mellacheruvu, D.; Kong, A.T.; Nesvizhskii, A.I. Philosopher: A versatile toolkit for shotgun proteomics data analysis. Nat. Methods 2020, 17, 869–870. [Google Scholar] [CrossRef]
- Consortium, P.G.S. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189. [Google Scholar]
- Salanoubat, M.; Genin, S.; Artiguenave, F.; Gouzy, J.; Mangenot, S.; Arlat, M.; Billault, A.; Brottier, P.; Camus, J.; Cattolico, L. Genome sequence of the plant pathogen Ralstonia solanacearum. Nature 2002, 415, 497–502. [Google Scholar] [CrossRef] [Green Version]
- Poimala, A.; Vainio, E.J. Complete genome sequence of a novel toti-like virus from the plant-pathogenic oomycete Phytophthora cactorum. Arch. Virol. 2020, 165, 1679–1682. [Google Scholar] [CrossRef]
- Seo, S.; Pokhrel, A.; Coleman, J.J. The genome sequence of five genotypes of Fusarium oxysporum f. sp. vasinfectum: A resource for studies on Fusarium wilt of cotton. Mol. Plant-Microbe Interact. 2020, 33, 138–140. [Google Scholar] [CrossRef] [Green Version]
- Baroncelli, R.; Pensec, F.; Da Lio, D.; Boufleur, T.R.; Vicente, I.; Sarrocco, S.; Picot, A.; Baraldi, E.; Sukno, S.; Thon, M.R. Complete Genome Sequence of the plant pathogenic fungus Colletotrichum lupini. Mol. Plant-Microbe Interact. 2021, 34, 1461–1464. [Google Scholar] [CrossRef]
- Song, G.; Hsu, P.Y.; Walley, J.W. Assessment and refinement of sample preparation methods for deep and quantitative plant proteome profiling. Proteomics 2018, 18, 1800220. [Google Scholar] [CrossRef] [PubMed]
- Hussein, R.A.; El-Anssary, A.A. Plants secondary metabolites: The key drivers of the pharmacological actions of medicinal plants. Herb. Med. 2019, 1, 13. [Google Scholar]
- Shimizu, M.; Wariishi, H. Development of a sample preparation method for fungal proteomics. FEMS Microbiol. Lett. 2005, 247, 17–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balotf, S.; Wilson, R.; Tegg, R.S.; Nichols, D.S.; Wilson, C.R. Optimisation of sporosori purification and protein extraction techniques for the biotrophic protozoan plant pathogen Spongospora subterranea. Molecules 2020, 25, 3109. [Google Scholar] [CrossRef]
- Horvatić, A.; Kuleš, J.; Guillemin, N.; Galan, A.; Mrljak, V.; Bhide, M. High-throughput proteomics and the fight against pathogens. Mol. Biosyst. 2016, 12, 2373–2384. [Google Scholar] [CrossRef] [PubMed]
- Patole, C.; Bindschedler, L.V. Plant proteomics: A guide to improve the proteome coverage. In Advances in Biological Science Research; Elsevier: Amsterdam, The Netherlands, 2019; pp. 45–67. [Google Scholar]
- Wu, X.; Gong, F.; Wang, W. Protein extraction from plant tissues for 2DE and its application in proteomic analysis. Proteomics 2014, 14, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Charmont, S.; Jamet, E.; Pont-Lezica, R.; Canut, H. Proteomic analysis of secreted proteins from Arabidopsis thaliana seedlings: Improved recovery following removal of phenolic compounds. Phytochemistry 2005, 66, 453–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, T. Physical and chemical cell disruption for the recovery of intracellular proteins. Bioprocess Technol. 1991, 12, 57–83. [Google Scholar]
- Wang, W.; Tai, F.; Chen, S. Optimizing protein extraction from plant tissues for enhanced proteomics analysis. J. Sep. Sci. 2008, 31, 2032–2039. [Google Scholar] [CrossRef] [PubMed]
- Thiellement, H.; Zivy, M.; Damerval, C.; Méchin, V. Plant Proteomics; Human Press: Totowa, NJ, USA, 2007. [Google Scholar]
- Niu, L.; Yuan, H.; Gong, F.; Wu, X.; Wang, W. Protein extraction methods shape much of the extracted proteomes. Front. Plant Sci. 2018, 9, 802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Acero, F.J.; Carbú, M.; Garrido, C.; Vallejo, I.; Cantoral, J.M. Proteomic advances in phytopathogenic fungi. Curr. Proteom. 2007, 4, 79–88. [Google Scholar] [CrossRef]
- Makino, A.; Mae, T.; Ohira, K. Changes in photosynthetic capacity in rice leaves from emergence through senescence. Analysis from ribulose-1, 5-bisphosphate carboxylase and leaf conductance. Plant Cell Physiol. 1984, 25, 511–521. [Google Scholar]
- Koroleva, O.A.; Bindschedler, L.V. Efficient strategies for analysis of low abundance proteins in plant proteomics. In Sample Preparation in Biological Mass Spectrometry; Springer: Berlin/Heidelberg, Germany, 2011; pp. 381–409. [Google Scholar]
- Righetti, P.G.; Boschetti, E. Low-abundance plant protein enrichment with peptide libraries to enlarge proteome coverage and related applications. Plant Sci. 2020, 290, 110302. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Wang, Y.; Agrawal, G.K.; Rakwal, R.; Jo, I.H.; Bang, K.H.; Kim, S.T. Time to dig deep into the plant proteome: A hunt for low-abundance proteins. Front. Plant Sci. 2015, 6, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widjaja, I.; Naumann, K.; Roth, U.; Wolf, N.; Mackey, D.; Dangl, J.L.; Scheel, D.; Lee, J. Combining subproteome enrichment and Rubisco depletion enables identification of low abundance proteins differentially regulated during plant defense. Proteomics 2009, 9, 138–147. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, P.; Xing, Z.; Jin, S.; Chen, Z.; Liu, L.; Constantino, N.; Wang, X.; Shi, W.; Yuan, J.S. Application of an improved proteomics method for abundant protein cleanup: Molecular and genomic mechanisms study in plant defense. Mol. Cell. Proteom. 2013, 12, 3431–3442. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, H.B.; Natarajan, S.S. A rapid method for depletion of Rubisco from soybean (Glycine max) leaf for proteomic analysis of lower abundance proteins. Phytochemistry 2009, 70, 1958–1964. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-Q.; Jensen, O.N.; Møller, I.M.; Hebelstrup, K.H.; Rogowska-Wrzesinska, A. Evaluation of sample preparation methods for mass spectrometry-based proteomic analysis of barley leaves. Plant Methods 2018, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bahmani, M.; O’Lone, C.E.; Juhász, A.; Nye-Wood, M.; Dunn, H.; Edwards, I.B.; Colgrave, M.L. Application of mass spectrometry-based proteomics to barley research. J. Agric. Food Chem. 2021, 69, 8591–8609. [Google Scholar] [CrossRef] [PubMed]
- Ball, B.; Bermas, A.; Carruthers-Lay, D.; Geddes-McAlister, J. Mass spectrometry-based proteomics of fungal pathogenesis, host–fungal interactions, and antifungal development. J. Fungi 2019, 5, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Shen, X.; Chen, D.; Sun, L. Toward a universal sample preparation method for denaturing top-down proteomics of complex proteomes. J. Proteome Res. 2020, 19, 3315–3325. [Google Scholar] [CrossRef] [PubMed]
- Feist, P.; Hummon, A.B. Proteomic challenges: Sample preparation techniques for microgram-quantity protein analysis from biological samples. Int. J. Mol. Sci. 2015, 16, 3537–3563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wither, M.J.; Hansen, K.C.; Reisz, J.A. Mass spectrometry-based bottom-up proteomics: Sample preparation, LC-MS/MS analysis, and database query strategies. Curr. Protoc. Protein Sci. 2016, 86, 11–20. [Google Scholar] [CrossRef]
- Wiśniewski, J.R. Filter-aided sample preparation for proteome analysis. In Microbial Proteomics; Springer: Berlin/Heidelberg, Germany, 2018; pp. 3–10. [Google Scholar]
- Zougman, A.; Selby, P.J.; Banks, R.E. Suspension trapping (STrap) sample preparation method for bottom-up proteomics analysis. Proteomics 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.S.; Moggridge, S.; Müller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.J.; Garnett, S.; Blackburn, J.M.; Soares, N.C. Comparative reevaluation of FASP and enhanced FASP methods by LC–MS/MS. J. Proteome Res. 2015, 14, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Dubiak, K.M.; Huber, P.W.; Dovichi, N.J. Miniaturized filter-aided sample preparation (MICRO-FASP) method for high throughput, ultrasensitive proteomics sample preparation reveals proteome asymmetry in Xenopus laevis embryos. Anal. Chem. 2020, 92, 5554–5560. [Google Scholar] [CrossRef]
- Lipecka, J.; Chhuon, C.; Bourderioux, M.; Bessard, M.A.; van Endert, P.; Edelman, A.; Guerrera, I.C. Sensitivity of mass spectrometry analysis depends on the shape of the filtration unit used for filter aided sample preparation (FASP). Proteomics 2016, 16, 1852–1857. [Google Scholar] [CrossRef] [PubMed]
- Yeung, Y.G.; Stanley, E.R. Rapid detergent removal from peptide samples with ethyl acetate for mass spectrometry analysis. Curr. Protoc. Protein Sci. 2010, 59, 11–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- HaileMariam, M.; Eguez, R.V.; Singh, H.; Bekele, S.; Ameni, G.; Pieper, R.; Yu, Y. S-Trap, an ultrafast sample-preparation approach for shotgun proteomics. J. Proteome Res. 2018, 17, 2917–2924. [Google Scholar] [CrossRef]
- Hayoun, K.; Gaillard, J.-C.; Pible, O.; Alpha-Bazin, B.; Armengaud, J. High-throughput proteotyping of bacterial isolates by double barrel chromatography-tandem mass spectrometry based on microplate paramagnetic beads and phylopeptidomics. J. Proteom. 2020, 226, 103887. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Lozano, M.A.; Koopmans, F.; Paliukhovich, I.; Smit, A.B.; Li, K.W. A fast and economical sample preparation protocol for interaction proteomics analysis. Proteomics 2019, 19, 1900027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, K.R.; Schroll, M.M.; Hummon, A.B. Comparison of in-solution, FASP, and S-trap based digestion methods for bottom-up proteomic studies. J. Proteome Res. 2018, 17, 2480–2490. [Google Scholar] [CrossRef] [PubMed]
- Johnston, H.E.; Yadav, K.; Kirkpatrick, J.M.; Biggs, G.S.; Oxley, D.; Kramer, H.B.; Samant, R.S. Solvent Precipitation SP3 (SP4) enhances recovery for proteomics sample preparation without magnetic beads. bioRxiv 2021. [Google Scholar] [CrossRef]
- Mikulášek, K.; Konečná, H.; Potěšil, D.; Holánková, R.; Havliš, J.; Zdráhal, Z. SP3 Protocol for Proteomic Plant Sample Preparation Prior LC-MS/MS. Front. Plant Sci. 2021, 12, 369. [Google Scholar] [CrossRef] [PubMed]
- Stoyan Stoychev, P.N.; Mamputhaa, S.; Khumaloa, F.; Gerber, I.; van der Westhuyzena, C.; Jordaan, J. Universal Unbiased Pre-MS Clean-Up Using Magnetic HILIC Microparticles for SPE. Available online: https://resynbio.com/hilic/ (accessed on 25 February 2021).
- Lewandowska, D.; Zhang, R.; Colas, I.; Uzrek, N.; Waugh, R. Application of a sensitive and reproducible label-free proteomic approach to explore the proteome of individual meiotic-phase barley anthers. Front. Plant Sci. 2019, 10, 393. [Google Scholar] [CrossRef]
- Balotf, S.; Wilson, R.; Tegg, R.S.; Nichols, D.S.; Wilson, C.R. Quantitative proteomics provides an insight into germination-related proteins in the obligate biotrophic plant pathogen Spongospora subterranea. Environ. Microbiol. Rep. 2021, 13, 521–532. [Google Scholar] [CrossRef]
- Schubert, O.T.; Röst, H.L.; Collins, B.C.; Rosenberger, G.; Aebersold, R. Quantitative proteomics: Challenges and opportunities in basic and applied research. Nat. Protoc. 2017, 12, 1289–1294. [Google Scholar] [CrossRef] [PubMed]
- Elmore, J.M.; Griffin, B.D.; Walley, J.W. Advances in functional proteomics to study plant-pathogen interactions. Curr. Opin. Plant Biol. 2021, 63, 102061. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.C.; Russell, J.D.; Bailey, D.J.; Westphall, M.S.; Coon, J.J. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteom. 2012, 11, 1475–1488. [Google Scholar] [CrossRef] [Green Version]
- Borràs, E.; Sabidó, E. What is targeted proteomics? A concise revision of targeted acquisition and targeted data analysis in mass spectrometry. Proteomics 2017, 17, 1700180. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Costa, C.; Martínez-Bartolomé, S.; McClatchy, D.B.; Saviola, A.J.; Yu, N.-K.; Yates, J.R., III. Impact of the Identification Strategy on the Reproducibility of the DDA and DIA Results. J. Proteome Res. 2020, 19, 3153–3161. [Google Scholar] [CrossRef]
- Davies, V.; Wandy, J.; Weidt, S.; Van Der Hooft, J.J.; Miller, A.; Daly, R.; Rogers, S. Rapid Development of Improved Data-Dependent Acquisition Strategies. Anal. Chem. 2021, 93, 5676–5683. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Teo, G.C.; Kong, A.T.; Haynes, S.E.; Avtonomov, D.M.; Geiszler, D.J.; Nesvizhskii, A.I. Identification of modified peptides using localization-aware open search. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Verheggen, K.; Ræder, H.; Berven, F.S.; Martens, L.; Barsnes, H.; Vaudel, M. Anatomy and evolution of database search engines—A central component of mass spectrometry based proteomic workflows. Mass Spectrom. Rev. 2020, 39, 292–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruderer, R.; Bernhardt, O.M.; Gandhi, T.; Xuan, Y.; Sondermann, J.; Schmidt, M.; Gomez-Varela, D.; Reiter, L. Optimization of experimental parameters in data-independent mass spectrometry significantly increases depth and reproducibility of results. Mol. Cell. Proteom. 2017, 16, 2296–2309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.-X.; Zhang, Y.; Fernie, A.R.; Liu, Y.-G.; Zhu, F.-Y. SWATH-MS-based proteomics: Strategies and applications in plants. Trends Biotechnol. 2021, 39, 433–437. [Google Scholar] [CrossRef]
- Zhang, F.; Ge, W.; Ruan, G.; Cai, X.; Guo, T. Data-independent acquisition mass spectrometry-based proteomics and software tools: A glimpse in 2020. Proteomics 2020, 20, 1900276. [Google Scholar] [CrossRef]
- Gotti, C.; Roux-Dalvai, F.; Joly-Beauparlant, C.; Mangnier, L.; Leclercq, M.; Droit, A. Extensive and accurate benchmarking of DIA acquisition methods and software tools using a complex proteomic standard. J. Proteome Res. 2021, 20, 4801–4814. [Google Scholar] [CrossRef]
- Pino, L.K.; Just, S.C.; MacCoss, M.J.; Searle, B.C. Acquiring and analyzing data independent acquisition proteomics experiments without spectrum libraries. Mol. Cell. Proteom. 2020, 19, 1088–1103. [Google Scholar] [CrossRef] [Green Version]
- Sinitcyn, P.; Hamzeiy, H.; Salinas Soto, F.; Itzhak, D.; McCarthy, F.; Wichmann, C.; Steger, M.; Ohmayer, U.; Distler, U.; Kaspar-Schoenefeld, S. MaxDIA enables library-based and library-free data-independent acquisition proteomics. Nat. Biotechnol. 2021, 39, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.C.; Aebersold, R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2018, 14, e8126. [Google Scholar] [CrossRef]
- Meier, F.; Beck, S.; Grassl, N.; Lubeck, M.; Park, M.A.; Raether, O.; Mann, M. Parallel accumulation-serial fragmentation (PASEF): Multiplying sequencing speed and sensitivity by synchronized scans in a trapped ion mobility device. J. Proteome Res. 2015, 14, 5378–5387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Lima, F.; Kaplan, D.; Park, M. Note: Integration of trapped ion mobility spectrometry with mass spectrometry. Rev. Sci. Instrum. 2011, 82, 126106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelmann, K.; Silveira, J.A.; Ridgeway, M.E.; Park, M.A. Fundamentals of trapped ion mobility spectrometry. J. Am. Soc. Mass Spectrom. 2014, 26, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Charkow, J.; Röst, H.L. Trapped Ion Mobility Spectrometry Reduces Spectral Complexity in Mass Spectrometry-Based Proteomics. Anal. Chem. 2021, 93, 16751–16758. [Google Scholar] [CrossRef]
- Jin, J.-J.; Duan, S.-N.; Qi, Y.-Z.; Zhen, W.-C.; Jun, M. Identification of proteins associated with Fusarium crown rot resistance in wheat using label-free quantification analysis. J. Integr. Agric. 2021, 20, 3209–3221. [Google Scholar] [CrossRef]
- Anand, S.; Samuel, M.; Ang, C.-S.; Keerthikumar, S.; Mathivanan, S. Label-based and label-free strategies for protein quantitation. In Proteome Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2017; pp. 31–43. [Google Scholar]
- Ünlü, M.; Morgan, M.E.; Minden, J.S. Difference gel electrophoresis. A single gel method for detecting changes in protein extracts. Electrophoresis 1997, 18, 2071–2077. [Google Scholar] [CrossRef] [PubMed]
- Arruda, S.C.C.; de Sousa Barbosa, H.; Azevedo, R.A.; Arruda, M.A.Z. Two-dimensional difference gel electrophoresis applied for analytical proteomics: Fundamentals and applications to the study of plant proteomics. Analyst 2011, 136, 4119–4126. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Hao, G.; Hatt, S.; Wang, Z.; Francis, F. Host plant adaptability and proteomic differences of diverse Rhopalosiphum maidis (Fitch) lineages. Arch. Insect Biochem. Physiol. 2022, 109, e21853. [Google Scholar] [CrossRef]
- Bindschedler, L.V.; Cramer, R. Quantitative plant proteomics. Proteomics 2011, 11, 756–775. [Google Scholar] [CrossRef]
- Bindschedler, L.V.; Palmblad, M.; Cramer, R. Hydroponic isotope labelling of entire plants (HILEP) for quantitative plant proteomics; an oxidative stress case study. Phytochemistry 2008, 69, 1962–1972. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, H.; Berke, L.; Heck, A.J.; Mohammed, S.; Scheres, B.; Menke, F.L. Quantitative phosphoproteomics after auxin-stimulated lateral root induction identifies an SNX1 protein phosphorylation site required for growth. Mol. Cell. Proteom. 2013, 12, 1158–1169. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Li, L.; Liao, F.; Chen, W. iTRAQ-based quantitative proteomic analysis reveals NtGNL1-dependent regulatory network underlying endosome trafficking for pollen tube polar growth. Plant Physiol. Biochem. 2021, 161, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Jiang, W.; Liu, X.; Duan, Y.; Xiang, L.; Wang, Y.; Jiang, Y.; Shen, X.; Chen, X.; Yin, C. ITRAQ-based quantitative proteomic analysis of Fusarium moniliforme (Fusarium verticillioides) in response to Phloridzin inducers. Proteome Sci. 2021, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.-M.; Peng, G.; Zhao, J.-H.; Wang, G.-D.; Zhang, H.-M.; Cao, W.-L.; Xiang, X.; Zhang, Y.-F.; Rong, H.; Chen, Z.-X. iTRAQ-based quantitative proteomics analysis of defense responses triggered by the pathogen Rhizoctonia solani infection in rice. J. Integr. Agric. 2022, 21, 139–152. [Google Scholar] [CrossRef]
- Wang, F.-X.; Luo, Y.-M.; Ye, Z.-Q.; Cao, X.; Liang, J.-N.; Wang, Q.; Wu, Y.; Wu, J.-H.; Wang, H.-Y.; Zhang, M. iTRAQ-based proteomics analysis of autophagy-mediated immune responses against the vascular fungal pathogen Verticillium dahliae in Arabidopsis. Autophagy 2018, 14, 598–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, A.; Tang, Y.; Shi, X.; Jia, M.; Zhu, J.; Yan, X.; Chen, H.; Gu, Y. Proximity labeling proteomics reveals critical regulators for inner nuclear membrane protein degradation in plants. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Domon, B.; Aebersold, R. Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 2010, 28, 710–721. [Google Scholar] [CrossRef]
- Balotf, S.; Wilson, R.; Tegg, R.S.; Nichols, D.S.; Wilson, C.R. In Planta Transcriptome and Proteome Profiles of Spongospora subterranea in Resistant and Susceptible Host Environments Illuminates Regulatory Principles Underlying Host–Pathogen Interaction. Biology 2021, 10, 840. [Google Scholar] [CrossRef] [PubMed]
- Hassett, K.; Ellwood, S.R.; Zulak, K.G.; Muria-Gonzalez, M.J. Analysis of apoplastic proteins expressed during net form net blotch of barley. J. Plant Dis. Prot. 2020, 127, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Cheah, B.H.; Lin, H.-H.; Chien, H.-J.; Liao, C.-T.; Liu, L.-Y.D.; Lai, C.-C.; Lin, Y.-F.; Chuang, W.-P. SWAtH-MS-based quantitative proteomics reveals a uniquely intricate defense response in Cnaphalocrocis medinalis-resistant rice. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.-T.; Wang, K.-H.; Chang, W.-H.; Yang, J.-C.; Yeh, C.-F.; Cheng, K.-T.; Hung, S.-C.; Chen, Y.-R. Application of data-independent acquisition approach to study the proteome change from early to later phases of tomato pathogenesis responses. Int. J. Mol. Sci. 2019, 20, 863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, K.-T.; Hsu, Y.; Yeh, C.-F.; Chang, C.-H.; Chang, W.-H.; Chen, Y.-R. Quantitative Proteomics Reveals the Dynamic Regulation of the Tomato Proteome in Response to Phytophthora infestans. Int. J. Mol. Sci. 2021, 22, 4174. [Google Scholar] [CrossRef]
- Zhang, X.-Q.; Bai, L.; Sun, H.-B.; Yang, C.; Cai, B.-Y. Transcriptomic and proteomic analysis revealed the effect of funneliformis mosseae in soybean roots differential expression genes and proteins. J. Proteome Res. 2020, 19, 3631–3643. [Google Scholar] [CrossRef] [PubMed]
- Castillejo, M.-Á.; Fondevilla-Aparicio, S.; Fuentes-Almagro, C.; Rubiales, D. Quantitative analysis of target peptides related to resistance against Ascochyta blight (Peyronellaea pinodes) in pea. J. Proteome Res. 2020, 19, 1000–1012. [Google Scholar] [CrossRef] [PubMed]
- Kerr, E.D.; Phung, T.K.; Caboche, C.H.; Fox, G.P.; Platz, G.J.; Schulz, B.L. The intrinsic and regulated proteomes of barley seeds in response to fungal infection. Anal. Biochem. 2019, 580, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, Z.; Iqbal, M.S.; Hashem, A.; Abd_Allah, E.F.; Ansari, M.I. Plant defense responses to biotic stress and its interplay with fluctuating dark/light conditions. Front. Plant Sci. 2021, 12, 297. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Triplett, L.; Chen, X.-L. Emerging Roles of Posttranslational Modifications in Plant-Pathogenic Fungi and Bacteria. Annu. Rev. Phytopathol. 2021, 59, 99–124. [Google Scholar] [CrossRef]
- He, Z.; Huang, T.; Ao, K.; Yan, X.; Huang, Y. Sumoylation, phosphorylation, and acetylation fine-tune the turnover of plant immunity components mediated by ubiquitination. Front. Plant Sci. 2017, 8, 1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Sanchez, C.; Li, H.; Chen, S. Recent advances and challenges in plant phosphoproteomics. Proteomics 2015, 15, 1127–1141. [Google Scholar] [CrossRef] [PubMed]
- Batalha, I.L.; Lowe, C.R.; Roque, A.C. Platforms for enrichment of phosphorylated proteins and peptides in proteomics. Trends Biotechnol. 2012, 30, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Melo-Braga, M.N.; Larsen, M.R.; Jørgensen, H.J.; Palmisano, G. Battle through signaling between wheat and the fungal pathogen Septoria tritici revealed by proteomics and phosphoproteomics. Mol. Cell. Proteom. 2013, 12, 2497–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Q.; Zhang, T.; Zhang, A.; Lin, C.; Kong, W.; Chen, S. Proteomics and phosphoproteomics revealed molecular networks of stomatal immune responses. Planta 2020, 252, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Thurston, G.; Regan, S.; Rampitsch, C.; Xing, T. Proteomic and phosphoproteomic approaches to understand plant–pathogen interactions. Physiol. Mol. Plant Pathol. 2005, 66, 3–11. [Google Scholar] [CrossRef]
- Xing, T.; Laroche, A. Revealing plant defense signaling: Getting more sophisticated with phosphoproteomics. Plant Signal. Behav. 2011, 6, 1469–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roustan, V.; Weckwerth, W. Quantitative phosphoproteomic and system-level analysis of TOR inhibition unravel distinct organellar acclimation in Chlamydomonas reinhardtii. Front. Plant Sci. 2018, 9, 1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Schneider, J.D.; Zhu, N.; Chen, S. Identification of MAPK substrates using quantitative phosphoproteomics. In Plant Pattern Recognition Receptors; Springer: Berlin/Heidelberg, Germany, 2017; pp. 133–142. [Google Scholar]
- Song, G.; Montes, C.; Walley, J.W. Quantitative profiling of protein abundance and phosphorylation state in plant tissues using tandem mass tags. In Plant Proteomics; Springer: Berlin/Heidelberg, Germany, 2020; pp. 147–156. [Google Scholar]
- Fíla, J.; Honys, D. Enrichment techniques employed in phosphoproteomics. Amino Acids 2012, 43, 1025–1047. [Google Scholar] [CrossRef] [Green Version]
- Low, T.Y.; Mohtar, M.A.; Lee, P.Y.; Omar, N.; Zhou, H.; Ye, M. Widening the bottleneck of phosphoproteomics: Evolving strategies for phosphopeptide enrichment. Mass Spectrom. Rev. 2021, 40, 309–333. [Google Scholar] [CrossRef]
- Qiu, W.; Evans, C.A.; Landels, A.; Pham, T.K.; Wright, P.C. Phosphopeptide enrichment for phosphoproteomic analysis—A tutorial and review of novel materials. Anal. Chim. Acta 2020, 1129, 158–180. [Google Scholar] [CrossRef]
- Wolschin, F.; Wienkoop, S.; Weckwerth, W. Enrichment of phosphorylated proteins and peptides from complex mixtures using metal oxide/hydroxide affinity chromatography (MOAC). Proteomics 2005, 5, 4389–4397. [Google Scholar] [CrossRef]
- Zhang, B.; Shao, L.; Wang, J.; Zhang, Y.; Guo, X.; Peng, Y.; Cao, Y.; Lai, Z. Phosphorylation of ATG18a by BAK1 suppresses autophagy and attenuates plant resistance against necrotrophic pathogens. Autophagy 2021, 17, 2093–2110. [Google Scholar] [CrossRef]
- Kadota, Y.; Liebrand, T.W.; Goto, Y.; Sklenar, J.; Derbyshire, P.; Menke, F.L.; Torres, M.A.; Molina, A.; Zipfel, C.; Coaker, G. Quantitative phosphoproteomic analysis reveals common regulatory mechanisms between effector-and PAMP-triggered immunity in plants. New Phytol. 2019, 221, 2160–2175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shi, Y.; Zhao, L.; Wei, F.; Feng, Z.; Feng, H. Phosphoproteomics profiling of cotton (Gossypium hirsutum L.) roots in response to Verticillium dahliae inoculation. ACS Omega 2019, 4, 18434–18443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Wei, Y.; Xu, L.; Wu, K.-C.; Yang, L.; Shi, C.-N.; Yang, G.-Y.; Chen, D.; Yu, F.-F.; Xie, Q. A Bunyavirus-inducible ubiquitin ligase targets RNA polymerase IV for degradation during viral pathogenesis in rice. Mol. Plant 2020, 13, 836–850. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-L.; Xie, X.; Wu, L.; Liu, C.; Zeng, L.; Zhou, X.; Luo, F.; Wang, G.-L.; Liu, W. Proteomic analysis of ubiquitinated proteins in rice (Oryza sativa) after treatment with pathogen-associated molecular pattern (PAMP) elicitors. Front. Plant Sci. 2018, 9, 1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubb, L.E.; Derbyshire, P.; Dunning, K.E.; Zipfel, C.; Menke, F.L.; Monaghan, J. Large-scale identification of ubiquitination sites on membrane-associated proteins in Arabidopsis thaliana seedlings. Plant Physiol. 2021, 185, 1483–1488. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, Y.; Du, M.; Liang, X.; Fan, F.; Huang, G.; Zou, Y.; Bai, J.; Lu, D. The calcium-dependent protein kinase CPK28 is targeted by the ubiquitin ligases ATL31 and ATL6 for proteasome-mediated degradation to fine-tune immune signaling in Arabidopsis. Plant Cell 2021, koab242. [Google Scholar] [CrossRef]
- Song, G.; Olatunji, D.; Montes, C.; Clark, N.M.; Pu, Y.; Kelley, D.R.; Walley, J.W. Quantitative proteomics reveals extensive lysine ubiquitination in the Arabidopsis root proteome and uncovers novel transcription factor stability states. bioRxiv 2021. [Google Scholar] [CrossRef]
- Walley, J.W.; Shen, Z.; McReynolds, M.R.; Schmelz, E.A.; Briggs, S.P. Fungal-induced protein hyperacetylation in maize identified by acetylome profiling. Proc. Natl. Acad. Sci. USA 2018, 115, 210–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, B.; Yang, Q.; Li, D.; Liang, W.; Song, L. Proteome-wide analysis of lysine acetylation in the plant pathogen Botrytis cinerea. Sci. Rep. 2016, 6, 29313. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Lv, B.; Tan, L.; Yang, Q.; Liang, W. Acetylome analysis reveals the involvement of lysine acetylation in diverse biological processes in Phytophthora sojae. Sci. Rep. 2016, 6, 29897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Wu, C. Comparative acetylome analysis reveals the potential roles of lysine acetylation for DON biosynthesis in Fusarium graminearum. BMC Genom. 2019, 20, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, X.; Li, Y.; Hu, Z.; Lin, Y.; Zheng, B.; Ding, J. Poplar acetylome profiling reveals lysine acetylation dynamics in seasonal bud dormancy release. Plant Cell Environ. 2021, 44, 1830–1845. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote) omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.D.; Goode, R.J.; Huang, C.; Powell, D.R.; Schittenhelm, R.B. LFQ-analyst: An easy-to-use interactive web platform to analyze and visualize label-free proteomics data preprocessed with MaxQuant. J. Proteome Res. 2019, 19, 204–211. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; Van Den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [Green Version]
- Handler, D.C.; Cheng, F.; Shathili, A.M.; Haynes, P.A. PeptideWitch–A software package to produce high-stringency proteomics data visualizations from label-free shotgun proteomics data. Proteomes 2020, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Pedro, H.; Maheswari, U.; Urban, M.; Irvine, A.G.; Cuzick, A.; McDowall, M.D.; Staines, D.M.; Kulesha, E.; Hammond-Kosack, K.E.; Kersey, P.J. PhytoPath: An integrative resource for plant pathogen genomics. Nucleic Acids Res. 2016, 44, D688–D693. [Google Scholar] [CrossRef] [Green Version]
- Takeya, M.; Yamasaki, F.; Uzuhashi, S.; Aoki, T.; Sawada, H.; Nagai, T.; Tomioka, K.; Tomooka, N.; Sato, T.; Kawase, M. NIASGBdb: NIAS Genebank databases for genetic resources and plant disease information. Nucleic Acids Res. 2010, 39, D1108–D1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, L.; Vizcaíno, J.A. A golden age for working with public proteomics data. Trends Biochem. Sci. 2017, 42, 333–341. [Google Scholar] [CrossRef] [Green Version]
- Craig, R.; Cortens, J.P.; Beavis, R.C. Open source system for analyzing, validating, and storing protein identification data. J. Proteome Res. 2004, 3, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Lam, H.; Aebersold, R. PeptideAtlas: A resource for target selection for emerging targeted proteomics workflows. EMBO Rep. 2008, 9, 429–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vizcaíno, J.A.; Csordas, A.; Del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Xi, L.; Zhang, Z.; Schulze, W.X. PhosPhAt 4.0: An Updated Arabidopsis Database for Searching Phosphorylation Sites and Kinase-Target Interactions. In Plant Phosphoproteomics; Springer: Berlin/Heidelberg, Germany, 2021; pp. 189–202. [Google Scholar]
- Pinski, A.; Roujol, D.; Pouzet, C.; Bordes, L.; San Clemente, H.; Hoffmann, L.; Jamet, E. Comparison of mass spectrometry data ad bioinformatics predictions to assess the bona fide localization of proteins identified in cell wall proteomics studies. Plant Sci. 2021, 310, 110979. [Google Scholar] [CrossRef]
- Dong, A.-Y.; Wang, Z.; Huang, J.-J.; Song, B.-A.; Hao, G.-F. Bioinformatic tools support decision-making in plant disease management. Trends Plant Sci. 2021, 26, 953–967. [Google Scholar] [CrossRef]
- Ctortecka, C.; Mechtler, K. The rise of single-cell proteomics. Anal. Sci. Adv. 2021, 2, 84–94. [Google Scholar] [CrossRef]
- Yu, C.; Huang, L. Cross-linking mass spectrometry (XL-MS): An emerging technology for interactomics and structural biology. Anal. Chem. 2018, 90, 144. [Google Scholar] [CrossRef]
- Iacobucci, C.; Götze, M.; Sinz, A. Cross-linking/mass spectrometry to get a closer view on protein interaction networks. Curr. Opin. Biotechnol. 2020, 63, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Piersimoni, L.; Kastritis, P.L.; Arlt, C.; Sinz, A. Cross-Linking Mass Spectrometry for Investigating Protein Conformations and Protein–Protein Interactions—A Method for All Seasons. Chem. Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yu, F.; Hu, Q.; Wang, T.; Yu, L.; Du, S.; Yu, W.; Li, N. Development of in planta chemical cross-Linking-Based quantitative interactomics in arabidopsis. J. Proteome Res. 2018, 17, 3195–3213. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balotf, S.; Wilson, R.; Tegg, R.S.; Nichols, D.S.; Wilson, C.R. Shotgun Proteomics as a Powerful Tool for the Study of the Proteomes of Plants, Their Pathogens, and Plant–Pathogen Interactions. Proteomes 2022, 10, 5. https://doi.org/10.3390/proteomes10010005
Balotf S, Wilson R, Tegg RS, Nichols DS, Wilson CR. Shotgun Proteomics as a Powerful Tool for the Study of the Proteomes of Plants, Their Pathogens, and Plant–Pathogen Interactions. Proteomes. 2022; 10(1):5. https://doi.org/10.3390/proteomes10010005
Chicago/Turabian StyleBalotf, Sadegh, Richard Wilson, Robert S. Tegg, David S. Nichols, and Calum R. Wilson. 2022. "Shotgun Proteomics as a Powerful Tool for the Study of the Proteomes of Plants, Their Pathogens, and Plant–Pathogen Interactions" Proteomes 10, no. 1: 5. https://doi.org/10.3390/proteomes10010005