
Partial Immunoblotting of 2D-Gels: A Novel Method to Identify Post-Translationally Modified Proteins Exemplified for the Myelin Acetylome

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Myelin Sample Preparation
2.3. Expression and Purification of Lysine-Acetylated Proteins
2.4. Isoelectric Focusing
2.5. Gel Electrophoresis
2.6. Partial Transfer
2.7. Immunodetection
2.8. Data Analysis
2.9. Protein Identification
2.10. Cryoimmuno Electron Microscopy
2.11. Estimation of Transfer Efficiency
3. Results
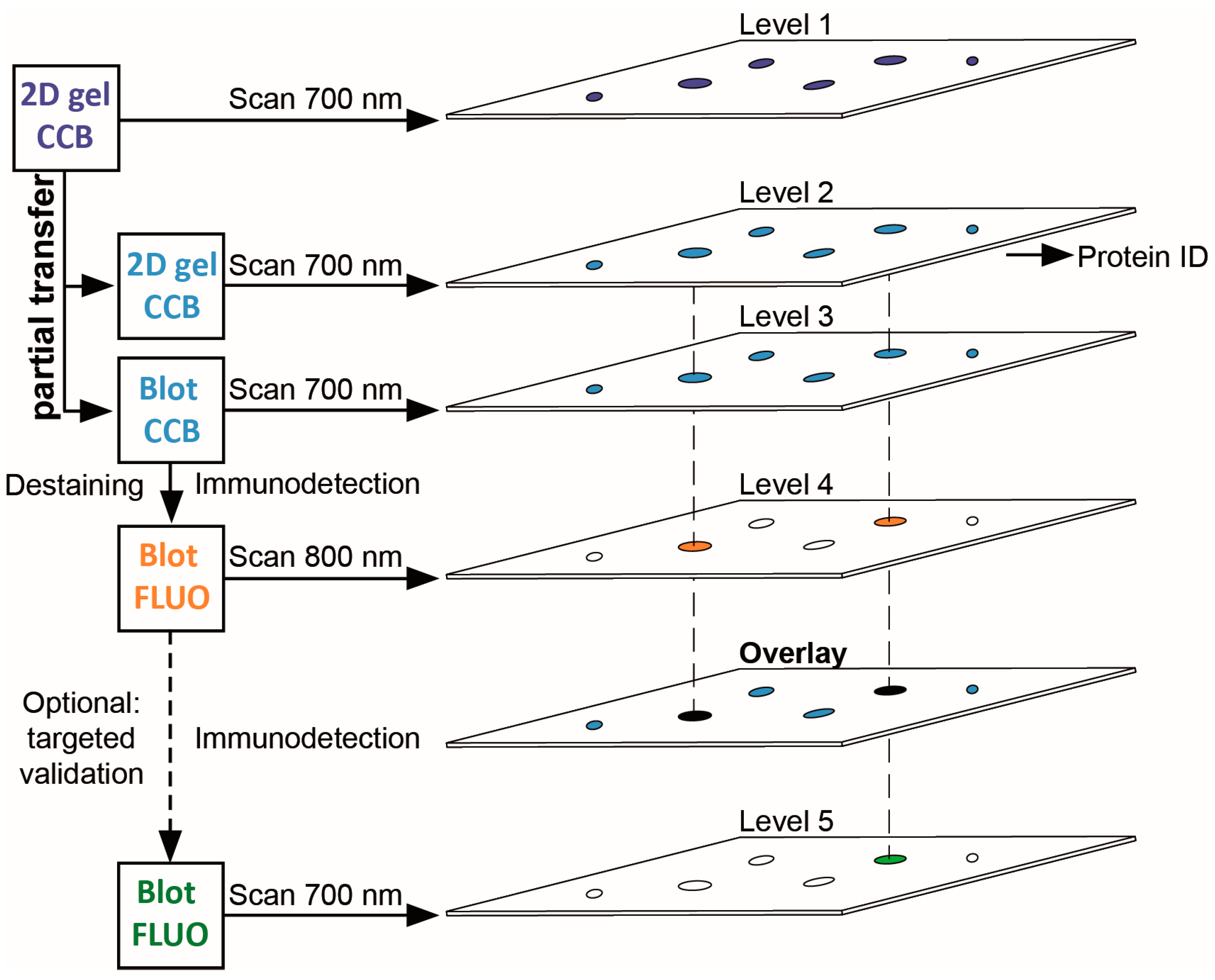
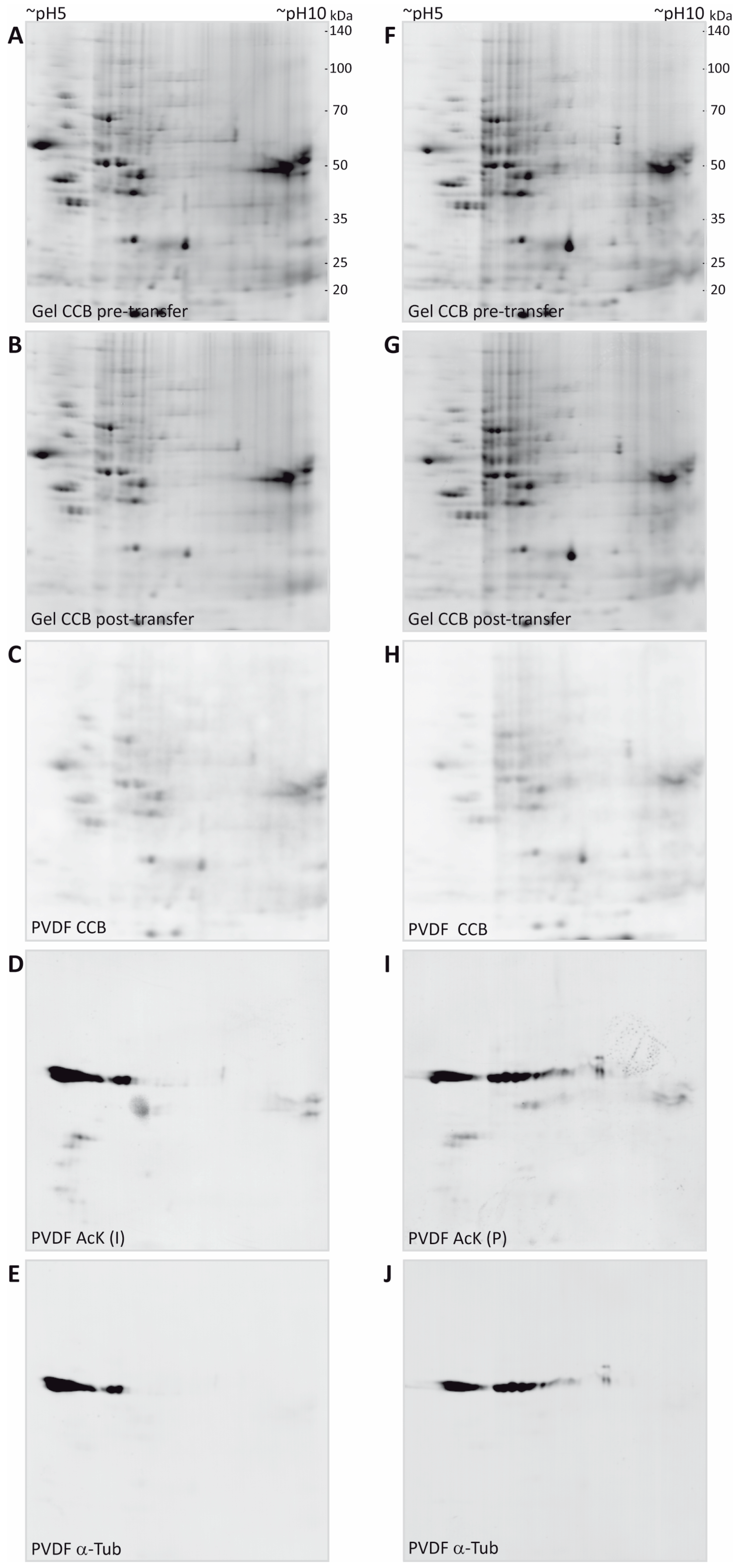
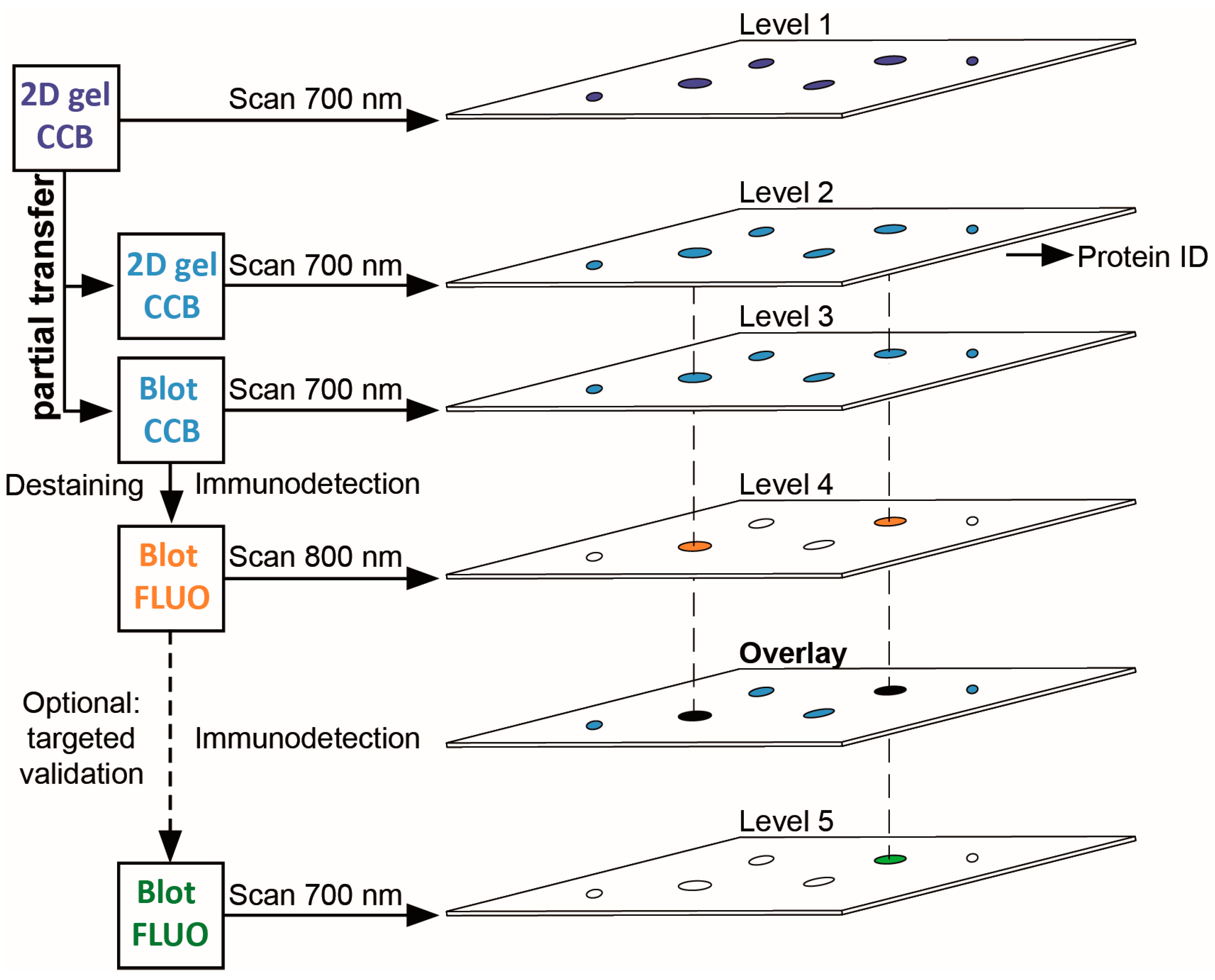
3.1. A Multi-Level Experimental Design for PTM Screening by 2D Gel Immunoblotting
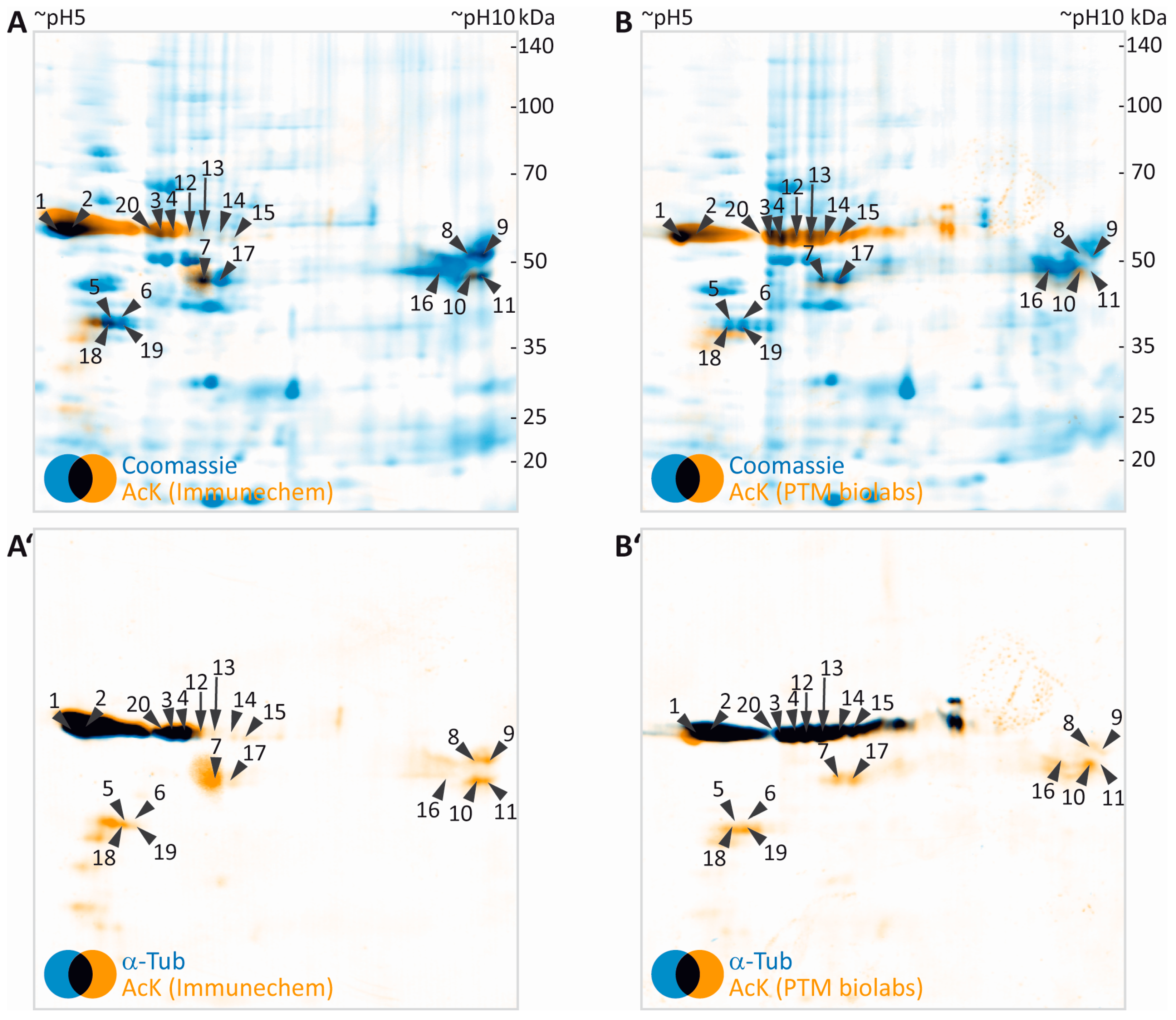
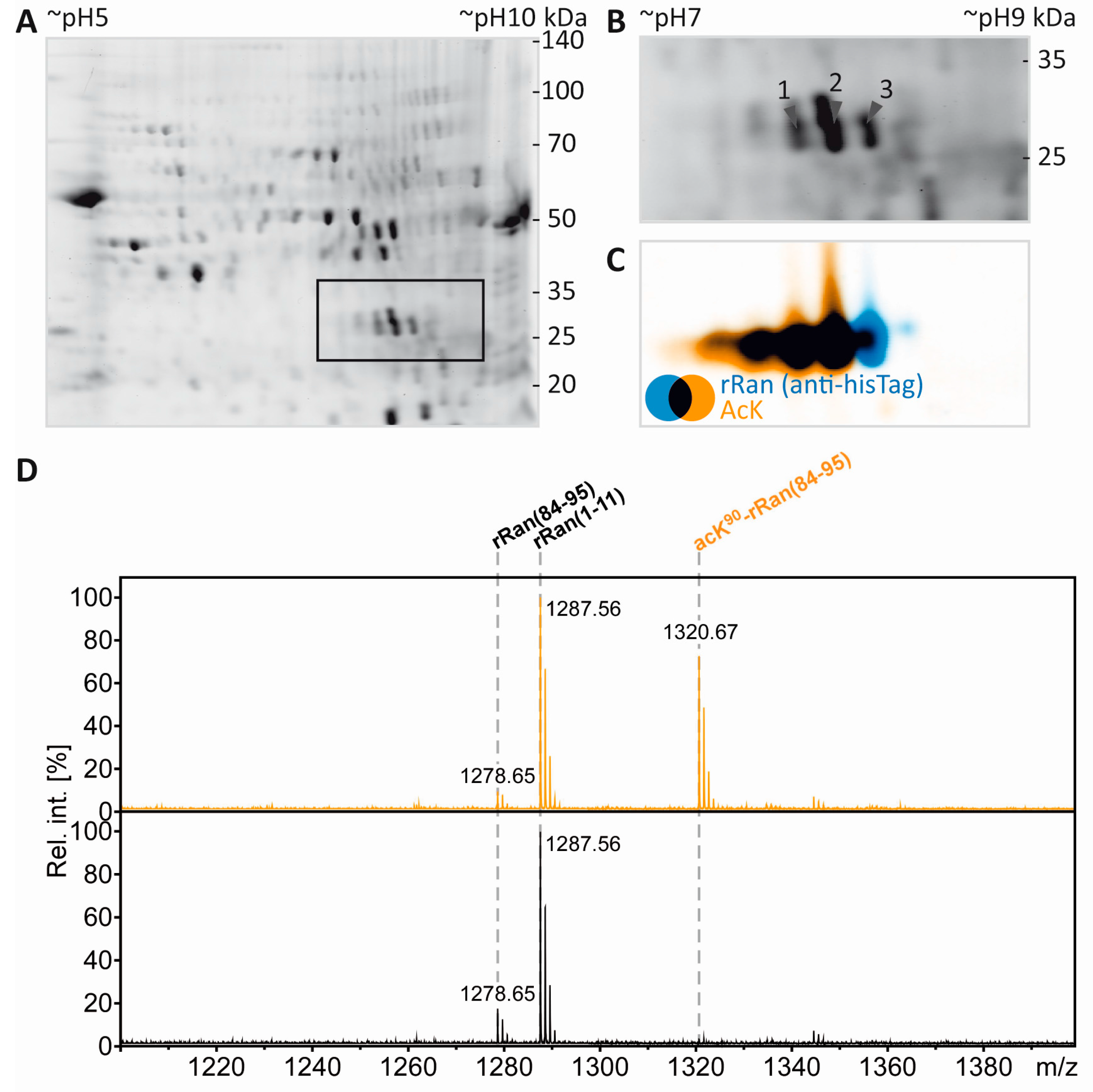
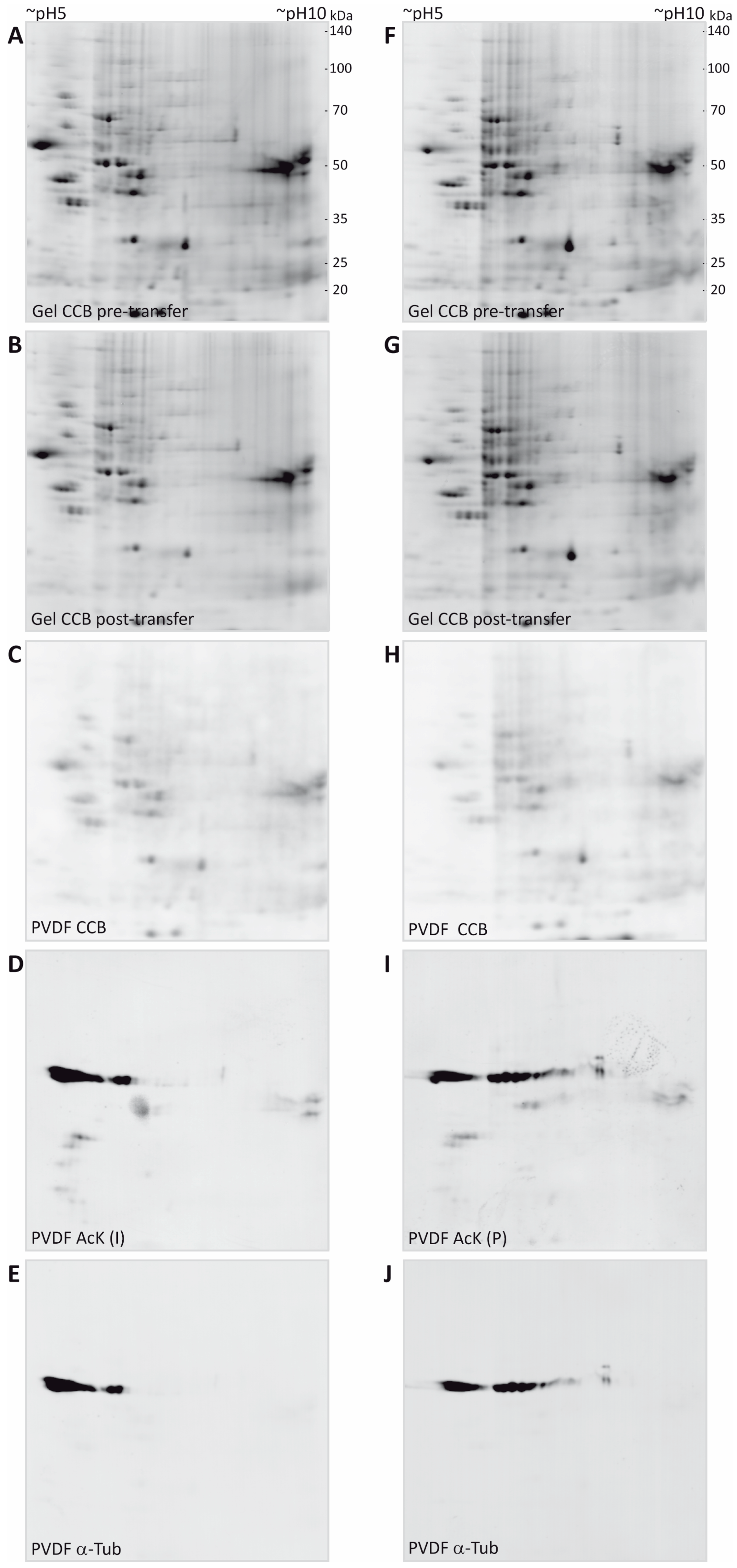
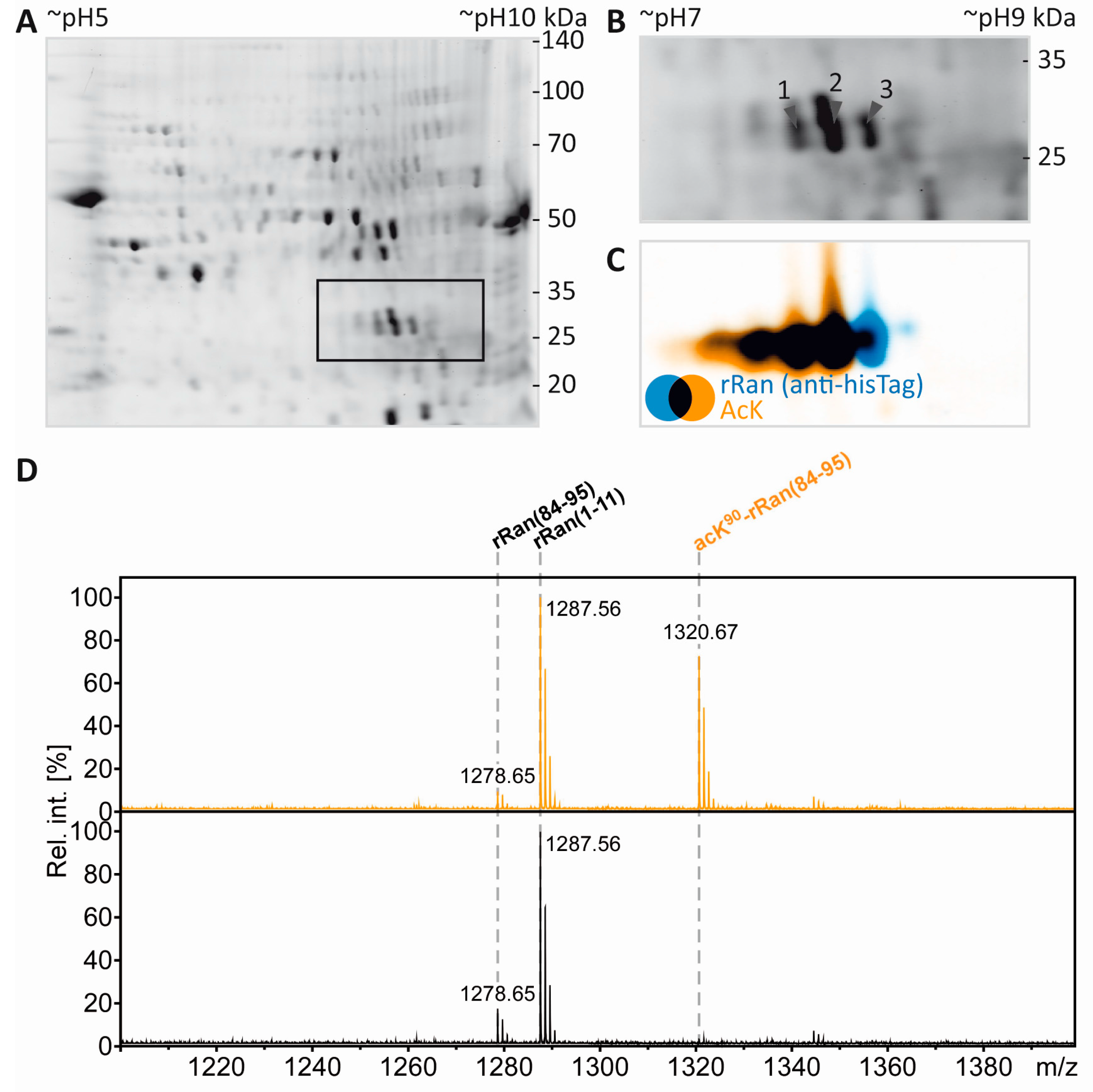
3.2. Immunodetection of Acetylated Myelin Proteins after Partial Transfer
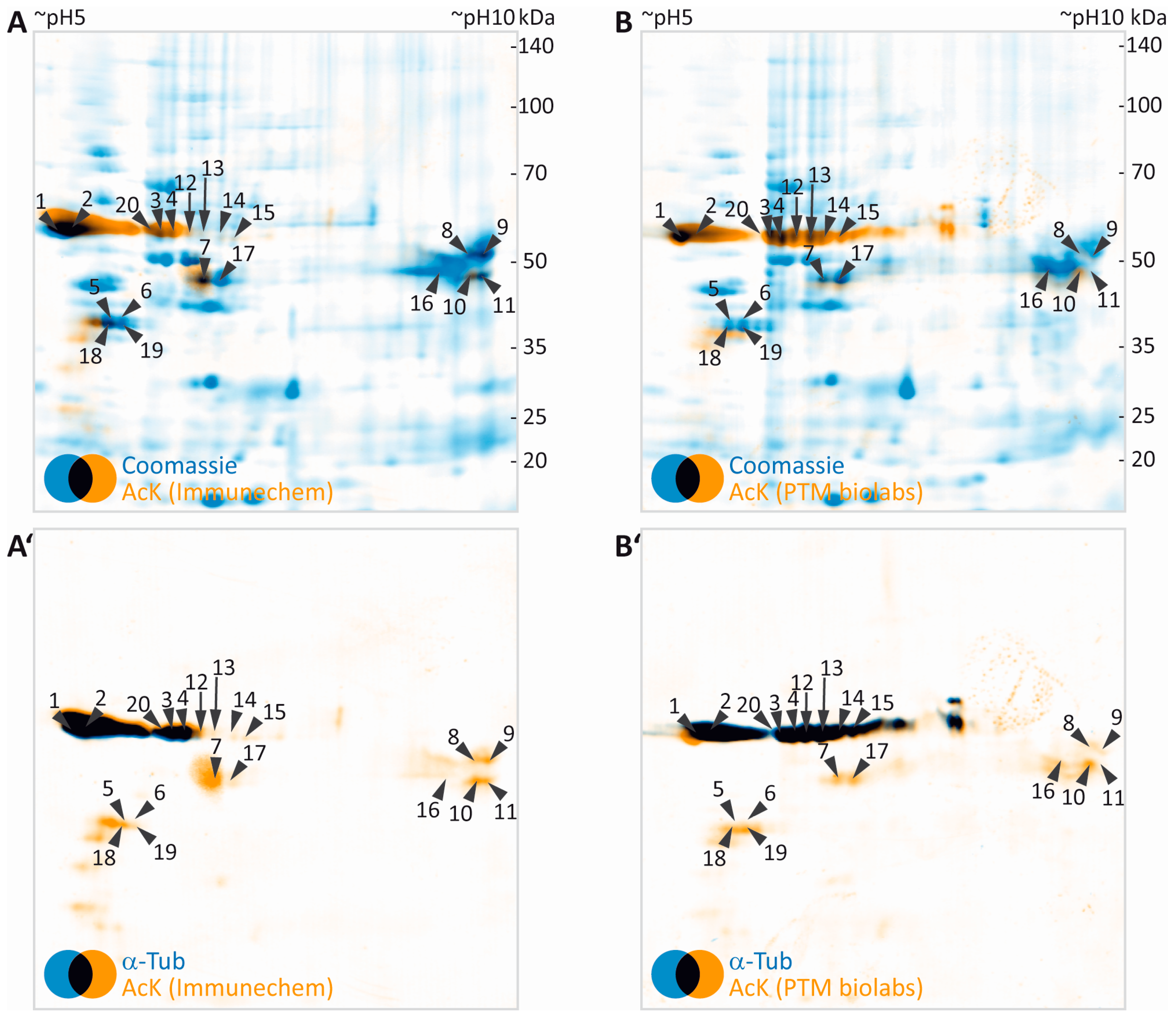
3.3. Identification of Potentially Acetylated Myelin Proteins
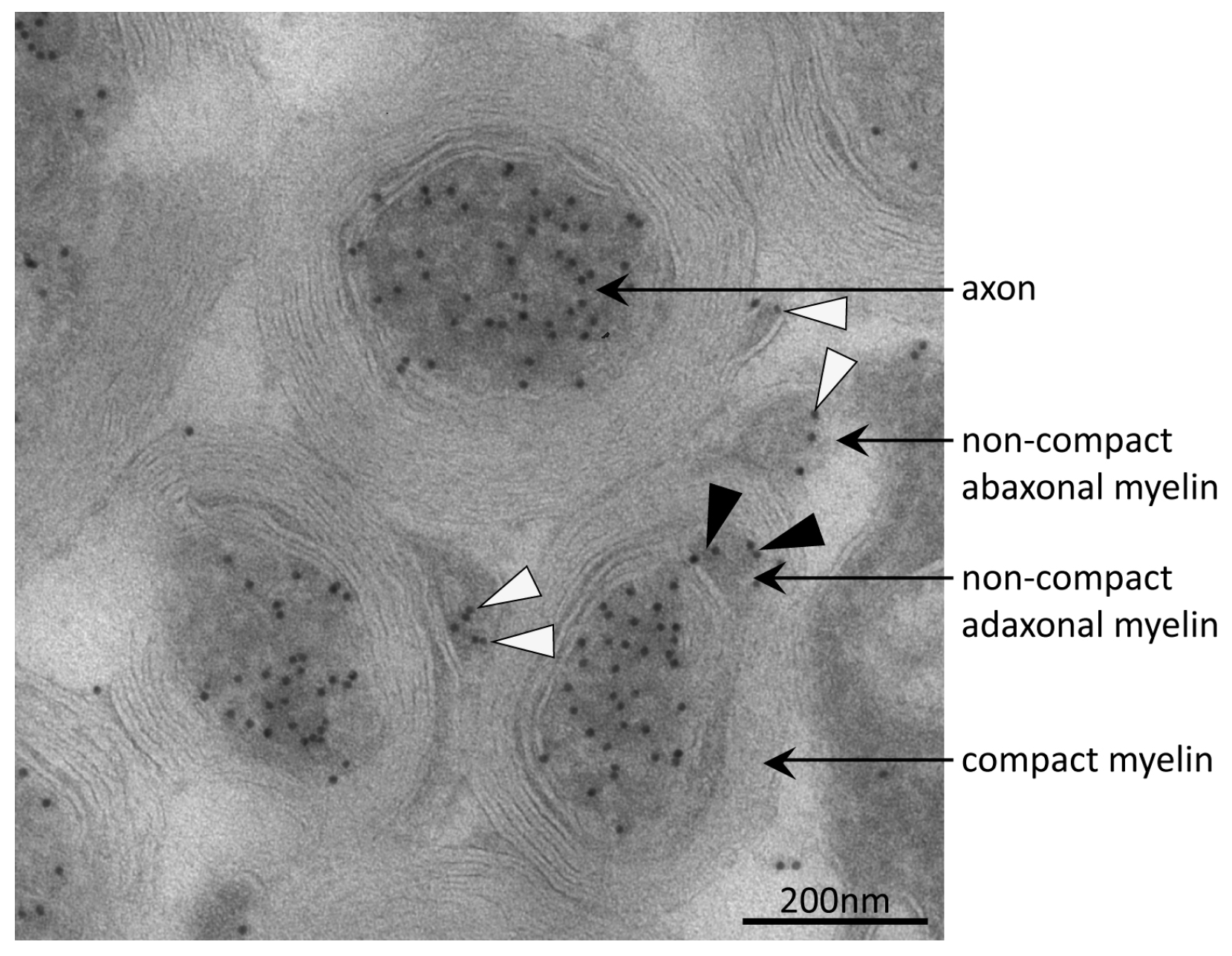
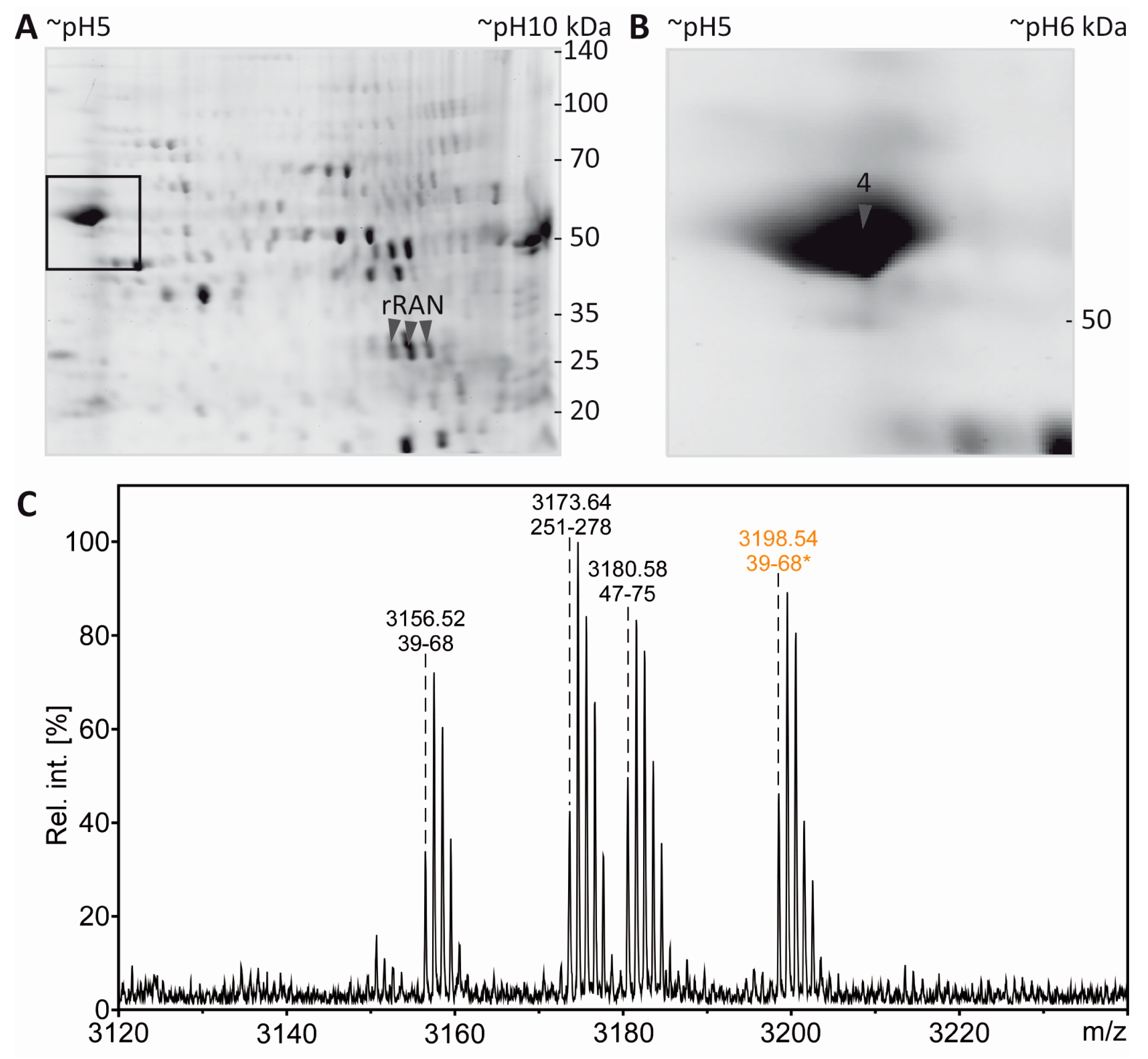
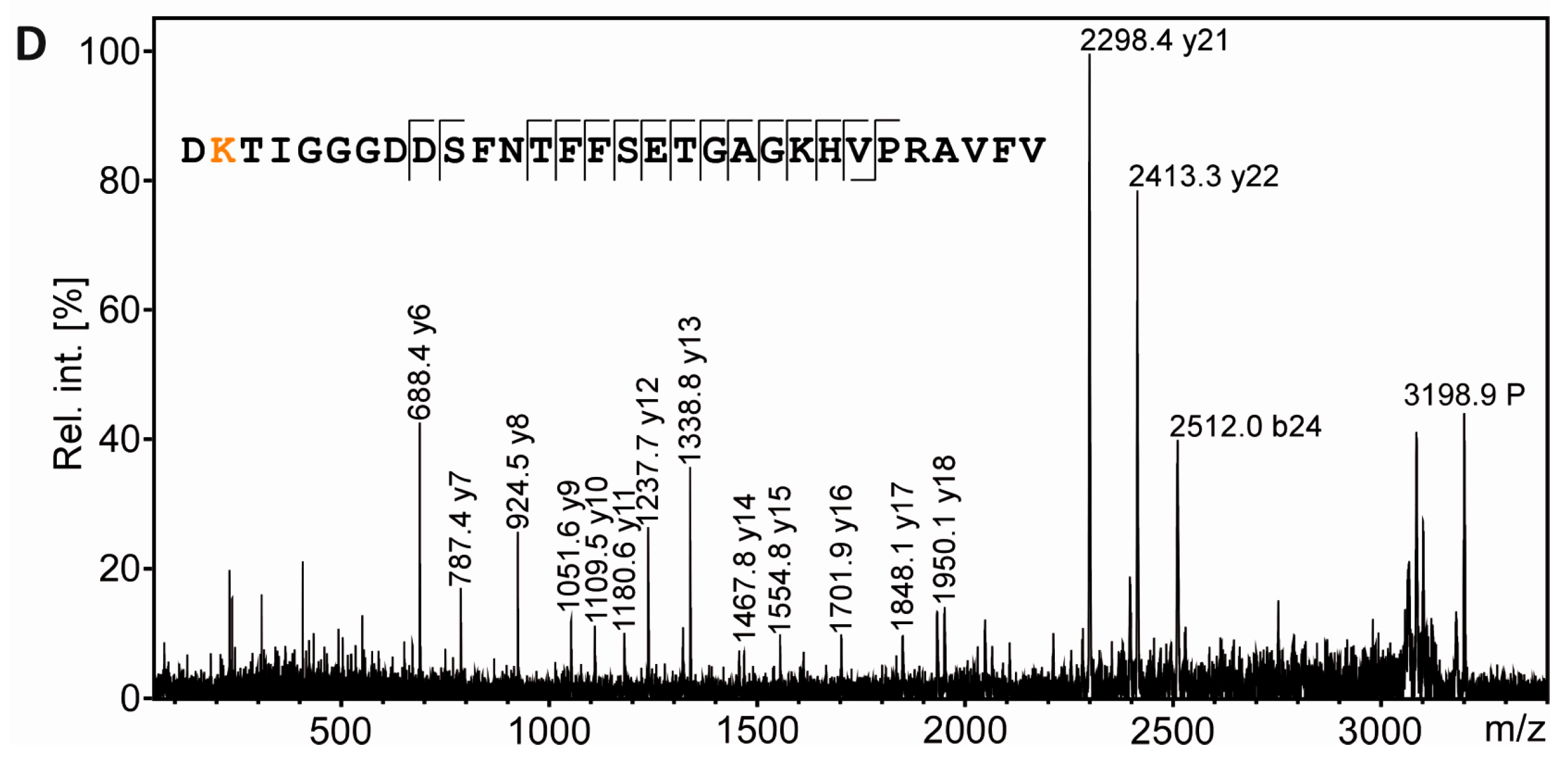
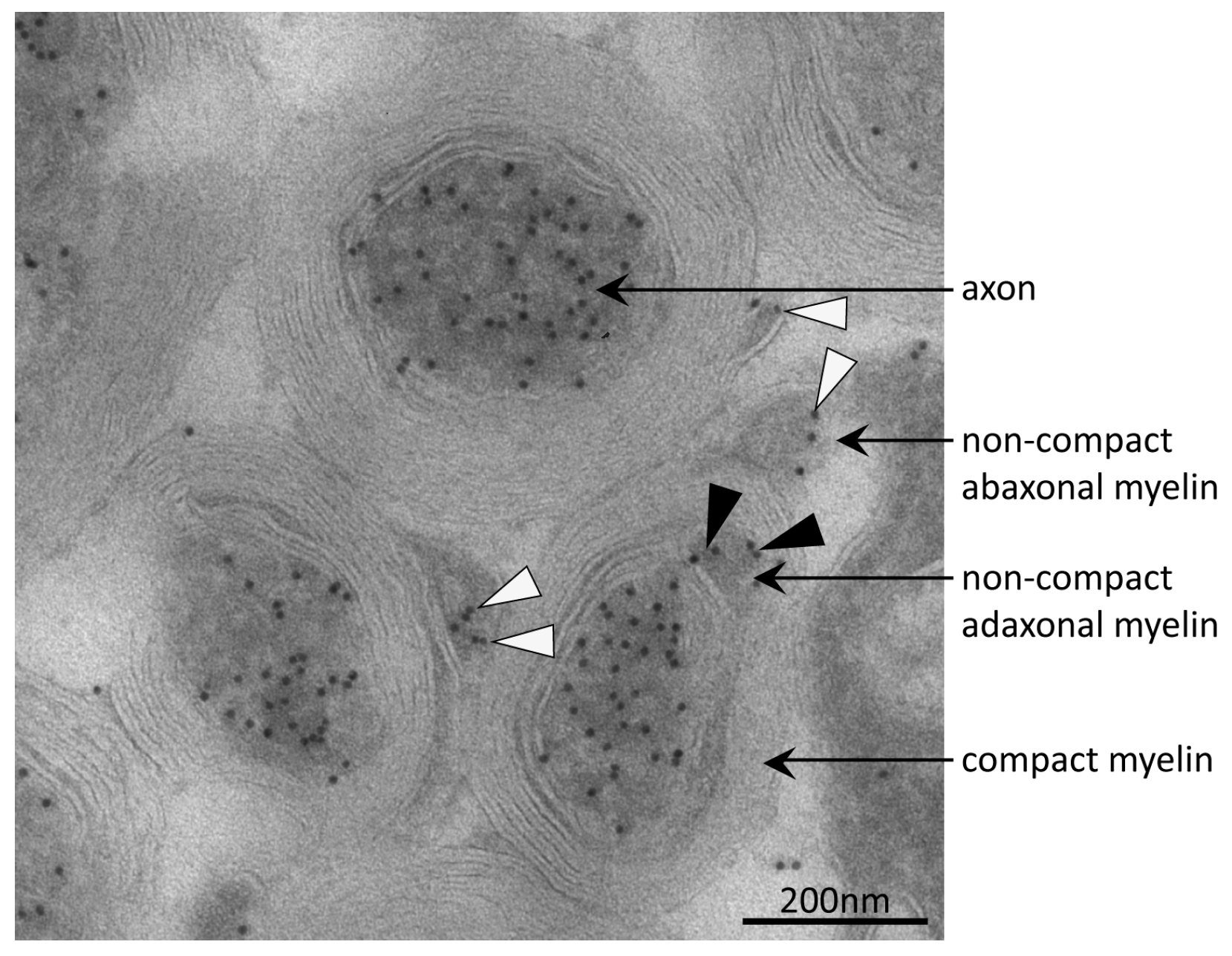
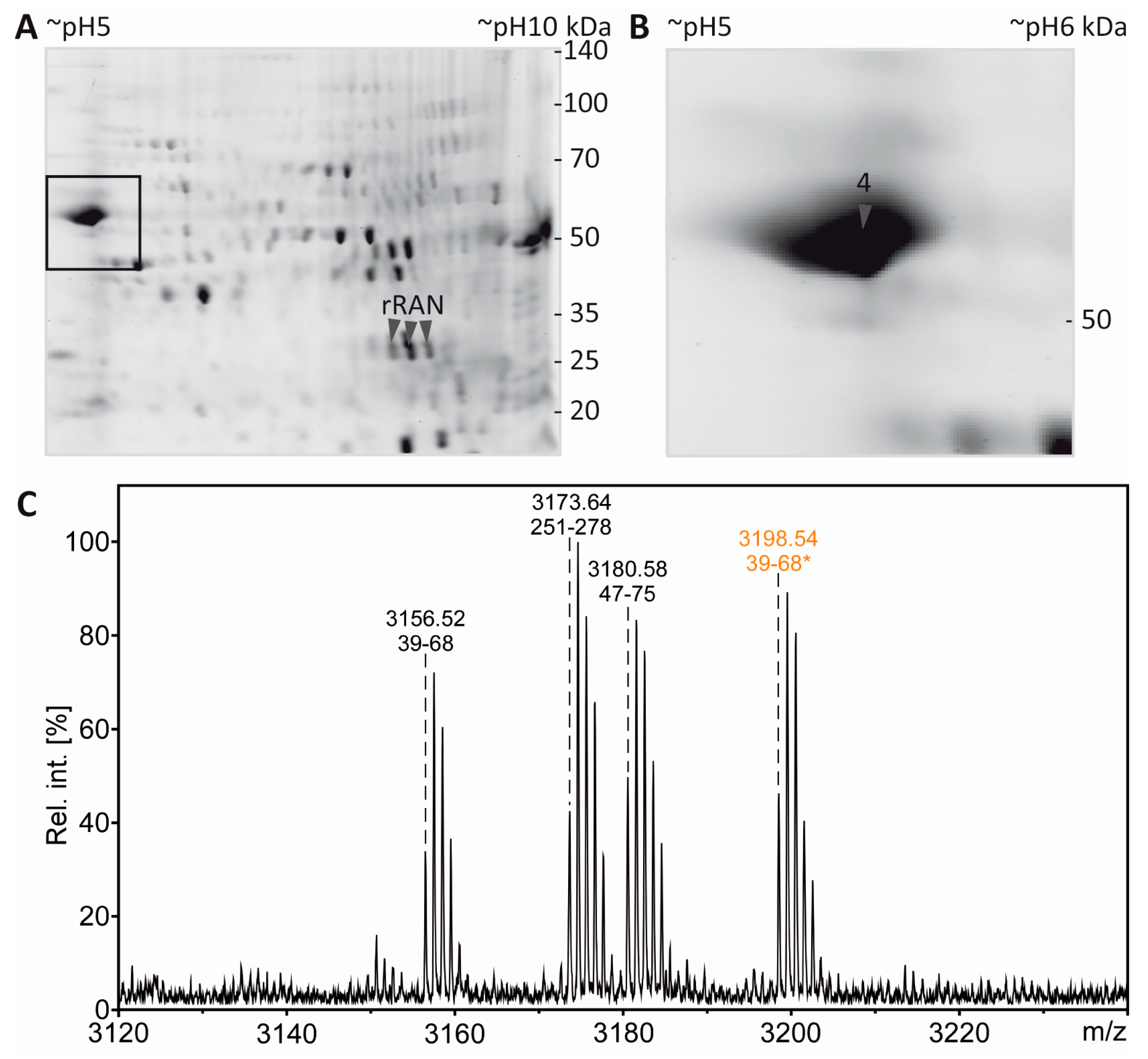
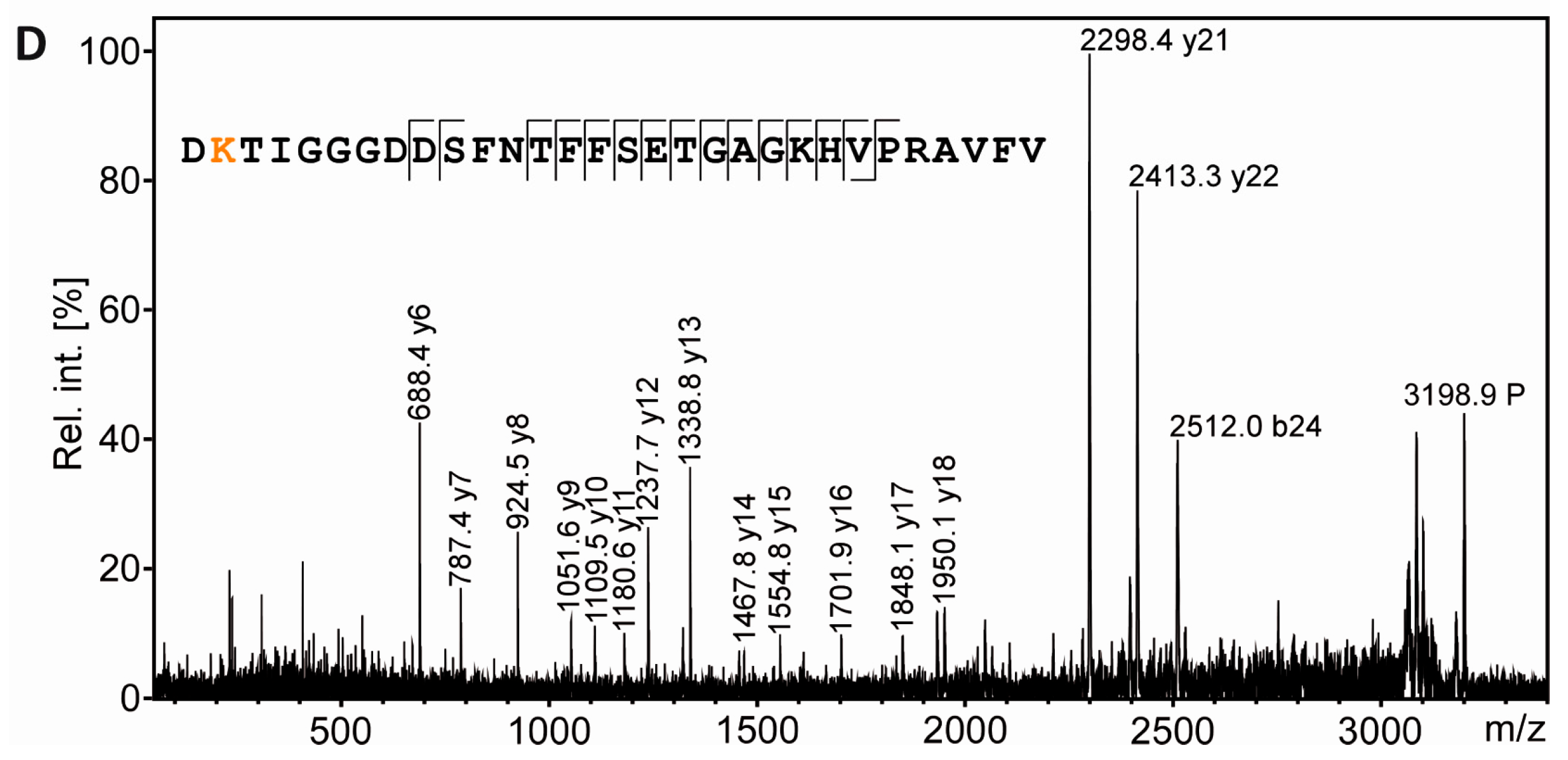
3.4. Tubulin Acetylation in Non-Compact Myelin
3.5. Separation of Proteoforms: The Benefit of Including Internal Control Proteins
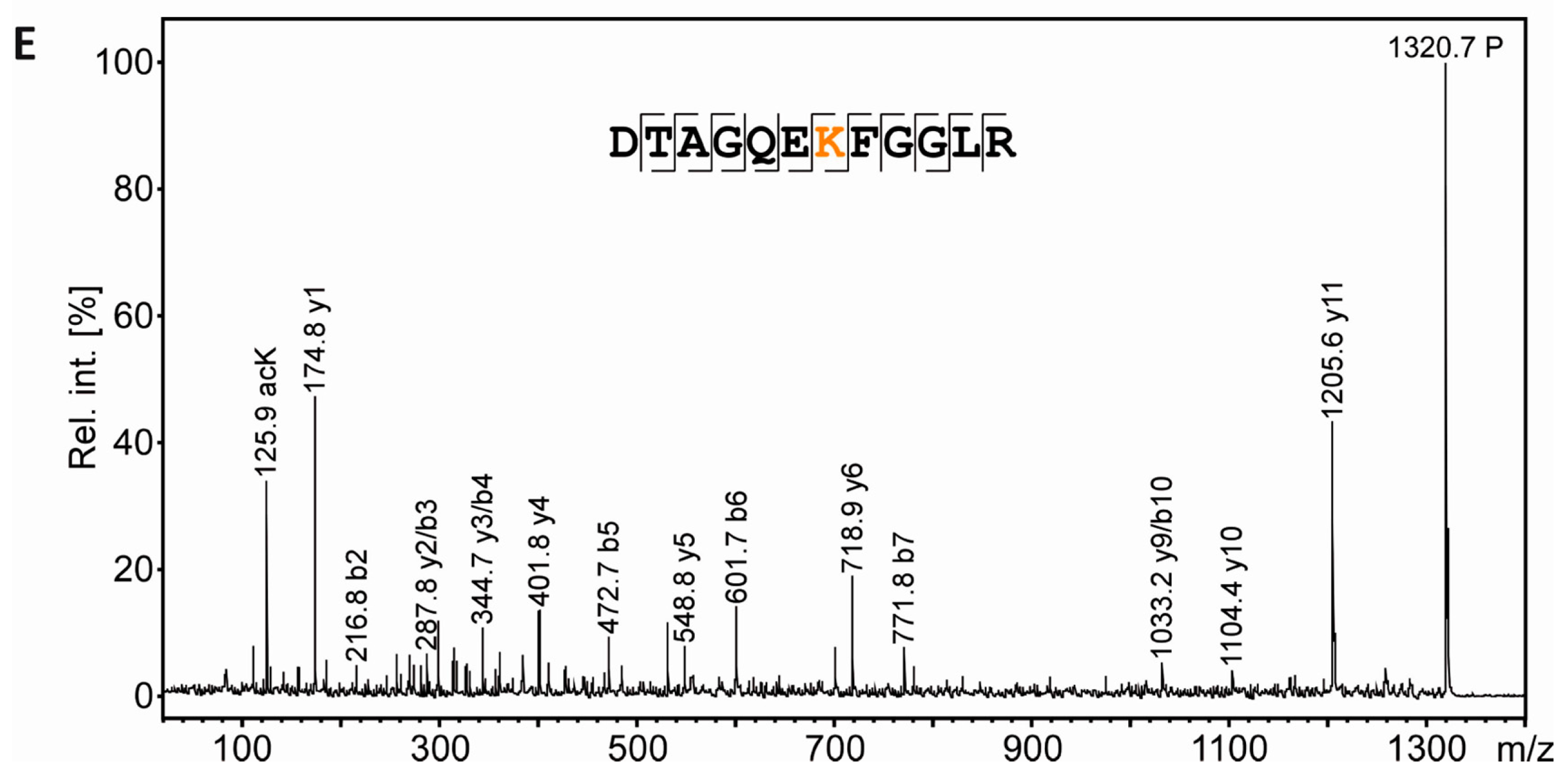
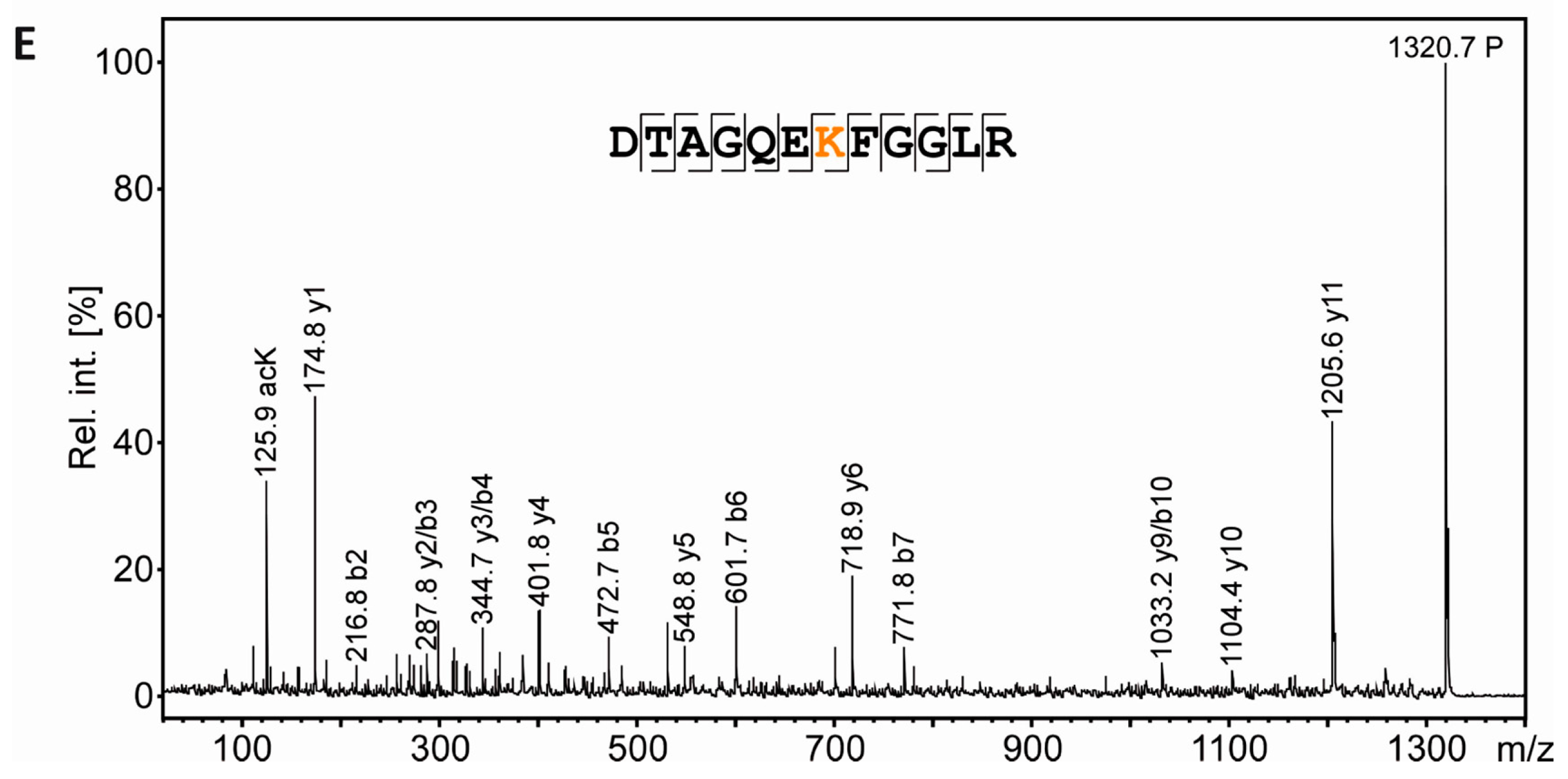
3.6. Mass Spectrometric Validation of AcK40 in α-Tubulin
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PTM | post-translational modification |
| 2D | two-dimensional |
| PVDF | Polyvinylidene difluoride |
| SERPA | serological proteome analysis |
| CNS | central nervous system |
| MS | mass spectrometry |
| TBS | Tris buffered saline |
| PBS | Phosphate buffered saline |
| IEF | isoelectric focusing |
| CCB | colloidal Coomassie |
| AcK | acetylated lysine |
| SDS | Sodium dodecyl sulfate |
| LDS | Lithium dodecyl sulfate |
| 2DE | 2D gel electrophoresis |
| SDS-PAGE | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
References
- Pagel, O.; Loroch, S.; Sickmann, A.; Zahedi, R.P. Current strategies and findings in clinically relevant post-translational modification-specific proteomics. Expert Rev. Proteom. 2015, 12, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villen, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef] [PubMed]
- Nave, K.A.; Werner, H.B. Myelination of the nervous system: Mechanisms and functions. Annu. Rev. Cell Dev. Biol. 2014, 30, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Jahn, O.; Tenzer, S.; Werner, H.B. Myelin proteomics: Molecular anatomy of an insulating sheath. Mol. Neurobiol. 2009, 40, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Norton, W.T.; Poduslo, S.E. Myelination in rat brain—Method of myelin isolation. J. Neurochem. 1973, 21, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Lees, M.B.; Paxman, S.A. Myelin proteins from different regions of central nervous system. J. Neurochem. 1974, 23, 825–831. [Google Scholar] [CrossRef] [PubMed]
- De Monasterio-Schrader, P.; Jahn, O.; Tenzer, S.; Wichert, S.P.; Patzig, J.; Werner, H.B. Systematic approaches to central nervous system myelin. Cell. Mol. Life Sci. 2012, 69, 2879–2894. [Google Scholar] [CrossRef] [PubMed]
- Dagley, L.F.; White, C.A.; Liao, Y.; Shi, W.; Smyth, G.K.; Orian, J.M.; Emili, A.; Purcell, A.W. Quantitative proteomic profiling reveals novel region-specific markers in the adult mouse brain. Proteomics 2014, 14, 241–261. [Google Scholar] [CrossRef] [PubMed]
- Gat-Viks, I.; Geiger, T.; Barbi, M.; Raini, G.; Elroy-Stein, O. Proteomics-level analysis of myelin formation and regeneration in a mouse model for Vanishing White Matter disease. J. Neurochem. 2015, 134, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, G.; Awasthi, A.; Belkaid, W.; De Faria, O., Jr.; Liazoghli, D.; Colman, D.R.; Dhaunchak, A.S. Lipidome and proteome map of myelin membranes. J. Neurosci. Res. 2013, 91, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Manrique-Hoyos, N.; Jurgens, T.; Gronborg, M.; Kreutzfeldt, M.; Schedensack, M.; Kuhlmann, T.; Schrick, C.; Bruck, W.; Urlaub, H.; Simons, M.; et al. Late motor decline after accomplished remyelination: Impact for progressive multiple sclerosis. Ann. Neurol. 2012, 71, 227–244. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.M.; Marta, C.B.; Claycomb, R.J.; Han, D.K.; Rasband, M.N.; Coetzee, T.; Pfeiffer, S.E. Proteomic mapping provides powerful insights into functional myelin biology. Proc. Natl. Acad. Sci. USA 2004, 101, 4643–4648. [Google Scholar] [CrossRef] [PubMed]
- Vanrobaeys, F.; Van Coster, R.; Dhondt, G.; Devreese, B.; Van Beeumen, J. Profiling of myelin proteins by 2D-gel electrophoresis and multidimensional liquid chromatography coupled to MALDI TOF-TOF mass spectrometry. J. Proteome Res. 2005, 4, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.B.; Kuhlmann, K.; Shen, S.; Uecker, M.; Schardt, A.; Dimova, K.; Orfaniotou, F.; Dhaunchak, A.; Brinkmann, B.G.; Mobius, W.; et al. Proteolipid protein is required for transport of sirtuin 2 into CNS myelin. J. Neurosci. 2007, 27, 7717–7730. [Google Scholar] [CrossRef] [PubMed]
- Dhaunchak, A.S.; Huang, J.K.; De Faria Junior, O.; Roth, A.D.; Pedraza, L.; Antel, J.P.; Bar-Or, A.; Colman, D.R. A proteome map of axoglial specializations isolated and purified from human central nervous system. Glia 2010, 58, 1949–1960. [Google Scholar] [CrossRef] [PubMed]
- Beniac, D.R.; Wood, D.D.; Palaniyar, N.; Ottensmeyer, F.P.; Moscarello, M.A.; Harauz, G. Cryoelectron microscopy of protein-lipid complexes of human myelin basic protein charge isomers differing in degree of citrullination. J. Struct. Biol. 2000, 129, 80–95. [Google Scholar] [CrossRef] [PubMed]
- Bizzozero, O.A.; Malkoski, S.P.; Mobarak, C.; Bixler, H.A.; Evans, J.E. Mass-spectrometric analysis of myelin proteolipids reveals new features of this family of palmitoylated membrane proteins. J. Neurochem. 2002, 81, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Harauz, G.; Ladizhansky, V.; Boggs, J.M. Structural polymorphism and multifunctionality of myelin basic protein. Biochemistry 2009, 48, 8094–8104. [Google Scholar] [CrossRef] [PubMed]
- Harauz, G.; Musse, A.A. A tale of two citrullines—Structural and functional aspects of myelin basic protein deimination in health and disease. Neurochem. Res. 2007, 32, 137–158. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Fu, Z.; Yang, J.; Troncosco, J.; Everett, A.D.; Van Eyk, J.E. Identification and characterization of citrulline-modified brain proteins by combining HCD and CID fragmentation. Proteomics 2013, 13, 2682–2691. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Mastronardi, F.G.; Wood, D.D.; Lubman, D.M.; Zand, R.; Moscarello, M.A. Multiple sclerosis: An important role for post-translational modifications of myelin basic protein in pathogenesis. Mol. Cell. Proteom. 2003, 2, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Laender, H.; Schulz, G.; Wolburg, H.; Nave, K.; Schulz, J.; Simons, M. Palmitoylation is a sorting determinant for transport to the myelin membrane. J. Cell Sci. 2005, 118, 2415–2423. [Google Scholar] [CrossRef] [PubMed]
- Tranquill, L.R.; Cao, L.; Ling, N.C.; Kalbacher, H.; Martin, R.M.; Whitaker, J.N. Enhanced T cell responsiveness to citrulline-containing myelin basic protein in multiple sclerosis patients. Mult. Scler. 2000, 6, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Sedzik, J.; Jastrzebski, J.P.; Grandis, M. Glycans of myelin proteins. J. Neurosci. Res. 2015, 93, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, F.; Sato, Y.; Tohyama, K.; Akagi, T.; Hashikawa, T.; Nagakura-Takagi, Y.; Sekine, Y.; Morita, N.; Baba, H.; Suzuki, Y.; et al. Opalin, a transmembrane sialylglycoprotein located in the central nervous system myelin paranodal loop membrane. J. Biol. Chem. 2008, 283, 20830–20840. [Google Scholar] [CrossRef] [PubMed]
- Ishii, A.; Dutta, R.; Wark, G.M.; Hwang, S.I.; Han, D.K.; Trapp, B.D.; Pfeiffer, S.E.; Bansal, R. Human myelin proteome and comparative analysis with mouse myelin. Proc. Natl. Acad. Sci. USA 2009, 106, 14605–14610. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, B.; Tang, J.; Cao, Q.; Wu, Y.; Wu, C.; Guo, J.; Ling, E.A.; Liang, F. Sirtuin 2, a mammalian homolog of yeast silent information regulator-2 longevity regulator, is an oligodendroglial protein that decelerates cell differentiation through deacetylating α-tubulin. J. Neurosci. 2007, 27, 2606–2616. [Google Scholar] [CrossRef] [PubMed]
- Southwood, C.M.; Peppi, M.; Dryden, S.; Tainsky, M.A.; Gow, A. Microtubule deacetylases, SirT2 and HDAC6, in the nervous system. Neurochem. Res. 2007, 32, 187–195. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 2003, 11, 437–444. [Google Scholar] [CrossRef]
- von Stechow, L.; Francavilla, C.; Olsen, J.V. Recent findings and technological advances in phosphoproteomics for cells and tissues. Expert Rev. Proteom. 2015, 12, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhao, W.; Yang, J.S.; Cheng, Z.; Luo, H.; Lu, Z.; Tan, M.; Gu, W.; Zhao, Y. Quantitative acetylome analysis reveals the roles of SIRT1 in regulating diverse substrates and cellular pathways. Mol. Cell. Proteom. 2012, 11, 1048–1062. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Lundby, A.; Lage, K.; Weinert, B.T.; Bekker-Jensen, D.B.; Secher, A.; Skovgaard, T.; Kelstrup, C.D.; Dmytriyev, A.; Choudhary, C.; Lundby, C.; et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012, 2, 419–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, Y.; Rardin, M.J.; Carrico, C.; He, W.; Sahu, A.K.; Gut, P.; Najjar, R.; Fitch, M.; Hellerstein, M.; Gibson, B.W.; et al. SIRT5 regulates both cytosolic and mitochondrial protein malonylation with glycolysis as a major target. Mol. Cell 2015, 59, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Sol, E.M.; Wagner, S.A.; Weinert, B.T.; Kumar, A.; Kim, H.S.; Deng, C.X.; Choudhary, C. Proteomic investigations of lysine acetylation identify diverse substrates of mitochondrial deacetylase Sirt3. PLoS ONE 2012, 7, e50545. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, S.; Liu, N.; Zhang, Y.; Liu, M.; Li, D.; Seto, E.; Yao, T.P.; Shui, W.; Zhou, J. Proteomic identification and functional characterization of MYH9, Hsc70, and DNAJA1 as novel substrates of HDAC6 deacetylase activity. Protein Cell 2015, 6, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Burnham-Marusich, A.R.; Plechaty, A.M.; Berninsone, P.M. Size-matched alkyne-conjugated cyanine fluorophores to identify differences in protein glycosylation. Electrophoresis 2014, 35, 2621–2625. [Google Scholar] [CrossRef] [PubMed]
- Cieniewski-Bernard, C.; Dupont, E.; Deracinois, B.; Lambert, M.; Bastide, B. Multiplexed detection of O-GlcNAcome, phosphoproteome, and whole proteome within the same gel. Front. Endocrinol. (Lausanne) 2014, 5, 184. [Google Scholar] [CrossRef] [PubMed]
- Marondedze, C.; Lilley, K.; Thomas, L. Comparative gel-based phosphoproteomics in response to signaling molecules. Methods Mol. Biol. 2013, 1016, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Nagai, K.; Arito, M.; Takakuwa, Y.; Ooka, S.; Sato, T.; Kurokawa, M.S.; Okamoto, K.; Uchida, T.; Suematsu, N.; Kato, T. Altered posttranslational modification on U1 small nuclear ribonucleoprotein 68k in systemic autoimmune diseases detected by 2D Western blot. Electrophoresis 2012, 33, 2028–2035. [Google Scholar] [CrossRef] [PubMed]
- Zong, C.; Young, G.W.; Wang, Y.; Lu, H.; Deng, N.; Drews, O.; Ping, P. Two-dimensional electrophoresis-based characterization of post-translational modifications of mammalian 20S proteasome complexes. Proteomics 2008, 8, 5025–5037. [Google Scholar] [CrossRef] [PubMed]
- Fritz, K.S.; Galligan, J.J.; Hirschey, M.D.; Verdin, E.; Petersen, D.R. Mitochondrial acetylome analysis in a mouse model of alcohol-induced liver injury utilizing SIRT3 knockout mice. J. Proteome Res. 2012, 11, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Li, J.; Guo, T.; Yang, H.; Li, M.; Sang, S.; Li, X.; Desiderio, D.M.; Zhan, X. Nitroproteins in human astrocytomas discovered by gel electrophoresis and tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2015, 26, 2062–2076. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Parrott, A.M.; Liu, T.; Beuve, A.; Li, H. Functional proteomics approaches for the identification of transnitrosylase and denitrosylase targets. Methods 2013, 62, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Kniemeyer, O.; Ebel, F.; Kruger, T.; Bacher, P.; Scheffold, A.; Luo, T.; Strassburger, M.; Brakhage, A.A. Immunoproteomics of Aspergillus for the development of biomarkers and immunotherapies. Proteom. Clin. Appl. 2016, 10, 910–921. [Google Scholar] [CrossRef] [PubMed]
- Jahn, O.; Tenzer, S.; Bartsch, N.; Patzig, J.; Werner, H.B. Myelin proteome analysis: Methods and implications for the myelin cytoskeleton. In The Cytoskeleton: Imaging, Isolation, and Interaction; Dermietzel, R., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 335–353. [Google Scholar]
- Wessel, D.; Flügge, U.-I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984, 138, 141–143. [Google Scholar] [CrossRef]
- Neumann, H.; Hancock, S.M.; Buning, R.; Routh, A.; Chapman, L.; Somers, J.; Owen-Hughes, T.; van Noort, J.; Rhodes, D.; Chin, J.W. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol. Cell 2009, 36, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Jahn, O.; Hesse, D.; Reinelt, M.; Kratzin, H.D. Technical innovations for the automated identification of gel-separated proteins by MALDI-TOF mass spectrometry. Anal. Bioanal. Chem. 2006, 386, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Martens, H.; Hassouna, I.; Oliveira, B.; Erck, C.; Zafeiriou, M.P.; Peteri, U.K.; Hesse, D.; Gerhart, S.; Altas, B.; et al. Widespread expression of erythropoietin receptor in brain and its induction by injury. Mol. Med. 2015, 21, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.B.; Kramer-Albers, E.M.; Strenzke, N.; Saher, G.; Tenzer, S.; Ohno-Iwashita, Y.; De Monasterio-Schrader, P.; Mobius, W.; Moser, T.; Griffiths, I.R.; et al. A critical role for the cholesterol-associated proteolipids PLP and M6B in myelination of the central nervous system. Glia 2013, 61, 567–586. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.R.; Churchward, M.A.; Butt, R.H.; Coorssen, J.R. Assessing detection methods for gel-based proteomic analyses. J. Proteome Res. 2007, 6, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Wehr, N.B.; Levine, R.L. Quantitation of protein on gels and blots by infrared fluorescence of Coomassie blue and Fast Green. Anal. Biochem. 2006, 350, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Patzig, J.; Erwig, M.S.; Tenzer, S.; Kusch, K.; Dibaj, P.; Mobius, W.; Goebbels, S.; Schaeren-Wiemers, N.; Nave, K.A.; Werner, H.B. Septin/anillin filaments scaffold central nervous system myelin to accelerate nerve conduction. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Edgar, J.M.; McLaughlin, M.; Werner, H.B.; McCulloch, M.C.; Barrie, J.A.; Brown, A.; Faichney, A.B.; Snaidero, N.; Nave, K.A.; Griffiths, I.R. Early ultrastructural defects of axons and axon-glia junctions in mice lacking expression of Cnp1. Glia 2009, 57, 1815–1824. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Ohayon, D.; McKenzie, I.A.; Sinclair-Wilson, A.; Wright, J.L.; Fudge, A.D.; Emery, B.; Li, H.; Richardson, W.D. Rapid production of new oligodendrocytes is required in the earliest stages of motor-skill learning. Nat. Neurosci. 2016, 19, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Bouchut, A.; Chawla, A.R.; Jeffers, V.; Hudmon, A.; Sullivan, W.J., Jr. Proteome-wide lysine acetylation in cortical astrocytes and alterations that occur during infection with brain parasite toxoplasma gondii. PLoS ONE 2015, 10, e0117966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebert, A.S.; Dittenhafer-Reed, K.E.; Yu, W.; Bailey, D.J.; Selen, E.S.; Boersma, M.D.; Carson, J.J.; Tonelli, M.; Balloon, A.J.; Higbee, A.J.; et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 2013, 49, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Holper, S.; Nolte, H.; Bober, E.; Braun, T.; Kruger, M. Dissection of metabolic pathways in the db/db mouse model by integrative proteome and acetylome analysis. Mol. Biosyst. 2015, 11, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Masri, S.; Patel, V.R.; Eckel-Mahan, K.L.; Peleg, S.; Forne, I.; Ladurner, A.G.; Baldi, P.; Imhof, A.; Sassone-Corsi, P. Circadian acetylome reveals regulation of mitochondrial metabolic pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 3339–3344. [Google Scholar] [CrossRef] [PubMed]
- Rardin, M.J.; Newman, J.C.; Held, J.M.; Cusack, M.P.; Sorensen, D.J.; Li, B.; Schilling, B.; Mooney, S.D.; Kahn, C.R.; Verdin, E.; et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of sirt3 in metabolic pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 6601–6606. [Google Scholar] [CrossRef] [PubMed]
- Franzel, B.; Fischer, F.; Steegborn, C.; Wolters, D.A. Proteinase k improves quantitative acylation studies. Proteomics 2015, 15, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.V.; Lee, J.E.; Sweredoski, M.J.; Yang, S.J.; Jeon, S.J.; Harrison, J.S.; Yim, J.H.; Lee, S.G.; Handa, H.; Kuhlman, B.; et al. Glutamine triggers acetylation-dependent degradation of glutamine synthetase via the thalidomide receptor cereblon. Mol. Cell 2016, 61, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Svinkina, T.; Gu, H.; Silva, J.C.; Mertins, P.; Qiao, J.; Fereshetian, S.; Jaffe, J.D.; Kuhn, E.; Udeshi, N.D.; Carr, S.A. Deep, quantitative coverage of the lysine acetylome using novel anti-acetyl-lysine antibodies and an optimized proteomic workflow. Mol. Cell. Proteom. 2015, 14, 2429–2440. [Google Scholar] [CrossRef] [PubMed]
- Sadoul, K.; Khochbin, S. The growing landscape of tubulin acetylation: Lysine 40 and many more. Biochem. J. 2016, 473, 1859–1868. [Google Scholar] [CrossRef] [PubMed]
- Yu, I.; Garnham, C.P.; Roll-Mecak, A. Writing and reading the tubulin code. J. Biol. Chem. 2015, 290, 17163–17172. [Google Scholar] [CrossRef] [PubMed]
- Lunn, K.F.; Baas, P.W.; Duncan, I.D. Microtubule organization and stability in the oligodendrocyte. J. Neurosci. 1997, 17, 4921–4932. [Google Scholar] [PubMed]
- Sherman, D.L.; Brophy, P.J. Mechanisms of axon ensheathment and myelin growth. Nat. Rev. Neurosci. 2005, 6, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; O’connor, L.T.; Yu, W.; Baas, P.W.; Duncan, I.D. Microtubule alterations in cultured taiep rat oligodendrocytes lead to deficits in myelin membrane formation. J. Neurocytol. 1999, 28, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Mischerikow, N.; Heck, A.J. Targeted large-scale analysis of protein acetylation. Proteomics 2011, 11, 571–589. [Google Scholar] [CrossRef] [PubMed]
- Cong, W.T.; Hwang, S.Y.; Jin, L.T.; Choi, J.K. Detection of proteins on blots using Direct Blue 71. In The Protein Protocols Handbook, 3rd ed.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 729–735. [Google Scholar]
- Hong, H.Y.; Yoo, G.S.; Choi, J.K. Direct Blue 71 staining of proteins bound to blotting membranes. Electrophoresis 2000, 21, 841–845. [Google Scholar] [CrossRef]
- Welinder, C.; Ekblad, L. Coomassie staining as loading control in Western blot analysis. J. Proteome Res. 2011, 10, 1416–1419. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, V.; De, P.K. Western blot of proteins from Coomassie-stained polyacrylamide gels. Anal. Biochem. 1996, 234, 102–104. [Google Scholar] [CrossRef] [PubMed]
- Bifulco, M.; Laezza, C.; Stingo, S.; Wolff, J. 2′,3′-Cyclic nucleotide 3’-phosphodiesterase: A membrane-bound, microtubule-associated protein and membrane anchor for tubulin. Proc. Natl. Acad. Sci. USA 2002, 99, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.; Fifková, E. Distribution of acetylated α-tubulin in brain. Cell Tissue Res. 1991, 265, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Dubey, J.; Ratnakaran, N.; Koushika, S.P. Neurodegeneration and microtubule dynamics: Death by a thousand cuts. Front. Cell. Neurosci. 2015, 9, 343. [Google Scholar] [CrossRef] [PubMed]
- Czopka, T.; Ffrench-Constant, C.; Lyons, D.A. Individual oligodendrocytes have only a few hours in which to generate new myelin sheaths in vivo. Dev. Cell 2013, 25, 599–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, K.M.; Psachoulia, K.; Tripathi, R.B.; Dunn, S.J.; Cossell, L.; Attwell, D.; Tohyama, K.; Richardson, W.D. Oligodendrocyte dynamics in the healthy adult CNS: Evidence for myelin remodeling. Neuron 2013, 77, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Yeung, M.S.; Zdunek, S.; Bergmann, O.; Bernard, S.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Brundin, L.; et al. Dynamics of oligodendrocyte generation and myelination in the human brain. Cell 2014, 159, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Funfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Mobius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Nave, K.A. Myelination and support of axonal integrity by glia. Nature 2010, 468, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Mobius, W.; Goetze, B.; Jahn, H.M.; Huang, W.; et al. Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron 2016, 91, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, A.; Kondo, Y.; Song, J.; Liu, S.; Compston, A.; Black, J.A.; Waxman, S.G.; Duncan, I.D. Slowly progressive axonal degeneration in a rat model of chronic, nonimmune-mediated demyelination. J. Neuropathol. Exp. Neurol. 2010, 69, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- LeDizet, M.; Piperno, G. Identification of an acetylation site of Chlamydomonas α-tubulin. Proc. Natl. Acad. Sci. USA 1987, 84, 5720–5724. [Google Scholar] [CrossRef] [PubMed]
- Weinert, B.T.; Wagner, S.A.; Horn, H.; Henriksen, P.; Liu, W.R.; Olsen, J.V.; Jensen, L.J.; Choudhary, C. Proteome-wide mapping of the Drosophila acetylome demonstrates a high degree of conservation of lysine acetylation. Sci. Signal. 2011, 4, ra48. [Google Scholar] [CrossRef] [PubMed]
- Giansanti, P.; Tsiatsiani, L.; Low, T.Y.; Heck, A.J. Six alternative proteases for mass spectrometry-based proteomics beyond trypsin. Nat. Protoc. 2016, 11, 993–1006. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Xiong, Y.; Ren, Y.; Zhang, L.; He, X.; Wang, X.; Liu, M.; Li, D.; Shui, W.; Zhou, J. Proteomic profiling and functional characterization of multiple post-translational modifications of tubulin. J. Proteome Res. 2015, 14, 3292–3304. [Google Scholar] [CrossRef] [PubMed]
- Couto, N.; Martins, J.; Lourenco, A.M.; Pomba, C.; Varela Coelho, A. Identification of vaccine candidate antigens of Staphylococcus pseudintermedius by whole proteome characterization and serological proteomic analyses. J. Proteom. 2016, 133, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Kolata, J.; Bode, L.G.; Holtfreter, S.; Steil, L.; Kusch, H.; Holtfreter, B.; Albrecht, D.; Hecker, M.; Engelmann, S.; van Belkum, A.; et al. Distinctive patterns in the human antibody response to Staphylococcus aureus bacteremia in carriers and non-carriers. Proteomics 2011, 11, 3914–3927. [Google Scholar] [CrossRef] [PubMed]
- Vu, C.H.; Kolata, J.; Stentzel, S.; Beyer, A.; Gesell Salazar, M.; Steil, L.; Pane-Farre, J.; Ruhmling, V.; Engelmann, S.; Gotz, F.; et al. Adaptive immune response to lipoproteins of Staphylococcus aureus in healthy subjects. Proteomics 2016, 16, 2667–2677. [Google Scholar] [CrossRef] [PubMed]
- Dutoit-Lefevre, V.; Dubucquoi, S.; Launay, D.; Sobanski, V.; Dussart, P.; Chafey, P.; Broussard, C.; Duban-Deweer, S.; Vermersch, P.; Prin, L.; et al. An optimized fluorescence-based bidimensional immunoproteomic approach for accurate screening of autoantibodies. PLoS ONE 2015, 10, e0132142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloppot, P.; Selle, M.; Kohler, C.; Stentzel, S.; Fuchs, S.; Liebscher, V.; Muller, E.; Kale, D.; Ohlsen, K.; Broker, B.M.; et al. Microarray-based identification of human antibodies against Staphylococcus aureus antigens. Proteom. Clin. Appl. 2015, 9, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Selle, M.; Hertlein, T.; Oesterreich, B.; Klemm, T.; Kloppot, P.; Muller, E.; Ehricht, R.; Stentzel, S.; Broker, B.M.; Engelmann, S.; et al. Global antibody response to Staphylococcus aureus live-cell vaccination. Sci. Rep. 2016, 6, 24754. [Google Scholar] [CrossRef] [PubMed]
- Stentzel, S.; Sundaramoorthy, N.; Michalik, S.; Nordengrun, M.; Schulz, S.; Kolata, J.; Kloppot, P.; Engelmann, S.; Steil, L.; Hecker, M.; et al. Specific serum IgG at diagnosis of Staphylococcus aureus bloodstream invasion is correlated with disease progression. J. Proteom. 2015, 128, 1–7. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | UniProtKB Accession | Protein Name | MW (Da) | CpI | Spot | PMF Score Gel A/Gel B | PMF Coverage [%] Gel A/Gel B | No. of Peptides Sequenced Gel A/Gel B | Cumulative MS/MS Score Gel A/Gel B | References for Previous Identification of Lysine Acetylation |
|---|---|---|---|---|---|---|---|---|---|---|
| Atp5b | P56480 | ATP synthase subunit beta, mitochondrial | 56,265 | 5.1 | 1 | 212/224 | 66/58 | 3/4 | 227/231 | [33,34,43,58,59,60,61,62] |
| Cnp | P16330 | 2′,3′-cyclic-nucleotide 3′-phosphodiesterase | 47,493 | 9.1 | 8 | 72/61 | 37/26 | 2/- | 94/- | [34,58,59,60,63] |
| 9 | 218/147 | 55/39 | 3/2 | 262/106 | ||||||
| 16 | 246/147 | 59/46 | 4/2 | 353/107 | ||||||
| Enpp6 | Q8BGN3 | Ectonucleotide pyrophosphatase/phosphodiesterase family member 6 | 50,928 | 6.9 | 13 | -/67 | -/29 | -/1 | -/29 | [34,63] |
| 14 | 72/50 | 21/15 | 2/1 | 58/30 | ||||||
| 15 | 102/- | 32/- | 3/1 | 77/30 | ||||||
| Glul | P15105 | Glutamine synthetase | 42,834 | 6.6 | 7 | 127/143 | 28/32 | 2/4 | 61/183 | [34,43,59,60,63,64,65] |
| 17 | 163/197 | 50/52 | 4/4 | 281/236 | ||||||
| Gnb1 | P62874 | Guanine nucleotide-binding protein G(I)/G(S)/G(T) subunit beta-1 | 38,151 | 5.6 | 5 | 73/98 | 27/34 | -/2 | -/43 | [34] |
| 6 | 90/130 | 27/41 | -/3 | -/102 | ||||||
| Gnb2 | P62880 | Guanine nucleotide-binding protein G(I)/G(S)/G(T) subunit beta-2 | 38,048 | 5.6 | 18 | 132/92 | 48/32 | 2/1 | 67/29 | - |
| 19 | 102/167 | 44/55 | 3/3 | 130/122 | ||||||
| Got2 | P05202 | Aspartate aminotransferase, mitochondrial | 47,780 | 9.1 | 10 | 93/105 | 34/35 | -/1 | -/33 | [33,34,43,58,59,60,61,62,65] |
| 11 | 107/- | 39/- | 3/- | 50/- | ||||||
| Sept8 | Q8CHH9 | Septin-8 | 50,123 | 5.7 | 20 | 96/98 | 33/33 | 1/- | 28/- | [34,65] |
| Tuba1a | P69369 | Tubulin alpha-1A chain | 50,788 | 4.9 | 1 | 132/83 | 51/36 | 4/4 | 73/130 | [33,58,66] |
| 3 | 70/90 | 24/33 | 1/1 | 42/41 | ||||||
| 20 | 61/60 | 21/20 | 1/2 | 42/60 | ||||||
| Tuba1c | P68373 | Tubulin alpha-1C chain | 50,562 | 5.0 | 4 | 76/76 | 33/33 | 1/1 | 36/36 | [33,58,59,66] |
| Tubb4a | Q9D6F9 | Tubulin beta-4A chain | 50,010 | 4.8 | 1 | 105/74 | 43/26 | 2/- | 42/- | [33,58,59,66] |
| 4 | 88/147 | 26/50 | -/1 | -/32 | ||||||
| 12 | -/141 | -/40 | -/1 | -/27 | ||||||
| 13 | -/147 | -/46 | -/1 | -/28 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kusch, K.; Uecker, M.; Liepold, T.; Möbius, W.; Hoffmann, C.; Neumann, H.; Werner, H.B.; Jahn, O. Partial Immunoblotting of 2D-Gels: A Novel Method to Identify Post-Translationally Modified Proteins Exemplified for the Myelin Acetylome. Proteomes 2017, 5, 3. https://doi.org/10.3390/proteomes5010003
Kusch K, Uecker M, Liepold T, Möbius W, Hoffmann C, Neumann H, Werner HB, Jahn O. Partial Immunoblotting of 2D-Gels: A Novel Method to Identify Post-Translationally Modified Proteins Exemplified for the Myelin Acetylome. Proteomes. 2017; 5(1):3. https://doi.org/10.3390/proteomes5010003
Chicago/Turabian StyleKusch, Kathrin, Marina Uecker, Thomas Liepold, Wiebke Möbius, Christian Hoffmann, Heinz Neumann, Hauke B. Werner, and Olaf Jahn. 2017. "Partial Immunoblotting of 2D-Gels: A Novel Method to Identify Post-Translationally Modified Proteins Exemplified for the Myelin Acetylome" Proteomes 5, no. 1: 3. https://doi.org/10.3390/proteomes5010003