In-Depth Proteomic Characterization of Classical and Non-Classical Monocyte Subsets

 , , , , ,
, , , , ,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects and Samples
2.2. Purification of PB Monocyte Subsets
2.3. Cell Lysis
2.4. Sample Processing and Mass Spectrometry Analysis
2.5. Shotgun Data Analysis
2.6. Functional Analysis
3. Results and Discussion
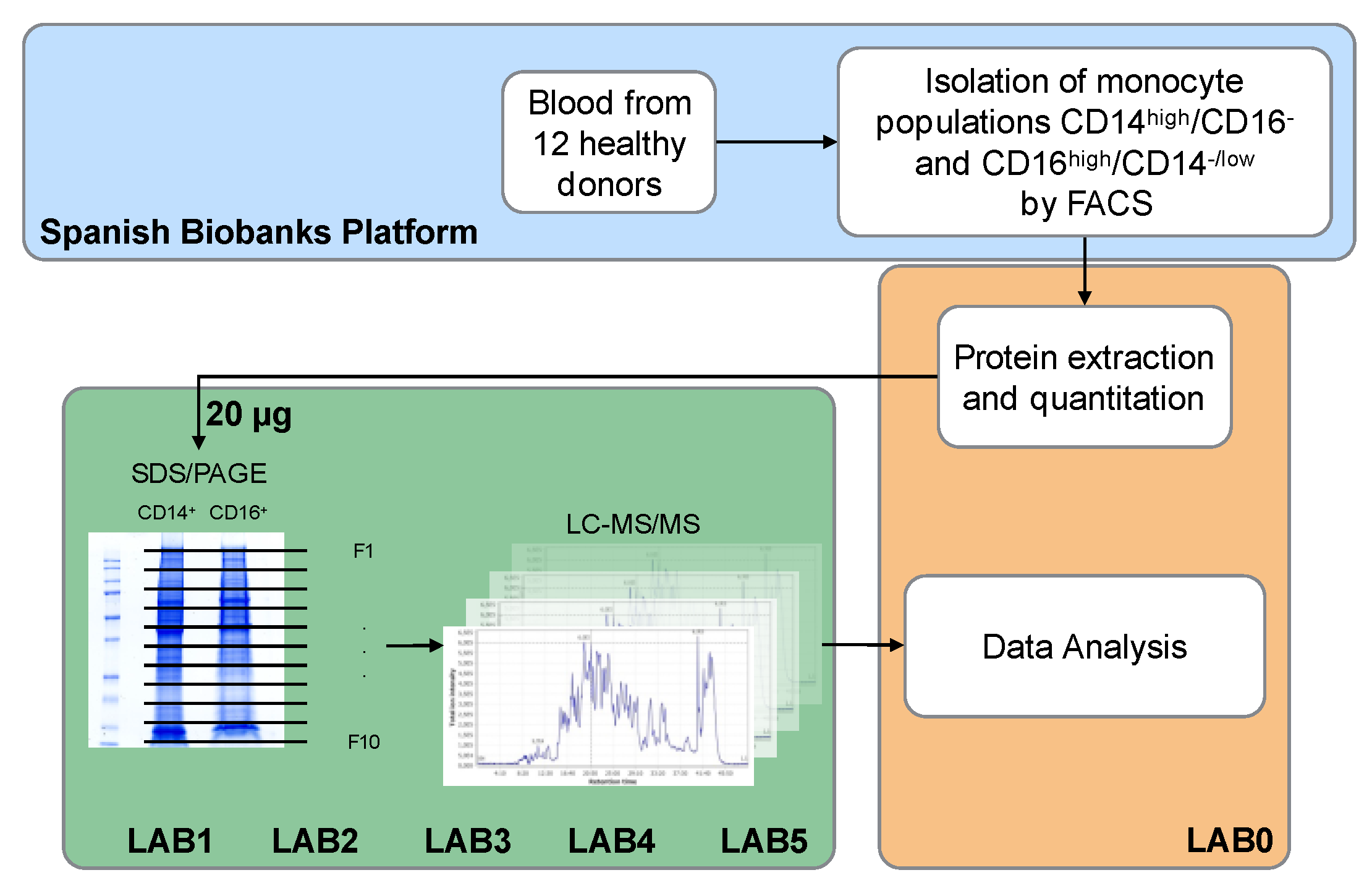
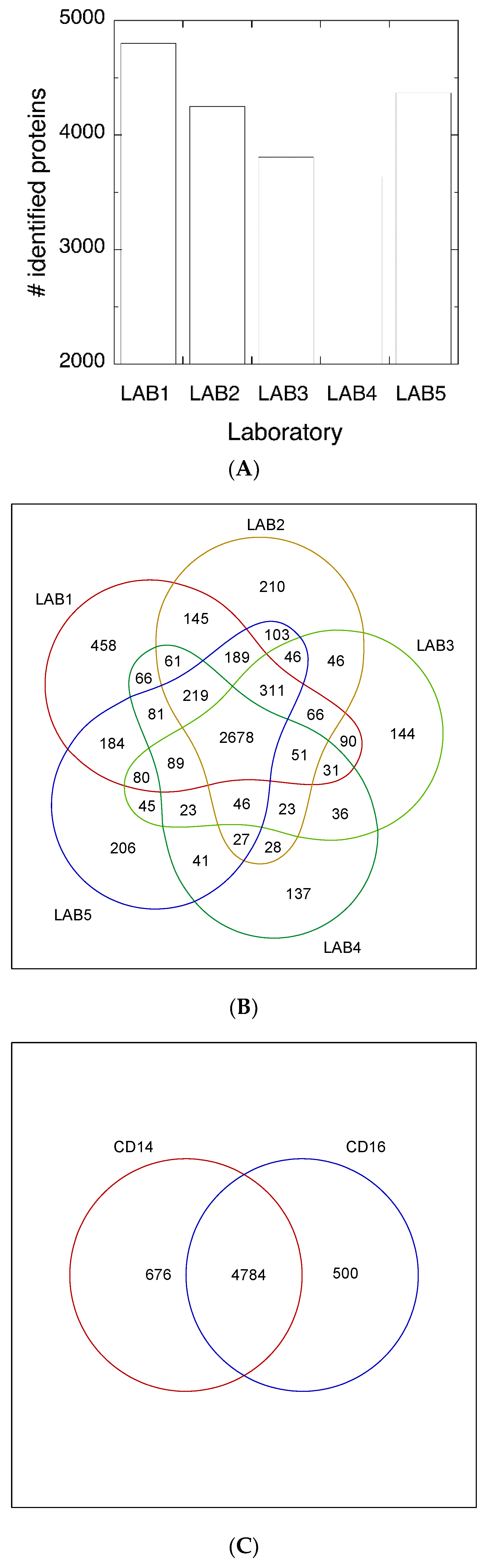
3.1. Experimental Design
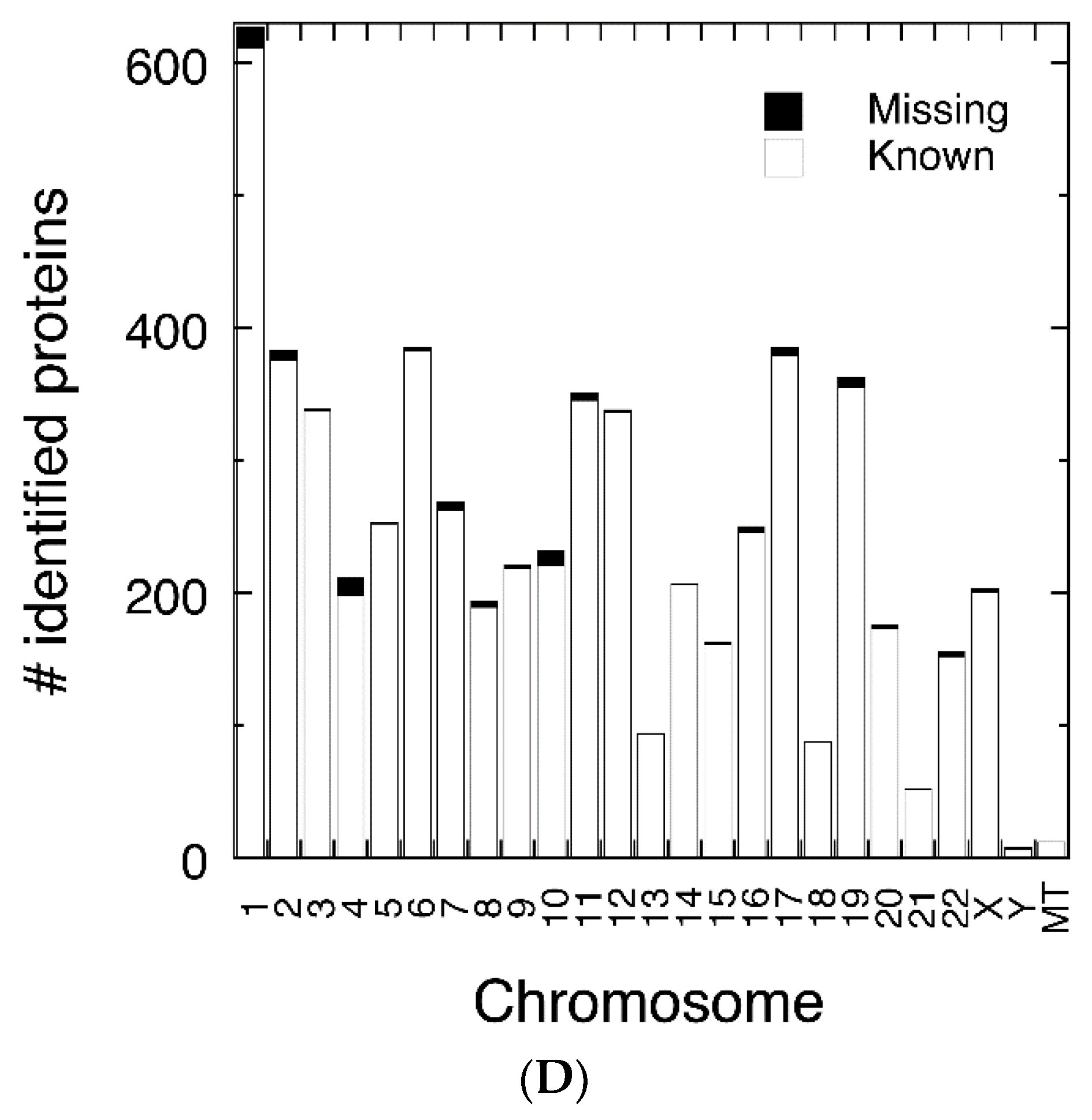
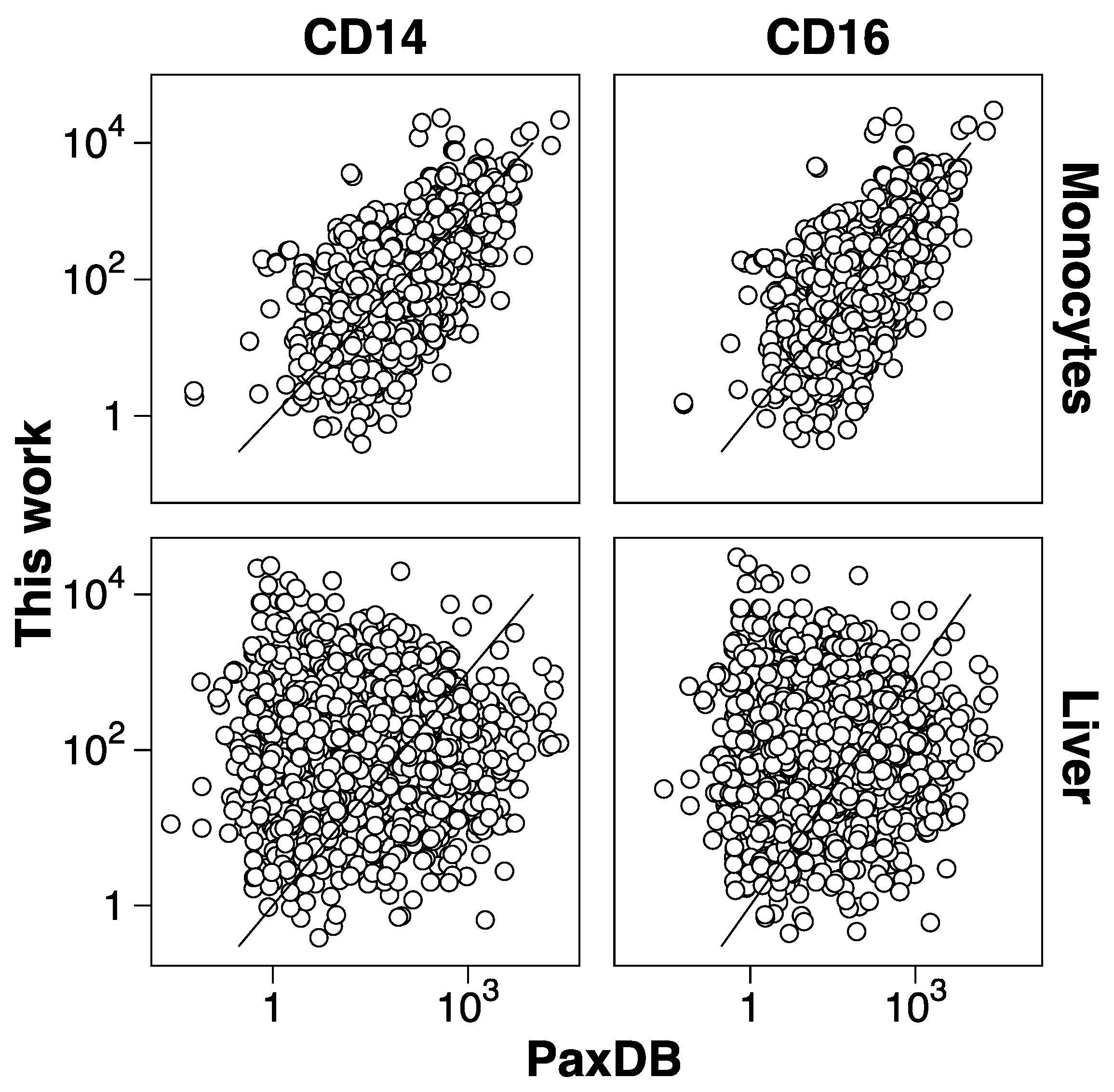
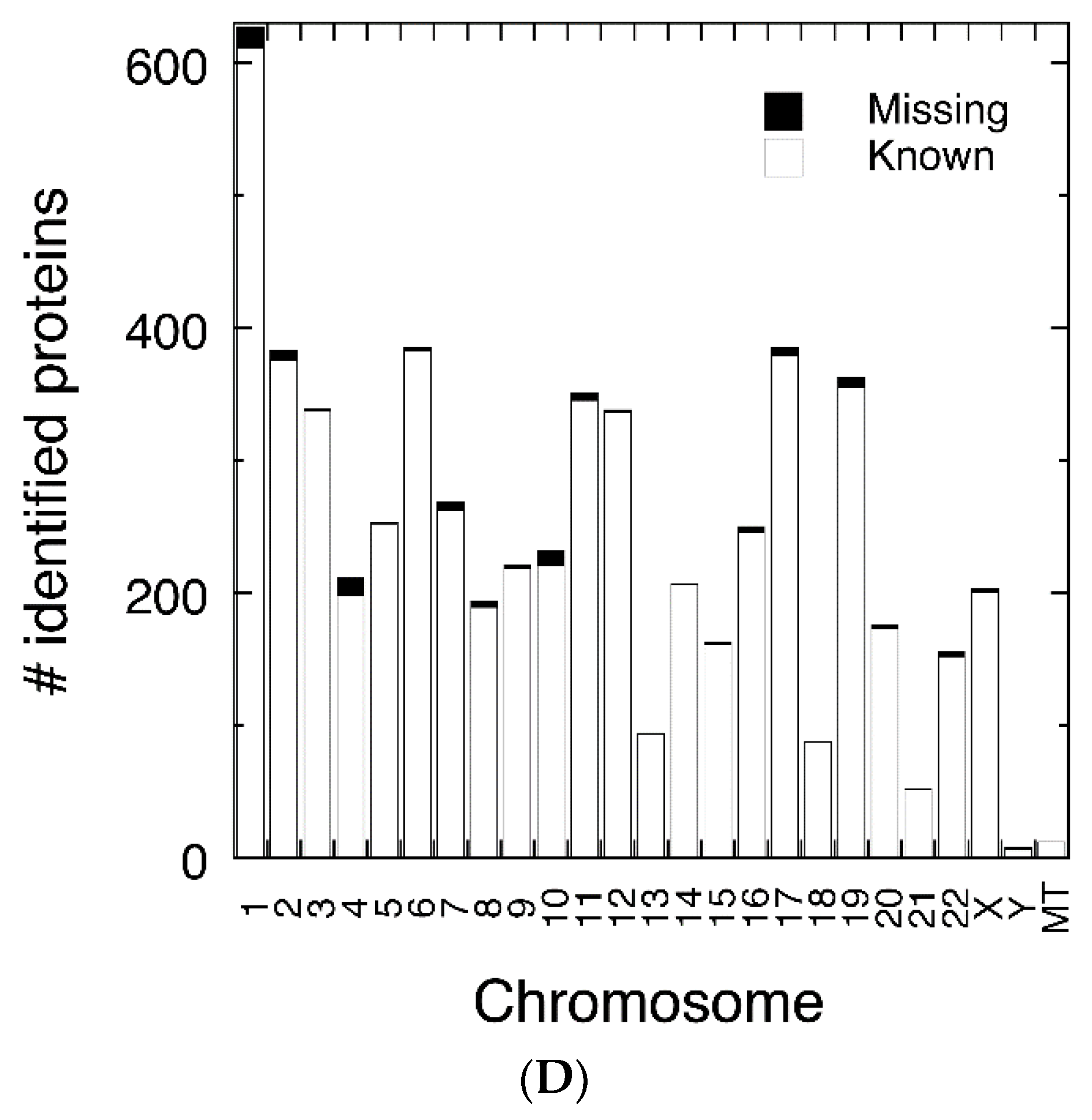
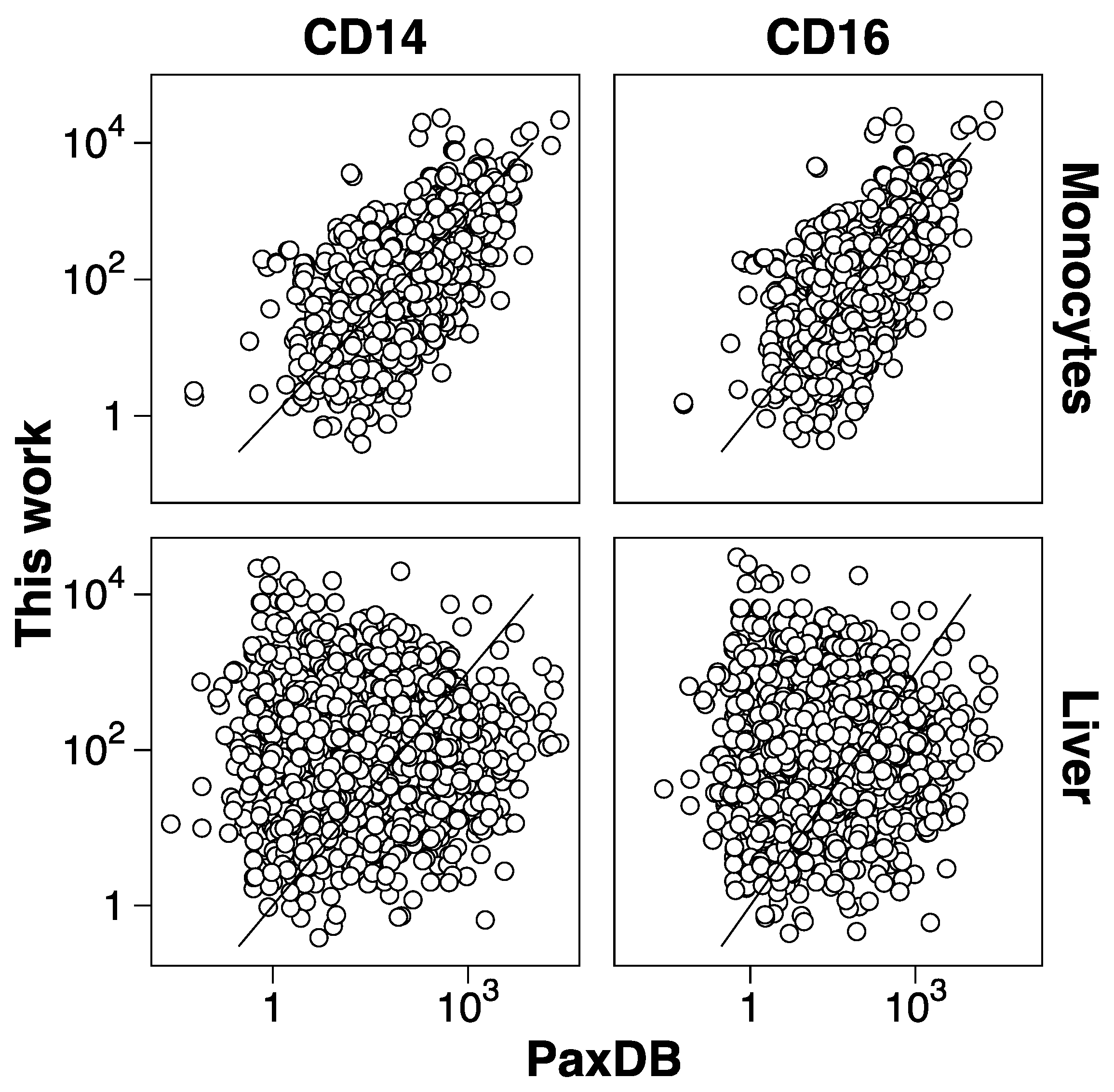
3.2. Protein Abundance
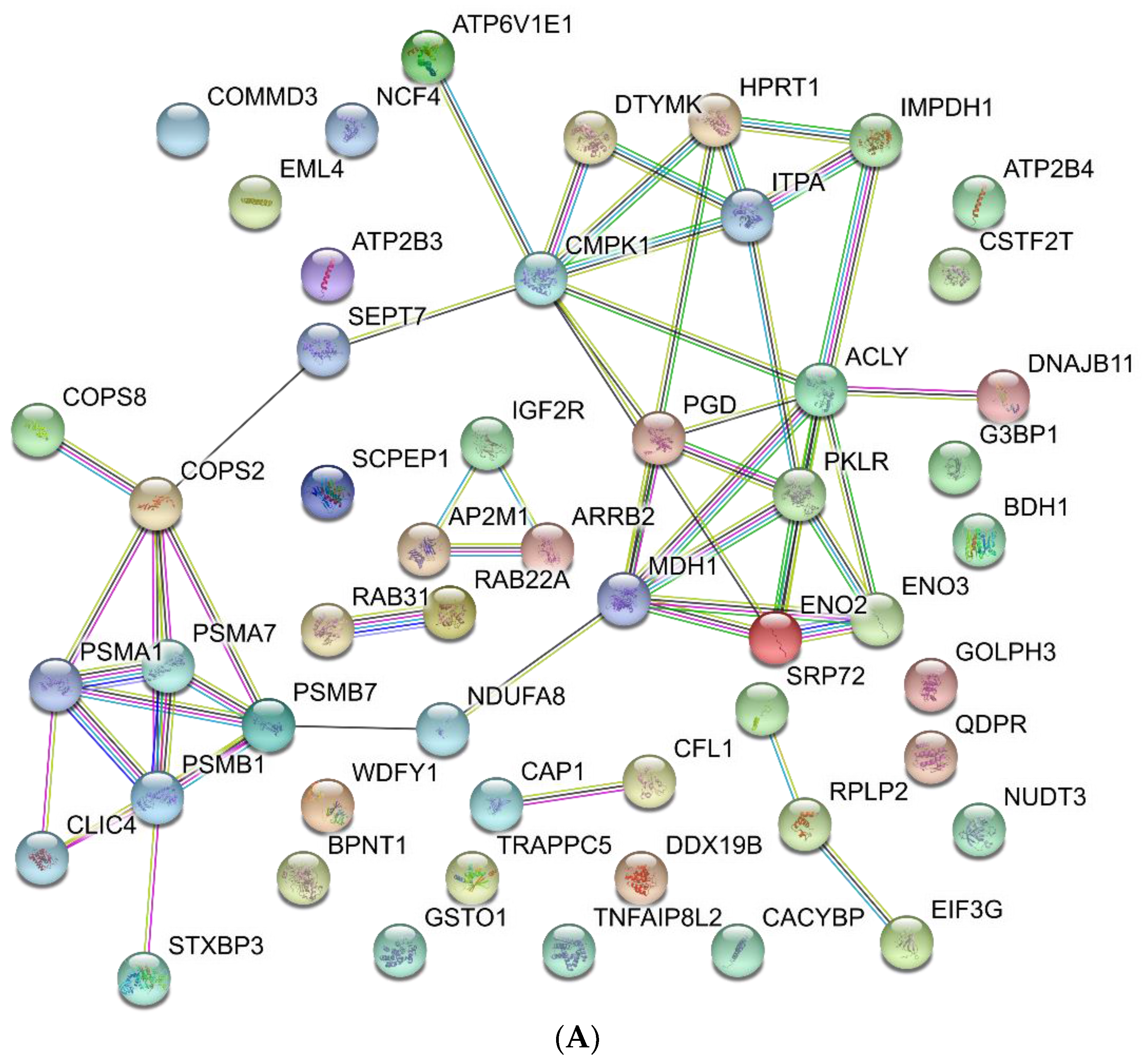
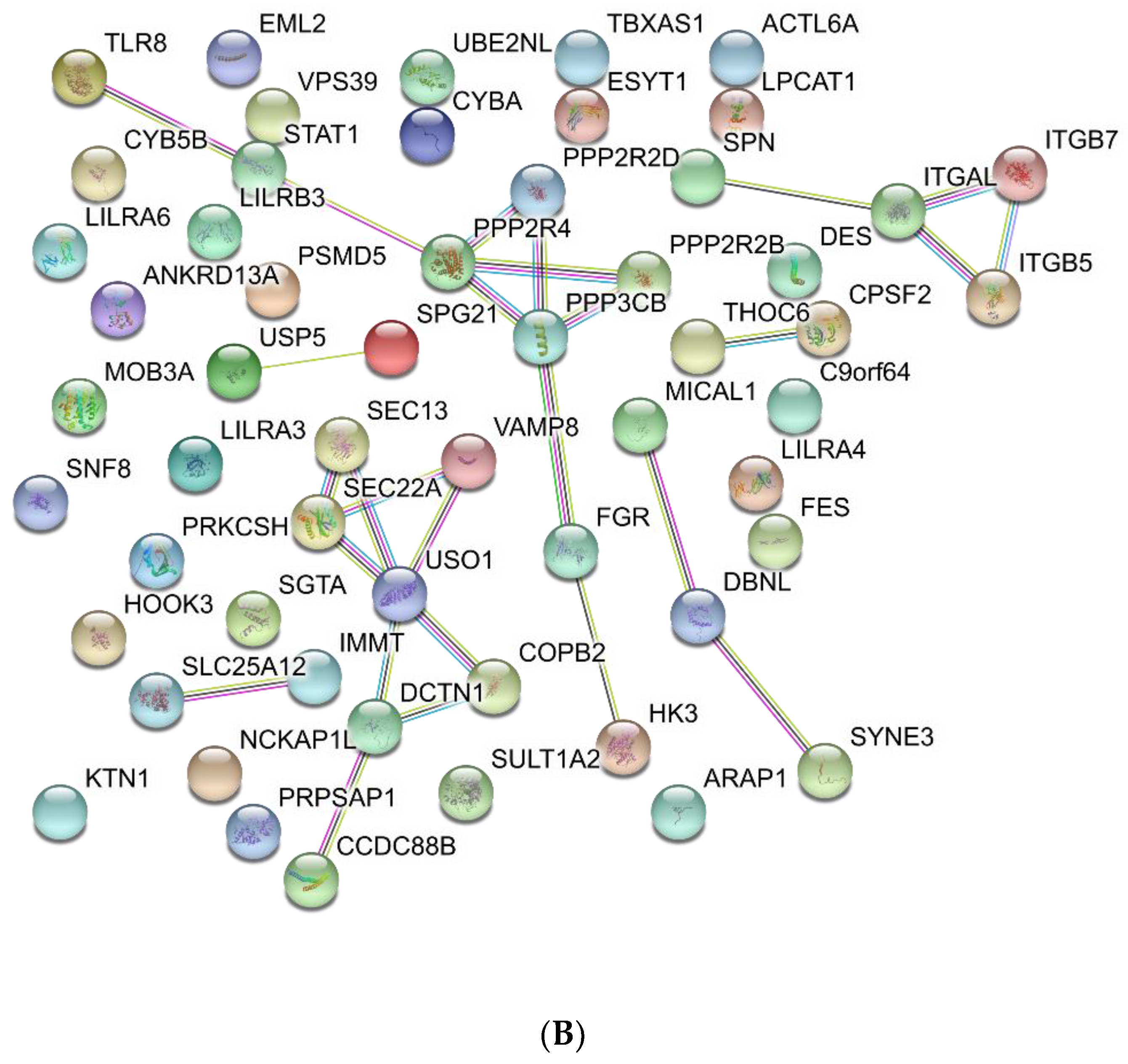
3.3. Functional Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van Furth, R.; Cohn, Z.A. The origin and kinetics of mononuclear phagocytes. J. Exp. Med. 1968, 128, 415–435. [Google Scholar] [CrossRef] [PubMed]
- Passlick, B.; Flieger, D.; Ziegler-Heitbrock, H.W. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood 1989, 74, 2527–2534. [Google Scholar] [PubMed]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.J.; Liu, Y.J.; MacPherson, G.; Randolph, G.J.; et al. Nomenclature of monocytes and dendritic cells in blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef] [PubMed]
- Belge, K.U.; Dayyani, F.; Horelt, A.; Siedlar, M.; Frankenberger, M.; Frankenberger, B.; Espevik, T.; Ziegler-Heitbrock, L. The proinflammatory CD14+CD16+DR++ monocytes are a major source of TNF. J. Immunol. 2002, 168, 3536–3542. [Google Scholar] [CrossRef] [PubMed]
- Cros, J.; Cagnard, N.; Woollard, K.; Patey, N.; Zhang, S.Y.; Senechal, B.; Puel, A.; Biswas, S.K.; Moshous, D.; Picard, C.; et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity 2010, 33, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.L.; Tai, J.J.; Wong, W.C.; Han, H.; Sem, X.; Yeap, W.H.; Kourilsky, P.; Wong, S.C. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 2011, 118, e16–e31. [Google Scholar] [CrossRef] [PubMed]
- Fingerle, G.; Pforte, A.; Passlick, B.; Blumenstein, M.; Strobel, M.; Ziegler-Heitbrock, H.W. The novel subset of CD14+/CD16+ blood monocytes is expanded in sepsis patients. Blood 1993, 82, 3170–3176. [Google Scholar] [PubMed]
- Grip, O.; Bredberg, A.; Lindgren, S.; Henriksson, G. Increased subpopulations of CD16(+) and CD56(+) blood monocytes in patients with active Crohn's disease. Inflamm. Bowel Dis. 2007, 13, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, H.W. Heterogeneity of human blood monocytes: The CD14+ CD16+ subpopulation. Immunol. Today 1996, 17, 424–428. [Google Scholar] [CrossRef]
- Wong, K.L.; Yeap, W.H.; Tai, J.J.; Ong, S.M.; Dang, T.M.; Wong, S.C. The three human monocyte subsets: Implications for health and disease. Immunol. Res. 2012, 53, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, L.; Yu, C.; Yang, X.F.; Wang, H. Monocyte and macrophage differentiation: Circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark. Res. 2014, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Fingerle-Rowson, G.; Auers, J.; Kreuzer, E.; Fraunberger, P.; Blumenstein, M.; Ziegler-Heitbrock, L.H. Expansion of CD14+CD16+ monocytes in critically ill cardiac surgery patients. Inflammation 1998, 22, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Zawada, A.M.; Rogacev, K.S.; Rotter, B.; Winter, P.; Marell, R.R.; Fliser, D.; Heine, G.H. SuperSAGE evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood 2011, 118, e50–e61. [Google Scholar] [CrossRef] [PubMed]
- Anbazhagan, K.; Duroux-Richard, I.; Jorgensen, C.; Apparailly, F. Transcriptomic network support distinct roles of classical and non-classical monocytes in human. Int. Rev. Immunol. 2014, 33, 470–489. [Google Scholar] [CrossRef] [PubMed]
- Weiner, L.M.; Li, W.; Holmes, M.; Catalano, R.B.; Dovnarsky, M.; Padavic, K.; Alpaugh, R.K. Phase I trial of recombinant macrophage colony-stimulating factor and recombinant gamma-interferon: Toxicity, monocytosis, and clinical effects. Cancer Res. 1994, 54, 4084–4090. [Google Scholar] [PubMed]
- Archakov, A.; Zgoda, V.; Kopylov, A.; Naryzhny, S.; Chernobrovkin, A.; Ponomarenko, E.; Lisitsa, A. Chromosome-centric approach to overcoming bottlenecks in the Human Proteome Project. Expert Rev. Proteom. 2012, 9, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Legrain, P.; Aebersold, R.; Archakov, A.; Bairoch, A.; Bala, K.; Beretta, L.; Bergeron, J.; Borchers, C.H.; Corthals, G.L.; Costello, C.E.; et al. The human proteome project: Current state and future direction. Mol. Cell. Proteom. 2011, 10, M111.009993. [Google Scholar] [CrossRef] [PubMed]
- Omenn, G.S. The strategy, organization, and progress of the HUPO Human Proteome Project. J. Proteom. 2014, 100, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Paik, Y.K.; Jeong, S.K.; Omenn, G.S.; Uhlen, M.; Hanash, S.; Cho, S.Y.; Lee, H.J.; Na, K.; Choi, E.Y.; Yan, F.; et al. The Chromosome-Centric Human Proteome Project for cataloging proteins encoded in the genome. Nat. Biotechnol. 2012, 30, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Paik, Y.K.; Omenn, G.S.; Uhlen, M.; Hanash, S.; Marko-Varga, G.; Aebersold, R.; Bairoch, A.; Yamamoto, T.; Legrain, P.; Lee, H.J.; et al. Standard guidelines for the chromosome-centric human proteome project. J. Proteome Res. 2012, 11, 2005–2013. [Google Scholar] [CrossRef] [PubMed]
- Cantor, D.I.; Nice, E.C.; Baker, M.S. Recent findings from the Human Proteome Project: Opening the mass spectrometry toolbox to advance cancer diagnosis, surveillance and treatment. Expert Rev. Proteom. 2015, 12, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Almeida, J.; Bueno, C.; Alguero, M.C.; Sanchez, M.L.; de Santiago, M.; Escribano, L.; Diaz-Agustin, B.; Vaquero, J.M.; Laso, F.J.; San Miguel, J.F.; et al. Comparative analysis of the morphological, cytochemical, immunophenotypical, and functional characteristics of normal human peripheral blood lineage(-)/CD16(+)/HLA-DR(+)/CD14(-/lo) cells, CD14(+) monocytes, and CD16(-) dendritic cells. Clin. Immunol. 2001, 100, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Zor, T.; Selinger, Z. Linearization of the Bradford protein assay increases its sensitivity: Theoretical and experimental studies. Anal. Biochem. 1996, 236, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Jensen, O.N.; Podtelejnikov, A.V.; Sagliocco, F.; Wilm, M.; Vorm, O.; Mortensen, P.; Shevchenko, A.; Boucherie, H.; Mann, M. Linking genome and proteome by mass spectrometry: Large-scale identification of yeast proteins from two dimensional gels. Proc. Natl. Acad. Sci. USA 1996, 93, 14440–14445. [Google Scholar] [CrossRef] [PubMed]
- Lane, L.; Argoud-Puy, G.; Britan, A.; Cusin, I.; Duek, P.D.; Evalet, O.; Gateau, A.; Gaudet, P.; Gleizes, A.; Masselot, A.; et al. neXtProt: A knowledge platform for human proteins. Nucleic Acids Res. 2012, 40, D76–D83. [Google Scholar] [CrossRef] [PubMed]
- Reiter, L.; Claassen, M.; Schrimpf, S.P.; Jovanovic, M.; Schmidt, A.; Buhmann, J.M.; Hengartner, M.O.; Aebersold, R. Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Mol. Cell. Proteom. 2009, 8, 2405–2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto, G.; Aloria, K.; Osinalde, N.; Fullaondo, A.; Arizmendi, J.M.; Matthiesen, R. PAnalyzer: A software tool for protein inference in shotgun proteomics. BMC Bioinf. 2012, 13, 288. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, A.C.; Parmely, T.J.; Tomomori-Sato, C.; Sato, S.; Zhu, D.; Conaway, R.C.; Conaway, J.W.; Florens, L.; Washburn, M.P. Quantitative proteomic analysis of distinct mammalian Mediator complexes using normalized spectral abundance factors. Proc. Natl. Acad. Sci. USA 2006, 103, 18928–18933. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.; Schrimpf, S.; Hengartner, M.O.; Lercher, M.J.; von Mering, C. Shotgun proteomics data from multiple organisms reveals remarkable quantitative conservation of the eukaryotic core proteome. Proteomics 2010, 10, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Buil, A.; Collins, B.C.; Gillet, L.C.; Blum, L.C.; Cheng, L.Y.; Vitek, O.; Mouritsen, J.; Lachance, G.; Spector, T.D.; et al. Quantitative variability of 342 plasma proteins in a human twin population. Mol. Syst. Biol. 2015, 11, 786. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Barderas, M.G.; Tunon, J.; Darde, V.M.; De la Cuesta, F.; Duran, M.C.; Jimenez-Nacher, J.J.; Tarin, N.; Lopez-Bescos, L.; Egido, J.; Vivanco, F. Circulating human monocytes in the acute coronary syndrome express a characteristic proteomic profile. J. Proteome Res. 2007, 6, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Daswani, B.; Gupta, M.K.; Gavali, S.; Desai, M.; Sathe, G.J.; Patil, A.; Parte, P.; Sirdeshmukh, R.; Khatkhatay, M.I. Monocyte Proteomics Reveals Involvement of Phosphorylated HSP27 in the Pathogenesis of Osteoporosis. Dis. Markers 2015, 2015, 196589. [Google Scholar] [CrossRef] [PubMed]
- Pabst, M.J.; Pabst, K.M.; Handsman, D.B.; Beranova-Giorgianni, S.; Giorgianni, F. Proteome of monocyte priming by lipopolysaccharide, including changes in interleukin-1beta and leukocyte elastase inhibitor. Proteome Sci. 2008, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Polati, R.; Castagna, A.; Bossi, A.; Campostrini, N.; Zaninotto, F.; Timperio, A.M.; Zolla, L.; Olivieri, O.; Corrocher, R.; Girelli, D. High resolution preparation of monocyte-derived macrophages (MDM) protein fractions for clinical proteomics. Proteome Sci. 2009, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Tarasova, N.K.; Ytterberg, A.J.; Lundberg, K.; Zhang, X.M.; Harris, R.A.; Zubarev, R.A. Proteomics Reveals a Role for Attachment in Monocyte Differentiation into Efficient Proinflammatory Macrophages. J. Proteome Res. 2015, 14, 3940–3947. [Google Scholar] [CrossRef] [PubMed]
- Tarasova, N.K.; Ytterberg, A.J.; Lundberg, K.; Zhang, X.M.; Harris, R.A.; Zubarev, R.A. Establishing a Proteomics-Based Monocyte Assay To Assess Differential Innate Immune Activation Responses. J. Proteome Res. 2016, 15, 2337–2345. [Google Scholar] [CrossRef] [PubMed]
- Eligini, S.; Brioschi, M.; Fiorelli, S.; Tremoli, E.; Banfi, C.; Colli, S. Human monocyte-derived macrophages are heterogenous: Proteomic profile of different phenotypes. J. Proteom. 2015, 124, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Shipman, M.; Lubick, K.; Fouchard, D.; Guram, R.; Grieco, P.; Jutila, M.; Dratz, E.A. Proteomic and systems biology analysis of monocytes exposed to securinine, a GABA(A) receptor antagonist and immune adjuvant. PLoS ONE 2012, 7, e41278. [Google Scholar] [CrossRef] [PubMed]
- Maes, E.; Landuyt, B.; Mertens, I.; Schoofs, L. Interindividual variation in the proteome of human peripheral blood mononuclear cells. PLoS ONE 2013, 8, e61933. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Tan, Y.C.; Wong, W.C.; Sem, X.; Zhang, H.; Han, H.; Ong, S.M.; Wong, K.L.; Yeap, W.H.; Sze, S.K.; et al. The CD14(+/low)CD16(+) monocyte subset is more susceptible to spontaneous and oxidant-induced apoptosis than the CD14(+)CD16(−) subset. Cell Death Dis. 2010, 1, e95. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhang, H.; Wong, W.C.; Sem, X.; Han, H.; Ong, S.M.; Tan, Y.C.; Yeap, W.H.; Gan, C.S.; Ng, K.Q.; et al. Identification of novel functional differences in monocyte subsets using proteomic and transcriptomic methods. J. Proteome Res. 2009, 8, 4028–4038. [Google Scholar] [CrossRef] [PubMed]
- Bosch, L.J.W.; de Wit, M.; Pham, T.V.; Coupé, V.M.H.; Hiemstra, A.C.; Piersma, S.R.; Oudgenoeg, G.; Scheffer, G.L.; Mongera, S.; Sive Droste, J.T.; et al. Novel Stool-Based Protein Biomarkers for Improved Colorectal Cancer Screening: A Case-Control Study. Ann. Intern. Med. 2017, 167, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Wegler, C.; Gaugaz, F.Z.; Andersson, T.B.; Wisniewski, J.R.; Busch, D.; Gröer, C.; Oswald, S.; Norén, A.; Weiss, F.; Hammer, H.S.; et al. Variability in Mass Spectrometry-based Quantification of Clinically Relevant Drug Transporters and Drug Metabolizing Enzymes. Mol. Pharm. 2017, 14, 3142–3151. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Weiss, M.; Simonovic, M.; Haertinger, G.; Schrimpf, S.P.; Hengartner, M.O.; von Mering, C. PaxDb, a database of protein abundance averages across all three domains of life. Mol. Cell. Proteom. 2012, 11, 492–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, B.; O'Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker | Fluorochrome | Clone | Source |
|---|---|---|---|
| CD3 | APC-H7 | SK7 | BD Bioscience 1 |
| CD14 | FITC | 47-3D6 | Immunostep 2 |
| CD16 | PE-Cy7 | 3G8 | BD Bioscience 1 |
| CD19 | APC | A3B1 | Immunostep 2 |
| CD33 | PerCP-Cy5.5 | P67.6 | BD Bioscience 1 |
| CD45 | PO | HI30 | Immunostep 2 |
| CD56 | PE | C5.9 | Cytognos 3 |
| HLA-DR | PB | L243 | Biolegend 4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segura, V.; Valero, M.L.; Cantero, L.; Muñoz, J.; Zarzuela, E.; García, F.; Aloria, K.; Beaskoetxea, J.; Arizmendi, J.M.; Navajas, R.; et al. In-Depth Proteomic Characterization of Classical and Non-Classical Monocyte Subsets. Proteomes 2018, 6, 8. https://doi.org/10.3390/proteomes6010008
Segura V, Valero ML, Cantero L, Muñoz J, Zarzuela E, García F, Aloria K, Beaskoetxea J, Arizmendi JM, Navajas R, et al. In-Depth Proteomic Characterization of Classical and Non-Classical Monocyte Subsets. Proteomes. 2018; 6(1):8. https://doi.org/10.3390/proteomes6010008
Chicago/Turabian StyleSegura, Víctor, M. Luz Valero, Laura Cantero, Javier Muñoz, Eduardo Zarzuela, Fernando García, Kerman Aloria, Javier Beaskoetxea, Jesús M. Arizmendi, Rosana Navajas, and et al. 2018. "In-Depth Proteomic Characterization of Classical and Non-Classical Monocyte Subsets" Proteomes 6, no. 1: 8. https://doi.org/10.3390/proteomes6010008
APA StyleSegura, V., Valero, M. L., Cantero, L., Muñoz, J., Zarzuela, E., García, F., Aloria, K., Beaskoetxea, J., Arizmendi, J. M., Navajas, R., Paradela, A., Díez, P., Dégano, R. M., Fuentes, M., Orfao, A., García Montero, A., Garin-Muga, A., Corrales, F. J., & Sánchez del Pino, M. M. (2018). In-Depth Proteomic Characterization of Classical and Non-Classical Monocyte Subsets. Proteomes, 6(1), 8. https://doi.org/10.3390/proteomes6010008