From Synapse to Function: A Perspective on the Role of Neuroproteomics in Elucidating Mechanisms of Drug Addiction

{kind=link}
{kind=link}
Abstract
:1. Drug Addiction: A Dysregulation of Plasticity in Motivational Circuitry
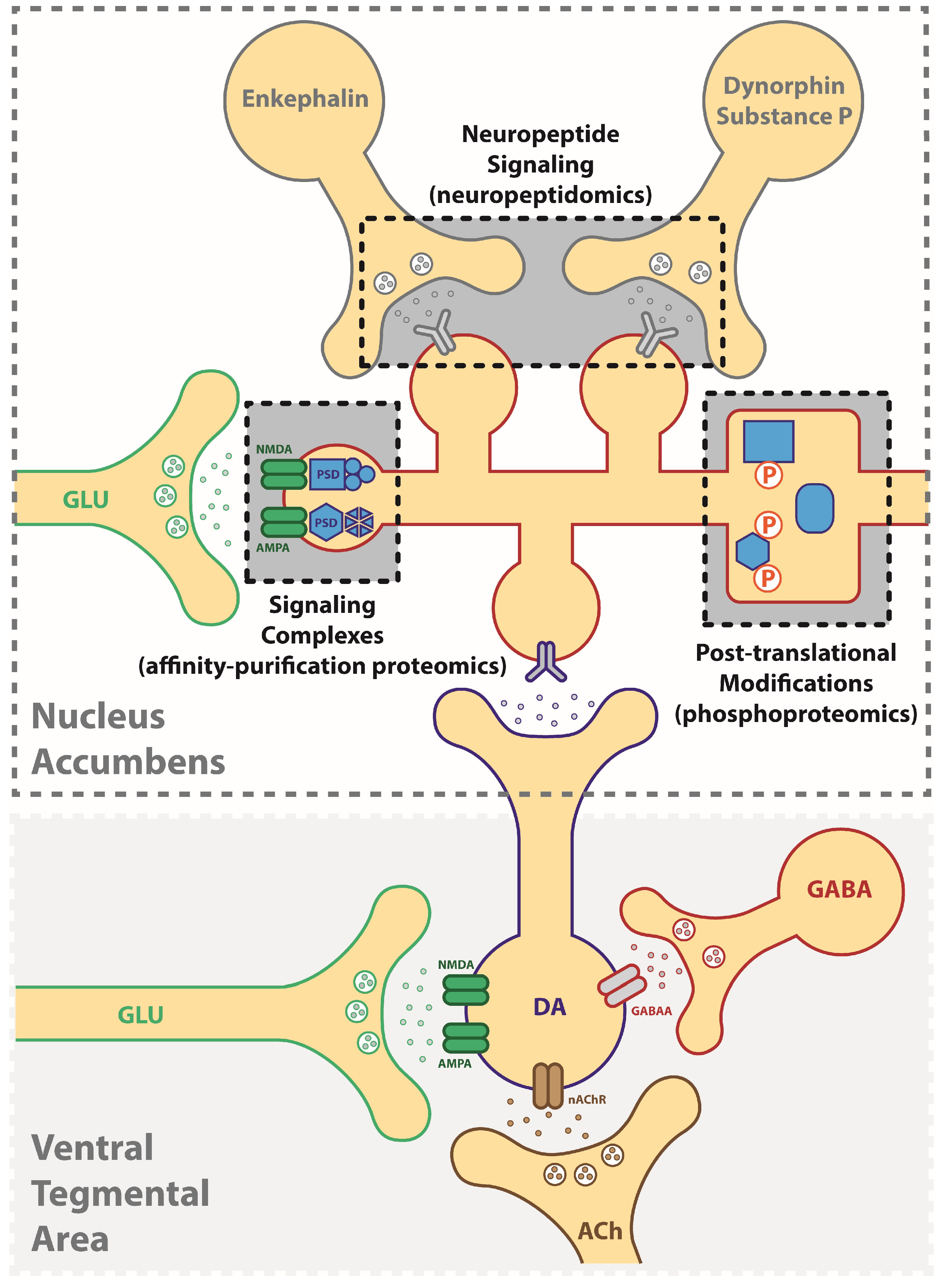
2. Identification of Druggable Targets for Treating Addiction Using a “Neuroproteomics” Approach
3. Proteomics: Identifying Druggable Targets from Changes in Protein Expression
3.1. Current Limitations of Proteomics in Addiction Research
3.2. Identification of Important Changes in Expression
3.3. Confirmation of Targets Identified by Proteomics
4. Affinity-Based Proteomics: Protein Interactome as an Approach for Targeting Mechanisms of Addiction
5. Phosphoproteomics: Signaling-Driven Phosphorylation States Underlying Addictive Behaviors
6. Neuropeptides: Peptide Signals Driving Neuroplasticity in Addiction
6.1. Where Are Neuropeptides Located in the CNS?
6.2. Which Neuropeptides Get Released for Extracellular Signaling in the CNS?
6.3. How do We Elucidate Neuropeptide Bioactivity?
7. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hasin, D.S.; O’Brien, C.P.; Auriacombe, M.; Borges, G.; Bucholz, K.; Budney, A.; Compton, W.M.; Crowley, T.; Ling, W.; Petry, N.M.; et al. DSM-5 criteria for substance use disorders: recommendations and rationale. Am. J. Psychiatry 2013, 170, 834–851. [Google Scholar] [CrossRef] [PubMed]
- Koob, G.F.; Volkow, N.D. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry 2016, 3, 760–773. [Google Scholar] [CrossRef]
- Awasaki, Y.; Nishida, N.; Sasaki, S.; Sato, S. Dopamine D(1) antagonist SCH23390 attenuates self-administration of both cocaine and fentanyl in rats. Environ. Toxicol. Pharmacol. 1997, 3, 115–122. [Google Scholar] [CrossRef]
- Koob, G.F.; Le, H.T.; Creese, I. The D1 dopamine receptor antagonist SCH 23390 increases cocaine self-administration in the rat. Neurosci. Lett. 1987, 79, 315–320. [Google Scholar] [CrossRef]
- Wise, R.A. D1- and D2-Type Contributions to Psychomotor Sensitization and Reward: Implications for Pharmacological Treatment Strategies. Clin. Neuropharmacol. 1995, 18, S74–S83. [Google Scholar] [CrossRef]
- Robinson, T.E.; Berridge, K.C. The incentive sensitization theory of addiction: some current issues. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 3137–3146. [Google Scholar] [CrossRef] [Green Version]
- Russo, S.J.; Nestler, E.J. The brain reward circuitry in mood disorders. Nat. Rev. Neurosci. 2013, 14, 609–625. [Google Scholar] [CrossRef] [Green Version]
- Scofield, M.D.; Heinsbroek, J.A.; Gipson, C.D.; Kupchik, Y.M.; Spencer, S.; Smith, A.C.; Roberts-Wolfe, D.; Kalivas, P.W. The Nucleus Accumbens: Mechanisms of Addiction across Drug Classes Reflect the Importance of Glutamate Homeostasis. Pharmacol. Rev. 2016, 68, 816–871. [Google Scholar] [CrossRef] [Green Version]
- Gorini, G.; Harris, R.A.; Mayfield, R.D. Proteomic approaches and identification of novel therapeutic targets for alcoholism. Neuropsychopharmacology 2014, 39, 104–130. [Google Scholar] [CrossRef] [PubMed]
- Abul-Husn, N.S.; Devi, L.A. Neuroproteomics of the synapse and drug addiction. J. Pharmacol. Exp. Ther. 2006, 318, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Kobeissy, F.H.; Zhang, Z.; Sadasivan, S.; Gold, M.S.; Wang, K.K. Methods in drug abuse neuroproteomics: methamphetamine psychoproteome. Methods Mol. Biol. 2009, 566, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Wolters, D.A.; Washburn, M.P.; Yates, J.R., 3rd. An automated multidimensional protein identification technology for shotgun proteomics. Anal. Chem. 2001, 73, 5683–5690. [Google Scholar] [CrossRef]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.C.; Yates, J.R., 3rd. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef]
- Andrade, E.C.; Krueger, D.D.; Nairn, A.C. Recent advances in neuroproteomics. Curr. Opin. Mol. Ther. 2007, 9, 270–281. [Google Scholar]
- Craft, G.E.; Chen, A.; Nairn, A.C. Recent advances in quantitative neuroproteomics. Methods 2013, 61, 186–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, K.J.; Wu, C.C. Advances in neuromembrane proteomics: efforts towards a comprehensive analysis of membrane proteins in the brain. Brief. Funct. Genomic. 2007, 6, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Hosp, F.; Mann, M. A Primer on Concepts and Applications of Proteomics in Neuroscience. Neuron 2017, 96, 558–571. [Google Scholar] [CrossRef]
- Lull, M.E.; Freeman, W.M.; VanGuilder, H.D.; Vrana, K.E. The use of neuroproteomics in drug abuse research. Drug Alcohol Depend. 2010, 107, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Graybiel, A.M.; Grafton, S.T. The striatum: where skills and habits meet. Cold Spring Harb. Perspect. Biol. 2015, 7, a021691. [Google Scholar] [CrossRef]
- Yin, H.H.; Knowlton, B.J.; Balleine, B.W. Lesions of dorsolateral striatum preserve outcome expectancy but disrupt habit formation in instrumental learning. Eur. J. Neurosci. 2004, 19, 181–189. [Google Scholar] [CrossRef]
- Yin, H.H.; Ostlund, S.B.; Knowlton, B.J.; Balleine, B.W. The role of the dorsomedial striatum in instrumental conditioning. Eur. J. Neurosci. 2005, 22, 513–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.R.; Rubio, F.J.; Bossert, J.M.; Marchant, N.J.; Fanous, S.; Hou, X.; Shaham, Y.; Hope, B.T. Detection of molecular alterations in methamphetamine-activated Fos-expressing neurons from a single rat dorsal striatum using fluorescence-activated cell sorting (FACS). J. Neurochem. 2014, 128, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Z.; Savas, J.N. Uncovering Discrete Synaptic Proteomes to Understand Neurological Disorders. Proteomes 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Salling, M.C.; Faccidomo, S.P.; Li, C.; Psilos, K.; Galunas, C.; Spanos, M.; Agoglia, A.E.; Kash, T.L.; Hodge, C.W. Moderate Alcohol Drinking and the Amygdala Proteome: Identification and Validation of Calcium/Calmodulin Dependent Kinase II and AMPA Receptor Activity as Novel Molecular Mechanisms of the Positive Reinforcing Effects of Alcohol. Biol. Psychiatry 2016, 79, 430–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reissner, K.J.; Uys, J.D.; Schwacke, J.H.; Comte-Walters, S.; Rutherford-Bethard, J.L.; Dunn, T.E.; Blumer, J.B.; Schey, K.L.; Kalivas, P.W. AKAP signaling in reinstated cocaine seeking revealed by iTRAQ proteomic analysis. J. Neurosci. 2011, 31, 5648–5658. [Google Scholar] [CrossRef]
- Rauniyar, N.; Yates, J.R., 3rd. Isobaric labeling-based relative quantification in shotgun proteomics. J. Proteome Res. 2014, 13, 5293–5309. [Google Scholar] [CrossRef]
- Rauniyar, N.; McClatchy, D.B.; Yates, J.R., 3rd. Stable isotope labeling of mammals (SILAM) for in vivo quantitative proteomic analysis. Methods 2013, 61, 260–268. [Google Scholar] [CrossRef]
- Lull, M.E.; Erwin, M.S.; Morgan, D.; Roberts, D.C.; Vrana, K.E.; Freeman, W.M. Persistent proteomic alterations in the medial prefrontal cortex with abstinence from cocaine self-administration. Proteomics Clin. Appl. 2009, 3, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Nimitvilai, S.; Uys, J.D.; Woodward, J.J.; Randall, P.K.; Ball, L.E.; Williams, R.W.; Jones, B.C.; Lu, L.; Grant, K.A.; Mulholland, P.J. Orbitofrontal Neuroadaptations and Cross-Species Synaptic Biomarkers in Heavy-Drinking Macaques. J. Neurosci. 2017, 37, 3646–3660. [Google Scholar] [CrossRef]
- Baker, M. Reproducibility crisis: Blame it on the antibodies. Nature 2015, 521, 274–276. [Google Scholar] [CrossRef] [Green Version]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salling, M.C.; Hodge, C.J.; Psilos, K.E.; Eastman, V.R.; Faccidomo, S.P.; Hodge, C.W. Cue-induced reinstatement of alcohol-seeking behavior is associated with increased CaMKII T286 phosphorylation in the reward pathway of mice. Pharmacol. Biochem. Behav. 2017, 163, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faccidomo, S.; Reid, G.T.; Agoglia, A.E.; Ademola, S.A.; Hodge, C.W. CaMKII inhibition in the prefrontal cortex specifically increases the positive reinforcing effects of sweetened alcohol in C57BL/6J mice. Behav. Brain. Res. 2016, 298, 286–290. [Google Scholar] [CrossRef] [Green Version]
- Cannady, R.; Fisher, K.R.; Graham, C.; Crayle, J.; Besheer, J.; Hodge, C.W. Potentiation of amygdala AMPA receptor activity selectively promotes escalated alcohol self-administration in a CaMKII-dependent manner. Addict. Biol. 2017, 22, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Bowers, M.S.; McFarland, K.; Lake, R.W.; Peterson, Y.K.; Lapish, C.C.; Gregory, M.L.; Lanier, S.M.; Kalivas, P.W. Activator of G protein signaling 3: a gatekeeper of cocaine sensitization and drug seeking. Neuron 2004, 42, 269–281. [Google Scholar] [CrossRef]
- Chen, Z.G.; Liu, X.; Wang, W.; Geng, F.; Gao, J.; Gan, C.L.; Chai, J.R.; He, L.; Hu, G.; Zhou, H.; et al. Dissociative role for dorsal hippocampus in mediating heroin self-administration and relapse through CDK5 and RhoB signaling revealed by proteomic analysis. Addict. Biol. 2017, 22, 1731–1742. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A.M.; Stuber, G.D. Optogenetic strategies to dissect the neural circuits that underlie reward and addiction. Cold Spring Harb Perspect Med. 2012, 2. [Google Scholar] [CrossRef]
- Saunders, B.T.; Richard, J.M.; Janak, P.H. Contemporary approaches to neural circuit manipulation and mapping: focus on reward and addiction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140210. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Li, Z.; Pei, L.; Le, A.D.; Liu, F. The alpha7nACh-NMDA receptor complex is involved in cue-induced reinstatement of nicotine seeking. J. Exp. Med. 2012, 209, 2141–2147. [Google Scholar] [CrossRef]
- Wills, T.A.; Baucum, A.J., 2nd; Holleran, K.M.; Chen, Y.; Pasek, J.G.; Delpire, E.; Tabb, D.L.; Colbran, R.J.; Winder, D.G. Chronic intermittent alcohol disrupts the GluN2B-associated proteome and specifically regulates group I mGlu receptor-dependent long-term depression. Addict. Biol. 2017, 22, 275–290. [Google Scholar] [CrossRef]
- Paulo, J.A.; Brucker, W.J.; Hawrot, E. Proteomic analysis of an α7 nicotinic acetylcholine receptor interactome. J. Proteome Res. 2009, 8, 1849–1858. [Google Scholar] [CrossRef] [PubMed]
- McClure-Begley, T.D.; Esterlis, I.; Stone, K.L.; Lam, T.T.; Grady, S.R.; Colangelo, C.M.; Lindstrom, J.M.; Marks, M.J.; Picciotto, M.R. Evaluation of the Nicotinic Acetylcholine Receptor-Associated Proteome at Baseline and Following Nicotine Exposure in Human and Mouse Cortex. eNeuro 2016, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.B.; Wilson, R.S.; Lam, T.T.; Nairn, A.C.; Picciotto, M.R. Evaluation of the Phosphoproteome of Mouse Alpha 4/Beta 2-Containing Nicotinic Acetylcholine Receptors In Vitro and In Vivo. Proteomes 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- McClatchy, D.B.; Yu, N.K.; Martinez-Bartolome, S.; Patel, R.; Pelletier, A.R.; Lavalle-Adam, M.; Powell, S.B.; Roberto, M.; Yates, J.R. Structural Analysis of Hippocampal Kinase Signal Transduction. ACS Chem. Neurosci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nestler, E.J. Molecular basis of long-term plasticity underlying addiction. Nat. Rev. Neurosci. 2001, 2, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.; Graham, D.L.; Whisler, K.N.; Self, D.W. Phosphorylation of GluR1, ERK, and CREB during spontaneous withdrawal from chronic heroin self-administration. Synapse 2009, 63, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Park, J. Phosphorylation of the AMPAR-TARP Complex in Synaptic Plasticity. Proteomes 2018, 6. [Google Scholar] [CrossRef]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [Green Version]
- McClatchy, D.B.; Savas, J.N.; Martinez-Bartolome, S.; Park, S.K.; Maher, P.; Powell, S.B.; Yates, J.R., 3rd. Global quantitative analysis of phosphorylation underlying phencyclidine signaling and sensorimotor gating in the prefrontal cortex. Mol. Psychiatry 2016, 21, 205–215. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: the road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruse, C.I.; McClatchy, D.B.; Lu, B.; Cociorva, D.; Motoyama, A.; Park, S.K.; Yates, J.R., 3rd. Motif-specific sampling of phosphoproteomes. J. Proteome Res. 2008, 7, 2140–2150. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Dephoure, N.; Haas, W.; Huttlin, E.L.; Zhai, B.; Sowa, M.E.; Gygi, S.P. Correct interpretation of comprehensive phosphorylation dynamics requires normalization by protein expression changes. Mol. Cell. Proteomics 2011, 10, M111–009654. [Google Scholar] [CrossRef] [PubMed]
- Ubersax, J.A.; Ferrell, J.E., Jr. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell. Biol. 2007, 8, 530–541. [Google Scholar] [CrossRef] [PubMed]
- McClatchy, D.B.; Dong, M.Q.; Wu, C.C.; Venable, J.D.; Yates, J.R., 3rd. 15N metabolic labeling of mammalian tissue with slow protein turnover. J. Proteome Res. 2007, 6, 2005–2010. [Google Scholar] [CrossRef] [PubMed]
- Rich, M.T.; Abbott, T.B.; Chung, L.; Gulcicek, E.E.; Stone, K.L.; Colangelo, C.M.; Lam, T.T.; Nairn, A.C.; Taylor, J.R.; Torregrossa, M.M. Phosphoproteomic Analysis Reveals a Novel Mechanism of CaMKIIalpha Regulation Inversely Induced by Cocaine Memory Extinction versus Reconsolidation. J. Neurosci. 2016, 36, 7613–7627. [Google Scholar] [CrossRef]
- Tronson, N.C.; Taylor, J.R. Molecular mechanisms of memory reconsolidation. Nat. Rev. Neurosci. 2007, 8, 262–275. [Google Scholar] [CrossRef]
- Quirk, G.J.; Pare, D.; Richardson, R.; Herry, C.; Monfils, M.H.; Schiller, D.; Vicentic, A. Erasing fear memories with extinction training. J. Neurosci. 2010, 30, 14993–14997. [Google Scholar] [CrossRef]
- Natividad, L.A.; Steinman, M.Q.; Laredo, S.A.; Irimia, C.; Polis, I.Y.; Lintz, R.; Buczynski, M.W.; Martin-Fardon, R.; Roberto, M.; Parsons, L.H. Phosphorylation of calcium/calmodulin-dependent protein kinase II in the rat dorsal medial prefrontal cortex is associated with alcohol-induced cognitive inflexibility. Addict. Biol. 2017. [Google Scholar] [CrossRef]
- Baucum, A.J., 2nd; Shonesy, B.C.; Rose, K.L.; Colbran, R.J. Quantitative proteomics analysis of CaMKII phosphorylation and the CaMKII interactome in the mouse forebrain. ACS Chem. Neurosci. 2015, 6, 615–631. [Google Scholar] [CrossRef]
- Cai, D.; Lee, A.Y.; Chiang, C.M.; Kodadek, T. Peptoid ligands that bind selectively to phosphoproteins. Bioorg Med. Chem. Lett. 2011, 21, 4960–4964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiele, T.E. Neuropeptides and Addiction: An Introduction. Int. Rev. Neurobiol. 2017, 136, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Frieden, T.R.; Hyde, P.S.; Cha, S.S. Medication-assisted therapies--tackling the opioid-overdose epidemic. N. Engl. J. Med. 2014, 370, 2063–2066. [Google Scholar] [CrossRef] [PubMed]
- Sarnyai, Z.; Kovacs, G.L. Oxytocin in learning and addiction: From early discoveries to the present. Pharmacol. Biochem. Behav. 2014, 119, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, J.; Martins, J.; Baptista, S.; Ambrosio, A.F.; Silva, A.P. Effects of drugs of abuse on the central neuropeptide Y system. Addict. Biol. 2016, 21, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Schank, J.R.; Heilig, M. Substance P and the Neurokinin-1 Receptor: The New CRF. Int. Rev. Neurobiol. 2017, 136, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Roberto, M.; Spierling, S.R.; Kirson, D.; Zorrilla, E.P. Corticotropin-Releasing Factor (CRF) and Addictive Behaviors. Int. Rev. Neurobiol. 2017, 136, 5–51. [Google Scholar] [CrossRef]
- Douglass, J.; McKinzie, A.A.; Couceyro, P. PCR differential display identifies a rat brain mRNA that is transcriptionally regulated by cocaine and amphetamine. J. Neurosci. 1995, 15, 2471–2481. [Google Scholar] [CrossRef] [Green Version]
- Hook, V.; Funkelstein, L.; Lu, D.; Bark, S.; Wegrzyn, J.; Hwang, S.R. Proteases for processing proneuropeptides into peptide neurotransmitters and hormones. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 393–423. [Google Scholar] [CrossRef]
- Park, Y.; Kim, K.T. Short-term plasticity of small synaptic vesicle (SSV) and large dense-core vesicle (LDCV) exocytosis. Cell. Signal. 2009, 21, 1465–1470. [Google Scholar] [CrossRef]
- Fricker, L.D.; Devi, L.A. Orphan neuropeptides and receptors: Novel therapeutic targets. Pharmacol. Ther. 2018, 185, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Burbach, J.P. What are neuropeptides? Methods Mol. Biol. 2011, 789, 1–36. [Google Scholar] [CrossRef] [PubMed]
- Tuppy, H. The amino-acid sequence in oxytocin. Biochim. Biophys. Acta 1953, 11, 449–450. [Google Scholar] [CrossRef]
- Chang, M.M.; Leeman, S.E.; Niall, H.D. Amino-acid sequence of substance P. Nat. New Biol. 1971, 232, 86–87. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.; Smith, T.W.; Kosterlitz, H.W.; Fothergill, L.A.; Morgan, B.A.; Morris, H.R. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature 1975, 258, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.; Tachibana, S.; Lowney, L.I.; Hunkapiller, M.; Hood, L. Dynorphin-(1-13), an extraordinarily potent opioid peptide. Proc. Natl. Acad. Sci. USA 1979, 76, 6666–6670. [Google Scholar] [CrossRef]
- Spiess, J.; Rivier, J.; Rivier, C.; Vale, W. Primary structure of corticotropin-releasing factor from ovine hypothalamus. Proc. Natl. Acad. Sci. USA 1981, 78, 6517–6521. [Google Scholar] [CrossRef]
- Burbach, J.P. Neuropeptides from concept to online database www.neuropeptides.nl. Eur. J. Pharmacol. 2010, 626, 27–48. [Google Scholar] [CrossRef]
- OuYang, C.; Liang, Z.; Li, L. Mass spectrometric analysis of spatio-temporal dynamics of crustacean neuropeptides. Biochim. Biophys. Acta 2015, 1854, 798–811. [Google Scholar] [CrossRef] [Green Version]
- Romanova, E.V.; Sweedler, J.V. Peptidomics for the discovery and characterization of neuropeptides and hormones. Trends Pharmacol. Sci. 2015, 36, 579–586. [Google Scholar] [CrossRef] [Green Version]
- Verdonck, R.; De Haes, W.; Cardoen, D.; Menschaert, G.; Huhn, T.; Landuyt, B.; Baggerman, G.; Boonen, K.; Wenseleers, T.; Schoofs, L. Fast and Reliable Quantitative Peptidomics with labelpepmatch. J. Proteome Res. 2016, 15, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Secher, A.; Kelstrup, C.D.; Conde-Frieboes, K.W.; Pyke, C.; Raun, K.; Wulff, B.S.; Olsen, J.V. Analytic framework for peptidomics applied to large-scale neuropeptide identification. Nat. Commun. 2016, 7, 11436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.G.; Lone, A.M.; Saghatelian, A. Analysis of the proteolysis of bioactive peptides using a peptidomics approach. Nat. Protoc. 2013, 8, 1730–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hishimoto, A.; Nomaru, H.; Ye, K.; Nishi, A.; Lim, J.; Aguilan, J.T.; Nieves, E.; Kang, G.; Angeletti, R.H.; Hiroi, N. Molecular Histochemistry Identifies Peptidomic Organization and Reorganization Along Striatal Projection Units. Biol. Psychiatry 2016, 79, 415–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathis, V.; Kenny, P.J. From controlled to compulsive drug-taking: The role of the habenula in addiction. Neurosci. Biobehav. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Anapindi, K.D.B.; Rubakhin, S.S.; Wei, P.; Yu, Q.; Li, L.; Kenny, P.J.; Sweedler, J.V. Neuropeptidomics of the Rat Habenular Nuclei. J. Proteome Res. 2018, 17, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Wang, J.; Tian, Z.; Ma, F.; Dowell, J.; Bremer, Q.; Lu, G.; Baldo, B.; Li, L. Quantitative Mass Spectrometry Reveals Food Intake-Induced Neuropeptide Level Changes in Rat Brain: Functional Assessment of Selected Neuropeptides as Feeding Regulators. Mol. Cell. Proteomics 2017, 16, 1922–1937. [Google Scholar] [CrossRef] [PubMed]
- Buczynski, M.W.; Parsons, L.H. Quantification of brain endocannabinoid levels: methods, interpretations and pitfalls. Br. J. Pharmacol. 2010, 160, 423–442. [Google Scholar] [CrossRef] [Green Version]
- De Luca, M.A.; Buczynski, M.W.; Di Chiara, G. Loren Parsons’ contribution to addiction neurobiology. Addict. Biol. 2018. [Google Scholar] [CrossRef]
- Fridjonsdottir, E.; Nilsson, A.; Wadensten, H.; Andren, P.E. Brain Tissue Sample Stabilization and Extraction Strategies for Neuropeptidomics. Methods Mol. Biol 2018, 1719, 41–49. [Google Scholar] [CrossRef]
- Boren, M. Sample preservation through heat stabilization of proteins: principles and examples. Methods Mol. Biol. 2015, 1295, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Van Wanseele, Y.; De Prins, A.; De Bundel, D.; Smolders, I.; Van Eeckhaut, A. Challenges for the in vivo quantification of brain neuropeptides using microdialysis sampling and LC-MS. Bioanalysis 2016, 8, 1965–1985. [Google Scholar] [CrossRef] [PubMed]
- Haskins, W.E.; Watson, C.J.; Cellar, N.A.; Powell, D.H.; Kennedy, R.T. Discovery and neurochemical screening of peptides in brain extracellular fluid by chemical analysis of in vivo microdialysis samples. Anal. Chem. 2004, 76, 5523–5533. [Google Scholar] [CrossRef] [PubMed]
- Bernay, B.; Gaillard, M.C.; Guryca, V.; Emadali, A.; Kuhn, L.; Bertrand, A.; Detraz, I.; Carcenac, C.; Savasta, M.; Brouillet, E.; et al. Discovering new bioactive neuropeptides in the striatum secretome using in vivo microdialysis and versatile proteomics. Mol. Cell. Proteomics 2009, 8, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Wardman, J.H.; Berezniuk, I.; Di, S.; Tasker, J.G.; Fricker, L.D. ProSAAS-derived peptides are colocalized with neuropeptide Y and function as neuropeptides in the regulation of food intake. PLoS ONE 2011, 6, e28152. [Google Scholar] [CrossRef] [PubMed]
- Bobeck, E.N.; Gomes, I.; Pena, D.; Cummings, K.A.; Clem, R.L.; Mezei, M.; Devi, L.A. The BigLEN-GPR171 Peptide Receptor System Within the Basolateral Amygdala Regulates Anxiety-Like Behavior and Contextual Fear Conditioning. Neuropsychopharmacology 2017, 42, 2527–2536. [Google Scholar] [CrossRef] [PubMed]
- Wardman, J.H.; Gomes, I.; Bobeck, E.N.; Stockert, J.A.; Kapoor, A.; Bisignano, P.; Gupta, A.; Mezei, M.; Kumar, S.; Filizola, M.; et al. Identification of a small-molecule ligand that activates the neuropeptide receptor GPR171 and increases food intake. Sci. Signal. 2016, 9, ra55. [Google Scholar] [CrossRef]
- Chen, T.; Cai, Q.; Hong, Y. Intrathecal sensory neuron-specific receptor agonists bovine adrenal medulla 8-22 and (Tyr6)-gamma2-MSH-6-12 inhibit formalin-evoked nociception and neuronal Fos-like immunoreactivity in the spinal cord of the rat. Neuroscience 2006, 141, 965–975. [Google Scholar] [CrossRef]
- Barnea, G.; Strapps, W.; Herrada, G.; Berman, Y.; Ong, J.; Kloss, B.; Axel, R.; Lee, K.J. The genetic design of signaling cascades to record receptor activation. Proc. Natl. Acad. Sci. USA 2008, 105, 64–69. [Google Scholar] [CrossRef]
- Li, Z.; Tseng, P.Y.; Tiwari, V.; Xu, Q.; He, S.Q.; Wang, Y.; Zheng, Q.; Han, L.; Wu, Z.; Blobaum, A.L.; et al. Targeting human Mas-related G protein-coupled receptor X1 to inhibit persistent pain. Proc. Natl. Acad. Sci. USA 2017, 114, E1996–E2005. [Google Scholar] [CrossRef] [Green Version]
- Lansu, K.; Karpiak, J.; Liu, J.; Huang, X.P.; McCorvy, J.D.; Kroeze, W.K.; Che, T.; Nagase, H.; Carroll, F.I.; Jin, J.; et al. In silico design of novel probes for the atypical opioid receptor MRGPRX2. Nat. Chem. Biol. 2017, 13, 529–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Q.; Jiang, J.; Chen, T.; Hong, Y. Sensory neuron-specific receptor agonist BAM8-22 inhibits the development and expression of tolerance to morphine in rats. Behav. Brain Res. 2007, 178, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.L.; Zhang, Y.; Fu, D.W.; Zhang, W.; Liu, T.; Yoshikawa, H.; Awaga, K.; Xiong, R.G. Above-room-temperature magnetodielectric coupling in a possible molecule-based multiferroic: triethylmethylammonium tetrabromoferrate(III). J. Am. Chem. Soc. 2012, 134, 18487–18490. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.A.; Pande, H.; Paxton, R.J.; Shively, L.; Padma, A.; Simmer, R.L.; Todd, C.W.; Riggs, A.D.; Shively, J.E. Molecular cloning of a gene belonging to the carcinoembryonic antigen gene family and discussion of a domain model. Proc. Natl. Acad. Sci. USA 1987, 84, 2965–2969. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Natividad, L.A.; Buczynski, M.W.; McClatchy, D.B.; Yates, J.R., III. From Synapse to Function: A Perspective on the Role of Neuroproteomics in Elucidating Mechanisms of Drug Addiction. Proteomes 2018, 6, 50. https://doi.org/10.3390/proteomes6040050
Natividad LA, Buczynski MW, McClatchy DB, Yates JR III. From Synapse to Function: A Perspective on the Role of Neuroproteomics in Elucidating Mechanisms of Drug Addiction. Proteomes. 2018; 6(4):50. https://doi.org/10.3390/proteomes6040050
Chicago/Turabian StyleNatividad, Luis A., Matthew W. Buczynski, Daniel B. McClatchy, and John R. Yates, III. 2018. "From Synapse to Function: A Perspective on the Role of Neuroproteomics in Elucidating Mechanisms of Drug Addiction" Proteomes 6, no. 4: 50. https://doi.org/10.3390/proteomes6040050