
Imaging Effector Memory T-Cells Predicts Response to PD1-Chemotherapy Combinations in Colon Cancer
 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. [18F]AlF-NOTA-KCNA3P Radiochemistry
2.2. Tumour Implantation and Dosing Regimen
2.3. PET-CT Imaging
2.4. Flow Cytometry and Dimension Reduction Analysis
2.5. Statistical Analysis
3. Results
3.1. Evaluation of Treatment Efficacy Using Tumour Volumes
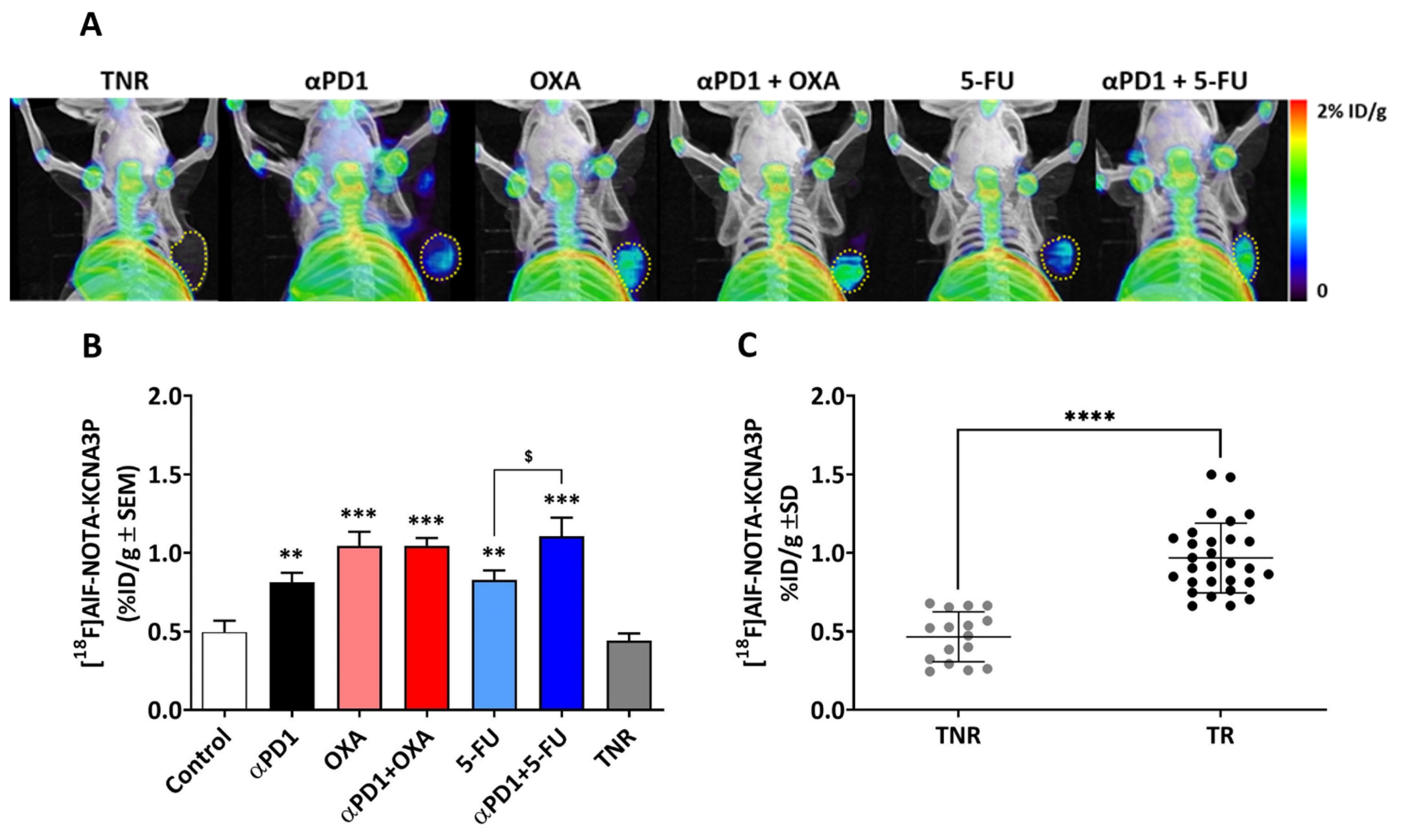
3.2. In Vivo PET Imaging with [18F]AlF-NOTA-KCNA3P
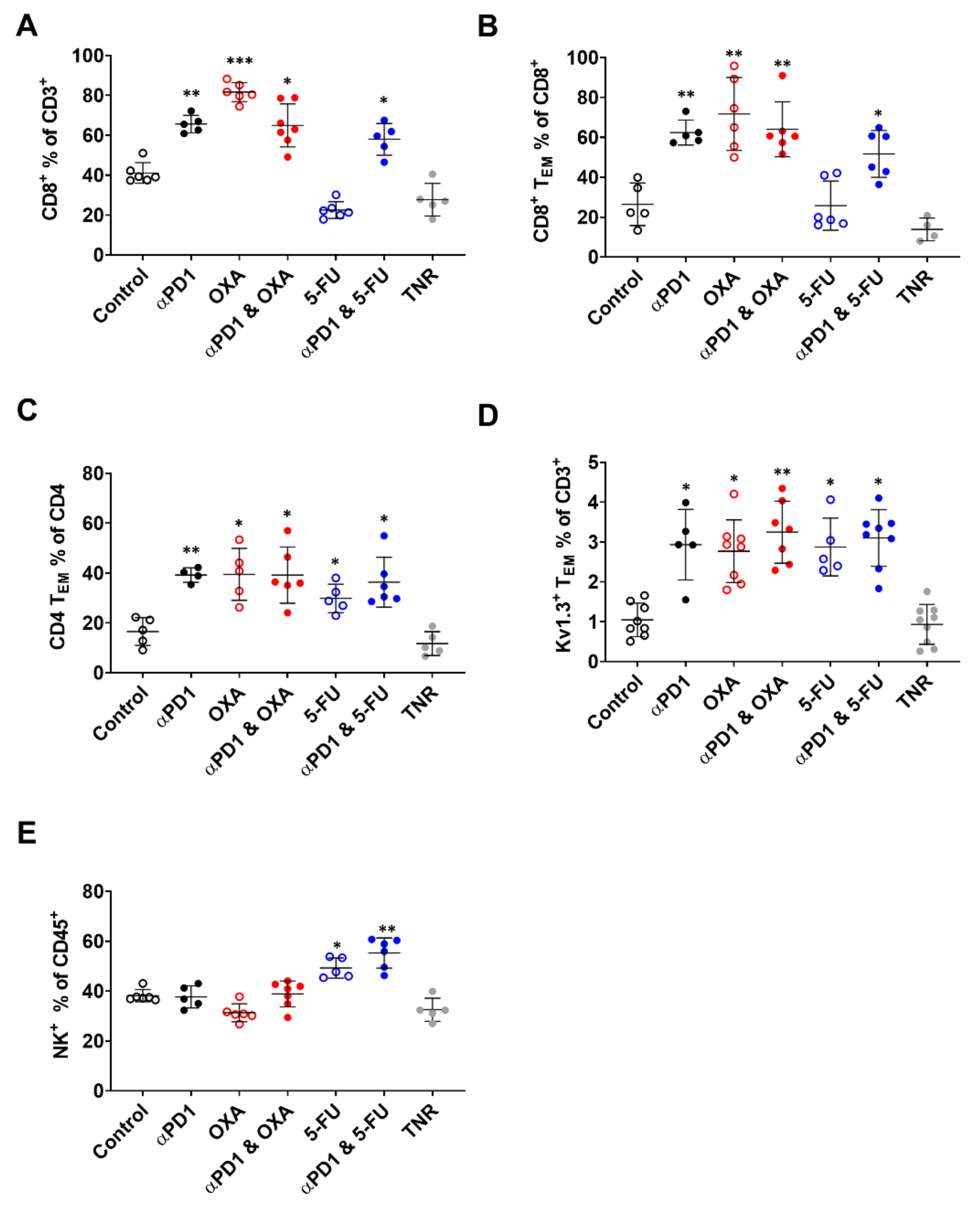
3.3. Tumour Infiltration of Effector Memory T-Cells Is Responsible for [18F]AlF-NOTA-KCNA3P Tumour Uptake
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Evrard, C.; Tachon, G.; Randrian, V.; Karayan-Tapon, L.; Tougeron, D. Microsatellite Instability: Diagnosis, Heterogeneity, Discordance, and Clinical Impact in Colorectal Cancer. Cancers 2019, 11, 1567. [Google Scholar] [CrossRef] [PubMed]
- Llosa, N.J.; Luber, B.; Siegel, N.; Awan, A.H.; Oke, T.; Zhu, Q.; Bartlett, B.R.; Aulakh, L.K.; Thompson, E.D.; Jaffee, E.M.; et al. Immunopathologic Stratification of Colorectal Cancer for Checkpoint Blockade Immunotherapy. Cancer Immunol. Res. 2019, 7, 1574–1579. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Du, W.; Yi, L.; Wu, S.; He, C.; Zhai, W.; Yue, C.; Sun, R.; Menk, A.V.; Delgoffe, G.M.; et al. Checkpoint molecules coordinately restrain hyperactivated effector T cells in the tumor microenvironment. Oncoimmunology 2020, 9, 1708064. [Google Scholar] [CrossRef]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef]
- Westdorp, H.; Sweep, M.W.D.; Gorris, M.A.J.; Hoentjen, F.; Boers-Sonderen, M.J.; van der Post, R.S.; van den Heuvel, M.M.; Piet, B.; Boleij, A.; Bloemendal, H.J.; et al. Mechanisms of Immune Checkpoint Inhibitor-Mediated Colitis. Front. Immunol. 2021, 12, 768957. [Google Scholar] [CrossRef]
- Schneider, B.J.; Naidoo, J.; Santomasso, B.D.; Lacchetti, C.; Adkins, S.; Anadkat, M.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; et al. Management of Immune-Related Adverse Events in Patients Treated With Immune Checkpoint Inhibitor Therapy: ASCO Guideline Update. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 4073–4126. [Google Scholar] [CrossRef]
- Khair, D.O.; Bax, H.J.; Mele, S.; Crescioli, S.; Pellizzari, G.; Khiabany, A.; Nakamura, M.; Harris, R.J.; French, E.; Hoffmann, R.M.; et al. Combining Immune Checkpoint Inhibitors: Established and Emerging Targets and Strategies to Improve Outcomes in Melanoma. Front. Immunol. 2019, 10, 453. [Google Scholar] [CrossRef]
- Goggi, J.L.; Hartimath, S.V.; Xuan, T.Y.; Khanapur, S.; Jieu, B.; Chin, H.X.; Ramasamy, B.; Cheng, P.; Rong, T.J.; Fong, Y.F.; et al. Granzyme B PET Imaging of Combined Chemotherapy and Immune Checkpoint Inhibitor Therapy in Colon Cancer. Mol. Imaging Biol. MIB Off. Publ. Acad. Mol. Imaging 2021, 23, 714–723. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Garg, A.D.; Galluzzi, L.; Apetoh, L.; Baert, T.; Birge, R.B.; Bravo-San Pedro, J.M.; Breckpot, K.; Brough, D.; Chaurio, R.; Cirone, M.; et al. Molecular and Translational Classifications of DAMPs in Immunogenic Cell Death. Front. Immunol. 2015, 6, 588. [Google Scholar] [CrossRef]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef]
- Yang, H.; Ma, Y.; Chen, G.; Zhou, H.; Yamazaki, T.; Klein, C.; Pietrocola, F.; Vacchelli, E.; Souquere, S.; Sauvat, A.; et al. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology 2016, 5, e1149673. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L. Enhancing the anticancer effects of 5-fluorouracil: Current challenges and future perspectives. Biomed. J. 2015, 38, 111–116. [Google Scholar] [CrossRef]
- Orecchioni, S.; Talarico, G.; Labanca, V.; Calleri, A.; Mancuso, P.; Bertolini, F. Vinorelbine, cyclophosphamide and 5-FU effects on the circulating and intratumoural landscape of immune cells improve anti-PD-L1 efficacy in preclinical models of breast cancer and lymphoma. Br. J. Cancer 2018, 118, 1329–1336. [Google Scholar] [CrossRef]
- Opzoomer, J.W.; Sosnowska, D.; Anstee, J.E.; Spicer, J.F.; Arnold, J.N. Cytotoxic Chemotherapy as an Immune Stimulus: A Molecular Perspective on Turning Up the Immunological Heat on Cancer. Front. Immunol. 2019, 10, 1654. [Google Scholar] [CrossRef]
- Mami-Chouaib, F.; Blanc, C.; Corgnac, S.; Hans, S.; Malenica, I.; Granier, C.; Tihy, I.; Tartour, E. Resident memory T cells, critical components in tumor immunology. J. Immunother. Cancer 2018, 6, 87. [Google Scholar] [CrossRef]
- Principe, N.; Kidman, J.; Goh, S.; Tilsed, C.M.; Fisher, S.A.; Fear, V.S.; Forbes, C.A.; Zemek, R.M.; Chopra, A.; Watson, M.; et al. Tumor Infiltrating Effector Memory Antigen-Specific CD8(+) T Cells Predict Response to Immune Checkpoint Therapy. Front. Immunol. 2020, 11, 584423. [Google Scholar] [CrossRef]
- Ribas, A.; Shin, D.S.; Zaretsky, J.; Frederiksen, J.; Cornish, A.; Avramis, E.; Seja, E.; Kivork, C.; Siebert, J.; Kaplan-Lefko, P.; et al. PD-1 Blockade Expands Intratumoral Memory T Cells. Cancer Immunol. Res. 2016, 4, 194–203. [Google Scholar] [CrossRef]
- Jin, Y.; Tan, A.; Feng, J.; Xu, Z.; Wang, P.; Ruan, P.; Luo, R.; Weng, Y.; Peng, M. Prognostic Impact of Memory CD8(+) T Cells on Immunotherapy in Human Cancers: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 698076. [Google Scholar] [CrossRef]
- Lanzavecchia, A.; Sallusto, F. Regulation of T cell immunity by dendritic cells. Cell 2001, 106, 263–266. [Google Scholar] [CrossRef]
- Pages, F.; Berger, A.; Camus, M.; Sanchez-Cabo, F.; Costes, A.; Molidor, R.; Mlecnik, B.; Kirilovsky, A.; Nilsson, M.; Damotte, D.; et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N. Engl. J. Med. 2005, 353, 2654–2666. [Google Scholar] [CrossRef]
- Ong, S.T.; Bajaj, S.; Tanner, M.R.; Chang, S.C.; Krishnarjuna, B.; Ng, X.R.; Morales, R.A.V.; Chen, M.W.; Luo, D.; Patel, D.; et al. Modulation of Lymphocyte Potassium Channel KV1.3 by Membrane-Penetrating, Joint-Targeting Immunomodulatory Plant Defensin. ACS Pharmacol. Transl. Sci. 2020, 3, 720–736. [Google Scholar] [CrossRef]
- Nicolaou, S.A.; Neumeier, L.; Steckly, A.; Kucher, V.; Takimoto, K.; Conforti, L. Localization of Kv1.3 channels in the immunological synapse modulates the calcium response to antigen stimulation in T lymphocytes. J. Immunol. 2009, 183, 6296–6302. [Google Scholar] [CrossRef]
- Chimote, A.A.; Hajdu, P.; Sfyris, A.M.; Gleich, B.N.; Wise-Draper, T.; Casper, K.A.; Conforti, L. Kv1.3 Channels Mark Functionally Competent CD8+ Tumor-Infiltrating Lymphocytes in Head and Neck Cancer. Cancer Res. 2017, 77, 53–61. [Google Scholar] [CrossRef]
- Goggi, J.L.; Khanapur, S.; Ramasamy, B.; Hartimath, S.V.; Rong, T.J.; Cheng, P.; Tan, Y.X.; Yeo, X.Y.; Jung, S.; Goay, S.S.M.; et al. Imaging Kv1.3 Expressing Memory T Cells as a Marker of Immunotherapy Response. Cancers 2022, 14, 1217. [Google Scholar] [CrossRef]
- Tomayko, M.M.; Reynolds, C.P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Pardoll, D.M. Immune checkpoint inhibitors: Making immunotherapy a reality for the treatment of lung cancer. Cancer Immunol. Res. 2013, 1, 85–91. [Google Scholar] [CrossRef]
- Emens, L.A.; Middleton, G. The interplay of immunotherapy and chemotherapy: Harnessing potential synergies. Cancer Immunol. Res. 2015, 3, 436–443. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodriguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Heinhuis, K.M.; Ros, W.; Kok, M.; Steeghs, N.; Beijnen, J.H.; Schellens, J.H.M. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann. Oncol. 2019, 30, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Newton, H.S.; Gawali, V.S.; Chimote, A.A.; Lehn, M.A.; Palackdharry, S.M.; Hinrichs, B.H.; Jandarov, R.; Hildeman, D.; Janssen, E.M.; Wise-Draper, T.M.; et al. PD1 blockade enhances K+ channel activity, Ca2+ signaling, and migratory ability in cytotoxic T lymphocytes of patients with head and neck cancer. J. Immunother. Cancer 2020, 8, e000844. [Google Scholar] [CrossRef] [PubMed]
- Fabian, K.P.; Wolfson, B.; Hodge, J.W. From Immunogenic Cell Death to Immunogenic Modulation: Select Chemotherapy Regimens Induce a Spectrum of Immune-Enhancing Activities in the Tumor Microenvironment. Front. Oncol. 2021, 11, 728018. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, M.K.; Clambey, E.T.; Kappler, J.W.; Marrack, P. CD4 memory T cells: What are they and what can they do? Semin. Immunol. 2009, 21, 53–61. [Google Scholar] [CrossRef]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulieres, D.; Tahara, M.; de Castro, G., Jr.; Psyrri, A.; Baste, N.; Neupane, P.; Bratland, A.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gumus, M.; Mazieres, J.; Hermes, B.; Cay Senler, F.; Csoszi, T.; Fulop, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef]
- Peng, H.; James, C.A.; Cullinan, D.R.; Hogg, G.D.; Mudd, J.L.; Zuo, C.; Takchi, R.; Caldwell, K.E.; Liu, J.; DeNardo, D.G.; et al. Neoadjuvant FOLFIRINOX Therapy Is Associated with Increased Effector T Cells and Reduced Suppressor Cells in Patients with Pancreatic Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 6761–6771. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Treatment Arm | [18F]AlF-NOTA-KCNA3P (%ID/g ± SD) |
|---|---|
| Control | 0.50 ± 0.19 |
| Treated Responders (TRs) αPD1 | 0.81 ± 0.17 ** |
| OXA | 1.05 ± 0.18 *** |
| αPD1 + OXA | 1.05 ± 0.11 *** |
| 5-FU | 0.83 ± 0.15 ** |
| αPD1 + 5-FU | 1.11 ± 0.31 ***,$ |
| Treated Non-Responders (TNRs) | 0.44 ± 0.14 |
| Tumour-Infiltrating Immune Cell Populations | |||||
|---|---|---|---|---|---|
| Treatment Arm | CD8+% of CD3+ | CD8+ TEM% of CD8+ | CD4+ TEM% of CD4+ | KV1.3+ TEM% of CD3+ | NK+% of CD45+ |
| Control | 41.12 ± 5.17 | 26.43 ± 10.69 | 16.54 ± 5.57 | 0.92 ± 0.40 | 38.16 ± 2.46 |
| TR αPD1 | 65.63 ± 4.36 ** | 62.42 ± 6.26 ** | 39.22 ± 2.91 ** | 2.63 ± 0.60 * | 37.70 ± 4.42 |
| OXA | 81.63 ± 4.75 *** | 71.70 ± 18.32 ** | 39.48 ± 10.44 * | 3.41 ± 0.72 * | 31.33 ± 3.60 |
| αPD1 + OXA | 64.96 ± 10.79 * | 64.06 ± 13.78 ** | 39.18 ± 11.26 * | 3.06 ± 0.83 ** | 38.89 ± 5.18 |
| 5-FU | 22.52 ± 4.21 | 25.76 ± 12.31 | 29.83 ± 5.72 | 2.67 ± 0.76 ** | 49.28 ± 4.05 * |
| αPD1 + 5-FU | 57.99 ± 7.96 * | 51.72 ± 11.73 * | 33.16 ± 5.08 | 3.49 ± 0.82 * | 55.31 ± 6.07 ** |
| TNR | 27.70 ± 8.18 | 13.85 ± 5.70 | 11.73 ± 4.78 | 0.83 ± 0.45 | 32.54 ± 4.65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goggi, J.L.; Khanapur, S.; Hartimath, S.V.; Ramasamy, B.; Cheng, P.; Chin, H.-X.; Tang, J.-R.; Hwang, Y.-Y.; Robins, E.G. Imaging Effector Memory T-Cells Predicts Response to PD1-Chemotherapy Combinations in Colon Cancer. Biomedicines 2022, 10, 2343. https://doi.org/10.3390/biomedicines10102343
Goggi JL, Khanapur S, Hartimath SV, Ramasamy B, Cheng P, Chin H-X, Tang J-R, Hwang Y-Y, Robins EG. Imaging Effector Memory T-Cells Predicts Response to PD1-Chemotherapy Combinations in Colon Cancer. Biomedicines. 2022; 10(10):2343. https://doi.org/10.3390/biomedicines10102343
Chicago/Turabian StyleGoggi, Julian L., Shivashankar Khanapur, Siddesh V. Hartimath, Boominathan Ramasamy, Peter Cheng, Hui-Xian Chin, Jun-Rong Tang, You-Yi Hwang, and Edward G. Robins. 2022. "Imaging Effector Memory T-Cells Predicts Response to PD1-Chemotherapy Combinations in Colon Cancer" Biomedicines 10, no. 10: 2343. https://doi.org/10.3390/biomedicines10102343