
Inflammation Related to Obesity in the Etiopathogenesis of Gastroenteropancreatic Neuroendocrine Neoplasms

 ,
, 
Abstract
:1. Introduction
2. Gastroenteropancreatic Neuroendocrine Tumors
3. Biochemical Diagnosis of GEP-NET
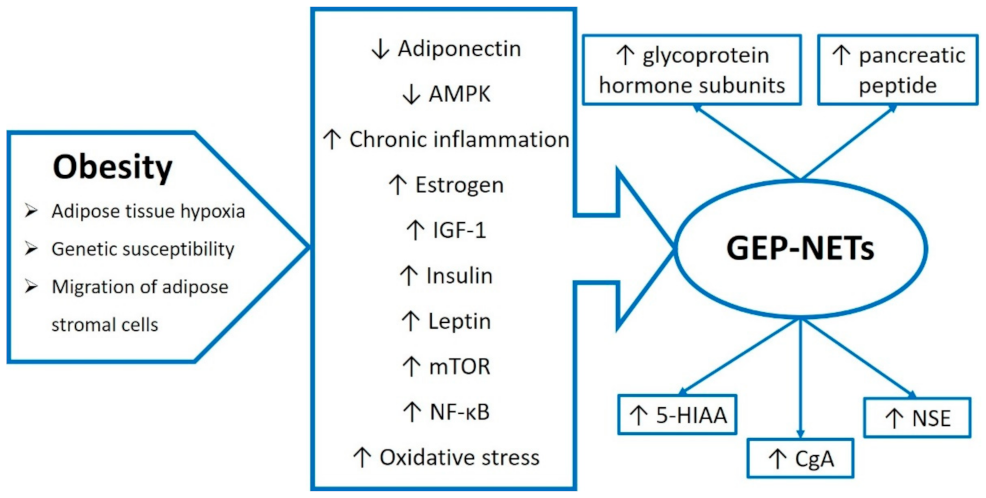
4. The Endocrine Role of Adipose Tissue and Inflammation in Obesity
5. Inflammation in Carcinogenesis
5.1. Interleukin 6
5.2. Tumor Necrosis Factor Alpha
5.3. Interleukin 17
5.4. Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand
5.5. Interleukin 10
5.6. Interleukin 12
5.7. Transforming Growth Factor β
6. Inflammatory Factors in GEP-NET
7. Obesity and Its Comorbidities and the Risk of GEP-NETs
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chang, M.-H.; You, S.-L.; Chen, C.-J.; Liu, C.-J.; Lee, C.-M.; Lin, S.-M.; Chu, H.-C.; Wu, T.-C.; Yang, S.-S.; Kuo, H.-S.; et al. Decreased Incidence of Hepatocellular Carcinoma in Hepatitis B Vaccinees: A 20-Year Follow-up Study. JNCI J. Natl. Cancer Inst. 2009, 101, 1348–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global Burden of Cancers Attributable to Infections in 2008: A Review and Synthetic Analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- de Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical Roles of the Immune System during Cancer Development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-W.; Karin, M. A Cytokine-Mediated Link between Innate Immunity, Inflammation, and Cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2020, 10, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Pi-Sunyer, X. The Medical Risks of Obesity. Postgrad. Med. 2009, 121, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Leocádio, P.C.L.; Oriá, R.B.; Crespo-Lopez, M.E.; Alvarez-Leite, J.I. Obesity: More Than an Inflammatory, an Infectious Disease? Front. Immunol. 2020, 10, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K. Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.; Colditz, G.A. Fresh Evidence Links Adiposity with Multiple Cancers. BMJ 2017, 356, j908. [Google Scholar] [CrossRef]
- Kolb, R.; Sutterwala, F.S.; Zhang, W. Obesity and Cancer: Inflammation Bridges the Two. Curr. Opin. Pharmacol. 2016, 29, 77–89. [Google Scholar] [CrossRef]
- Chawla, A.; Nguyen, K.D.; Goh, Y.P.S. Macrophage-Mediated Inflammation in Metabolic Disease. Nat. Rev. Immunol. 2011, 11, 738–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, N.S.; Catenacci, V.A.; Wyatt, H.R.; Hill, J.O. Obesity: Overview of an Epidemic. Psychiatr. Clin. North Am. 2011, 34, 717–732. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, Obesity, and Mortality from Cancer in a Prospectively Studied Cohort of U.S. Adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trofimiuk-Müldner, M.; Lewkowicz, E.; Wysocka, K.; Pach, D.; Kiełtyka, A.; Stefańska, A.; Sowa-Staszczak, A.; Tomaszewska, R.; Hubalewska-Dydejczyk, A. Epidemiologia Nowotworów Neuroendokrynnych Układu Pokarmowego w Krakowie i Powiecie Krakowskim w Latach 2007–2011. Endokrynol. Pol. 2017, 68, 42–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klöppel, G.; Perren, A.; Heitz, P.U. The Gastroenteropancreatic Neuroendocrine Cell System and Its Tumors: The WHO Classification. Ann. N. Y. Acad. Sci. 2004, 1014, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Chen, Q.; Xie, J.; Wang, J.; Lin, J.; Lu, J.; Cao, L.; Lin, M.; Tu, R.; Huang, Z.; et al. Incidence Trend and Conditional Survival Estimates of Gastroenteropancreatic Neuroendocrine Tumors: A Large Population-based Study. Cancer Med. 2018, 7, 3521–3533. [Google Scholar] [CrossRef]
- Asa, S.L.; Mete, O. Endocrine Pathology: Past, Present and Future. Pathology 2018, 50, 111–118. [Google Scholar] [CrossRef]
- Kos-Kudła, B.; Blicharz-Dorniak, J.; Handkiewicz-Junak, D.; Jarząb, B.; Jarząb, M.; Kunikowska, J.; Kuśnierz, K.; Król, R.; Królicki, L.; Krzakowski, M.; et al. Zalecenia Ogólne Dotyczące Postępowania w Nowotworach Neuroendokrynnych Układu Pokarmowego (Rekomendowane Przez Polską Sieć Guzów Neuroendokrynnych). Endokrynol. Pol. 2014, 64, 418–443. [Google Scholar] [CrossRef] [Green Version]
- Modlin, I.M.; Lye, K.D.; Kidd, M. A 5-Decade Analysis of 13,715 Carcinoid Tumors. Cancer 2003, 97, 934–959. [Google Scholar] [CrossRef] [PubMed]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients with Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335. [Google Scholar] [CrossRef] [PubMed]
- McMullen, T.; Al-Jahdali, A.; de Gara, C.; Ghosh, S.; McEwan, A.; Schiller, D. A Population-Based Study of Outcomes in Patients with Gastrointestinal Neuroendocrine Tumours. Can. J. Surg. 2017, 60, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.S.; Pritchard, D.M. Neuroendocrine tumours: What gastroenterologists need to know. Frontline Gastroenterol. 2022, 13, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Mottin, C.C.; Cruz, R.P.; Gomes Thomé, G.; Padoin, A.V. Carcinoid Tumors and Morbid Obesity. Obes. Surg. 2009, 19, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Frilling, A.; Åkerström, G.; Falconi, M.; Pavel, M.; Ramos, J.; Kidd, M.; Modlin, I.M. Neuroendocrine Tumor Disease: An Evolving Landscape. Endocr. Relat. Cancer 2012, 19, R163–R185. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Li, Q.; Kou, F.; Zhu, Y.; Lu, M.; Li, J.; Lu, Z.; Shen, L. Prognostic Value of Inflammation-Based Markers in Advanced or Metastatic Neuroendocrine Tumours. Curr. Oncol. 2019, 26, 4135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meeker, A.; Heaphy, C. Gastroenteropancreatic Endocrine Tumors. Mol. Cell. Endocrinol. 2014, 386, 101–120. [Google Scholar] [CrossRef] [PubMed]
- Kos-Kudła, B.; Čwikła, J.; Ruchała, M.; Dydejczyk, A.H.; Jarzab, B.; Krajewska, J.; Kamiński, G. Current Treatment Options for Gastroenteropancreatic Neuroendocrine Tumors with a Focus on the Role of Lanreotide. Wspolczesna Onkol. 2017, 21, 115–122. [Google Scholar] [CrossRef]
- Modlin, I.M.; Oberg, K.; Chung, D.C.; Jensen, R.T.; de Herder, W.W.; Thakker, R.V.; Caplin, M.; Delle Fave, G.; Kaltsas, G.A.; Krenning, E.P.; et al. Gastroenteropancreatic Neuroendocrine Tumours. Lancet Oncol. 2008, 9, 61–72. [Google Scholar] [CrossRef]
- Johnson, P.R.V. Gastroenteropancreatic Neuroendocrine (Carcinoid) Tumors in Children. Semin. Pediatr. Surg. 2014, 23, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Halperin, D.M.; Shen, C.; Dasari, A.; Xu, Y.; Chu, Y.; Zhou, S.; Shih, Y.-C.T.; Yao, J.C. Frequency of Carcinoid Syndrome at Neuroendocrine Tumour Diagnosis: A Population-Based Study. Lancet Oncol. 2017, 18, 525–534. [Google Scholar] [CrossRef]
- Crona, J.; Skogseid, B. Genetics of Neuroendocrine Tumors. Eur. J. Endocrinol. 2016, 174, R275–R290. [Google Scholar] [CrossRef] [Green Version]
- Cives, M.; Strosberg, J.R. Gastroenteropancreatic Neuroendocrine Tumors. CA Cancer J. Clin. 2018, 68, 471–487. [Google Scholar] [CrossRef]
- Kawasaki, K.; Fujii, M.; Sato, T. Gastroenteropancreatic Neuroendocrine Neoplasms: Genes, Therapies and Models. Dis. Model. Mech. 2018, 11, dmm029595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díez, M.; Teulé, A.; Salazar, R. Gastroenteropancreatic Neuroendocrine Tumors: Diagnosis and Treatment. Ann. Gastroenterol. 2013, 26, 29–36. [Google Scholar]
- Ramage, J.K.; Davies, A.H.G.; Ardill, J.; Bax, N.; Caplin, M.; Grossman, A.; Hawkins, R.; McNicol, A.M.; Reed, N.; Sutton, R.; et al. Guidelines for the Management of Gastroenteropancreatic Neuroendocrine (Including Carcinoid) Tumours. Gut 2005, 54, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Burks, M.L.; Bao, S. The 24-Hour Urinary 5-HIAA: A Simple Test with a Common Pitfall. AACE Clin. Case Rep. 2016, 2, e186–e188. [Google Scholar] [CrossRef]
- de Herder, W.W. Biochemistry of Neuroendocrine Tumours. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Oladejo, A. Gastroenteropancreatic Neuroendocrine Tumors (GEP-NETs)—Approach to Diagnosis and Managment. Ann. Ib. Postgrad. Med. 2011, 7, 29–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Risi, E.S.; Al-Essry, F.S.; Mula-Abed, W.-A.S. Chromogranin A as a Biochemical Marker for Neuroendocrine Tumors: A Single Center Experience at Royal Hospital, Oman. Oman Med. J. 2017, 32, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Helle, K.B.; Corti, A.; Metz-Boutigue, M.-H.; Tota, B. The Endocrine Role for Chromogranin A: A Prohormone for Peptides with Regulatory Properties. Cell. Mol. Life Sci. 2007, 64, 2863–2886. [Google Scholar] [CrossRef]
- Kaltsas, G.A.; Besser, G.M.; Grossman, A.B. The Diagnosis and Medical Management of Advanced Neuroendocrine Tumors. Endocr. Rev. 2004, 25, 458–511. [Google Scholar] [CrossRef] [PubMed]
- Glinicki, P.; Jeske, W. Chromogranin A (CgA)—Characteristic of the Currently Available Laboratory Methods and Conditions Which Can Influence the Results. Endokrynol. Pol. 2009, 60, 415–419. [Google Scholar] [PubMed]
- Yang, X.; Yang, Y.; Li, Z.; Cheng, C.; Yang, T.; Wang, C.; Liu, L.; Liu, S. Diagnostic Value of Circulating Chromogranin a for Neuroendocrine Tumors: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyubimova, N.V.; Churikova, T.K.; Kushlinskii, N.E. Chromogranin as a Biochemical Marker of Neuroendocrine Tumors. Bull. Exp. Biol. Med. 2016, 160, 702–704. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.C.; Hung, Y.S.; Hsu, J.T.; Chen, J.S.; Lu, C.H.; Hwang, T.L.; Rau, K.M.; Yeh, K.Y.; Chen, T.C.; Sun, C.F. Chromogranin a Is a Reliable Biomarker for Gastroenteropancreatic Neuroendocrine Tumors in an Asian Population of Patients. Neuroendocrinology 2012, 95, 344–350. [Google Scholar] [CrossRef]
- Oberg, K.; Stridsberg, M. Chromogranins as Diagnostic and Prognostic Markers in Neuroendocrine Tumours. Adv. Exp. Med. Biol. 2000, 482, 329–337. [Google Scholar] [CrossRef]
- Isgrò, M.A.; Bottoni, P.; Scatena, R. Neuron-Specific Enolase as a Biomarker: Biochemical and Clinical Aspects. Adv. Exp. Med. Biol. 2015, 867, 125–143. [Google Scholar] [CrossRef]
- Nobels, F.R.E.; Kwekkeboom, D.J.; Coopmans, W.; Schoenmakers, C.H.H.; Lindemans, J.; De Herder, W.W.; Krenning, E.P.; Bouillon, R.; Lamberts, S.W.J. Chromogranin A as Serum Marker for Neuroendocrine Neoplasia: Comparison with Neuron-Specific Enolase and the α-Subunit of Glycoprotein Hormones. J. Clin. Endocrinol. Metab. 1997, 82, 2622–2628. [Google Scholar] [CrossRef] [Green Version]
- Kershaw, E.E.; Flier, J.S. Adipose Tissue as an Endocrine Organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef]
- Coelho, M.; Oliveira, T.; Fernandes, R. State of the Art Paper Biochemistry of Adipose Tissue: An Endocrine Organ. Arch. Med. Sci. 2013, 2, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Berry, D.C.; Stenesen, D.; Zeve, D.; Graff, J.M. The Developmental Origins of Adipose Tissue. Development 2013, 140, 3939–3949. [Google Scholar] [CrossRef] [PubMed]
- Shamsi, F.; Tseng, Y.H.; Kahn, C.R. Adipocyte Microenvironment: Everybody in the Neighborhood Talks about the Temperature. Cell Metab. 2021, 33, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.B.; Chen, S.Y. White Adipose Tissue Browning and Obesity. J. Biomed. Res. 2017, 31, 1–2. [Google Scholar] [CrossRef]
- Cannon, B.; Nedergaard, J. Brown Adipose Tissue: Function and Physiological Significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Mottillo, E.P.; Granneman, J.G. Adipose Tissue Plasticity from WAT to BAT and in between. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 358–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinonen, S.; Rissanen, A. White Adipose Tissue Mitochondrial Metabolism in Health and in Obesity. Obes. Rev. 2019, 21, e12958. [Google Scholar] [CrossRef]
- Kivelä, R.; Alitalo, K. White Adipose Tissue Coloring by Intermittent Fasting. Cell Res. 2017, 27, 1300–1301. [Google Scholar] [CrossRef] [Green Version]
- WHO 2016, Fact Sheet: Obesity and Overweight. Available online: https://www.who.int/newsroom/fact-sheets/detail/obesity-and-overweight. (accessed on 1 July 2022).
- Gustafson, B. Adipose Tissue, Inflammation and Atherosclerosis. J. Atheroscler. Thromb. 2010, 17, 332–341. [Google Scholar] [CrossRef] [Green Version]
- Stępień, M.; Stępień, A.; Wlazeł, R.N.; Paradowski, M.; Banach, M.; Rysz, J. Obesity Indices and Inflammatory Markers in Obese Non-Diabetic Normo- and Hypertensive Patients: A Comparative Pilot Study. Lipids Health Dis. 2014, 13, 29. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.-M.; An, J. Cytokines, Inflammation, and Pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Castro, A.M.; Macedo-de la Concha, L.E.; Pantoja-Meléndez, C.A. Low-Grade Inflammation and Its Relation to Obesity and Chronic Degenerative Diseases. Rev. Médica Del Hosp. Gen. México 2017, 80, 101–105. [Google Scholar] [CrossRef]
- Slabbert, S.; De Ridder, H.J.; Underhay, C.; Kruger, S. Obesity as an Inflammatory Condition. Health SA Gesondheid 2006, 11, a220. [Google Scholar] [CrossRef]
- Wang, T.; He, C. Pro-Inflammatory Cytokines: The Link between Obesity and Osteoarthritis. Cytokine Growth Factor Rev. 2018, 44, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Higuchi, A.; Ohashi, K.; Oshima, Y.; Gokce, N.; Shibata, R.; Akasaki, Y.; Shimono, A.; Walsh, K. Sfrp5 Is an Anti-Inflammatory Adipokine That Modulates Metabolic Dysfunction in Obesity. Science 2010, 329, 454–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y. Obesity & Inflammation: The Linking Mechanism & the Complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef]
- Ekmen, N.; Helvaci, A.; Gunaldi, M.; Sasani, H.; Yildirmak, S.T. Leptin as an Important Link between Obesity and Cardiovascular Risk Factors in Men with Acute Myocardial Infarction. Indian Heart J. 2016, 68, 132–137. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Rui, L. Leptin Signaling and Leptin Resistance. Front. Med. 2013, 7, 207–222. [Google Scholar] [CrossRef]
- Janochova, K.; Haluzik, M.; Buzga, M. Visceral Fat and Insulin Resistance—What We Know? Biomed. Pap. 2019, 163, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Alzaim, I.; Hammoud, S.H.; Al-Koussa, H.; Ghazi, A.; Eid, A.H.; El-Yazbi, A.F. Adipose Tissue Immunomodulation: A Novel Therapeutic Approach in Cardiovascular and Metabolic Diseases. Front. Cardiovasc. Med. 2020, 7, 1–40. [Google Scholar] [CrossRef]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Frühbeck, G. Adipose Tissue Immunity and Cancer. Front. Physiol. 2013, 4, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte Dysfunctions Linking Obesity to Insulin Resistance and Type 2 Diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Lyon, C.J.; Bergin, S.; Caligiuri, M.A.; Hsueh, W.A. Obesity, Inflammation, and Cancer. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 421–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, S.; Hascall, V.C.; Markwald, R.R.; O’Brien, P.E.; Ghatak, S. Inflammation and Cancer. Wound Healing: Stem Cells Repair Restorations, Basic and Clinical Aspects; John Wiley & Sons: Hoboken, NJ, USA, 2018; Volume 420, pp. 239–274. [Google Scholar] [CrossRef]
- Bremnes, R.M.; Al-Shibli, K.; Donnem, T.; Sirera, R.; Al-Saad, S.; Andersen, S.; Stenvold, H.; Camps, C.; Busund, L.-T. The Role of Tumor-Infiltrating Immune Cells and Chronic Inflammation at the Tumor Site on Cancer Development, Progression, and Prognosis: Emphasis on Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2011, 6, 824–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prendergast, G.C. Immune Escape as a Fundamental Trait of Cancer: Focus on IDO. Oncogene 2008, 27, 3889–3900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Okada, F. Inflammation-Related Carcinogenesis: Current Findings in Epidemiological Trends, Causes and Mechanisms. Yonago Acta Med. 2014, 57, 65–72. [Google Scholar]
- Lan, T.; Chen, L.; Wei, X. Inflammatory Cytokines in Cancer: Comprehensive Understanding and Clinical Progress in Gene Therapy. Cells 2021, 10, 100. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Huang, S. Regulation of Metastases by Signal Transducer and Activator of Transcription 3 Signaling Pathway: Clinical Implications. Clin. Cancer Res. 2007, 13, 1362–1366. [Google Scholar] [CrossRef] [Green Version]
- Naugler, W.E.; Karin, M. NF-ΚB and Cancer—Identifying Targets and Mechanisms. Curr. Opin. Genet. Dev. 2008, 18, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Chonov, D.C.; Ignatova, M.M.K.; Ananiev, J.R.; Gulubova, M.V. IL-6 Activities in the Tumour Microenvironment. Part 1. Open Access Maced. J. Med. Sci. 2019, 7, 2391–2398. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Il-6 in Inflammation, Immunity, And Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Ting, E.Y.-C.; Yang, A.C.; Tsai, S.-J. Role of Interleukin-6 in Depressive Disorder. Int. J. Mol. Sci. 2020, 21, 2194. [Google Scholar] [CrossRef] [Green Version]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of Interleukin-6 in Cancer Progression and Therapeutic Resistance. Tumor Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef]
- Hirano, T. IL-6 in Inflammation, Autoimmunity and Cancer. Int. Immunol. 2021, 33, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic Inflammation and Cytokines in the Tumor Microenvironment. J. Immunol. Res. 2014, 2014, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.; Vallabhapurapu, S. Article IL-6 and Stat3 Are Required for Survival of Intestinal Epithelial Cells and Development of Colitis-Associated Cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Sethi, G. TNF: A Master Switch for Inflammation to Cancer. Front. Biosci. 2008, 13, 5094–5107. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lin, Y. Tumor Necrosis Factor and Cancer, Buddies or Foes? Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef] [Green Version]
- Lubecka-Macura, A.; Kohut, M. TNF Superfamily—Mechanisms of Action, Biologic Funtions and Therapeutic Possibilities. Gastroenterol. Rev. 2010, 6, 303–309. [Google Scholar] [CrossRef]
- Kim, J.J.; Lee, S.B.; Park, J.K.; Yoo, Y.D. TNF-α-Induced ROS Production Triggering Apoptosis Is Directly Linked to Romo1 and Bcl-XL. Cell Death Differ. 2010, 17, 1420–1434. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Yang, M.; Zhang, H.; Sun, Y.; Tao, Y.; Li, H.; Zhang, J.; Li, Y.; Yang, J. IL-17 Affects the Progression, Metastasis, and Recurrence of Laryngeal Cancer via the Inhibition of Apoptosis through Activation of the PI3K/AKT/FAS/FASL Pathways. J. Immunol. Res. 2020, 2020, 2953191. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, X.; Herjan, T.; Li, X. The Role of Interleukin-17 in Tumor Development and Progression. J. Exp. Med. 2020, 217, e20190297. [Google Scholar] [CrossRef] [PubMed]
- Falschlehner, C.; Schaefer, U.; Walczak, H. Following TRAIL’s Path in the Immune System. Immunology 2009, 127, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Thapa, B.; Remant, K.C.; Uludağ, H. TRAIL Therapy and Prospective Developments for Cancer Treatment. J. Control. Release 2020, 326, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.M.; Kong, W.Y.; Fang, C.-M.; Loh, H.-S.; Chuah, L.-H.; Abdullah, S.; Ngai, S.C. The TRAIL to Cancer Therapy: Hindrances and Potential Solutions. Crit. Rev. Oncol. Hematol. 2019, 143, 81–94. [Google Scholar] [CrossRef]
- LeBlanc, H.N.; Ashkenazi, A. Apo2L/TRAIL and Its Death and Decoy Receptors. Cell Death Differ. 2003, 10, 66–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Karstedt, S.; Montinaro, A.; Walczak, H. Exploring the TRAILs Less Travelled: TRAIL in Cancer Biology and Therapy. Nat. Rev. Cancer 2017, 17, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Garza, M.T.; Cruz-Vega, D.E.; Maldonado-Bernal, C. IL10 as Cancer Biomarker. In Translational Research in Cancer; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Qiao, J.; Liu, Z.; Dong, C.; Luan, Y.; Zhang, A.; Moore, C.; Fu, K.; Peng, J.; Wang, Y.; Ren, Z.; et al. Targeting Tumors with IL-10 Prevents Dendritic Cell-Mediated CD8+ T Cell Apoptosis. Cancer Cell 2019, 35, 901–915. [Google Scholar] [CrossRef] [PubMed]
- Kundu, N.; Fulton, A.M. Interleukin-10 Inhibits Tumor Metastasis, Downregulates MHC Class I, and Enhances NK Lysis. Cell. Immunol. 1997, 180, 55–61. [Google Scholar] [CrossRef]
- Oft, M. IL-10: Master Switch from Tumor-Promoting Inflammation to Antitumor Immunity. Cancer Immunol. Res. 2014, 2, 194–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, K.G.; Vrabel, M.R.; Mantooth, S.M.; Hopkins, J.J.; Wagner, E.S.; Gabaldon, T.A.; Zaharoff, D.A. Localized Interleukin-12 for Cancer Immunotherapy. Front. Immunol. 2020, 11, 575597. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Interleukin-12 and the Regulation of Innate Resistance and Adaptive Immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Mirlekar, B.; Pylayeva-Gupta, Y. IL-12 Family Cytokines in Cancer and Immunotherapy. Cancers 2021, 13, 167. [Google Scholar] [CrossRef]
- Fabregat, I.; Fernando, J.; Mainez, J.; Sancho, P. TGF-Beta Signaling in Cancer Treatment. Curr. Pharm. Des. 2014, 20, 2934–2947. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-Induced Epithelial to Mesenchymal Transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Peinado, H.; Quintanilla, M.; Cano, A. Transforming Growth Factor β-1 Induces Snail Transcription Factor in Epithelial Cell Lines. Mechanisms for Epithelial Mesenchymal Transitions. J. Biol. Chem. 2003, 278, 21113–21123. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Baker, D.; ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, A.; Wakabayashi, Y.; Mori, T. Cellular and Molecular Basis for the Regulation of Inflammation by TGF-β. J. Biochem. 2010, 147, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Berkovic, M.C.; Cacev, T.; Ivkovic, T.C.; Zjacic-Rotkvic, V.; Kapitanovic, S. New Insights into the Role of Chronic Inflammation and Cytokines in the Etiopathogenesis of Gastroenteropancreatic Neuroendocrine Tumors. Neuroendocrinology 2014, 99, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Girardi, D.M.; Silva, A.C.B.; Rêgo, J.F.M.; Coudry, R.A.; Riechelmann, R.P. Unraveling Molecular Pathways of Poorly Differentiated Neuroendocrine Carcinomas of the Gastroenteropancreatic System: A Systematic Review. Cancer Treat. Rev. 2017, 56, 28–35. [Google Scholar] [CrossRef]
- Mahečić, D.H.; Berković, M.C.; Zjačić-Rotkvić, V.; Čačev, T.; Kapitanović, S.; Ulamec, M. Inflammation-Related Cytokines and Their Roles in Gastroenteropancreatic Neuroendocrine Neoplasms. Bosn. J. Basic Med. Sci. 2020, 20, 445–450. [Google Scholar] [CrossRef]
- Berković, M.C.; Jokić, M.; Marout, J.; Radošević, S.; Zjačić-Rotkvić, V.; Kapitanović, S. IL-6-174 C/G Polymorphism in the Gastroenteropancreatic Neuroendocrine Tumors (GEP-NETs). Exp. Mol. Pathol. 2007, 83, 474–479. [Google Scholar] [CrossRef]
- Chung, Y.C.; Chang, Y.F. Serum Interleukin-6 Levels Reflect the Disease Status of Colorectal Cancer. J. Surg. Oncol. 2003, 83, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Macarthur, M.; Hold, G.L.; El-Omar, E.M. Inflammation and Cancer II. Role of Chronic Inflammation and Cytokine Gene Polymorphisms in the Pathogenesis of Gastrointestinal Malignancy. Am. J. Physiol. Liver Physiol. 2004, 286, G515–G520. [Google Scholar] [CrossRef] [PubMed]
- De Vita, F.; Romano, C.; Orditura, M.; Galizia, G.; Martinelli, E.; Lieto, E.; Catalano, G. Interleukin-6 Serum Level Correlates with Survival in Advanced Gastrointestinal Cancer Patients but Is Not an Independent Prognostic Indicator. J. Interf. Cytokine Res. 2001, 21, 45–52. [Google Scholar] [CrossRef]
- Berković, M.C.; Ivković, T.C.; Marout, J.; Zjačić-Rotkvić, V.; Kapitanović, S. Interleukin 1β Gene Single-Nucleotide Polymorphisms and Susceptibility to Pancreatic Neuroendocrine Tumors. DNA Cell Biol. 2012, 31, 531–536. [Google Scholar] [CrossRef] [Green Version]
- Duerr, E.M.; Mizukami, Y.; Ng, A.; Xavier, R.J.; Kikuchi, H.; Deshpande, V.; Warshaw, A.L.; Glickman, J.; Kulke, M.H.; Chung, D.C. Defining Molecular Classifications and Targets in Gastroenteropancreatic Neuroendocrine Tumors through DNA Microarray Analysis. Endocr. Relat. Cancer 2008, 15, 243–256. [Google Scholar] [CrossRef]
- Oh, S.A.; Li, M.O. TGF-β: Guardian of T Cell Function. J. Immunol. 2013, 191, 3973–3979. [Google Scholar] [CrossRef]
- Leu, F.P.; Nandi, M.; Niu, C. The Effect of Transforming Growth Factor β on Human Neuroendocrine Tumor BON Cell Proliferation and Differentiation Is Mediated through Somatostatin Signaling. Mol. Cancer Res. 2008, 6, 1029–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, J.; Chen, W.; Zhu, H.J. The Immune Suppressive Function of Transforming Growth Factor-β (TGF-β) in Human Diseases. Growth Factors 2015, 33, 92–101. [Google Scholar] [CrossRef]
- Wimmel, A.; Wiedenmann, B.; Rosewicz, S. Autocrine Growth Inhibition by Transforming Growth Factor β-1 (TGFβ-1) in Human Neuroendocrine Tumour Cells. Gut 2003, 52, 1308–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köseoğlu, H.; Duzenli, T.; Sezikli, M. Gastric Neuroendocrine Neoplasms: A Review. World J. Clin. Cases 2021, 9, 7973–7985. [Google Scholar] [CrossRef]
- Roberto, G.A.; Rodrigues, C.M.B.; Peixoto, R.D.A.; Younes, R.N. Gastric Neuroendocrine Tumor: A Practical Literature Review. World J. Gastrointest. Oncol. 2020, 12, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.F.; El-Salhy, M.; Melgar, S.; Hammarström, M.L.; Danielsson, Å. Neuroendocrine Changes in Colon of Mice with a Disrupted IL-2 Gene. Clin. Exp. Immunol. 2000, 120, 424–433. [Google Scholar] [CrossRef]
- Pavel, M.E.; Hassler, G.; Baum, U.; Hahn, E.G.; Lohmann, T.; Schuppan, D. Circulating of Angiogenic Cytokines Can Predict Tumour Progression and Prognosis in Neuroendocrine Carcinomas. Clin. Endocrinol. 2005, 62, 434–443. [Google Scholar] [CrossRef]
- Hussain, F.; Wang, J.; Ahmed, R.; Guest, S.K.; Lam, E.W.F.; Stamp, G.; El-Bahrawy, M. The Expression of IL-8 and IL-8 Receptors in Pancreatic Adenocarcinomas and Pancreatic Neuroendocrine Tumours. Cytokine 2010, 49, 134–140. [Google Scholar] [CrossRef]
- Cigrovski Berković, M.; Čačev, T.; Catela Ivković, T.; Marout, J.; Ulamec, M.; Zjačić-Rotkvić, V.; Kapitanović, S. High VEGF Serum Values Are Associated with Locoregional Spread of Gastroenteropancreatic Neuroendocrine Tumors (GEP-NETs). Mol. Cell. Endocrinol. 2016, 425, 61–68. [Google Scholar] [CrossRef]
- Berardi, R.; Torniai, M.; Partelli, S.; Rubini, C.; Pagliaretta, S.; Savini, A.; Polenta, V.; Santoni, M.; Giampieri, R.; Onorati, S.; et al. Impact of Vascular Endothelial Growth Factor (VEGF) and Vascular Endothelial Growth Factor Receptor (VEGFR) Single Nucleotide Polymorphisms on Outcome in Gastroenteropancreatic Neuroendocrine Neoplasms. PLoS ONE 2018, 13, e0197035. [Google Scholar] [CrossRef]
- Kulahci, O.; Koseci, T. The Correlation of the Neutrophil-Lymphocyte Ratio and the Platelet-Lymphocyte Ratio with Pathological Findings in Neuroendocrine Tumors. Cureus 2021, 13, e17164. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Liu, C.; Cheng, H.; Jin, K.; Guo, M.; Lu, Y.; Long, J.; Xu, J.; Ni, Q.; Chen, J.; et al. Neutrophil-Lymphocyte Ratio Predicts Survival in Pancreatic Neuroendocrine Tumors. Oncol. Lett. 2017, 13, 2454–2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, Z.; Liu, L.; Zheng, Y.; Jiang, W.; Zhao, P.; Fang, W.; Wang, W. Predictive Value of Preoperative Peripheral Blood Neutrophil/Lymphocyte Ratio for Lymph Node Metastasis in Patients of Resectable Pancreatic Neuroendocrine Tumors: A Nomogram-Based Study. World J. Surg. Oncol. 2017, 15, 108. [Google Scholar] [CrossRef]
- Pozza, A.; Pauletti, B.; Scarpa, M.; Ruffolo, C.; Bassi, N.; Massani, M. Prognostic Role of Neutrophil-to-Lymphocyte Ratio and Platelet-to-Lymphocyte Ratio in Patients with Midgut Neuroendocrine Tumors Undergoing Resective Surgery. Int. J. Colorectal Dis. 2019, 34, 1849–1856. [Google Scholar] [CrossRef]
- Giannetta, E.; La Salvia, A.; Rizza, L.; Muscogiuri, G.; Campione, S.; Pozza, C.; Colao, A.A.L.; Faggiano, A. Are Markers of Systemic Inflammatory Response Useful in the Management of Patients with Neuroendocrine Neoplasms? Front. Endocrinol. 2021, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Berković, M.C.; Jokić, M.; Marout, J.; Radošević, S.; Zjačić-Rotkvić, V.; Kapitanović, S. IL-2 −330 T/G SNP and Serum Values—Potential New Tumor Markers in Neuroendocrine Tumors of the Gastrointestinal Tract and Pancreas (GEP-NETs). J. Mol. Med. 2010, 88, 423–429. [Google Scholar] [CrossRef]
- Zhou, B.; Zhan, C.; Wu, J.; Liu, J.; Zhou, J.; Zheng, S. Prognostic Significance of Preoperative Neutrophil-to-Lymphocyte Ratio in Surgically Resectable Pancreatic Neuroendocrine Tumors. Med. Sci. Monit. 2017, 23, 5574–5588. [Google Scholar] [CrossRef] [Green Version]
- Hruby, A.; Hu, F.B. The Epidemiology of Obesity: A Big Picture. Pharmacoeconomics 2015, 33, 673–689. [Google Scholar] [CrossRef] [PubMed]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory Mechanisms in Obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, E.J.; LeRoith, D. Obesity and Diabetes: The Increased Risk of Cancer and Cancer-Related Mortality. Physiol. Rev. 2015, 95, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Seo, B.R.; Bhardwaj, P.; Choi, S.; Gonzalez, J.; Eguiluz, R.C.A.; Wang, K.; Mohanan, S.; Morris, P.G.; Du, B.; Zhou, X.K.; et al. Obesity-Dependent Changes in Interstitial ECM Mechanics Promote Breast Tumorigenesis. Sci. Transl. Med. 2015, 7, 301ra130. [Google Scholar] [CrossRef] [Green Version]
- Roberts, D.L.; Dive, C.; Renehan, A.G. Biological Mechanisms Linking Obesity and Cancer Risk: New Perspectives. Annu. Rev. Med. 2010, 61, 301–316. [Google Scholar] [CrossRef]
- Keshishian, A.; Hamilton, J.; Hwang, L.; Petrosyan, M. Carcinoid Tumor and Bariatric Surgery. Obes. Surg. 2002, 12, 874–875. [Google Scholar] [CrossRef]
- Katz, L.H.; Levi, Z.; Twig, G.; Kark, J.D.; Leiba, A.; Derazne, E.; Liphshiz, I.; Keinan-Boker, L.; Eisenstein, S.; Afek, A. Risk Factors Associated with Gastroenteropancreatic Neuroendocrine Tumors in a Cohort of 2.3 Million Israeli Adolescents. Int. J. Cancer 2018, 143, 1876–1883. [Google Scholar] [CrossRef] [Green Version]
- Hassan, M.M.; Phan, A.; Li, D.; Dagohoy, C.G.; Leary, C.; Yao, J.C. Risk Factors Associated with Neuroendocrine Tumors: A U.S.-Based Case-Control Study. Int. J. Cancer 2008, 123, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, H.; Nair, J. Chronic Inflammation and Oxidative Stress in the Genesis and Perpetuation of Cancer: Role of Lipid Peroxidation, DNA Damage, and Repair. Langenbeck’s Arch. Surg. 2006, 391, 499–510. [Google Scholar] [CrossRef]
- Al-Goblan, A.S.; Al-Alfi, M.A.; Khan, M.Z. Mechanism Linking Diabetes Mellitus and Obesity. Diabetes Metab. Syndr. Obes. Targets Ther. 2014, 7, 587–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, A.P.; Santos, A.C.; Castro, C.; Raposo, L.; Pereira, S.S.; Torres, I.; Henrique, R.; Cardoso, H.; Monteiro, M.P. Visceral Obesity and Metabolic Syndrome Are Associated with Well-Differentiated Gastroenteropancreatic Neuroendocrine Tumors. Cancers 2018, 10, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, A.P.; Castro, C.; Antunes, L.; Henrique, R.; Helena Cardoso, M.; Monteiro, M.P. Disseminated Well-Differentiated Gastro-Entero- Pancreatic Tumors Are Associated with Metabolic Syndrome. J. Clin. Med. 2019, 8, 1479. [Google Scholar] [CrossRef] [Green Version]
- Leoncini, E.; Carioli, G.; La Vecchia, C.; Boccia, S.; Rindi, G. Risk Factors for Neuroendocrine Neoplasms: A Systematic Review and Meta-Analysis. Ann. Oncol. 2016, 27, 68–81. [Google Scholar] [CrossRef]
- Zhan, H.X.; Cong, L.; Zhao, Y.P.; Zhang, T.P.; Chen, G. Risk Factors for the Occurrence of Insulinoma: A Case-Control Study. Hepatobiliary Pancreat. Dis. Int. 2013, 12, 324–328. [Google Scholar] [CrossRef]
- Halfdanarson, T.R.; Bamlet, W.R.; McWilliams, R.R.; Hobday, T.J.; Burch, P.A.; Rabe, K.G.; Petersen, G.M. Risk Factors for Pancreatic Neuroendocrine Tumors a Clinic-Based Case-Control Study. Pancreas 2014, 43, 1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, A.J.; Hollenbeck, A.R.; Park, Y. A Large Prospective Study of Risk Factors for Adenocarcinomas and Malignant Carcinoid Tumors of the Small Intestine. Cancer Causes Control 2013, 24, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Glazer, E.; Stanko, K.; Ong, E.; Guerrero, M. Decreased Inpatient Mortality in Obese Patients with Abdominal Nets. Endocr. Pract. 2014, 20, 1309–1314. [Google Scholar] [CrossRef]
- Abdel-Rahman, O.; Ghosh, S.; Morrish, D. Impact of Baseline Body Mass Index on the Outcomes of Patients with Neuroendocrine Neoplasms. J. Endocrinol. Invest. 2022, 45, 1683–1688. [Google Scholar] [CrossRef]
- Pereira, S.S.; Pereira, R.; Santos, A.P.; Costa, M.M.; Morais, T.; Sampaio, P.; Machado, B.; Afonso, L.P.; Henrique, R.; Monteiro, M.P. Higher IL-6 Peri-Tumoural Expression Is Associated with Gastro-Intestinal Neuroendocrine Tumour Progression. Pathology 2019, 51, 593–599. [Google Scholar] [CrossRef]
- Yao, J.C.; Hassan, M.; Phan, A.; Dagohoy, C.; Leary, C.; Mares, J.E.; Abdalla, E.K.; Fleming, J.B.; Vauthey, J.-N.; Rashid, A.; et al. One Hundred Years After “Carcinoid”: Epidemiology of and Prognostic Factors for Neuroendocrine Tumors in 35,825 Cases in the United States. J. Clin. Oncol. 2008, 26, 3063–3072. [Google Scholar] [CrossRef] [Green Version]
- Malik, V.S.; Willet, W.C.; Hu, F.B. Nearly a Decade on—Trends, Risk Factors and Policy Implications in Global Obesity. Nat. Rev. Endocrinol. 2020, 16, 615–616. [Google Scholar] [CrossRef]
- Cigrovski, M.; Herman, D.; Tomai, V.; Hrabar, D.; Zjai-Rotkvi, V. The Association of Chronic Inflammation and Gastroenteropancreatic Neuroendocrine Tumors (GEP-NETs). In Neuroendocrine Tumor; InTech: London, UK, 2012. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Type of Tumor | Characteristics of Subjects | Parameters | References |
|---|---|---|---|
| GEP-NET | 101 patients with GEP-NET and 150 controls (healthy volunteers); analysis of single nucleotide polymorphisms (SNPs) | IL-2-330 T/G, statistically significant association between high-expression GG genotype and high-expression G-allele and risk of GEP-NET | [139] |
| GEP-NET | 101 patients with GEP-NET and 20 controls (healthy volunteers) | ↑IL-2 (highest in patients with functional GEP-NET); IL-2 more specific in detecting GEP-NET than CgA and 5-HIAA | [139] |
| GEP-NEN | 43 patients with GEP-NENs | ↑TNF-α (correlation with tumor to higher grade and proliferation rates, expression positively correlated with death); ↑IL-6 with tumor grade (not statistically relevant); ↓IL-2, ↓IL-1β | [116] |
| GEP-NET | 80 patients with GEP-NETs and 162 healthy controls; SNP IL-6-174 C/G and IL-6 serum values | ↑IL-6-174 CG/GG-NF-pNET (statistically significant with functional pNET); ↑IL-6 values serum: correlated significantly GG IL-6-174 genotype in all GEP-NETs, significantly higher IL-6 in NF-pNET with functional-pNET and GI-NET | [117] |
| pNET | 60 patients with pNET and 60 healthy controls; IL-1β SNP (IL-1β-511-C/T, +3954C/T) and IL-1β serum values | High expression IL-1β-511-C/T in functional pNET (statistically significant); −511/+3954 CTCT: risk of developing functional pNET (statistically significant); −511/+3954 CTCC: risk of developing NF-pNET (statistically significant); IL-1β serum levels: undetectable | [121] |
| GEP-NET | Human NET cell lines: BON (functional pNET, LCC-18 (NF-colorectal NET), QGP (NF-pNET), and cell lines BxPc3 and PANC-1 (human pancreatic carcinoma) | Expression TGF-β1 and its receptor TGF-βRI and TGF-βRII in three tested NET cell lines and in 67% of human NETs (possible inhibition of paracrine and autocrine growth by TGFβ1) | [126] |
| GEP-NET | 38 patients with NENs and 23 healthy controls | ↑VEGF vs controls (statistically significant, also a significant increase with tumor progression); ↑angiogenin vs controls (statistically significant); ↑IL-8 vs control (statistically significant); highest level IL-6 in patients which non-survivors compared to survivors | [130] |
| pNET | 52 cases of pancreatic adenocarcinoma and 52 with pNETs | IL-8, IL-8RA; IL-8RB expression in pNET (21%, 63%, 92%): no statistically significant correlation between the expression IL-8, IL-8RA; IL-8RB and tumor grade | [131] |
| GEP-NET | 145 patients with GEP-NETs and 150 healthy controls | ↑VEGF in GEP-NETs vs healthy controls (the main with metastatic tumors; statistically significant); ↑VEGF in patients with metastases compared to patients with non-metastatic tumor (statistically significant); VEGF-1145G allele more frequent in NF-GI-NET than in controls (not statistically significant); Higher sensitivity VEGF than 5-HIAA but lower than CgA in tumor detection (both pNET and GI-NET); higher VEGF expression in midgut tumors than in foregut tumors (statistically significant difference) | [132] |
| GEP-NET | 58 patients with GEP-NETs; the analysis of SNPs VEGFA, VEGFR2, and VEGFR3 | VEGF-A rs699947C, VEGF-A rs2010963GC, and VEGFR-3 rs307821C correlated with an increased risk of disease recurrence; | [133] |
| NET | 115 patients with NETs | A correlation between NLR cut-off value above 3.01 and tumor localization (the main in the lung), higher histological grade, high mitosis, a high Ki-67 proliferation index, distant metastasis, and lymphovascular invasion; no correlations in the case of PLR. | [134] |
| pNET | 172 patients with pNETs undergoing potentially curative resection (166 patients, curative resection and 6 patients, palliative surgery) | ↑NLR and ↑PLR significantly associated with shorter overall survival and disease-free survival. | [140] |
| pNET | 165 patients with pNET | A correlation statistically significant between NLR cut-off value >2.4 with tumor size, tumor-node metastasis (stage III and IV), and tumor grade; no correlation NLR with age, gender, location, vessel, and nerve invasion. | [135] |
| pNETs | 95 patients with resectable pNETs | ↑NLR is related with advanced tumor stage and higher grade (higher levels NLR in neuroendocrine carcinoma G3 than NET G1 and G2, higher in later stage, tumor thrombus and lymph node metastasis: statistically significant results; NLR independent risk factors for lymph node metastasis (significant differences between groups with and without lymph node metastasis) | [136] |
| GEP-NET | 48 patients with GEP-NETs (foregut, midgut, and hindgut) that underwent resective surgery (curative GI-NET) | NLR >2.6 predicted the presence of peritoneal metastasis (statistically significant); PLR and PWR tended to predict the presence of multifocal disease but statistically insignificant | [137] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Budek, M.; Nuszkiewicz, J.; Piórkowska, A.; Czuczejko, J.; Szewczyk-Golec, K. Inflammation Related to Obesity in the Etiopathogenesis of Gastroenteropancreatic Neuroendocrine Neoplasms. Biomedicines 2022, 10, 2660. https://doi.org/10.3390/biomedicines10102660
Budek M, Nuszkiewicz J, Piórkowska A, Czuczejko J, Szewczyk-Golec K. Inflammation Related to Obesity in the Etiopathogenesis of Gastroenteropancreatic Neuroendocrine Neoplasms. Biomedicines. 2022; 10(10):2660. https://doi.org/10.3390/biomedicines10102660
Chicago/Turabian StyleBudek, Marlena, Jarosław Nuszkiewicz, Anna Piórkowska, Jolanta Czuczejko, and Karolina Szewczyk-Golec. 2022. "Inflammation Related to Obesity in the Etiopathogenesis of Gastroenteropancreatic Neuroendocrine Neoplasms" Biomedicines 10, no. 10: 2660. https://doi.org/10.3390/biomedicines10102660