
Cancer Stem Cell Relationship with Pro-Tumoral Inflammatory Microenvironment

Immunology Division, Department of Internal Medicine and Hematology, Semmelweis University, 1085 Budapest, Hungary
*
Author to whom correspondence should be addressed.
Biomedicines 2023, 11(1), 189; https://doi.org/10.3390/biomedicines11010189
Submission received: 21 December 2022
/
Revised: 5 January 2023
/
Accepted: 10 January 2023
/
Published: 11 January 2023
(This article belongs to the Collection Recent Advances in Cancer Stem Cells)
Abstract
:Inflammatory processes and cancer stem cells (CSCs) are increasingly recognized as factors in the development of tumors. Emerging evidence indicates that CSCs are associated with cancer properties such as metastasis, treatment resistance, and disease recurrence. However, the precise interaction between CSCs and the immune microenvironment remains unexplored. Although evasion of the immune system by CSCs has been extensively studied, new research demonstrates that CSCs can also control and even profit from the immune response. This review provides an overview of the reciprocal interplay between CSCs and tumor-infiltrating immune cells, collecting pertinent data about how CSCs stimulate leukocyte reprogramming, resulting in pro-tumor immune cells that promote metastasis, chemoresistance, tumorigenicity, and even a rise in the number of CSCs. Tumor-associated macrophages, neutrophils, Th17 and regulatory T cells, mesenchymal stem cells, and cancer-associated fibroblasts, as well as the signaling pathways involved in these pro-tumor activities, are among the immune cells studied. Although cytotoxic leukocytes have the potential to eliminate CSCs, immune evasion mechanisms in CSCs and their clinical implications are also known. We intended to compile experimental findings that provide direct evidence of interactions between CSCs and the immune system and CSCs and the inflammatory milieu. In addition, we aimed to summarize key concepts in order to comprehend the cross-talk between CSCs and the tumor microenvironment as a crucial process for the effective design of anti-CSC therapies.
1. Introduction
The presence of cancer stem cells (CSCs) has been confirmed in several tumors. They play a fundamental role in tumor development, progression, metastasis, and relapse. Through their self-renewal capacity and differentiation potential, CSCs are able to maintain tumor heterogeneity. In addition, they are able to activate resistance mechanisms (e.g., altering the expression of drug export systems, reducing cell division, promoting epithelial-to-mesenchymal transition (EMT), increasing resistance to hypoxia through angiogenesis, being able to induce immune escape by reducing tumor antigens, and influencing the composition of cytokines and growth factors that determine the inflammatory environment) [1,2].
Identification of CSCs is not a simple task, as the boundary between tumor cells and CSCs is often unclear. In recent years, a number of CSC markers have been identified that can help define the CSC phenotype (Table 1) [3,4,5]. However, these markers are expressed not only by CSCs but also by embryonic or adult stem cells, and they are rarely expressed on normal tissue [6].
In healthy tissues, stem cells are located in a defined compartment. The interrelationship between stem cells and their environment is essential for the maintenance of the stem cell phenotype. CSCs, like conventional stem cells, are located in a specialized environment. Chronic inflammation has been recognized as a hallmark of cancer since the seminal publications of Colotta [7], Hanahan, and Weinberg [8,9]. CSCs can influence their own capabilities to enable their own survival and progression, as well as their interaction with their inflammatory environment [10]. In this review article, we aim to provide a concise overview of the recent findings regarding the reciprocal effects between chronic inflammation and cancer stem cells and how they promote the maintenance of the stem cell phenotype and thus disease progression. We aim to bring together specific experimental results that provide direct evidence for interactions between CSCs and the immune system and between CSCs and the inflammatory microenvironment.
2. CSCs Influence Their Own Capabilities by Different Mechanisms
In the past, cancer was described as a heterogeneous mass of cells composed of distinct subgroups, and numerous tumor models have attempted to explain the underlying cellular heterogeneity. The hierarchical model of CSCs posits the existence of clusters of malignant cells with varying proliferative and differentiative capacities, with the highest hierarchical level and the highest oncogenic capacity being associated with stem cell properties and successive differentiation potentials [11]. In accordance with the dynamic CSC concept, a feedback control system can be established between CSCs and tumor progenitor cells, indicating that cancer progenitor cells can acquire stem properties in response to certain microenvironmental signals [12,13]. In this context, it is becoming evident that inflammatory circumstances collaborate to induce deregulations, mutations, cell fusion, and other phenomena, ultimately resulting in conditions that promote CSC.
In the early stages of cancer development, the cancer cells are under intense immunological attack, so that the immune system destroys them. However, during this process, cells that are less immunogenic and therefore almost invisible to the immune system may be selected. Such cells include CSCs. CSCs are able to modify their own properties to ensure their survival. One possible way for CSCs to survive is to exit the cell cycle and enter a dormant state. This dormant state is regulated by Nanog Homeobox (NANOG) through wingless-related integration site (Wnt)/β-catenin signaling [14]. Compared to tumor mass cells, CSCs are able to ensure their own survival by enhancing the deoxyribonucleic acid (DNA) repair mechanisms [15], reducing apoptosis [16], and increasing the expression of certain drug efflux pumps (e.g., multi-drug resistance 1 /MDR1/, ATP binding cassette subfamily G member 2 /ABCG2/) [17].
To further ensure their survival, CSCs can also evade the innate immune system in several ways [18]. Crucially, in glioblastoma, melanoma, and colorectal cancer, they are able to downregulate the expression of major histocompatibility complex (MHC) I and II molecules [19,20,21]. They are also able to convert a subset of immature dendritic cells (DCs) into transforming growth factor (TGF)β-secreting cells, which ultimately leads to the expansion of regulatory T cells (Tregs) in lymphoid organs [22]. They are also capable of reducing the activity of natural killer (NK) cells in several ways, e.g., by decreasing the expression of natural killer group 2, member D (NKG2D) ligands in glioblastoma and breast cancer [19,23]; by decreasing the expression of ligands for NK cell activating receptors such as NKp44, NKp30, NKp46, and CD16 [24,25]; or by increasing the expression of NK cell inhibitory receptor ligands [24,25]. In lung, pancreatic, and hepatocellular cancers, they are also able to inhibit their own phagocytosis by enhancing CD47 (“don’t hurt me”) signaling via SIRPα on tumor-associated macrophages (TAMs) [26,27,28,29]. In addition, they are resistant to apoptosis-inducing T and NK cells and chemotherapy. According to the results from prostate cancer, this is mainly achieved by increasing the expression of CD200, CD95/FasL, B-cell lymphoma 2 (Bcl2), B-cell lymphoma-extra large (Bcl-xL), and survivin [30,31]. Survivor CSCs are also capable of retaining their stemness. Through prolonged CD95/Fas stimulation, they promote signal transducer and activator of transcription (STAT)1 activation by type I interferons (IFNs) [32], which in turn enhances the expression of stem-like markers (Figure 1) [33].
Despite the promising results, CSCs’ biology and immunomodulatory ability are not yet entirely understood. Understanding the mechanisms that guide the plasticity and phenotype of CSCs, such as immunogenicity, proliferative activity, differentiation, or migration during tumoral development, as well as the recognition of CSC-specific markers, are the main drawbacks for CSC-targeted anti-cancer therapies, which aim to eradicate the tumor completely and effectively prevent relapses.
3. The Mutual Role of TME and CSCs in Immunomodulation and Stem Cell Niche Maintenance
The heterogeneous (i.e., differences in immune cell infiltration and the amount of necrotic tumor cells, interstitial pressure, genetic and epigenetic alterations) and location dependent (i.e., tumor periphery vs. tumor core) tumor microenvironment (TME) is consisting of stroma, extracellular matrix, vasculature, immune cells, and different signaling molecules and pathways (i.e., Notch-, Wnt-, and Hedgehog-pathways) [34,35]. Crosstalk between CSCs and cells in the TME is variable and extensive, involving interconnections between CSCs, tumor stromal cells, and non-CSCs. It is assumed that CSCs inhabit a particular sub-compartment of TME known as the CSC niche. A favorable microenvironment and the absence of specific stimuli that affect cell proliferation keep CSCs quiescent [36]. CSCs survive tumor eradication in quiescence but do not lose their malignant potential, orchestrating the transition to the escape phase. According to acute leukemia studies, repeated tumor growth is triggered by the aggressive and slowly dividing CSC clone [37]. During the escape phase, CSCs secrete cytokines, chemokines, and soluble factors to blunt and alter immune functions and develop immune tolerance in order to create a pro-tumor niche [38]. Tregs, TAMs, and myeloid-derived suppressor cells (MDSCs) are the main organizers of this process, as they mainly enhance the formation of an immune-tolerant TME by secreting interleukin (IL)10, TGFβ, and prostaglandins, as found in colorectal cancer (CRC) [39,40,41,42]. They also inhibit the secretion of IL12 by DCs, block the efficient Th1 response, and inhibit NK, natural killer T (NKT), and effector T cells [40,41,42,43]. During the further development of TME, the formation of angiogenesis-promoting N2-polarized tumor-associated neutrophil granulocytes (TANs) is enhanced through immunosuppressive factors and cytokines (e.g., TGFβ) [44].
In cancer patients, a so-called emergency myelopoiesis is observed, whereby TAMs and MDSCs proliferate in abundance, leading to an abnormal overgrowth of tumor-supporting myeloid cells [45,46]. In addition to the local immune cell dysregulation that occurs in TME, cancers also alter the differentiation of bone marrow progenitors through systemic effects, thereby affecting the extent, composition, and specific functions of hemopoiesis [47,48]. Myeloid cells that have been transferred from the bone marrow to the periphery are transported to the tumor, where they encounter extreme conditions (e.g., hypoxia, low pH, low glucose, and inflammatory signals). The altered microenvironment further enhances their reprogramming towards a pro-tumor phenotype [43,49]. CSCs promote the differentiation of immature myeloid cells by secreting inflammatory mediators (e.g., IL10, IL13) [50]. In addition, granulocyte colony-stimulating factor (G-CSF), C-C motif chemokine ligand (CCL)2, CCL15, and chemokine (C-X-C motif) ligand (CXCL)12 produced by CSCs recruit additional MDSCs to the TME in colon cancers [51,52]. Experimental results in pancreatic cancer have demonstrated that monocyte-derived MDSCs (M-MDSCs) promote CSC expansion and the expression of genes related to the epithelial-to-mesenchymal transition [53]. Similarly, in CRC, granulocyte-derived MDSCs (G-MDSCs) promote CSC formation via exosomes, especially within hypoxic microenvironments [54].
In addition, in uterus, breast, and hepatocellular cancers MDSCs can promote the emergence and maintenance of the CSC phenotype in several ways (e.g., C-terminal binding protein 2 /CtBP2/ inhibition by micro-ribonucleic acid (miRNA)10 1; increased prostaglandin E2 (PGE2) production; increased IL6 and nitric oxide (NO) production by involving the STAT3 signaling) [55,56,57,58]. TAMs also increase the number of CSCs through the induction of the STAT3/IL6 pathway in liver cancer [57] and promote the self-renewal, tumorigenicity, chemoresistance, and migration of CSCs via interferon-stimulated gene 15 (ISG15) secretion in pancreas and nasopharyngeal cancers [59,60]. M2-polarized TAMs promote CSC proliferation and invasion in liver cancer through the secretion of TGFβ, platelet-derived growth factor (PDGF), CXCL12, and IL8 [61,62]. They also stimulate angiogenesis and maintain stem cell properties throughout VEGF production in breast cancer [63,64]. This suggests that TAMs promote tumor progression by supporting the CSC niche [58,65].
Tregs are also an important component of the TME. Tregs are essentially immunosuppressive and act against tissue damage caused by inflammation [66]. In tumors, however, Tregs suppress the anti-tumor effect of tumor-infiltrating immune cells, thereby promoting tumor escape. The functions of Tregs and their polarization between “anti-tumor” and “pro-tumor” states are regulated by complex molecular and cellular interactions. The binding of semaphorin-4a (Sema-4a) expressed on immune cells to neuropilin-1 (Nrp-1), a receptor for Tregs, enhances the survival and immunosuppressive activity of Tregs. The Nrp-1/Sema-4a pathway is absolutely required for the protection and prolongation of Treg survival in TME [66,67]. Other T cell types can interconvert between phenotypes as well. IL17 producing CD4+ Th17 and Th2 cell are able to switch to IFNγ producing ones via epigenetic, metabolic, and cytokine signaling pathways [68]. Furthermore, in non-small cell lung cancer (NSCLC), memory (CD4+ forkhead box P3/Foxp3/+ CD25high CD27+ CD45RA−) Tregs can also be transformed into Th17-like phenotype, expressing C-C Motif Chemokine Receptor (CCR)6 and IL17 [69,70].
Mesenchymal stem cells (MSCs) can promote the chemoresistance of CSCs both directly and indirectly. In breast, colon, and gastric cancers, as well as in glioma, and acute lymphoid leukemia, they contribute to CSC survival during various anti-cancer treatments by secreting fatty acids, exosomes, chemoattractants and growth factors, and by cell–cell contact via miRNA upregulation [71,72,73,74,75,76,77,78,79]. Based on the results in gastric cancer, lung, liver, and ovarian cancers, cancer-associated fibroblasts (CAFs) derived from MSCs, fibroblasts, or epithelial cells also promote EMT and the survival of the CSCs’ stem cell phenotype throughout paracrine actions (IL6, IL1β, and CXCL12 secretion) and via insulin-like growth factor 1 receptor (IGF1R), TGFβ, STAT1, and nuclear factor-κB (NF-kB) pathways [80,81,82,83,84,85].
Besides the positive effects of TME on CSCs, activated CSCs provide favorable conditions for the M2 polarization of TAMs and their pro-tumorigenic effect as well [40]. Several different factors in gliomas, glioblastomas, and ovarian cancers may play a role in this process, such as periostin (an extracellular matrix protein), colony stimulating factors, CCL2, cyclooxygenase 2 (COX2), or TGFβ [86,87,88,89,90]. CSCs contribute to the development of their own vascular network through vascular endothelial growth factor (VEGF) production [63,64], and promote their own stem cell development through the activation of Wnt-signaling via the interaction between CSCs and TAMs [91].
In addition to the action of pro-tumorigenic cytokines via the paracrine pathway, the CSC-derived secretome also plays an important role in the establishment and maintenance of TME. Glioblastoma CSC-derived exosomes can stimulate M2 polarization, programmed death-ligand 1 (PD-L1) expression, and the production of monocyte chemotactic protein 3 (MCP-3) and CXCL1, which promote myeloid cell recruitment, through the STAT3 pathway in glioblastoma [92]. Using the same secretome in glioma, circulating monocytes produce increased IL10 and arginase 1 (Arg1), decrease Human Leukocyte Antigen DR isotype (HLA-DR) expression, and thereby transform into M-MDSC-like cells [93]. In breast cancer, exosomes containing TGFβ, complement component 1q (C1q), and semaphorins also promote M2-directed (immunosuppressive) polarization and differentiation of the M-MDSCs [94]. Maturation of DCs and T cell responses can be inhibited by HLA-G-containing extracellular vesicles, which favor renal tumor cell immune escape mechanisms [95]. Exosomes of CRC-derived CSCs increase neutrophil granulocyte lifespan and promote the formation of pro-tumorigenic phenotype TANs by increasing the IL1β expression [96]. Melanoma CSCs can educate neutrophils to support cancer progression in several ways, such as neutrophil recruitment via TGF-β, IL6, and IL8, enhancing N2-polarization by the activation of ERK, STAT3, and P38 pathways, as well as the overexpression of CXCR2 and NF-kB [97]. MSC-derived secretome of TME also favors the tumorigenic inflammatory response of TAMs by decreasing pro-inflammatory and increasing anti-inflammatory cytokine production [98]. According to the results from liver, gastric, renal, and thyroid cancers, exosomes derived from CSCs can influence apoptosis, angiogenesis, EMT, and metastasis formation by modulating the expression of p53, Bcl2, VEGF, angiopoietin1, TGFβ, matrix metalloproteinase (MMP)2 and MMP9, as well as by displaying a pro-tumor miRNA profile [99,100,101,102,103].
Along with the paracrine effects of the cytokines and the secretome-mediated TME formation possibilities, TAMs interact with CSCs through direct cell–cell contact. The binding of CD90/Thy-1 of TAMs and ephrin-A receptor 4 (EphA4) expressed on the surface of breast CSCs induces IL6, IL18, and granulocyte-macrophage colony-stimulating factor (GM-CSF) production, which promote the maintenance of a stem cell-like microenvironment [104].
CSCs may exhibit potent angiogenic properties and contribute to the recruitment of blood vessels during cancer. Vascular endothelial cells (ECs) provide CSCs with supportive signals through cell-to-cell interactions [105]. Under the impact of TGFβ, glioblastoma CSCs are able to generate pericytes that enable neovascularization and cancer progression [106]. CSCs produce the angiogenic molecules VEGF and CXCL12 to stimulate EC angiogenesis. ECs, in turn, secrete stemness-maintaining substances such as NO and osteopontin, and stimulate Notch signaling [107]. Anti-VEGF medication that inhibits ECs can, surprisingly, also be tumorigenic. The anti-VEGF medication may create hypoxia within the TME, which unexpectedly induces VEGF within the TME via a negative feedback loop [108]. This hypoxic environment can also inhibit CSC differentiation, increase cell treatment resistance, and boost stem-like characteristics in non-CSCs [108,109]. Moreover, ECs upregulate capillary morphogenesis gene 2 (CMG2) protein to increase stemness, invasion, and metastasis of CSCs via activating the Wnt/β-catenin pathway observed in gastric cancer [110] (Figure 2).
Future new therapeutic techniques will require a comprehensive understanding of the relationship between TME and CSCs. In this direction, researchers have previously identified CSCs as the major source of cancer relapse and chemotherapeutic drug resistance in numerous solid tumors. In addition to this cell subpopulation, TAMs, TANs, CAFs, T cell subsets, and other immune cells, as well as their secretome, participate in interactions that can benefit or hinder the fight against cancer. As a result, targeting the cellular or secretome components of the TME provides a potential cancer therapeutic approach.
4. The CSC-TME Crosstalk in Highly Inflammatory Cancers
The link between inflammation and the development of cancer is rather complex [111]. In the case of acute inflammation following tumor antigen uptake or activation by a Toll-like receptor (TLR) agonist, mature DCs may regulate the anti-tumor immune response by modulating the inflammatory response through various mechanisms (e.g., cross-presentation of tumor antigens and priming of tumor-specific CD8+ T cells, polarization of immune cells towards the anti-tumor phenotype, recruitment of NK cells, thereby maintaining the T cell response) [111,112]. If the acute inflammation is not resolved, it is prolonged over time and transforms into chronic inflammation. In this microenvironment, cancer cells (including CSCs) can hijack DCs, thereby preventing the presentation of tumor antigens, and in addition, they can recruit a variety of immunosuppressive cells (e.g., MDSCs, Tregs, M2-TAMs, and N2-TANs) by producing cytokines, chemokines, and inflammatory mediators. The resulting environment is rich in pro-angiogenic and pro-tumorigenic factors and prevents innate immunity and the T cell response from exerting anti-tumor effects [111,112].
The degree of inflammation may vary depending on the type of tumor. Some tumor types are specifically inflammatory, such as liver, gastric, colorectal, or breast cancer [66,113].
CSC niches have been identified in a number of human cancer types, such as esophageal [114], gastric [115], colorectal [116], liver [117], pancreatic [118], breast [119], ovarian [120], prostate [121], renal [122], brain [123], head and neck [124], lung [125], or melanoma [126]. Numerous studies have examined the similarities and variations between the habitats of various malignancies, as well as the effect of cancer-specific microenvironments on the establishment and growth of CSCs [127,128,129,130,131,132,133]. In order to gain a better understanding of the interaction between tumors and CSC niches, as well as the role of inflammatory milieu in the maintenance of various CSCs, we will now examine some of the most significant, highly inflammatory cancers.
In the microenvironment of the liver, CSCs promote pro-tumor TME formation in several ways, such as the production of the tissue inhibitor of metalloproteinase 1 (TIMP1) or the activation of the hepatocyte-derived growth factor/hepatocyte-derived growth factor receptor (HGF/HGFR) system by the hypoxia-induced activation of HIF1. Kupffer cells and neutrophils enhance tumor necrosis factor (TNF)α, IL1, and MMP9 production as well [134]. The production of growth factors (e.g., epidermal growth factor receptor /EGFR/, VEGF, PDGF, and stromal cell-derived factor 1 /SDF1/), TNF, and other angiogenic factors also promotes the growth and survival of CSCs and their resistance to radiotherapy and chemotherapy [135,136,137,138]. A number of surface markers are known for CSCs in liver cancer (e.g., CD133, CD90, CD24, CD13, epithelial cell adhesion molecule /EpCAM/, aldehyde dehydrogenase /ALDH/, and hepatic progenitor cell marker OV-6). The expression of stem cell markers confers different properties to CSCs. In CD90+ cells, genes associated with inflammation and drug resistance are upregulated, whereas CD133+ cells are resistant to apoptosis and radiotherapy through activation of the Ak strain transforming/Protein kinase B (Akt/PKB) pathway [139,140].
Gastric CSCs have been identified in several cell lineages and are characterized by several stem cell markers (e.g., CD44, ALDH, CD54, CD24, CD71, CD326, CD49f, CD54, CD90, CD133, SRY-box transcription factor 2 /SOX2/, octamer-binding transcription factor 4 /OCT4/, NANOG) [141,142,143]. Infection with Helicobacter pylori has been shown to favor the development of gastric cancer CSCs in animal models [141,142,143]. The chronic inflammation caused by such an infection is also helpful for the development and maintenance of the CSC niche [141,142,143].
CD44, CD133, CD166, leucine-rich repeat-containing G-protein coupled receptor 5 (Lgr5), ALDH1, EpCAM, and other more general markers such as NANOG, SOX2, OCT4, CD51, CD24, CD26, and CD29 are used to identify colorectal CSCs [144]. The expression of CD133, OCT4, and NANOG in colitis-associated cancers (CACs) are significantly lower than in sporadic CRC [144]. Additionally, although recent research identifies the Lgr5+ stem cells as the possible cells of origin for the formation of mice adenoma and human CRC [145,146,147], the proportion of Lgr5+ CSCs in CAC is one third less than in CRC [148]. This confirms that the molecular pathogenesis of CAC is distinct from that of sporadic CRCs, as genomic alterations appear to be directly connected to the effects of chronic inflammation and repetitive mucosal injury in inflammatory bowel diseases [149]. Using an azoxymethane/dextran sodium sulfate (AOM-DSS) mouse model of CAC, it was demonstrated that DSS isolates colonic epithelial stem cells from both the stem cell niche and the Wnt signaling-supporting basal lamina. In doing so, the DSS stops the stem cell program. Within ex vivo circumstances, niche damage caused by a progressively increasing dose of DSS promoted the formation of Wnt-independent dysplastic organoids. These organoids contain tenfold more Lgr5+ colonic epithelial stem cells and have orthologous Wnt mutations to human CRC driver mutations. These suggest that CRC is formed by the niche injury-induced outgrowth of normally suppressed mutant stem cells [150]. Deletion of the aryl hydrocarbon receptor (AhR) increases the number of Lgr5+ stem cells and enhances their organoid-initiating capacity. In a colorectal inflammatory tumor model, AhR knockdown in intestinal epithelial cells increases basal stem cell and crypt injury-induced cell proliferation by upregulating forkhead box M1 (FoxM1) signaling and promotes colitis-associated carcinogenesis [151].
Different populations of breast cancer CSCs can give rise to different tumor cell lines. Breast CSCs that are CD44+/CD24− are known to be associated with intra-tumoral inflammation and tumor-infiltrating CD4+ T cells [152]. However, the genotype of the new cells may not resemble the genetic profile of the original CSCs, which may indicate the development of mutations [153]. The negative feedback balance between the tumor suppressor breast cancer gene 1 (BRCA1) and the transcription factor SNAI2 gene (Slug) is a key element to maintaining normal tumor growth and determining TME’s stem cell concentration [154,155]. Overexpression of the SOX family promotes EMT and upregulates the expression of the enhancer of zeste homolog 2 (EZH2), which plays an important role in the histone methylation of several genes and can activate the Raf-1 proto-oncogene, serine/threonine kinase (Raf1)/β-catenin pathway [156,157].
5. Emergence of CSC Phenotype without TME
Interestingly, the activation of the TLR9 inflammatory signaling pathway, which is part of the innate immune system, can result in the CSC phenotype without the presence of TME. Regarding cell-free DNA (cfDNA), it has been shown that the structure and origin of cfDNA influence its biological effects on cancer cells [158]. Using an in vitro cellular model that lacked both the TME and the immune system of the tumor-bearing host, we investigated the pathobiological effects of self-DNA administration in HT29 colon cancer cells. We provided evidence [159] for a close existing interplay between TLR9 signaling and the autophagy response, which had significant effects on tumor cell survival in HT29 cells treated with intact or modified self-DNA. Interestingly, we also found colonosphere formation with a strong cytoplasmatic CD133 immunoreactivity in artificially hypermethylated DNA-treated HT29 cells. We further discovered [160] that the combined use of tumorous self-DNA and IGF1R inhibition displays anti-proliferative properties that can be suppressed by inhibiting TLR9 signaling. Autophagy induced by self-DNA and IGF1R inhibitor also resulted in the survival of CD133-positive HT29 stem-like cancer cells, which may play a role in the CRC recurrence. Since HT29 cancer cells are wildtype regarding Kirsten rat sarcoma virus (K-Ras) mutation, it cannot be ruled out that this observed phenomenon is partly mediated by the RAS/extracellular-signal-regulated kinase (ERK) and phosphoinositide 3-kinase (PI3K)/Akt pathways, with a close connection to the pro-inflammatory factors like IL17, IL22, and IL23 [161]. However, it should not be overlooked that while HT29 cells are able to express CD133, there are colon cancer cells (e.g., HCT15, LS180, SW480, DLD1, and COLO205) that do not express CD133 [162].
6. Utilization of the Inflammatory Process in Cancer Therapy
Theoretically, a number of new ways to boost the immune response against tumors seem to be able to control inflammation caused by cancer.
Local inflammation generated by irradiation or oncolytic viruses can stimulate an anti-cancer innate immune response by activating nucleic acid receptors (TLR9, cyclic GMP–AMP synthase /cGAS/-stimulator of interferon genes /STING/, or RIG-I like receptor /RLR/), followed by a type I IFN response [158]. The PI3K/Akt/mammalian target of rapamycin (mTOR) pathway is one of the major signaling pathways in CSCs involved in stemness maintenance, proliferation, differentiation, EMT, migration, and autophagy. Therefore, inhibiting the PI3K/Akt/mTOR pathway may also be a promising targeted cancer treatment method [163].
Restricting the infiltration and function of immunosuppressive cells (e.g., MDSCs, Tregs, M2-TAMs, and N2-TANs) may restore immune surveillance by blocking inflammatory pathways [111]. Immunotherapeutic strategies such as immune checkpoint blockage, monoclonal antibodies, vaccination, CD8+ T cell treatment, and activation of innate immune responses such as NK cells, cytokine-induced killer cells, can be used to target CSCs [164,165]. Loss of cancer antigen expression, activation of oncolytic pathways, and promotion of an immunosuppressive milieu and (epi)genetic modifications that diminish their identification by the immune system are just a few of the ways that CSCs have developed to evade a potential attack from the immune system. These immunotherapeutic techniques have the potential to increase CSCs’ sensitivity to chemotherapy and/or radiotherapy. Immune checkpoint blockage strategies, for instance, can inhibit the immunosuppressive activity of CSCs and other immunosuppressive cells inside the TME. CSCs and cancer cells generate PD-L1, whereas Tregs express its receptor PD-1 [107,166,167]. CSCs also distribute PD-1 to their respective specialization [165]. PD-L1 could lead to the depletion and malfunction of effector T cells [168] and prevent CSCs from evading the immune system. Importance is placed on employing immunotherapeutic strategies targeting specific CSC markers and antigens that are preferentially expressed by the cells [165].
Recent studies on the epigenetic regulation of CSCs by histone lysine methyltransferase and histone demethylase inhibitors have received significant attention [169,170,171]. Moreover, because signaling pathways play key roles in stimulating the proliferation of CSCs, maintaining the phenotype of CSCs, and in embryonic development, therapeutic strategies targeting these pathways have been discovered. Included among these signaling pathways are NF-kB, Janus kinase (JAK)-STAT, and TGFβ/suppressor of mothers against decapentaplegic (Smad). Specifically, addressing epigenetic alterations in signaling networks has emerged as a potential tumor therapy research approach. Tocilizumab, for instance, blocks IL6/STAT3 signaling and reduces the cancer/inflammation epigenetic IL6/STAT3/NF-kB positive feedback loop, which is of immense therapeutic utility for patients with resistant triple-negative breast cancer [172]. In addition, activation of the NF-kB pathway in pancreatic cancer stem cells is dependent on methylation of the downstream regulatory gene SOX9, and DNA methyltransferase inhibitors may represent a novel therapeutic option for pancreatic cancer treatment [173].
Besides targeting CAF-derived components, depleting pro-tumor CAFs or transforming them into dormant or anti-tumor cells are all potential anti-cancer therapeutic strategies as well [174,175,176,177,178,179]. The capacity of CAFs to confer stemness to cancer cells renders this treatment possibility intriguing. Compared to epithelial cancer cells, immunological cells, and endothelial cells, CAFs are more positively connected with gene sets associated with poor prognosis, providing support for targeting CAFs as a viable therapeutic route. However, the variety of CAFs needs the identification of more specific markers, as there are CAFs that inhibit tumor growth. Intriguingly, the tumor devoid of myofibroblasts displayed improved spheroid formation, indicating a higher proportion of CSCs. Indeed, the identification of CAF subtypes demonstrates that they promote or inhibit tumor progression in a tissue-dependent way, supporting the necessity for additional research into CAF-specific indicators [180]. Reeducating pro-tumor CAFs into a state of quiescence or even anti-tumor CAFs is an attractive concept. Given that vitamins (e.g., all-trans retinoic acid or the vitamin D metabolite 1α, 25-dihydroxyvitamin D3) are essential for healthy tissue and their toxicity is relatively lower compared to chemotherapy, reusing vitamin analogs to reconfigure stimulated fibroblasts into a quiescent state may be a clinically viable therapeutic strategy [177,178]. Reprogramming the fibroblasts using growth factors, as evidenced by the flexibility of CAFs, is another technique for rewiring the fibroblast population. TGFβ inhibits an IL1-induced phenotype and drives the fibroblast to acquire a myofibroblastic phenotype with less carcinogenesis, including reduced expression of markers supporting cancer stemness, such as IL6 and CXCL12 [179]. This justifies the option to convert tumor-promoting fibroblasts into tumor-restraining fibroblasts. To account for the cancer cell’s potential to activate tumor-promoting CAFs, inhibitors may be utilized to circumvent cancer cell-mediated CAF activation.
Anti-VEGF resistance causes unsuccessful cancer treatment and recurrence. (Epi)genetic changes cause acquired resistance in cancer cells [181]. Tumor ECs have epigenetic changes that contribute to anti-angiogenic treatment resistance. Anti-resistance therapy may include many anti-angiogenic substances or anti-angiogenic drugs combined with other treatments. Intussusceptive microvascular formation, vasculogenic mimicry, and vascular co-option are anti-angiogenic treatment resistance mechanisms. Angiogenesis and immune cells interact, which is why anti-angiogenic and immunological checkpoint drugs work so well together. Pan-omics profiling improves clinical outcomes and fights anti-angiogenesis drug resistance [181].
It is necessary to develop reliable tests for measuring stem cell function in human specimens. Identification of CSCs and tracking of anti-CSC treatment effectiveness in clinical samples rely mostly on surface markers at present. Due to the constraints of marker-based selection and the flexibility of the CSC state, it is crucial to optimize functional assays as a validation of self-renewal to eradicate all subclones of CSC [182]. Window-of-opportunity trials, in which surgery follows targeted therapy, provide an opportunity for comprehensive evaluation of the therapy response, including comparisons of the CSC rate, stemness indicators, genetic signatures, and functional assays (e.g., xenotransplantation or surrogate in vitro assays) with the diagnostic sample. Specifically, the determination of CSC genetic signatures can aid in patient stratification (risk assessment), identification of therapy response (surrogate markers), and/or differential diagnosis (identifying who is most likely to be responsive to which medications). Before biomarkers can be used regularly in clinical practice, they must be validated through clinical research [182,183]. This research must take into account a number of important factors, such as scientific reasoning, clinical trial design, marker evaluation methods, cost, and feasibility.
Even if the entire remaining combination of immune-evasive strategies is successfully targeted by experimental therapies, novel, as-yet-unknown mechanisms are likely to emerge to thwart therapeutic efforts; consequently, a continually changing, thorough knowledge of TME biology is essential for preparing for the future. New discoveries in basic biology will unquestionably lead to ways of surpassing tumor development, which seeks to elude pharmacologic and biologic therapy, as well as more effective strategies for eradicating CSCs. Future research should focus on integrating these therapies into combination immunotherapy regimens and limiting the effects of these approaches to the site of action in order to minimize systemic pro-inflammatory effects. Also, in the field of gene therapy, monoclonal antibodies against cytokines or cell-based medicines may work well with small molecule-based targeted therapies to kill cancer cells (including CSCs) for a long time. More precise and personalized approaches need to be tested in well-designed clinical trials. However, this is challenging because the relationship between CSCs and inflammatory TME is extremely complex; it is therefore almost impossible to identify a single or small number of therapeutic targets whose manipulation will exclusively result in a beneficial therapeutic effect. Hence, it is obvious that more research is required to improve cancer treatment strategies based on targeting tumor-promoting inflammation.
7. Conclusions
CSCs are capable of altering their own properties in a variety of ways to preserve their stem cell phenotype, resist different therapies, and evade the immune system’s anti-tumor attack. Through immune escape mechanisms, they are not only able to hide themselves from the immune system but also to influence the anti-tumor immune elimination mechanisms in a way that is favorable to them. By manipulating their own capabilities, CSCs have the potential to develop entirely novel anti-cancer treatments and methods to prevent disease recurrence.
It is clear that there is an intense and complex multi-level relationship between the TME and CSCs. CSCs are able to develop an inflammatory niche that allows them to persist and divide on their own. They maintain an intense relationship with the cellular elements of the TME, reprogramming them into cells for the survival and proliferation of CSCs. In turn, the reprogrammed TME cells enhance the survival and proliferation of CSCs and thereby facilitate their own survival and function. If we can understand all the elements of the cross-talk between CSCs and TMEs, the development of a number of novel and targeted anti-cancer therapies will become possible.
Tumors can also be distinguished by their characteristic inflammatory infiltration. In the case of tumors with a highly inflammatory character, stemness markers play a major role, as they not only affect the stem cell phenotype of CSCs but also play a major role in the maintenance and regulation of pro-tumor inflammation. A better understanding of CSC markers and their relationship to inflammation could also serve as a starting point for potential anti-cancer therapies.
It is also a very important observation that the CSC phenotype can be expressed without TME simply by triggering certain signaling pathways involved in inflammation. This will allow the development of potential anti-CSC phenotype treatment strategies in vitro.
Many efforts have been made over the past few decades to uncover the mechanisms through which inflammation promotes carcinogenesis. Few of these studies attempt to propose a theoretical hypothesis and advance our understanding of the underlying laws governing inflammation-induced carcinogenesis, whereas the majority of these investigations give segmental and fragmentary evidence. The theory of “Cancer Evo-Dev” began by reviewing research findings on hepatitis B-induced hepatocarcinogenesis, then moved on to other inflammation-related carcinogenesis [184]. This new idea not only aids in the comprehension of the mechanisms by which inflammation promotes the development of malignancies, but it also lays the groundwork for the creation of targeted cancer prevention and treatment. However, it should not be forgotten that modulation of the inflammatory immune response can be a double-edged weapon that, under inappropriate conditions, can cause CSCs to survive, divide, and spread, and thus cause cancer progression or recurrence. In view of these findings, we believe that further experimental investigation of the relationship between the inflammatory microenvironment and cancer stem cells is warranted.
Author Contributions
F.S. and G.M. drafted the manuscript and designed the figure. F.S. and G.M. wrote, discussed and reviewed the final manuscript. All authors contributed to the article and approved the submitted version. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhang, S.; Yang, X.; Wang, L.; Zhang, C. Interplay between inflammatory tumor microenvironment and cancer stem cells. Oncol. Lett. 2018, 16, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Bruno, A.; Gallo, C.; Pajardi, G.; Noonan, D.M.; Dallaglio, K. Cancer stem cells and the tumor microenvironment: Interplay in tumor heterogeneity. Connect. Tissue Res. 2015, 56, 414–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef] [PubMed]
- Lee-Theilen, M.; Fadini, D.D.; Hadhoud, J.R.; van Dongen, F.; Kroll, G.; Rolle, U.; Fiegel, H.C. Hepatoblastoma Cancer Stem Cells Express PD-L1, Reveal Plasticity and Can Emerge upon Chemotherapy. Cancers 2022, 14, 5825. [Google Scholar] [CrossRef] [PubMed]
- Gzil, A.; Zarębska, I.; Bursiewicz, W.; Antosik, P.; Grzanka, D.; Szylberg, Ł. Markers of pancreatic cancer stem cells and their clinical and therapeutic implications. Mol. Biol. Rep. 2019, 46, 6629–6645. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.T.; Ryu, C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Kise, K.; Kinugasa-Katayama, Y.; Takakura, N. Tumor microenvironment for cancer stem cells. Adv. Drug Deliv. Rev. 2016, 99 Pt B, 197–205. [Google Scholar] [CrossRef]
- Gasch, C.; Ffrench, B.; O’Leary, J.J.; Gallagher, M.F. Catching moving targets: Cancer stem cell hierarchies, therapy-resistance & considerations for clinical intervention. Mol. Cancer 2017, 16, 43. [Google Scholar]
- Rich, J.N. Cancer stem cells: Understanding tumor hierarchy and heterogeneity. Medicine 2016, 95 (Suppl. 1), S2–S7. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonde, A.K.; Tischler, V.; Kumar, S.; Soltermann, A.; Schwendener, R.A. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 2012, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Blanpain, C.; Mohrin, M.; Sotiropoulou, P.A.; Passegué, E. DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell 2011, 8, 16–29. [Google Scholar] [CrossRef]
- Begicevic, R.R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef] [Green Version]
- Pastò, A.; Consonni, F.M.; Sica, A. Influence of Innate Immunity on Cancer Cell Stemness. Int. J. Mol. Sci. 2020, 21, 3352. [Google Scholar] [CrossRef]
- Di Tomaso, T.; Mazzoleni, S.; Wang, E.; Sovena, G.; Clavenna, D.; Franzin, A.; Mortini, P.; Ferrone, S.; Doglioni, C.; Marincola, F.M.; et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin. Cancer Res. 2010, 16, 800–813. [Google Scholar] [CrossRef] [Green Version]
- Schatton, T.; Schütte, U.; Frank, N.Y.; Zhan, Q.; Hoerning, A.; Robles, S.C.; Zhou, J.; Hodi, F.S.; Spagnoli, G.C.; Murphy, G.F.; et al. Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Res. 2010, 70, 697–708. [Google Scholar] [CrossRef] [Green Version]
- Volonté, A.; Di Tomaso, T.; Spinelli, M.; Todaro, M.; Sanvito, F.; Albarello, L.; Bissolati, M.; Ghirardelli, L.; Orsenigo, E.; Ferrone, S.; et al. Cancer-initiating cells from colorectal cancer patients escape from T cell-mediated immunosurveillance in vitro through membrane-bound IL-4. J. Immunol. 2014, 192, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Shang, B.; Liu, Y.; Jiang, S.J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Wang, Q.; Wang, Z.; Jiang, J.; Yu, S.C.; Ping, Y.F.; Yang, J.; Xu, S.L.; Ye, X.Z.; Xu, C.; et al. Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Res. 2014, 74, 5746–5757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruttel, V.S.; Wischhusen, J. Cancer stem cell immunology: Key to understanding tumorigenesis and tumor immune escape? Front. Immunol. 2014, 5, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drukker, M.; Katz, G.; Urbach, A.; Schuldiner, M.; Markel, G.; Itskovitz-Eldor, J.; Reubinoff, B.; Mandelboim, O.; Benvenisty, N. Characterization of the expression of MHC proteins in human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2002, 99, 9864–9869. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhang, L.; Yang, L.; Li, H.; Li, R.; Yu, J.; Yang, L.; Wei, F.; Yan, C.; Sun, Q.; et al. Anti-CD47 Antibody As a Targeted Therapeutic Agent for Human Lung Cancer and Cancer Stem Cells. Front. Immunol. 2017, 8, 404. [Google Scholar] [CrossRef] [Green Version]
- Cioffi, M.; Trabulo, S.; Hidalgo, M.; Costello, E.; Greenhalf, W.; Erkan, M.; Kleeff, J.; Sainz, B.; Heeschen, C., Jr. Inhibition of CD47 Effectively Targets Pancreatic Cancer Stem Cells via Dual Mechanisms. Clin. Cancer Res. 2015, 21, 2325–2337. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.K.; Cheung, V.C.; Lu, P.; Lau, E.Y.; Ma, S.; Tang, K.H.; Tong, M.; Lo, J.; Ng, I.O. Blockade of CD47-mediated cathepsin S/protease-activated receptor 2 signaling provides a therapeutic target for hepatocellular carcinoma. Hepatology 2014, 60, 179–191. [Google Scholar] [CrossRef]
- Uger, R.; Johnson, L. Blockade of the CD47-SIRPα axis: A promising approach for cancer immunotherapy. Expert Opin. Biol. Ther. 2020, 20, 5–8. [Google Scholar] [CrossRef]
- Abdullah, L.N.; Chow, E.K. Mechanisms of chemoresistance in cancer stem cells. Clin. Transl. Med. 2013, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.; Cozzi, P.; Hao, J.; Duan, W.; Graham, P.; Kearsley, J.; Li, Y. Cancer stem cells in prostate cancer chemoresistance. Curr. Cancer Drug Targets 2014, 14, 225–240. [Google Scholar] [CrossRef]
- Qadir, A.S.; Ceppi, P.; Brockway, S.; Law, C.; Mu, L.; Khodarev, N.N.; Kim, J.; Zhao, J.C.; Putzbach, W.; Murmann, A.E.; et al. CD95/Fas Increases Stemness in Cancer Cells by Inducing a STAT1-Dependent Type I Interferon Response. Cell Rep. 2017, 18, 2373–2386. [Google Scholar] [CrossRef]
- Ceppi, P.; Hadji, A.; Kohlhapp, F.J.; Pattanayak, A.; Hau, A.; Liu, X.; Liu, H.; Murmann, A.E.; Peter, M.E. CD95 and CD95L promote and protect cancer stem cells. Nat. Commun. 2014, 5, 5238. [Google Scholar] [CrossRef] [Green Version]
- Relation, T.; Dominici, M.; Horwitz, E.M. Concise Review: An (Im)Penetrable Shield: How the Tumor Microenvironment Protects Cancer Stem Cells. Stem Cells 2017, 35, 1123–1130. [Google Scholar] [CrossRef] [Green Version]
- Ju, F.; Atyah, M.M.; Horstmann, N.; Gul, S.; Vago, R.; Bruns, C.J.; Zhao, Y.; Dong, Q.Z.; Ren, N. Characteristics of the cancer stem cell niche and therapeutic strategies. Stem Cell Res. Ther. 2022, 13, 233. [Google Scholar] [CrossRef]
- Maccalli, C.; Rasul, K.I.; Elawad, M.; Ferrone, S. The role of cancer stem cells in the modulation of anti-tumor immune responses. Semin. Cancer Biol. 2018, 53, 189–200. [Google Scholar] [CrossRef]
- Shlush, L.I.; Chapal-Ilani, N.; Adar, R.; Pery, N.; Maruvka, Y.; Spiro, A.; Shouval, R.; Rowe, J.M.; Tzukerman, M.; Bercovich, D.; et al. Cell lineage analysis of acute leukemia relapse uncovers the role of replication-rate heterogeneity and microsatellite instability. Blood 2012, 120, 603–612. [Google Scholar] [CrossRef] [Green Version]
- Vahidian, F.; Duijf, P.H.G.; Safarzadeh, E.; Derakhshani, A.; Baghbanzadeh, A.; Baradaran, B. Interactions between cancer stem cells, immune system and some environmental components: Friends or foes? Immunol. Lett. 2019, 208, 19–29. [Google Scholar] [CrossRef]
- De Vlaeminck, Y.; González-Rascón, A.; Goyvaerts, C.; Breckpot, K. Cancer-Associated Myeloid Regulatory Cells. Front. Immunol. 2016, 7, 113. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, A.; Guarneri, V.; Gennari, A. Myelopoiesis, metabolism and therapy: A crucial crossroads in cancer progression. Cell Stress 2019, 3, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Strauss, L.; Sangaletti, S.; Consonni, F.M.; Szebeni, G.; Morlacchi, S.; Totaro, M.G.; Porta, C.; Anselmo, A.; Tartari, S.; Doni, A.; et al. RORC1 Regulates Tumor-Promoting “Emergency” Granulo-Monocytopoiesis. Cancer Cell 2015, 28, 253–269. [Google Scholar] [CrossRef] [Green Version]
- Ueha, S.; Shand, F.H.; Matsushima, K. Myeloid cell population dynamics in healthy and tumor-bearing mice. Int. Immunopharmacol. 2011, 11, 783–788. [Google Scholar] [CrossRef]
- Velten, L.; Haas, S.F.; Raffel, S.; Blaszkiewicz, S.; Islam, S.; Hennig, B.P.; Hirche, C.; Lutz, C.; Buss, E.C.; Nowak, D.; et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol. 2017, 19, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Bronte, V. Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Investig. 2007, 117, 1155–1166. [Google Scholar] [CrossRef] [Green Version]
- Ravindran, S.; Rasool, S.; Maccalli, C. The Cross Talk between Cancer Stem Cells/Cancer Initiating Cells and Tumor Microenvironment: The Missing Piece of the Puzzle for the Efficient Targeting of these Cells with Immunotherapy. Cancer Microenviron. 2019, 12, 133–148. [Google Scholar] [CrossRef] [Green Version]
- Lau, E.Y.; Ho, N.P.; Lee, T.K. Cancer Stem Cells and Their Microenvironment: Biology and Therapeutic Implications. Stem Cells Int. 2017, 2017, 3714190. [Google Scholar] [CrossRef]
- Inamoto, S.; Itatani, Y.; Yamamoto, T.; Minamiguchi, S.; Hirai, H.; Iwamoto, M.; Hasegawa, S.; Taketo, M.M.; Sakai, Y.; Kawada, K. Loss of SMAD4 Promotes Colorectal Cancer Progression by Accumulation of Myeloid-Derived Suppressor Cells through the CCL15-CCR1 Chemokine Axis. Clin. Cancer Res. 2016, 22, 492–501. [Google Scholar] [CrossRef] [Green Version]
- Panni, R.Z.; Sanford, D.E.; Belt, B.A.; Mitchem, J.B.; Worley, L.A.; Goetz, B.D.; Mukherjee, P.; Wang-Gillam, A.; Link, D.C.; Denardo, D.G.; et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic Cancer. Cancer Immunol. Immunother. 2014, 63, 513–528. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yin, K.; Tian, J.; Xia, X.; Ma, J.; Tang, X.; Xu, H.; Wang, S. Granulocytic Myeloid-Derived Suppressor Cells Promote the Stemness of Colorectal Cancer Cells through Exosomal S100A9. Adv. Sci. 2019, 6, 1901278. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, H.; Mabuchi, S.; Yokoi, E.; Komura, N.; Kozasa, K.; Matsumoto, Y.; Kawano, M.; Takahashi, R.; Sasano, T.; Shimura, K.; et al. Prostaglandin E2 produced by myeloid-derived suppressive cells induces cancer stem cells in uterine cervical Cancer. Oncotarget 2018, 9, 36317–36330. [Google Scholar] [CrossRef] [Green Version]
- Peng, D.; Tanikawa, T.; Li, W.; Zhao, L.; Vatan, L.; Szeliga, W.; Wan, S.; Wei, S.; Wang, Y.; Liu, Y.; et al. Myeloid-Derived Suppressor Cells Endow Stem-like Qualities to Breast Cancer Cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res. 2016, 76, 3156–3165. [Google Scholar] [CrossRef] [Green Version]
- Wan, S.; Zhao, E.; Kryczek, I.; Vatan, L.; Sadovskaya, A.; Ludema, G.; Simeone, D.M.; Zou, W.; Welling, T.H. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 2014, 147, 1393–1404. [Google Scholar] [CrossRef] [Green Version]
- Jinushi, M.; Chiba, S.; Yoshiyama, H.; Masutomi, K.; Kinoshita, I.; Dosaka-Akita, H.; Yagita, H.; Takaoka, A.; Tahara, H. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc. Natl. Acad. Sci. USA 2011, 108, 12425–12430. [Google Scholar] [CrossRef] [Green Version]
- Sainz BJr Martín, B.; Tatari, M.; Heeschen, C.; Guerra, S. ISG15 is a critical microenvironmental factor for pancreatic cancer stem cells. Cancer Res. 2014, 74, 7309–7320. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.H.; Du, Y.; Han, P.; Wang, H.B.; Liang, F.Y.; Feng, G.K.; Zhou, A.J.; Cai, M.Y.; Zhong, Q.; Zeng, M.S.; et al. ISG15 predicts poor prognosis and promotes cancer stem cell phenotype in nasopharyngeal carcinoma. Oncotarget 2016, 7, 16910–16922. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.M.; Jing, Y.Y.; Yu, G.F.; Kou, X.R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.D.; Yang, Y.; Lu, Z.H.; et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elaimy, A.L.; Guru, S.; Chang, C.; Ou, J.; Amante, J.J.; Zhu, L.J.; Goel, H.L.; Mercurio, A.M. VEGF-neuropilin-2 signaling promotes stem-like traits in breast cancer cells by TAZ-mediated repression of the Rac GAP β2-chimaerin. Sci. Signal. 2018, 11, eaao6897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercurio, A.M. VEGF/Neuropilin Signaling in Cancer Stem Cells. Int. J. Mol. Sci. 2019, 20, 490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. Regulatory T cell subsets in human cancer: Are they regulating for or against tumor progression? Cancer Immunol. Immunother. 2014, 63, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Arneth, B. Tumor Microenvironment. Medicina (Kaunas) 2019, 56, 15. [Google Scholar] [CrossRef] [Green Version]
- Hirahara, K.; Poholek, A.; Vahedi, G.; Laurence, A.; Kanno, Y.; Milner, J.D.; O’Shea, J.J. Mechanisms underlying helper T-cell plasticity: Implications for immune-mediated disease. J. Allergy Clin. Immunol. 2013, 131, 1276–1287. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Bi, G.; Shan, G.; Jin, X.; Bian, Y.; Wang, Q. Tumor-Associated Regulatory T Cells in Non-Small-Cell Lung Cancer: Current Advances and Future Perspectives. J. Immunol. Res. 2022, 2022, 4355386. [Google Scholar] [CrossRef]
- Peck, A.; Mellins, E.D. Plasticity of T-cell phenotype and function: The T helper type 17 example. Immunology 2010, 129, 147–153. [Google Scholar] [CrossRef]
- Cuiffo, B.G.; Campagne, A.; Bell, G.W.; Lembo, A.; Orso, F.; Lien, E.C.; Bhasin, M.K.; Raimo, M.; Hanson, S.E.; Marusyk, A.; et al. MSC-regulated microRNAs converge on the transcription factor FOXP2 and promote breast cancer metastasis. Cell Stem Cell 2014, 15, 762–774. [Google Scholar] [CrossRef]
- Gwak, J.M.; Kim, H.J.; Kim, E.J.; Chung, Y.R.; Yun, S.; Seo, A.N.; Lee, H.J.; Park, S.Y. MicroRNA-9 is associated with epithelial-mesenchymal transition, breast cancer stem cell phenotype, and tumor progression in breast Cancer. Breast Cancer Res. Treat. 2014, 147, 39–49. [Google Scholar] [CrossRef]
- Frisbie, L.; Buckanovich, R.J.; Coffman, L. Carcinoma-Associated Mesenchymal Stem/Stromal Cells: Architects of the Pro-tumorigenic Tumor Microenvironment. Stem Cells 2022, 40, 705–715. [Google Scholar] [CrossRef]
- Roodhart, J.M.; Daenen, L.G.; Stigter, E.C.; Prins, H.J.; Gerrits, J.; Houthuijzen, J.M.; Gerritsen, M.G.; Schipper, H.S.; Backer, M.J.; van Amersfoort, M.; et al. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell 2011, 20, 370–383. [Google Scholar] [CrossRef] [Green Version]
- van der Velden, D.L.; Cirkel, G.A.; Houthuijzen, J.M.; van Werkhoven, E.; Roodhart, J.M.L.; Daenen, L.G.M.; Kaing, S.; Gerrits, J.; Verhoeven-Duif, N.M.; Grootscholten, C.; et al. Phase I study of combined indomethacin and platinum-based chemotherapy to reduce platinum-induced fatty acids. Cancer Chemother. Pharmacol. 2018, 81, 911–921. [Google Scholar] [CrossRef]
- Mallampati, S.; Leng, X.; Ma, H.; Zeng, J.; Li, J.; Wang, H.; Lin, K.; Lu, Y.; Yang, Y.; Sun, B.; et al. Tyrosine kinase inhibitors induce mesenchymal stem cell-mediated resistance in BCR-ABL+ acute lymphoblastic leukemia. Blood 2015, 125, 2968–2973. [Google Scholar] [CrossRef]
- Xue, B.Z.; Xiang, W.; Zhang, Q.; Wang, H.F.; Zhou, Y.J.; Tian, H.; Abdelmaksou, A.; Xue, J.; Sun, M.X.; Yi, D.Y.; et al. CD90low glioma-associated mesenchymal stromal/stem cells promote temozolomide resistance by activating FOXS1-mediated epithelial-mesenchymal transition in glioma cells. Stem Cell Res. Ther. 2021, 12, 394. [Google Scholar]
- Eliason, S.; Hong, L.; Sweat, Y.; Chalkley, C.; Cao, H.; Liu, Q.; Qi, H.; Xu, H.; Zhan, F.; Amendt, B.A. Extracellular vesicle expansion of PMIS-miR-210 expression inhibits colorectal tumour growth via apoptosis and an XIST/NME1 regulatory mechanism. Clin. Transl. Med. 2022, 12, e1037. [Google Scholar] [CrossRef]
- Ji, R.; Zhang, B.; Zhang, X.; Xue, J.; Yuan, X.; Yan, Y.; Wang, M.; Zhu, W.; Qian, H.; Xu, W. Exosomes derived from human mesenchymal stem cells confer drug resistance in gastric Cancer. Cell Cycle 2015, 14, 2473–2483. [Google Scholar] [CrossRef] [Green Version]
- Naito, Y.; Yoshioka, Y.; Ochiya, T. Intercellular crosstalk between cancer cells and cancer-associated fibroblasts via extracellular vesicles. Cancer Cell Int. 2022, 22, 367. [Google Scholar] [CrossRef]
- Kondo, R.; Sakamoto, N.; Harada, K.; Hashimoto, H.; Morisue, R.; Yanagihara, K.; Kinoshita, T.; Kojima, M.; Ishii, G. Cancer-associated fibroblast-dependent and -independent invasion of gastric cancer cells. J. Cancer Res. Clin. Oncol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.J.; Ho, C.C.; Chang, Y.L.; Chen, H.Y.; Lin, C.A.; Ling, T.Y.; Yu, S.L.; Yuan, S.S.; Chen, Y.J.; Lin, C.Y.; et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat. Commun. 2014, 5, 3472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, E.Y.; Lo, J.; Cheng, B.Y.; Ma, M.K.; Lee, J.M.; Ng, J.K.; Chai, S.; Lin, C.H.; Tsang, S.Y.; Ma, S.; et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 Signaling. Cell Rep. 2016, 15, 1175–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, T.S.; Hsu, C.C.; Pai, V.C.; Liao, W.Y.; Huang, S.S.; Tan, K.T.; Yen, C.J.; Hsu, S.C.; Chen, W.Y.; Shan, Y.S.; et al. Metronomic chemotherapy prevents therapy-induced stromal activation and induction of tumor-initiating cells. J. Exp. Med. 2016, 213, 2967–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilczyński, J.R.; Wilczyński, M.; Paradowska, E. Cancer Stem Cells in Ovarian Cancer-A Source of Tumor Success and a Challenging Target for Novel Therapies. Int. J. Mol. Sci. 2022, 23, 2496. [Google Scholar] [CrossRef]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010, 12, 1113–1125. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Cai, D.J.; Li, B. Ovarian cancer stem-like cells elicit the polarization of M2 macrophages. Mol. Med. Rep. 2015, 11, 4685–4693. [Google Scholar] [CrossRef] [Green Version]
- Nusblat, L.M.; Carroll, M.J.; Roth, C.M. Crosstalk between M2 macrophages and glioma stem cells. Cell. Oncol. 2017, 40, 471–482. [Google Scholar] [CrossRef]
- Kokubu, Y.; Tabu, K.; Muramatsu, N.; Wang, W.; Murota, Y.; Nobuhisa, I.; Jinushi, M.; Taga, T. Induction of protumoral CD11c(high) macrophages by glioma cancer stem cells through GM-CSF. Genes Cells 2016, 21, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Raghavan, S.; Mehta, P.; Xie, Y.; Lei, Y.L.; Mehta, G. Ovarian cancer stem cells and macrophages reciprocally interact through the WNT pathway to promote pro-tumoral and malignant phenotypes in 3D engineered microenvironments. J. Immunother. Cancer 2019, 7, 190. [Google Scholar] [CrossRef] [Green Version]
- Gabrusiewicz, K.; Li, X.; Wei, J.; Hashimoto, Y.; Marisetty, A.L.; Ott, M.; Wang, F.; Hawke, D.; Yu, J.; Healy, L.M.; et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology 2018, 7, e1412909. [Google Scholar] [CrossRef]
- Domenis, R.; Cesselli, D.; Toffoletto, B.; Bourkoula, E.; Caponnetto, F.; Manini, I.; Beltrami, A.P.; Ius, T.; Skrap, M.; Di Loreto, C.; et al. Systemic T Cells Immunosuppression of Glioma Stem Cell-Derived Exosomes Is Mediated by Monocytic Myeloid-Derived Suppressor Cells. PLoS ONE 2017, 12, e0169932. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Mandal, G.; Roy Chowdhury, S.; Purohit, S.; Payne, K.K.; Anadon, C.; Gupta, A.; Swanson, P.; Yu, X.; Conejo-Garcia, J.R.; et al. Exosomes Produced by Mesenchymal Stem Cells Drive Differentiation of Myeloid Cells into Immunosuppressive M2-Polarized Macrophages in Breast Cancer. J. Immunol. 2019, 203, 3447–3460. [Google Scholar] [CrossRef]
- Grange, C.; Tapparo, M.; Tritta, S.; Deregibus, M.C.; Battaglia, A.; Gontero, P.; Frea, B.; Camussi, G. Role of HLA-G and extracellular vesicles in renal cancer stem cell-induced inhibition of dendritic cell differentiation. BMC Cancer 2015, 15, 1009. [Google Scholar] [CrossRef] [Green Version]
- Hwang, W.L.; Lan, H.Y.; Cheng, W.C.; Huang, S.C.; Yang, M.H. Tumor stem-like cell-derived exosomal RNAs prime neutrophils for facilitating tumorigenesis of colon Cancer. J. Hematol. Oncol. 2019, 12, 10. [Google Scholar] [CrossRef] [Green Version]
- Anselmi, M.; Fontana, F.; Marzagalli, M.; Gagliano, N.; Sommariva, M.; Limonta, P. Melanoma Stem Cells Educate Neutrophils to Support Cancer Progression. Cancers 2022, 14, 3391. [Google Scholar] [CrossRef]
- Műzes, G.; Sipos, F. Mesenchymal Stem Cell-Derived Secretome: A Potential Therapeutic Option for Autoimmune and Immune-Mediated Inflammatory Diseases. Cells 2022, 11, 2300. [Google Scholar] [CrossRef]
- Alzahrani, F.A.; El-Magd, M.A.; Abdelfattah-Hassan, A.; Saleh, A.A.; Saadeldin, I.M.; El-Shetry, E.S.; Badawy, A.A.; Alkarim, S. Potential Effect of Exosomes Derived from Cancer Stem Cells and MSCs on Progression of DEN-Induced HCC in Rats. Stem Cells Int. 2018, 2018, 8058979. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.P.; Li, A.Q.; Jia, W.H.; Ye, S.; Van Eps, G.; Yu, J.M.; Yang, W.J. MicroRNA expression profiling in exosomes derived from gastric cancer stem-like cells. Oncotarget 2017, 8, 93839–93855. [Google Scholar] [CrossRef] [Green Version]
- Grange, C.; Tapparo, M.; Collino, F.; Vitillo, L.; Damasco, C.; Deregibus, M.C.; Tetta, C.; Bussolati, B.; Camussi, G. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res. 2011, 7, 5346–5356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Yang, G.; Zhao, D.; Wang, J.; Bai, Y.; Peng, Q.; Wang, H.; Fang, R.; Chen, G.; Wang, Z.; et al. CD103-positive CSC exosome promotes EMT of clear cell renal cell carcinoma: Role of remote MiR-19b-3p. Mol. Cancer 2019, 18, 86. [Google Scholar] [CrossRef] [PubMed]
- Hardin, H.; Helein, H.; Meyer, K.; Robertson, S.; Zhang, R.; Zhong, W.; Lloyd, R.V. Thyroid cancer stem-like cell exosomes: Regulation of EMT via transfer of lncRNAs. Lab. Investig. 2018, 98, 1133–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Clauser, K.R.; Tam, W.L.; Fröse, J.; Ye, X.; Eaton, E.N.; Reinhardt, F.; Donnenberg, V.S.; Bhargava, R.; Carr, S.A.; et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat. Cell Biol. 2014, 16, 1105–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasorella, A.; Benezra, R.; Iavarone, A. The ID proteins: Master regulators of cancer stem cells and tumour aggressiveness. Nat. Rev. Cancer 2014, 14, 77–91. [Google Scholar] [CrossRef]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Dianat-Moghadam, H.; Heidarifard, M.; Jahanban-Esfahlan, R.; Panahi, Y.; Hamishehkar, H.; Pouremamali, F.; Rahbarghazi, R.; Nouri, M. Cancer stem cells-emanated therapy resistance: Implications for liposomal drug delivery systems. J. Control. Re-lease 2018, 288, 62–83. [Google Scholar] [CrossRef]
- Lytle, N.K.; Barber, A.G.; Reya, T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 2018, 18, 669–680. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell. Physiol. 2019, 234, 8381–8395. [Google Scholar] [CrossRef]
- Ji, C.; Yang, L.; Yi, W.; Xiang, D.; Wang, Y.; Zhou, Z.; Qian, F.; Ren, Y.; Cui, W.; Zhang, X.; et al. Capillary morphogenesis gene 2 maintains gastric cancer stem-like cell phenotype by activating a Wnt/β-catenin pathway. Oncogene 2018, 37, 3953–3966. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Tiwari, A.; Trivedi, R.; Lin, S.Y. Tumor microenvironment: Barrier or opportunity towards effective cancer therapy. J. Biomed. Sci. 2022, 29, 83. [Google Scholar] [CrossRef]
- Kordes, C.; Häussinger, D. Hepatic stem cell niches. J Clin Invest 2013, 123, 1874–1880. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Wang, L.; Ge, D.; Tan, L.; Cao, B.; Fan, H.; Xue, L. Exosomal O-GlcNAc transferase from esophageal carcinoma stem cell promotes cancer immunosuppression through up-regulation of PD-1 in CD8+ T cells. Cancer Lett. 2021, 500, 98–106. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Ariyama, H.; Stancikova, J.; Sakitani, K.; Asfaha, S.; Renz, B.W.; Dubeykovskaya, Z.A.; Shibata, W.; Wang, H.; Westphalen, C.B.; et al. Mist1 Expressing Gastric Stem Cells Maintain the Normal and Neoplastic Gastric Epithelium and Are Supported by a Perivascular Stem Cell Niche. Cancer Cell 2015, 28, 800–814. [Google Scholar] [CrossRef] [Green Version]
- Sphyris, N.; Hodder, M.C.; Sansom, O.J. Subversion of Niche-Signalling Pathways in Colorectal Cancer: What Makes and Breaks the Intestinal Stem Cell. Cancers 2021, 13, 1000. [Google Scholar] [CrossRef]
- Li, Y.; Tang, T.; Lee, H.J.; Song, K. Selective Anti-Cancer Effects of Plasma-Activated Medium and Its High Efficacy with Cisplatin on Hepatocellular Carcinoma with Cancer Stem Cell Characteristics. Int. J. Mol. Sci. 2021, 22, 3956. [Google Scholar] [CrossRef]
- Nimmakayala, R.K.; Leon, F.; Rachagani, S.; Rauth, S.; Nallasamy, P.; Marimuthu, S.; Shailendra, G.K.; Chhonker, Y.S.; Chugh, S.; Chirravuri, R.; et al. Metabolic programming of distinct cancer stem cells promotes metastasis of pancreatic ductal adenocarcinoma. Oncogene 2021, 40, 215–231. [Google Scholar] [CrossRef]
- Lin, X.; Chen, W.; Wei, F.; Xie, X. TV-circRGPD6 Nanoparticle Suppresses Breast Cancer Stem Cell-Mediated Metastasis via the miR-26b/YAF2 Axis. Mol. Ther. 2021, 29, 244–262. [Google Scholar] [CrossRef]
- Jain, S.; Annett, S.L.; Morgan, M.P.; Robson, T. The Cancer Stem Cell Niche in Ovarian Cancer and Its Impact on Immune Surveillance. Int. J. Mol. Sci. 2021, 22, 4091. [Google Scholar] [CrossRef]
- Hagiwara, M.; Yasumizu, Y.; Yamashita, N.; Rajabi, H.; Fushimi, A.; Long, M.D.; Li, W.; Bhattacharya, A.; Ahmad, R.; Oya, M.; et al. MUC1-C Activates the BAF (mSWI/SNF) Complex in Prostate Cancer Stem Cells. Cancer Res. 2021, 81, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Fendler, A.; Bauer, D.; Busch, J.; Jung, K.; Wulf-Goldenberg, A.; Kunz, S.; Song, K.; Myszczyszyn, A.; Elezkurtaj, S.; Erguen, B.; et al. Inhibiting WNT and NOTCH in renal cancer stem cells and the implications for human patients. Nat. Commun. 2020, 11, 929. [Google Scholar] [CrossRef] [PubMed]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal Cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Billet, S.; Choudhury, D.; Cheng, R.; Haldar, S.; Fernandez, A.; Biondi, S.; Liu, Z.; Zhou, H.; Bhowmick, N.A. Bone marrow mesenchymal stem cells interact with head and neck squamous cell carcinoma cells to promote cancer progression and drug resistance. Neoplasia 2021, 23, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Raniszewska, A.; Vroman, H.; Dumoulin, D.; Cornelissen, R.; Aerts, J.G.J.V.; Domagała-Kulawik, J. PD-L1+ lung cancer stem cells modify the metastatic lymph-node immunomicroenvironment in nsclc patients. Cancer Immunol. Immunother. 2021, 70, 453–461. [Google Scholar] [CrossRef]
- Hsu, M.Y.; Yang, M.H.; Schnegg, C.I.; Hwang, S.; Ryu, B.; Alani, R.M. Notch3 signaling-mediated melanoma-endothelial crosstalk regulates melanoma stem-like cell homeostasis and niche morphogenesis. Lab. Investig. 2017, 97, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Lacina, L.; Plzak, J.; Kodet, O.; Szabo, P.; Chovanec, M.; Dvorankova, B.; Smetana, K., Jr. Cancer Microenvironment: What Can We Learn from the Stem Cell Niche. Int. J. Mol. Sci. 2015, 16, 24094–24110. [Google Scholar] [CrossRef]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef]
- Tabu, K.; Taga, T. Cancer ego-system in glioma: An iron-replenishing niche network systemically self-organized by cancer stem cells. Inflamm. Regen. 2022, 42, 54. [Google Scholar] [CrossRef]
- Kim, M.; Jo, K.W.; Kim, H.; Han, M.E.; Oh, S.O. Genetic heterogeneity of liver cancer stem cells. Anat. Cell Biol. 2022. [Google Scholar] [CrossRef]
- Pedersen, R.K.; Andersen, M.; Skov, V.; Kjær, L.; Hasselbalch, H.C.; Ottesen, J.T.; Stiehl, T. HSC niche dynamics in regeneration, pre-malignancy and cancer: Insights from mathematical modeling. Stem Cells 2022, sxac079. [Google Scholar] [CrossRef]
- Akindona, F.A.; Frederico, S.C.; Hancock, J.C.; Gilbert, M.R. Exploring the origin of the cancer stem cell niche and its role in anti-angiogenic treatment for glioblastoma. Front. Oncol. 2022, 12, 947634. [Google Scholar] [CrossRef]
- Luo, S.; Yang, G.; Ye, P.; Cao, N.; Chi, X.; Yang, W.H.; Yan, X. Macrophages Are a Double-Edged Sword: Molecular Crosstalk between Tumor-Associated Macrophages and Cancer Stem Cells. Biomolecules 2022, 12, 850. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Sakai, K.; Nakamura, T.; Matsumoto, K. Hepatocyte growth factor twenty years on: Much more than a growth factor. J. Gastroenterol. Hepatol. 2011, 26 (Suppl. 1), 188–202. [Google Scholar] [CrossRef] [Green Version]
- Lau, C.K.; Yang, Z.F.; Ho, D.W.; Ng, M.N.; Yeoh, G.C.; Poon, R.T.; Fan, S.T. An Akt/hypoxia-inducible factor-1alpha/platelet-derived growth factor-BB autocrine loop mediates hypoxia-induced chemoresistance in liver cancer cells and tumorigenic hepatic progenitor cells. Clin. Cancer Res. 2009, 15, 3462–3471. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Sun, W.; Shen, W.; Xia, M.; Chen, C.; Xiang, D.; Ning, B.; Cui, X.; Li, H.; Li, X.; et al. Long non-coding RNA DILC regulates liver cancer stem cells via IL-6/STAT3 axis. J. Hepatol. 2016, 64, 1283–1294. [Google Scholar] [CrossRef]
- Liu, G.; Yang, Z.F.; Sun, J.; Sun, B.Y.; Zhou, P.Y.; Zhou, C.; Guan, R.Y.; Wang, Z.T.; Yi, Y.; Qiu, S.J. The LINC00152/miR-205-5p/CXCL11 axis in hepatocellular carcinoma cancer-associated fibroblasts affects cancer cell phenotypes and tumor growth. Cell Oncol. 2022, 45, 1435–1449. [Google Scholar] [CrossRef]
- Huang, H.; Hu, M.; Li, P.; Lu, C.; Li, M. Mir-152 inhibits cell proliferation and colony formation of CD133(+) liver cancer stem cells by targeting KIT. Tumor Biol. 2015, 36, 921–928. [Google Scholar] [CrossRef]
- Sukowati, C.H.; Tiribelli, C. The biological implication of cancer stem cells in hepatocellular carcinoma: A possible target for future therapy. Expert Rev. Gastroenterol. Hepatol. 2013, 7, 749–757. [Google Scholar] [CrossRef]
- Singh, S.R. Gastric cancer stem cells: A novel therapeutic target. Cancer Lett. 2013, 338, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, P.; Xu, X.Y. Epithelial-mesenchymal transition and gastric cancer stem cell. Tumor Biol. 2017, 39, 1010428317698373. [Google Scholar] [CrossRef] [PubMed]
- Becerril-Rico, J.; Alvarado-Ortiz, E.; Toledo-Guzmán, M.E.; Pelayo, R.; Ortiz-Sánchez, E. The cross talk between gastric cancer stem cells and the immune microenvironment: A tumor-promoting factor. Stem Cell Res. Ther. 2021, 12, 498. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xia, L.; Wang, H.; Oyang, L.; Su, M.; Liu, Q.; Lin, J.; Tan, S.; Tian, Y.; Liao, Q.; et al. Cancer stem cells in progression of colorectal Cancer. Oncotarget 2017, 9, 33403–33415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasquez, E.G.; Nasreddin, N.; Valbuena, G.N.; Mulholland, E.J.; Belnoue-Davis, H.L.; Eggington, H.R.; Schenck, R.O.; Wouters, V.M.; Wirapati, P.; Gilroy, K.; et al. Dynamic and adaptive cancer stem cell population admixture in colorectal neoplasia. Cell Stem Cell 2022, 29, 1213–1228.e8. [Google Scholar] [CrossRef]
- Schepers, A.G.; Snippert, H.J.; Stange, D.E.; van den Born, M.; van Es, J.H.; van de Wetering, M.; Clevers, H. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science 2012, 337, 730–735. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.L.; Zeng, Z.; Adileh, M.; Jacobo, A.; Li, C.; Vakiani, E.; Hua, G.; Zhang, L.; Haimovitz-Friedman, A.; Fuks, Z.; et al. Logarithmic expansion of LGR5+ cells in human colorectal Cancer. Cell Signal. 2018, 42, 97–105. [Google Scholar] [CrossRef]
- Iwaya, M.; Ota, H.; Nakajima, T.; Uehara, T.; Riddell, R.; Conner, J. Most colitis associated carcinomas lack expression of LGR5: A preliminary study with implications for unique pathways of carcinogenesis compared to sporadic colorectal carcinoma. BMC Cancer 2021, 21, 119. [Google Scholar] [CrossRef]
- Yaeger, R.; Shah, M.A.; Miller, V.A.; Kelsen, J.R.; Wang, K.; Heins, Z.J.; Ross, J.S.; He, Y.; Sanford, E.; Yantiss, R.K.; et al. Genomic Alterations Observed in Colitis-Associated Cancers Are Distinct From Those Found in Sporadic Colorectal Cancers and Vary by Type of Inflammatory Bowel Disease. Gastroenterology 2016, 151, 278–287.e6. [Google Scholar] [CrossRef] [Green Version]
- Klingler, S.; Hsu, K.S.; Hua, G.; Martin, M.L.; Adileh, M.; Baslan, T.; Zhang, Z.; Paty, P.B.; Fuks, Z.; Brown, A.M.C.; et al. Disruption of the crypt niche promotes outgrowth of mutated colorectal tumor stem cells. J. Clin. Investig. Insight. 2022, 7, e153793. [Google Scholar] [CrossRef]
- Han, H.; Davidson, L.A.; Fan, Y.Y.; Goldsby, J.S.; Yoon, G.; Jin, U.H.; Wright, G.A.; Landrock, K.K.; Weeks, B.R.; Wright, R.C.; et al. Loss of aryl hydrocarbon receptor potentiates FoxM1 signaling to enhance self-renewal of colonic stem and progenitor cells. EMBO J. 2020, 39, e104319. [Google Scholar] [CrossRef]
- Jeong, Y.J.; Oh, H.K.; Park, S.H.; Bong, J.G. Association between inflammation and cancer stem cell phenotype in breast Cancer. Oncol. Lett. 2018, 15, 2380–2386. [Google Scholar] [CrossRef]
- Guo, W. Concise review: Breast cancer stem cells: Regulatory networks, stem cell niches, and disease relevance. Stem Cells Transl. Med. 2014, 3, 942–948. [Google Scholar] [CrossRef]
- Proia, T.A.; Keller, P.J.; Gupta, P.B.; Klebba, I.; Jones, A.D.; Sedic, M.; Gilmore, H.; Tung, N.; Naber, S.P.; Schnitt, S.; et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell 2011, 8, 149–163. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.Q.; Li, X.Y.; Hu, C.Y.; Ford, M.; Kleer, C.G.; Weiss, S.J. Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc. Natl. Acad. Sci. USA 2012, 109, 16654–16659. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, N.; Tiwari, V.K.; Waldmeier, L.; Balwierz, P.J.; Arnold, P.; Pachkov, M.; Meyer-Schaller, N.; Schübeler, D.; van Nimwegen, E.; Christofori, G. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell 2013, 23, 768–783. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.J.; Yang, J.Y.; Xia, W.; Chen, C.T.; Xie, X.; Chao, C.H.; Woodward, W.A.; Hsu, J.M.; Hortobagyi, G.N.; Hung, M.C. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-β-catenin signaling. Cancer Cell 2011, 19, 86–100. [Google Scholar] [CrossRef] [Green Version]
- Műzes, G.; Bohusné Barta, B.; Szabó, O.; Horgas, V.; Sipos, F. Cell-Free DNA in the Pathogenesis and Therapy of Non-Infectious Inflammations and Tumors. Biomedicines 2022, 10, 2853. [Google Scholar] [CrossRef]
- Sipos, F.; Kiss, A.L.; Constantinovits, M.; Tulassay, Z.; Műzes, G. Modified Genomic Self-DNA Influences In Vitro Survival of HT29 Tumor Cells via TLR9- and Autophagy Signaling. Pathol. Oncol. Res. 2019, 25, 1505–1517. [Google Scholar] [CrossRef]
- Sipos, F.; Bohusné Barta, B.; Simon, Á.; Nagy, L.; Dankó, T.; Raffay, R.E.; Petővári, G.; Zsiros, V.; Wichmann, B.; Sebestyén, A.; et al. Survival of HT29 Cancer Cells Is Affected by IGF1R Inhibition via Modulation of Self-DNA-Triggered TLR9 Signaling and the Autophagy Response. Pathol. Oncol. Res. 2022, 28, 1610322. [Google Scholar] [CrossRef]
- Qi, Y.; Zou, H.; Zhao, X.; Kapeleris, J.; Monteiro, M.; Li, F.; Xu, Z.P.; Deng, Y.; Wu, Y.; Tang, Y.; et al. Inhibition of colon cancer K-RasG13D mutation reduces cancer cell proliferation but promotes stemness and inflammation via RAS/ERK pathway. Front. Pharmacol. 2022, 13, 996053. [Google Scholar] [CrossRef] [PubMed]
- Muraro, M.G.; Mele, V.; Däster, S.; Han, J.; Heberer, M.; Cesare Spagnoli, G.; Iezzi, G. CD133+, CD166+CD44+, and CD24+CD44+ phenotypes fail to reliably identify cell populations with cancer stem cell functional features in established human colorectal cancer cell lines. Stem Cells Transl. Med. 2012, 1, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Karami Fath, M.; Ebrahimi, M.; Nourbakhsh, E.; Zia Hazara, A.; Mirzaei, A.; Shafieyari, S.; Salehi, A.; Hoseinzadeh, M.; Payandeh, Z.; Barati, G. PI3K/Akt/mTOR signaling pathway in cancer stem cells. Pathol. Res. Pract. 2022, 237, 154010. [Google Scholar] [CrossRef] [PubMed]
- Nengroo, M.A.; Verma, A.; Datta, D. Cytokine chemokine network in tumor microenvironment: Impact on CSC properties and therapeutic applications. Cytokine 2022, 156, 155916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tang, D.G.; Rycaj, K. Cancer stem cells: Regulation programs, immunological properties and immunotherapy. Semin Cancer Biol. 2018, 52, 94–106. [Google Scholar] [CrossRef]
- Mortezaee, K. Human hepatocellular carcinoma: Protection by melatonin. J. Cell. Physiol. 2018, 233, 6486–6508. [Google Scholar] [CrossRef]
- Zappasodi, R.; Budhu, S.; Hellmann, M.D.; Postow, M.A.; Senbabaoglu, Y.; Manne, S.; Gasmi, B.; Liu, C.; Zhong, H.; Li, Y.; et al. Non-conventional Inhibitory CD4+Foxp3-PD-1hi T Cells as a Biomarker of Immune Checkpoint Blockade Activity. Cancer Cell 2018, 33, 1017–1032.e7. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Tang, F.; Liu, M.; Su, J.; Zhang, Y.; Wu, W.; Devenport, M.; Lazarski, C.A.; Zhang, P.; Wang, X.; et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res. 2018, 28, 416–432. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Ren, Y.; Meng, L.; Li, L.; Beatson, R.; Deng, J.; Zhang, T.; Liu, J.; Han, X. Epigenetic Signaling of Cancer Stem Cells During Inflammation. Front. Cell Dev. Biol. 2021, 9, 772211. [Google Scholar] [CrossRef]
- Pang, X.; Tang, Y.L.; Liang, X.H. Transforming growth factor-β signaling in head and neck squamous cell carcinoma: Insights into cellular responses. Oncol. Lett. 2018, 16, 4799–4806. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018, 62, 50–60. [Google Scholar] [CrossRef]
- Alraouji, N.N.; Al-Mohanna, F.H.; Ghebeh, H.; Arafah, M.; Almeer, R.; Al-Tweigeri, T.; Aboussekhra, A. Tocilizumab potentiates cisplatin cytotoxicity and targets cancer stem cells in triple-negative breast Cancer. Mol. Carcinog. 2020, 59, 1041–1051. [Google Scholar] [CrossRef]
- Sun, L.; Mathews, L.A.; Cabarcas, S.M.; Zhang, X.; Yang, A.; Zhang, Y.; Young, M.R.; Klarmann, K.D.; Keller, J.R.; Farrar, W.L. Epigenetic regulation of SOX9 by the NF-κB signaling pathway in pancreatic cancer stem cells. Stem Cells 2013, 31, 1454–1466. [Google Scholar] [CrossRef] [Green Version]
- Loh, J.J.; Ma, S. The Role of Cancer-Associated Fibroblast as a Dynamic Player in Mediating Cancer Stemness in the Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 727640. [Google Scholar] [CrossRef]
- Pienta, K.J.; Machiels, J.P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate Cancer. Investig. New Drugs 2013, 31, 760–768. [Google Scholar] [CrossRef]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal Cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar]
- Ferrer-Mayorga, G.; Gómez-López, G.; Barbáchano, A.; Fernández-Barral, A.; Peña, C.; Pisano, D.G.; Cantero, R.; Rojo, F.; Muñoz, A.; Larriba, M.J. Vitamin D receptor expression and associated gene signature in tumour stromal fibroblasts predict clinical outcome in colorectal Cancer. Gut 2017, 66, 1449–1462. [Google Scholar] [CrossRef] [Green Version]
- Kocher, H.M.; Basu, B.; Froeling, F.E.M.; Sarker, D.; Slater, S.; Carlin, D.; deSouza, N.M.; De Paepe, K.N.; Goulart, M.R.; Hughes, C.; et al. Phase I clinical trial repurposing all-trans retinoic acid as a stromal targeting agent for pancreatic Cancer. Nat. Commun. 2020, 11, 4841. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [Green Version]
- Galbo, P.M., Jr.; Zang, X.; Zheng, D. Molecular Features of Cancer-associated Fibroblast Subtypes and their Implication on Cancer Pathogenesis, Prognosis, and Immunotherapy Resistance. Clin. Cancer Res. 2021, 27, 2636–2647. [Google Scholar] [CrossRef]
- Ribatti, D.; Solimando, A.G.; Pezzella, F. The Anti-VEGF(R) Drug Discovery Legacy: Improving Attrition Rates by Breaking the Vicious Cycle of Angiogenesis in Cancer. Cancers 2021, 13, 3433. [Google Scholar] [CrossRef] [PubMed]
- Saygin, C.; Matei, D.; Majeti, R.; Reizes, O.; Lathia, J.D. Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem Cell 2019, 24, 25–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandrekar, S.J.; Sargent, D.J. Genomic advances and their impact on clinical trial design. Genome Med. 2009, 1, 69. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Deng, Y.; Li, Z.; Chen, Y.; Zhu, X.; Tan, X.; Cao, G. Cancer Evo-Dev: A Theory of Inflammation-Induced Oncogenesis. Front. Immunol. 2021, 12, 768098. [Google Scholar] [CrossRef]
Figure 1.
CSCs have multiple ways of ensuring their own survival. In addition to affecting basic cellular functions, they can also alter the anti-tumor function of the immune system. The figure was partly created with BioRender.com.
Figure 1.
CSCs have multiple ways of ensuring their own survival. In addition to affecting basic cellular functions, they can also alter the anti-tumor function of the immune system. The figure was partly created with BioRender.com.

Figure 2.
There is an intense association between CSCs and the inflammatory TME. CSCs are able to use immune-competent cells to their own advantage. At the same time, inflammatory TME cells promote the survival of CSCs. Green arrows indicate a stimulatory effect, while the red arrow indicates inhibition. The figure was partly created with BioRender.com.
Figure 2.
There is an intense association between CSCs and the inflammatory TME. CSCs are able to use immune-competent cells to their own advantage. At the same time, inflammatory TME cells promote the survival of CSCs. Green arrows indicate a stimulatory effect, while the red arrow indicates inhibition. The figure was partly created with BioRender.com.


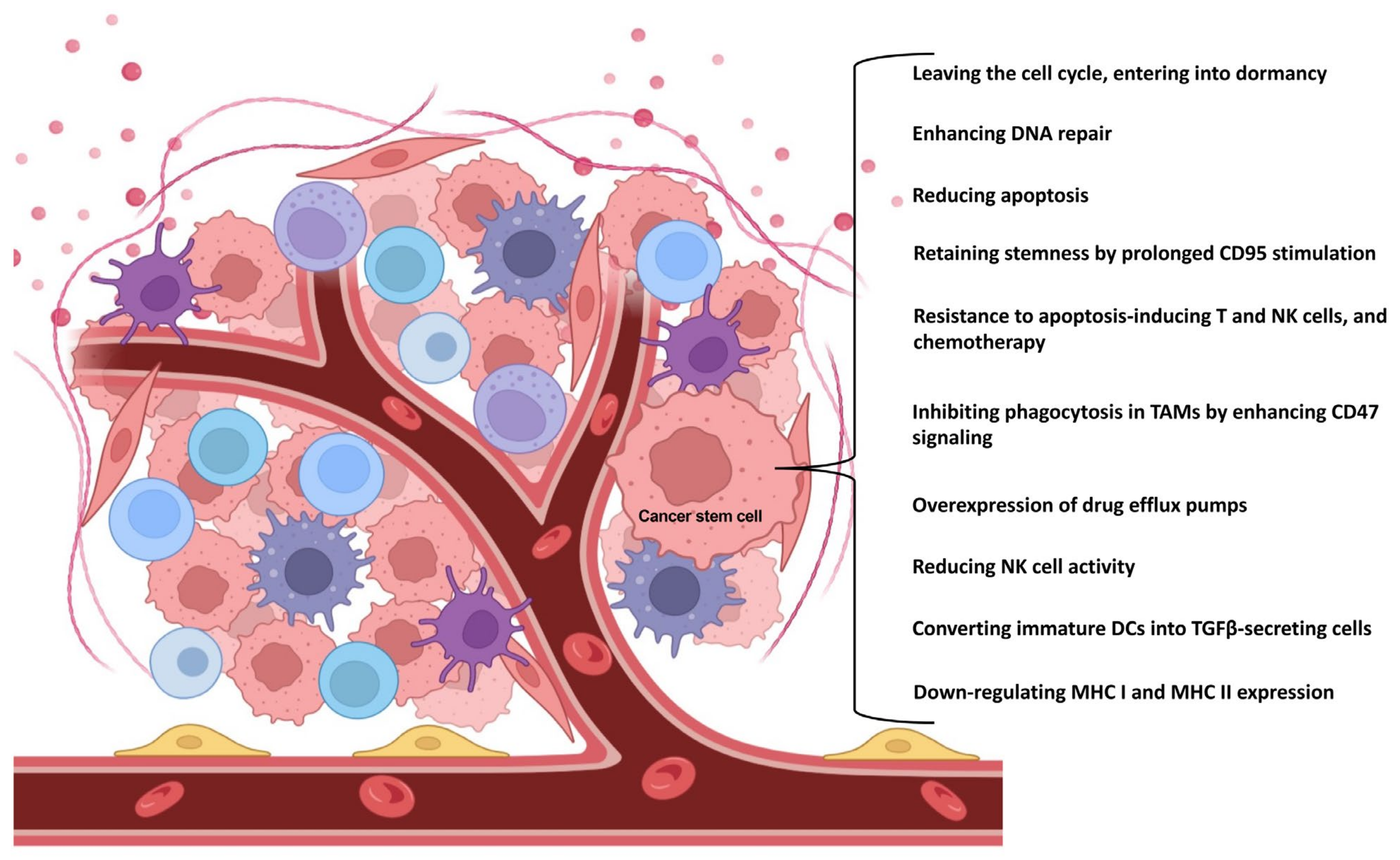{kind=link}
{kind=link}
Table 1.
In the most commonly investigated cancers, CSC-related markers have been revealed. It is important to note that several of these markers may also be expressed by healthy stem cells and normal tissue cells (for more information on this topic, see references [3,4,5,6]). EpCAM: Epithelial cell adhesion molecule; CXCR4: C-X-C chemokine receptor type 4; Lgr5: Leucine-rich repeat-containing G-protein coupled receptor 5; ProC-R: Protein C receptor; LINGO2: Leucine rich repeat and Ig domain containing 2; CLL-1: C-type lectin-like molecule-1; TIM3: T-cell immunoglobulin and mucin domain 3; IL1RAP: Interleukin 1 receptor accessory protein; OV-6: hepatic progenitor cell marker; ESA: epithelial surface antigen; DCLK1: Doublecortin-like kinase 1; ABCB1: ATP Binding Cassette Subfamily B Member 1; CHL1: close homolog of L1; TACSTD1: tumor-associated calcium signal transducer-1; ALDH: Aldehyde dehydrogenase; Nanog: Nanog Homeobox; Oct-3/4: Octamer-binding transcription factor 3/4; BMI-1: B-cell-specific Moloney murine leukemia virus integration site-1; SOX2: SRY-Box Transcription Factor 2; Letm1: Leucine zipper-EF-hand containing transmembrane protein 1; FOXO: Forkhead transcription factor family O; Sall4: Spalt-Like Transcription Factor 4; AFP: alpha-fetoprotein; KLF4: Kruppel-like factor 4; NES: neuroepithelial stem cell protein; TGM2: transglutaminase 2 gene.
Table 1.
In the most commonly investigated cancers, CSC-related markers have been revealed. It is important to note that several of these markers may also be expressed by healthy stem cells and normal tissue cells (for more information on this topic, see references [3,4,5,6]). EpCAM: Epithelial cell adhesion molecule; CXCR4: C-X-C chemokine receptor type 4; Lgr5: Leucine-rich repeat-containing G-protein coupled receptor 5; ProC-R: Protein C receptor; LINGO2: Leucine rich repeat and Ig domain containing 2; CLL-1: C-type lectin-like molecule-1; TIM3: T-cell immunoglobulin and mucin domain 3; IL1RAP: Interleukin 1 receptor accessory protein; OV-6: hepatic progenitor cell marker; ESA: epithelial surface antigen; DCLK1: Doublecortin-like kinase 1; ABCB1: ATP Binding Cassette Subfamily B Member 1; CHL1: close homolog of L1; TACSTD1: tumor-associated calcium signal transducer-1; ALDH: Aldehyde dehydrogenase; Nanog: Nanog Homeobox; Oct-3/4: Octamer-binding transcription factor 3/4; BMI-1: B-cell-specific Moloney murine leukemia virus integration site-1; SOX2: SRY-Box Transcription Factor 2; Letm1: Leucine zipper-EF-hand containing transmembrane protein 1; FOXO: Forkhead transcription factor family O; Sall4: Spalt-Like Transcription Factor 4; AFP: alpha-fetoprotein; KLF4: Kruppel-like factor 4; NES: neuroepithelial stem cell protein; TGM2: transglutaminase 2 gene.
| Cancer Stem Cells | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lung | Breast | Gastric | Colorectal | Liver | Pancreas | Melanoma | Glioblastoma | Prostate | Acute Myeloid Leukemia | Chronic Myeloid Leukemia | ||
| Surface CD markers | CD15 | + | ||||||||||
| CD24 | + | + | + | + | + | |||||||
| CD25 | + | + | ||||||||||
| CD26 | + | |||||||||||
| CD29 | + | |||||||||||
| CD33 | + | + | ||||||||||
| CD36 | + | |||||||||||
| CD44 (and variants) | + | + | + | + | + | + | + | + | ||||
| CD49f | + | |||||||||||
| CD61 | + | |||||||||||
| CD70 | + | |||||||||||
| CD87 | + | |||||||||||
| CD90 | + | + | + | + | + | |||||||
| CD117 | + | + | ||||||||||
| CD123 | + | + | ||||||||||
| CD133 | + | + | + | + | + | + | + | + | + | |||
| CD166 | + | |||||||||||
| CD271 | + | |||||||||||
| CD274 | + | |||||||||||
| Other surface markers | EpCAM | + | + | + | + | + | + | |||||
| CXCR4 | + | + | + | |||||||||
| Lgr5 | + | + | + | |||||||||
| ProC-R | + | |||||||||||
| LINGO2 | + | |||||||||||
| CLL-1 | + | |||||||||||
| TIM3 | + | |||||||||||
| IL1RAP | + | |||||||||||
| cell surface vimentin | + | |||||||||||
| OV-6 | + | |||||||||||
| ESA | + | |||||||||||
| DCLK1 | + | |||||||||||
| ABCB1 | + | |||||||||||
| CHL1 | + | |||||||||||
| TACSTD2 | + | |||||||||||
| Intracellular markers | ALDH | + | + | + | + | + | + | + | + | |||
| Nanog | + | + | + | + | + | + | + | + | ||||
| Oct-3/4 | + | + | + | + | + | + | + | + | ||||
| BMI-1 | + | |||||||||||
| SOX2 | + | + | + | + | + | + | + | + | ||||
| Letm1 | + | + | ||||||||||
| Musashi2 | + | |||||||||||
| FOXO | + | |||||||||||
| Sall4 | + | + | ||||||||||
| AFP | + | |||||||||||
| KLF4 | + | |||||||||||
| NES | + | |||||||||||
| TGM2 | + | |||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sipos, F.; Műzes, G. Cancer Stem Cell Relationship with Pro-Tumoral Inflammatory Microenvironment. Biomedicines 2023, 11, 189. https://doi.org/10.3390/biomedicines11010189
AMA Style
Sipos F, Műzes G. Cancer Stem Cell Relationship with Pro-Tumoral Inflammatory Microenvironment. Biomedicines. 2023; 11(1):189. https://doi.org/10.3390/biomedicines11010189
Chicago/Turabian StyleSipos, Ferenc, and Györgyi Műzes. 2023. "Cancer Stem Cell Relationship with Pro-Tumoral Inflammatory Microenvironment" Biomedicines 11, no. 1: 189. https://doi.org/10.3390/biomedicines11010189
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.