
Therapeutic Potential of Antioxidants and Hybrid TEMPOL Derivatives in Ocular Neurodegenerative Diseases: A Glimpse into the Future




Abstract
:1. Introduction
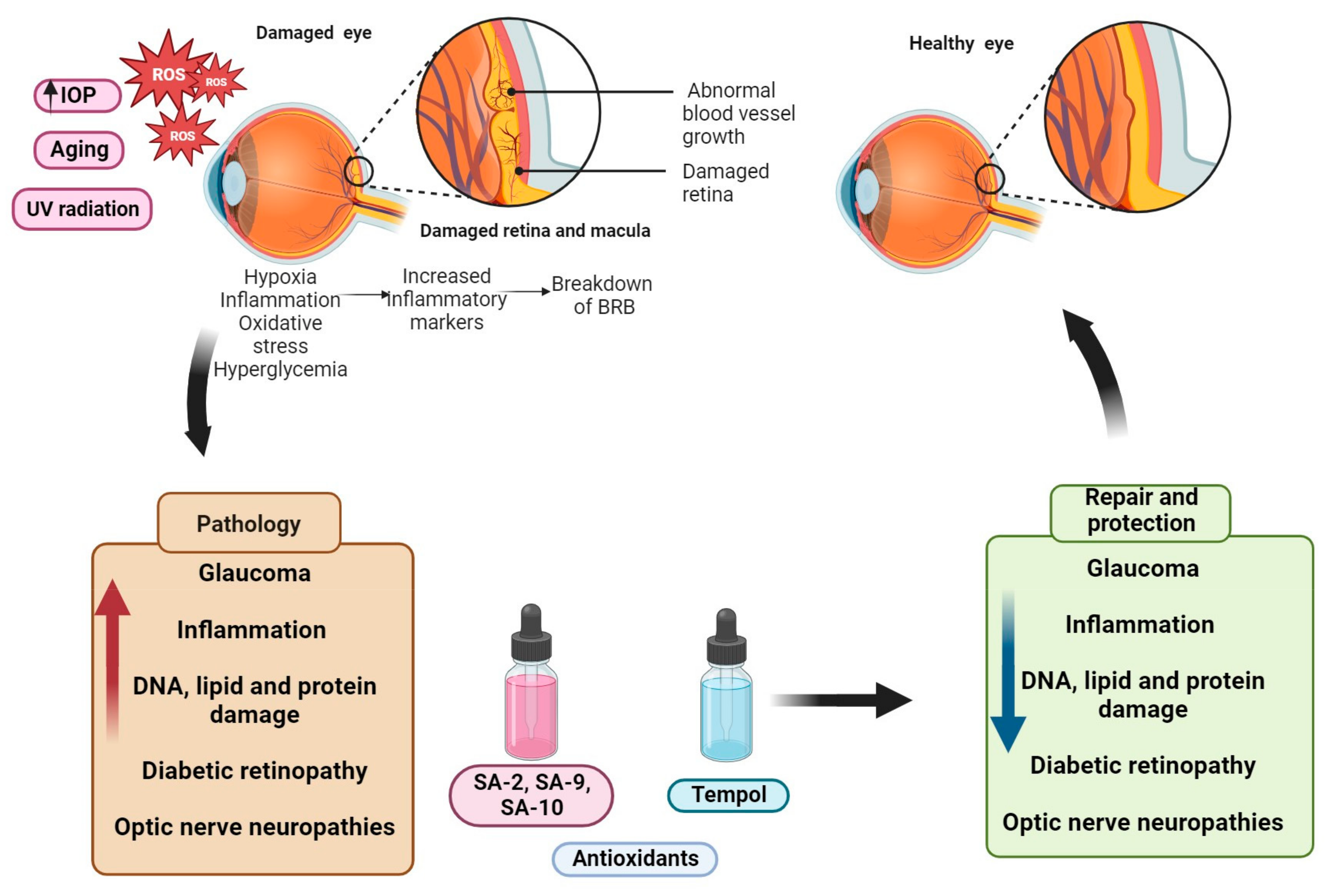
2. Mechanisms and Signaling Pathways Involved in Ocular Neurodegenerative Diseases
2.1. Oxidative Stress Pathways in Ocular Neurodegenerative Diseases
2.2. Oxidative Stress in Glaucoma
2.3. Oxidative Stress in Age-Related Macular Degeneration
2.4. Oxidative Stress in Diabetic Retinopathy
3. Some Recent Advancements in Glaucoma Therapy
3.1. Increasing AH outflow and Decreasing AH Formation
3.2. Alpha-2 Receptor Agonists
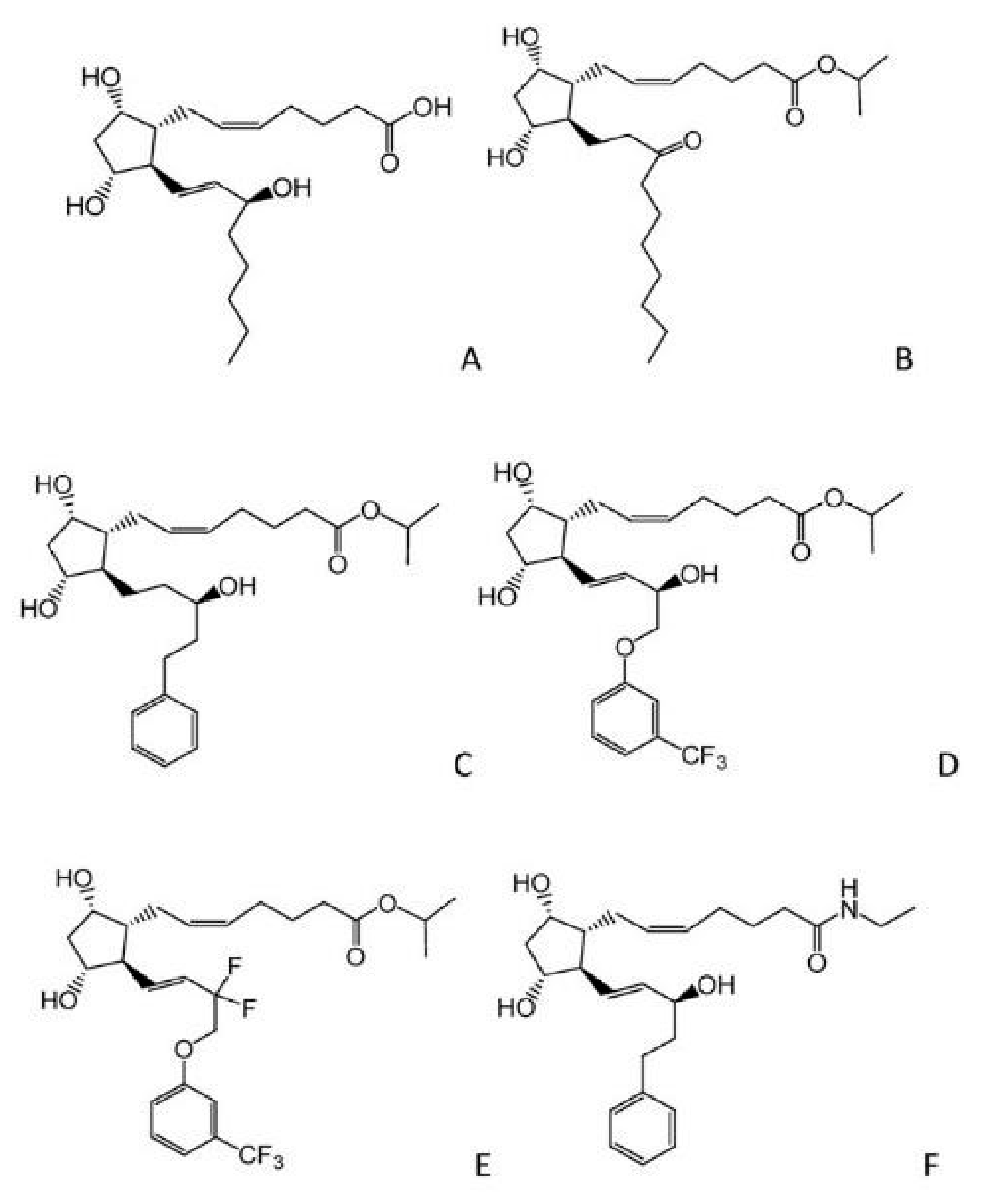
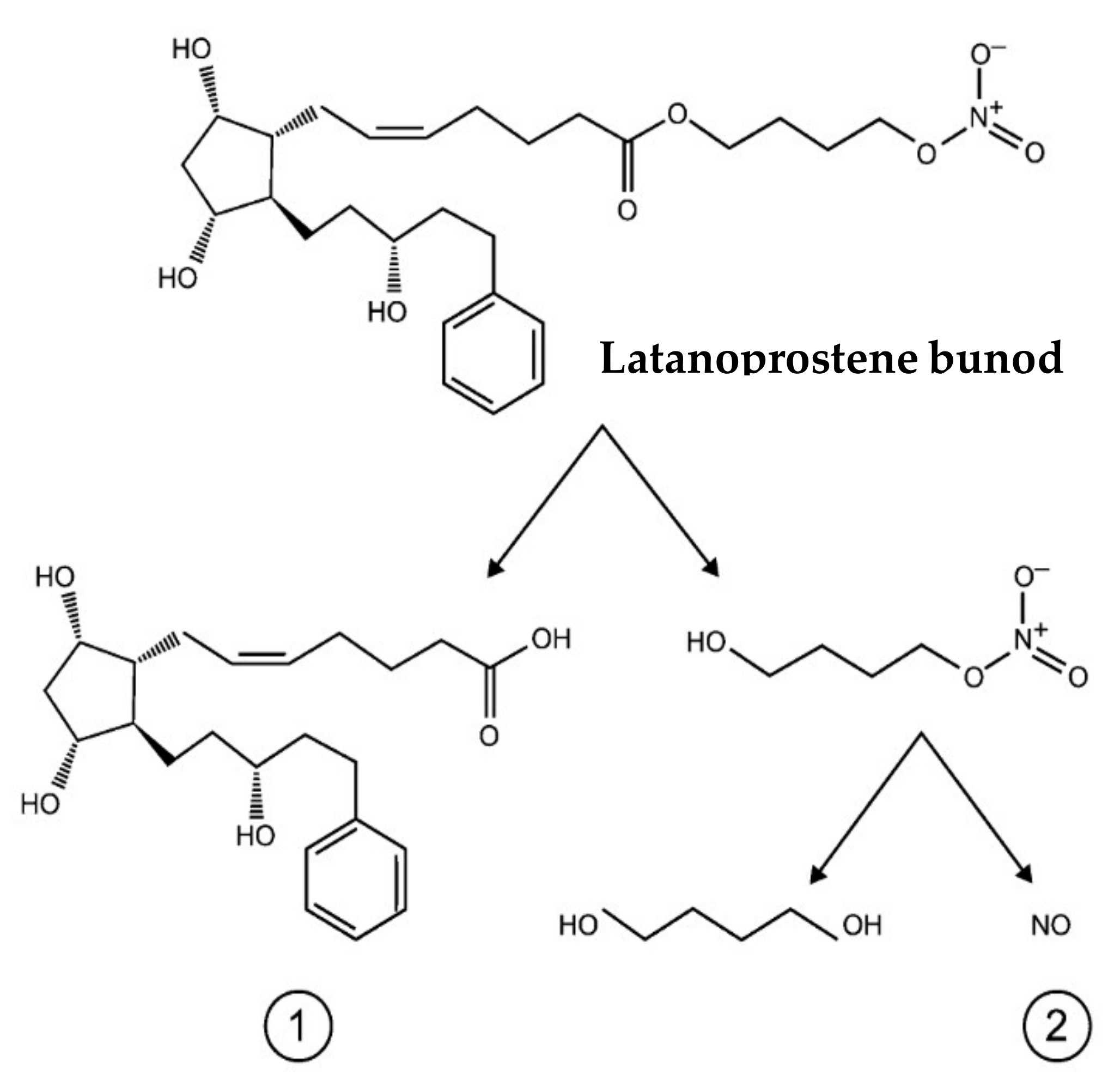
3.3. Nitric Oxide Donors
3.4. Rho Kinase Inhibitors
3.5. Antioxidant Therapy
3.6. Natural Antioxidants
3.7. Synthetic Antioxidants
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aslan, M.; Cort, A.; Yucel, I. Oxidative and nitrative stress markers in glaucoma. Free Radic. Biol. Med. 2008, 45, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Ster, A.M.; Popp, R.A.; Petrisor, F.M.; Stan, C.; Pop, V.I. The Role of Oxidative Stress and Vascular Insufficiency in Primary Open Angle Glaucoma. Clujul. Med. 2014, 87, 143–146. [Google Scholar] [PubMed]
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Ung, L.; Pattamatta, U.; Carnt, N.; Wilkinson-Berka, J.L.; Liew, G.; White, A.J. Oxidative stress and reactive oxygen species: A review of their role in ocular disease. Clin. Sci. 2017, 131, 2865–2883. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Li, S.; Cao, W.; Sun, X. The Association of Oxidative Stress Status with Open-Angle Glaucoma and Exfoliation Glaucoma: A Systematic Review and Meta-Analysis. J. Ophthalmol. 2019, 2019, 1803619. [Google Scholar] [CrossRef] [PubMed]
- Kamel, K.; Farrell, M.; O’Brien, C. Mitochondrial dysfunction in ocular disease: Focus on glaucoma. Mitochondrion 2017, 35, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: Chronic diseases and aging. Arch. Toxicol. 2023, 97, 2499–2574. [Google Scholar] [CrossRef]
- Misra, A.; Ganesh, S.; Shahiwala, A.; Shah, S.P. Drug delivery to the central nervous system: A review. J. Pharm. Pharm. Sci. 2003, 6, 252–273. [Google Scholar]
- Quigley, H.A.; Broman, A.T. The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol. 2006, 90, 262–267. [Google Scholar] [CrossRef]
- Duarte, J.N. Neuroinflammatory Mechanisms of Mitochondrial Dysfunction and Neurodegeneration in Glaucoma. J. Ophthalmol. 2021, 2021, 4581909. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Kelsey, N.A.; Wilkins, H.M.; Linseman, D.A. Nutraceutical antioxidants as novel neuroprotective agents. Molecules 2010, 15, 7792–7814. [Google Scholar] [CrossRef] [PubMed]
- Potashkin, J.A.; Meredith, G.E.; Papp, L.V.; Lu, J.; Bolderson, E.; Boucher, D.; Singh, R.; Holmgren, A.; Khanna, K.K.; Keller, J.N.; et al. The role of oxidative stress in the dysregulation of gene expression and protein metabolism in neurodegenerative disease. Antioxid. Redox Signal. 2006, 8, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Tangvarasittichai, O.; Tangvarasittichai, S. Oxidative Stress, Ocular Disease and Diabetes Retinopathy. Curr. Pharm. Des. 2018, 24, 4726–4741. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Wax, M.B. Hypoxia-inducible factor 1alpha in the glaucomatous retina and optic nerve head. Arch. Ophthalmol. 2004, 122, 1348–1356. [Google Scholar] [CrossRef] [PubMed]
- Wareham, L.K.; Buys, E.S.; Sappington, R.M. The nitric oxide-guanylate cyclase pathway and glaucoma. Nitric Oxide 2018, 77, 75–87. [Google Scholar] [CrossRef]
- Jiang, S.; Moriarty-Craige, S.E.; Orr, M.; Cai, J.; Sternberg, P.; Jones, D.P. Oxidant-induced apoptosis in human retinal pigment epithelial cells: Dependence on extracellular redox state. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1054–1061. [Google Scholar] [CrossRef]
- Liang, Y.; Li, J.; Lin, Q.; Huang, P.; Zhang, L.; Wu, W.; Ma, Y. Research Progress on Signaling Pathway-Associated Oxidative Stress in Endothelial Cells. Oxidative Med. Cell Longev. 2017, 2017, 7156941. [Google Scholar] [CrossRef]
- Izzotti, A.; Longobardi, M.; Cartiglia, C.; Saccà, S.C. Mitochondrial damage in the trabecular meshwork occurs only in primary open-angle glaucoma and in pseudoexfoliative glaucoma. PLoS ONE 2011, 6, e14567. [Google Scholar] [CrossRef]
- Siegfried, C.J.; Shui, Y.-B.; Holekamp, N.M.; Bai, F.; Beebe, D.C. Oxygen distribution in the human eye: Relevance to the etiology of open-angle glaucoma after vitrectomy. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5731–5738. [Google Scholar] [CrossRef]
- Chan, E.; Liu, G.-S.; Dusting, G. Redox mechanisms in pathological angiogenesis in the retina: Roles for NADPH oxidase. Curr. Pharm. Des. 2015, 21, 5988–5998. [Google Scholar] [CrossRef]
- Amankwa, C.E.; Young, O.; DebNath, B.; Gondi, S.R.; Rangan, R.; Ellis, D.Z.; Zode, G.; Stankowska, D.L.; Acharya, S. Modulation of Mitochondrial Metabolic Parameters and Antioxidant Enzymes in Healthy and Glaucomatous Trabecular Meshwork Cells with Hybrid Small Molecule SA-2. Int. J. Mol. Sci. 2023, 24, 11557. [Google Scholar] [CrossRef]
- Ruan, Y.; Jiang, S.; Musayeva, A.; Gericke, A. Oxidative Stress and Vascular Dysfunction in the Retina: Therapeutic Strategies. Antioxidants 2020, 9, 761. [Google Scholar] [CrossRef]
- Chaphalkar, R.M.; Stankowska, D.L.; He, S.; Kodati, B.; Phillips, N.; Prah, J.; Yang, S.; Krishnamoorthy, R.R. Endothelin-1 Mediated Decrease in Mitochondrial Gene Expression and Bioenergetics Contribute to Neurodegeneration of Retinal Ganglion Cells. Sci. Rep. 2020, 10, 3571. [Google Scholar] [CrossRef]
- Mitchell, D.M.; Lovel, A.G.; Stenkamp, D.L. Dynamic changes in microglial and macrophage characteristics during degeneration and regeneration of the zebrafish retina. J. Neuroinflammation 2018, 15, 163. [Google Scholar] [CrossRef]
- Shibata, M.; Ishizaki, E.; Zhang, T.; Fukumoto, M.; Barajas-Espinosa, A.; Li, T.; Puro, D.G. Purinergic Vasotoxicity: Role of the Pore/Oxidant/K(ATP) Channel/Ca(2+) Pathway in P2X(7)-Induced Cell Death in Retinal Capillaries. Vision 2018, 2, 25. [Google Scholar] [CrossRef]
- McElnea, E.; Quill, B.; Docherty, N.; Irnaten, M.; Siah, W.; Clark, A.; O’brien, C.; Wallace, D. Oxidative stress, mitochondrial dysfunction and calcium overload in human lamina cribrosa cells from glaucoma donors. Mol. Vis. 2011, 17, 1182–1191. [Google Scholar]
- Tezel, G.; Wax, M.B. Increased production of tumor necrosis factor-alpha by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J. Neurosci. 2000, 20, 8693–8700. [Google Scholar] [CrossRef]
- Hanus, J.; Anderson, C.; Wang, S. RPE necroptosis in response to oxidative stress and in AMD. Ageing Res. Rev. 2015, 24, 286–298. [Google Scholar] [CrossRef]
- Lu, L.; Gu, X.; Hong, L.; Laird, J.; Jaffe, K.; Choi, J.; Crabb, J.; Salomon, R.G. Synthesis and structural characterization of carboxyethylpyrrole-modified proteins: Mediators of age-related macular degeneration. Bioorganic Med. Chem. 2009, 17, 7548–7561. [Google Scholar] [CrossRef]
- Totan, Y.; Yağcı, R.; Bardak, Y.; Özyurt, H.; Kendir, F.; Yılmaz, G.; Şahin, Ş.; Şahin Tığ, U. Oxidative macromolecular damage in age-related macular degeneration. Curr. Eye Res. 2009, 34, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Bokhary, K.; Aljaser, F.; Abudawood, M.; Tabassum, H.; Bakhsh, A.; Alhammad, S.; Aleyadhi, R.; Almajed, F.; Alsubki, R. Role of Oxidative Stress and Severity of Diabetic Retinopathy in Type 1 and Type 2 Diabetes. Ophthalmic Res. 2021, 64, 613–621. [Google Scholar] [CrossRef]
- Baynes, J.W.; Thorpe, S.R. Role of oxidative stress in diabetic complications: A new perspective on an old paradigm. Diabetes 1999, 48, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Ammar, D.A.; Hamweyah, K.M.; Kahook, M.Y. Antioxidants Protect Trabecular Meshwork Cells from Hydrogen Peroxide-Induced Cell Death. Transl. Vis. Sci. Technol. 2012, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Keller, K.E.; Acott, T.S. The Juxtacanalicular Region of Ocular Trabecular Meshwork: A Tissue with a Unique Extracellular Matrix and Specialized Function. J. Ocul. Biol. 2013, 1, 3. [Google Scholar]
- Armstrong, D.; Al-Awadi, F. Lipid peroxidation and retinopathy in streptozotocin-induced diabetes. Free. Radic. Biol. Med. 1991, 11, 433–436. [Google Scholar] [CrossRef]
- Bastos, A.d.S.; Graves, D.T.; Loureiro, A.P.d.M.; Júnior, C.R.; Corbi, S.C.T.; Frizzera, F.; Scarel-Caminaga, R.M.; Câmara, N.O.; Andriankaja, O.M.; Hiyane, M.I.; et al. Diabetes and increased lipid peroxidation are associated with systemic inflammation even in well-controlled patients. J. Diabetes Complicat. 2016, 30, 1593–1599. [Google Scholar] [CrossRef]
- Fatani, S.H.; Babakr, A.T.; NourEldin, E.M.; Almarzouki, A.A. Lipid peroxidation is associated with poor control of type-2 diabetes mellitus. Diabetes Metab. Syndr. Clin. Res. Rev. 2016, 10 (Suppl. S1), S64–S67. [Google Scholar] [CrossRef]
- Ito, F.; Sono, Y.; Ito, T. Measurement and Clinical Significance of Lipid Peroxidation as a Biomarker of Oxidative Stress: Oxidative Stress in Diabetes, Atherosclerosis, and Chronic Inflammation. Antioxidants 2019, 8, 72. [Google Scholar] [CrossRef] [PubMed]
- Stolz, J.; Alm, A. Latanoprost in the treatment of glaucoma. Clin. Ophthalmol. 2014, 8, 1967–1985. [Google Scholar] [CrossRef] [PubMed]
- Digiuni, M.; Fogagnolo, P.; Rossetti, L. A review of the use of latanoprost for glaucoma since its launch. Expert. Opin. Pharmacother. 2012, 13, 723–745. [Google Scholar] [CrossRef] [PubMed]
- Indication: For the reduction of intraocular pressure (IOP) in patients with open-angle glaucoma or ocular hypertension. In Pharmacoeconomic Review Report: Latanoprostene Bunod (Vyzulta); Bausch Health, Canada Inc.: Ottawa, ON, Canada, 2019.
- Impagnatiello, F.; Bastia, E.; Almirante, N.; Brambilla, S.; Duquesroix, B.; Kothe, A.C.; Bergamini, M.V.W. Prostaglandin analogues and nitric oxide contribution in the treatment of ocular hypertension and glaucoma. Br. J. Pharmacol. 2019, 176, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Latanoprostene Bunod Ophthalmic Solution 0.024%: A Review in Open-Angle Glaucoma and Ocular Hypertension. Drugs 2018, 78, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Masumoto, M.; Higa, R.; Wakakura, M.; Kohmoto, H.; Noguchi, K.; Tomita, G. Efficacy and safety of a switch to latanoprost 0.005% + timolol maleate 0.5% fixed combination eyedrops from latanoprost 0.005% monotherapy. Clin. Ophthalmol. 2012, 6, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Hernández, M.; Urcola, J.H.; Vecino, E. Retinal ganglion cell neuroprotection in a rat model of glaucoma following brimonidine, latanoprost or combined treatments. Exp. Eye Res. 2008, 86, 798–806. [Google Scholar] [CrossRef] [PubMed]
- López-Herrera, M.P.L.; Mayor-Torroglosa, S.; De Imperial, J.M.; Villegas-Pérez, M.P.; Vidal-Sanz, M. Transient ischemia of the retina results in altered retrograde axoplasmic transport: Neuroprotection with brimonidine. Exp. Neurol. 2002, 178, 243–258. [Google Scholar] [CrossRef] [PubMed]
- WoldeMussie, E.; Ruiz, G.; Wijono, M.; Wheeler, L.A. Neuroprotection of retinal ganglion cells by brimonidine in rats with laser-induced chronic ocular hypertension. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2849–2855. [Google Scholar]
- Gao, H.; Qiao, X.; Cantor, L.B.; WuDunn, D. Up-regulation of brain-derived neurotrophic factor expression by brimonidine in rat retinal ganglion cells. Arch. Ophthalmol. 2002, 120, 797–803. [Google Scholar] [CrossRef]
- Wheeler, L.; WoldeMussie, E.; Lai, R. Role of alpha-2 agonists in neuroprotection. Surv. Ophthalmol. 2003, 48 (Suppl. S1), S47–S51. [Google Scholar] [CrossRef]
- Ang, A.; Reddy, M.A.; Shepstone, L.; Broadway, D.C. Long term effect of latanoprost on intraocular pressure in normal tension glaucoma. Br. J. Ophthalmol. 2004, 88, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Turaçli, M.; Özden, R.; Gürses, M. The effect of betaxolol on ocular blood flow and visual fields in patients with normotension glaucoma. Eur. J. Ophthalmol. 1998, 8, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Andrés-Guerrero, V.; García-Feijoo, J. Nitric oxide-donating compounds for IOP lowering in glaucoma. Arch. Soc. Esp. Oftalmol. 2018, 93, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Cavet, M.E.; DeCory, H.H. The Role of Nitric Oxide in the Intraocular Pressure Lowering Efficacy of Latanoprostene Bunod: Review of Nonclinical Studies. J. Ocul. Pharmacol. Ther. 2018, 34, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Garhöfer, G.; Schmetterer, L. Nitric oxide: A drug target for glaucoma revisited. Drug Discov. Today 2019, 24, 1614–1620. [Google Scholar] [CrossRef] [PubMed]
- Cavet, M.E.; Vittitow, J.L.; Impagnatiello, F.; Ongini, E.; Bastia, E. Nitric oxide (NO): An emerging target for the treatment of glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5005–5015. [Google Scholar] [CrossRef] [PubMed]
- Shahidullah, M.; Mandal, A.; Wei, G.; Delamere, N.A. Nitric oxide regulation of Na, K-ATPase activity in ocular ciliary epithelium involves Src family kinase. J. Cell Physiol. 2014, 229, 343–352. [Google Scholar] [CrossRef]
- Kaufman, P.L. Latanoprostene bunod ophthalmic solution 0.024% for IOP lowering in glaucoma and ocular hypertension. Expert. Opin. Pharmacother. 2017, 18, 433–444. [Google Scholar] [CrossRef]
- Klimko, P.G.; Sharif, N.A. Discovery, characterization and clinical utility of prostaglandin agonists for the treatment of glaucoma. Br. J. Pharmacol. 2019, 176, 1051–1058. [Google Scholar] [CrossRef]
- Rao, P.V.; Deng, P.F.; Kumar, J.; Epstein, D.L. Modulation of aqueous humor outflow facility by the Rho kinase-specific inhibitor Y-27632. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1029–1037. [Google Scholar]
- Rao, V.P.; Epstein, D.L. Rho GTPase/Rho kinase inhibition as a novel target for the treatment of glaucoma. BioDrugs 2007, 21, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Kameda, T.; Inoue, T.; Inatani, M.; Fujimoto, T.; Honjo, M.; Kasaoka, N.; Inoue-Mochita, M.; Yoshimura, N.; Tanihara, H. The effect of Rho-associated protein kinase inhibitor on monkey Schlemm’s canal endothelial cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3092–3103. [Google Scholar] [CrossRef] [PubMed]
- Tanihara, H.; Inoue, T.; Yamamoto, T.; Kuwayama, Y.; Abe, H.; Fukushima, A.; Suganami, H.; Araie, M.; The K-115 Clinical Study Group. One-year clinical evaluation of 0.4% ripasudil (K-115) in patients with open-angle glaucoma and ocular hypertension. Acta Ophthalmol. 2016, 94, e26–e34. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Netarsudil Ophthalmic Solution 0.02%: First Global Approval. Drugs 2018, 78, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Neha, K.; Haider, R.; Pathak, A.; Yar, M.S. Medicinal prospects of antioxidants: A review. Eur. J. Med. Chem. 2019, 178, 687–704. [Google Scholar] [CrossRef] [PubMed]
- Amatore, D.; Celestino, I.; Brundu, S.; Galluzzi, L.; Coluccio, P.; Checconi, P.; Magnani, M.; Palamara, A.T.; Fraternale, A.; Nencioni, L. Glutathione increase by the n-butanoyl glutathione derivative (GSH-C4) inhibits viral replication and induces a predominant Th1 immune profile in old mice infected with influenza virus. FASEB BioAdvances 2019, 1, 296–305. [Google Scholar] [CrossRef]
- Brunetti, L.; Menghini, L.; Orlando, G.; Recinella, L.; Leone, S.; Epifano, F.; Lazzarin, F.; Chiavaroli, A.; Ferrante, C.; Vacca, M.; et al. Antioxidant effects of garlic in young and aged rat brain in vitro. J. Med. Food 2009, 12, 1166–1169. [Google Scholar] [CrossRef]
- Nassiri-Asl, M.; Farivar, T.N.; Abbasi, E.; Sadeghnia, H.R.; Sheikhi, M.; Lotfizadeh, M.; Bazahang, P. Effects of rutin on oxidative stress in mice with kainic acid-induced seizure. J. Integr. Med. 2013, 11, 337–342. [Google Scholar] [CrossRef]
- Carmona-Aparicio, L.; Cárdenas-Rodríguez, N.; Delgado-Lamas, G.; Pedraza-Chaverri, J.; Montesinos-Correa, H.; Rivera-Espinosa, L.; Torres-Espíndola, L.M.; Hernández, M.E.; López-Aceves, T.; Pérez-Lozano, D.L.; et al. Dose-Dependent Behavioral and Antioxidant Effects of Quercetin and Methanolic and Acetonic Extracts from Heterotheca inuloides on Several Rat Tissues following Kainic Acid-Induced Status Epilepticus. Oxid. Med. Cell Longev. 2019, 2019, 5287507. [Google Scholar] [CrossRef]
- Wilcox, C.S. Effects of tempol and redox-cycling nitroxides in models of oxidative stress. Pharmacol. Ther. 2010, 126, 119–145. [Google Scholar] [CrossRef]
- Leathem, A.; Simone, M.; Dennis, J.M.; Witting, P.K. The Cyclic Nitroxide TEMPOL Ameliorates Oxidative Stress but Not Inflammation in a Cell Model of Parkinson’s Disease. Antioxidants 2022, 11, 257. [Google Scholar] [CrossRef]
- Tiwari, A.; Tiwari, V.; Banik, B.K.; Sahoo, B.M. Mechanistic Role of Tempol: Synthesis, Catalysed Reactions and Therapeutic Potential. Med. Chem. 2023, 19, 859–878. [Google Scholar] [CrossRef]
- Chiarotto, G.B.; Drummond, L.; Cavarretto, G.; Bombeiro, A.L.; de Oliveira, A.L.R. Neuroprotective effect of tempol (4 hydroxy-tempo) on neuronal death induced by sciatic nerve transection in neonatal rats. Brain Res. Bull. 2014, 106, 1–8. [Google Scholar] [CrossRef]
- Rak, R.; Chao, D.L.; Pluta, R.M.; Mitchell, J.B.; Oldfield, E.H.; Watson, J.C. Neuroprotection by the stable nitroxide Tempol during reperfusion in a rat model of transient focal ischemia. J. Neurosurg. 2000, 92, 646–651. [Google Scholar] [CrossRef]
- Mustafa, A.G.; Bani-Ahmad, M.A.; Jaradat, A.Q.; Allouh, M.Z. Tempol protects blood proteins and lipids against peroxynitrite-mediated oxidative damage. Exp. Biol. Med. 2015, 240, 109–112. [Google Scholar] [CrossRef]
- Wilcox, C.S.; Pearlman, A. Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacol. Rev. 2008, 60, 418–469. [Google Scholar] [CrossRef]
- Wong, W.T.; Kam, W.; Cunningham, D.; Harrington, M.; Hammel, K.; Meyerle, C.B.; Cukras, C.; Chew, E.Y.; Sadda, S.R.; Ferris, F.L. Treatment of geographic atrophy by the topical administration of OT-551: Results of a phase II clinical trial. Investig. Ophthalmol. Vis. Sci. 2010, 51, 6131–6139. [Google Scholar] [CrossRef]
- Rodríguez-Yáñez, Y.; Bahena-Uribe, D.; Chávez-Munguía, B.; López-Marure, R.; González-Monroy, S.; Cisneros, B.; Albores, A. Commercial single-walled carbon nanotubes effects in fibrinolysis of human umbilical vein endothelial cells. Toxicol. Vitr. 2015, 29, 1201–1214. [Google Scholar] [CrossRef]
- Behar-Cohen, F.F.; Goureau, O.; D’Hermies, F.; Courtois, Y. Decreased intraocular pressure induced by nitric oxide donors is correlated to nitrite production in the rabbit eye. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1711–1715. [Google Scholar]
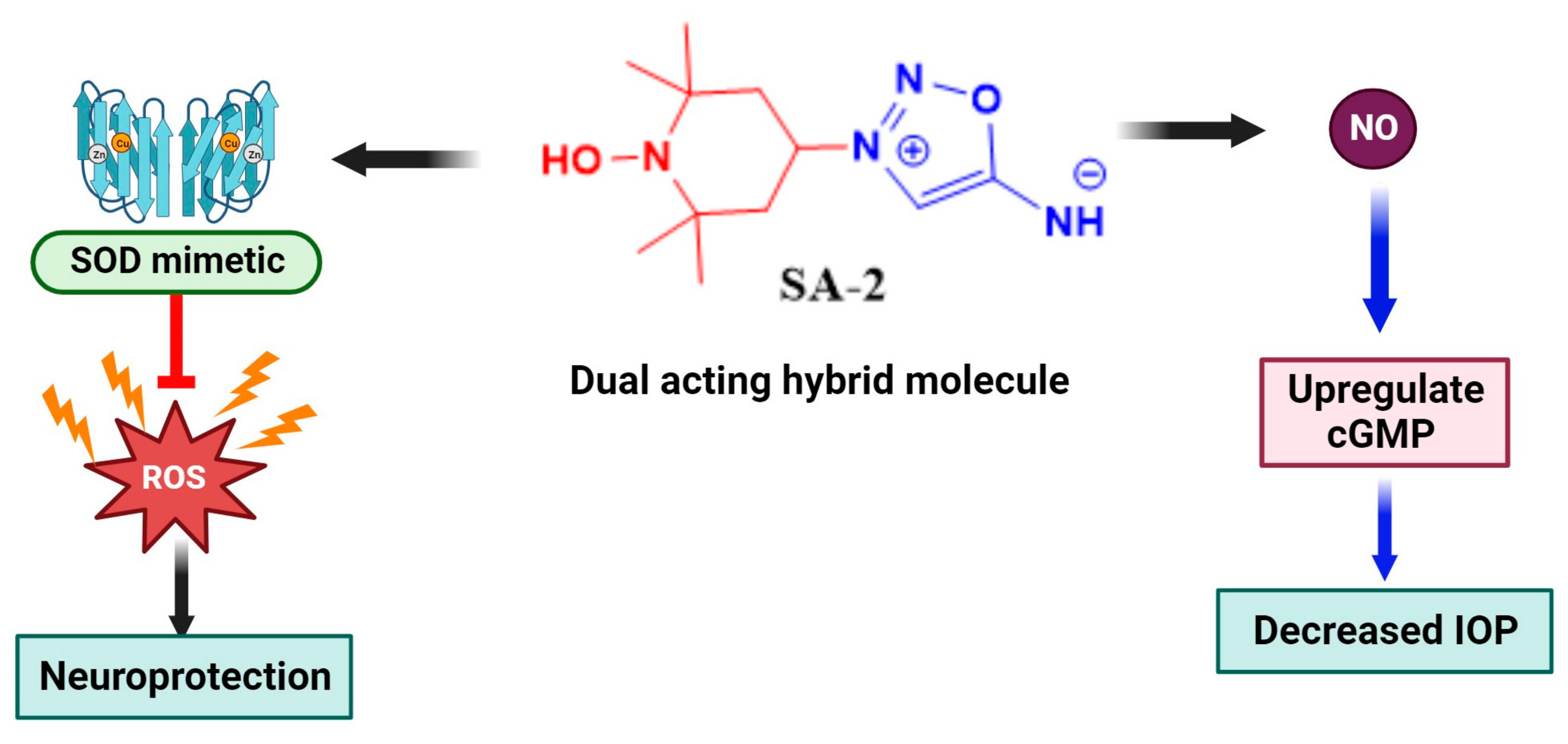
- Acharya, S.; Rogers, P.; Krishnamoorthy, R.R.; Stankowska, D.L.; Dias, H.V.R.; Yorio, T. Design and synthesis of novel hybrid sydnonimine and prodrug useful for glaucomatous optic neuropathy. Bioorg. Med. Chem. Lett. 2016, 26, 1490–1494. [Google Scholar] [CrossRef]
- Amankwa, C.E.; Gondi, S.R.; Dibas, A.; Weston, C.; Funk, A.; Nguyen, T.; Nguyen, K.T.; Ellis, D.Z.; Acharya, S. Novel Thiol Containing Hybrid Antioxidant-Nitric Oxide Donor Small Molecules for Treatment of Glaucoma. Antioxidants 2021, 10, 575. [Google Scholar] [CrossRef] [PubMed]
- Stankowska, D.L.; Krishnamoorthy, V.R.; Ellis, D.Z.; Krishnamoorthy, R.R. Neuroprotective effects of curcumin on endothelin-1 mediated cell death in hippocampal neurons. Nutr. Neurosci. 2017, 20, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Stankowska, D.L.; Millar, J.C.; Kodati, B.; Behera, S.; Chaphalkar, R.M.; Nguyen, T.; Krishnamoorthy, R.R.; Ellis, D.Z.; Acharya, S. Nanoencapsulated hybrid compound SA-2 with long-lasting intraocular pressure-lowering activity in rodent eyes. Mol. Vis. 2021, 27, 37–49. [Google Scholar] [PubMed]
- Hinkle, L.; Le, D.; Nguyen, T.; Tran, V.; Amankwa, C.E.; Weston, C.; Shen, H.; Nguyen, K.T.; Rahimi, M.; Acharya, S. Nano encapsulated novel compound SA-10 with therapeutic activity in both acute and chronic murine hindlimb ischemia models. Nanomedicine 2021, 35, 102400. [Google Scholar] [CrossRef] [PubMed]
- Pham, J.H.; Johnson, G.A.; Rangan, R.S.; Amankwa, C.E.; Acharya, S.; Stankowska, D.L. Neuroprotection of Rodent and Human Retinal Ganglion Cells In Vitro/Ex Vivo by the Hybrid Small Molecule SA-2. Cells 2022, 11, 3741. [Google Scholar] [CrossRef] [PubMed]
- Ardizzone, A.; Repici, A.; Capra, A.P.; De Gaetano, F.; Bova, V.; Casili, G.; Campolo, M.; Esposito, E. Efficacy of the Radical Scavenger, Tempol, to Reduce Inflammation and Oxidative Stress in a Murine Model of Atopic Dermatitis. Antioxidants 2023, 12, 1278. [Google Scholar] [CrossRef] [PubMed]
- Stankowska, D.L.; Dibas, A.; Li, L.; Zhang, W.; Krishnamoorthy, V.R.; Chavala, S.H.; Nguyen, T.P.; Yorio, T.; Ellis, D.Z.; Acharya, S. Hybrid Compound SA-2 is Neuroprotective in Animal Models of Retinal Ganglion Cell Death. Investig. Opthalmol. Vis. Sci. 2019, 60, 3064–3073. [Google Scholar] [CrossRef]
- Tezel, G.; Yang, X. Caspase-independent component of retinal ganglion cell death, in vitro. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4049–4059. [Google Scholar] [CrossRef]
- Jagadeesha, D.K.; Miller, F.J., Jr.; Bhalla, R.C. Inhibition of apoptotic signaling and neointimal hyperplasia by tempol and nitric oxide synthase following vascular injury. J. Vasc. Res. 2009, 46, 109–118. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins/Enzymes | Changes Occur during Neurodegeneration | Changes after Treatment with TEMPOL or Hybrid TEMPOL Derivatives | References |
|---|---|---|---|
| Total antioxidants, Superoxide Dismutase, Glutathione peroxidase, Catalase, |  |  | [22,72] |
| IL-1β, TNFα |  |  | [87] |
| Malondialdehyde, Lipid peroxidation, TBARS |  |  | [22,76,82] |
| RGC count (injury models), TM viability Bcl-2 |  |  | [74,75,84,86,88,89] |
| Caspase 3, 9, Bax, Bax/Bcl-xL ratio |  |  | [74,89,90] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amankwa, C.E.; Kodati, B.; Donkor, N.; Acharya, S. Therapeutic Potential of Antioxidants and Hybrid TEMPOL Derivatives in Ocular Neurodegenerative Diseases: A Glimpse into the Future. Biomedicines 2023, 11, 2959. https://doi.org/10.3390/biomedicines11112959
Amankwa CE, Kodati B, Donkor N, Acharya S. Therapeutic Potential of Antioxidants and Hybrid TEMPOL Derivatives in Ocular Neurodegenerative Diseases: A Glimpse into the Future. Biomedicines. 2023; 11(11):2959. https://doi.org/10.3390/biomedicines11112959
Chicago/Turabian StyleAmankwa, Charles E., Bindu Kodati, Nina Donkor, and Suchismita Acharya. 2023. "Therapeutic Potential of Antioxidants and Hybrid TEMPOL Derivatives in Ocular Neurodegenerative Diseases: A Glimpse into the Future" Biomedicines 11, no. 11: 2959. https://doi.org/10.3390/biomedicines11112959
APA StyleAmankwa, C. E., Kodati, B., Donkor, N., & Acharya, S. (2023). Therapeutic Potential of Antioxidants and Hybrid TEMPOL Derivatives in Ocular Neurodegenerative Diseases: A Glimpse into the Future. Biomedicines, 11(11), 2959. https://doi.org/10.3390/biomedicines11112959