

Adipokines as Clinically Relevant Therapeutic Targets in Obesity
 , , , ,
, , , ,
Abstract
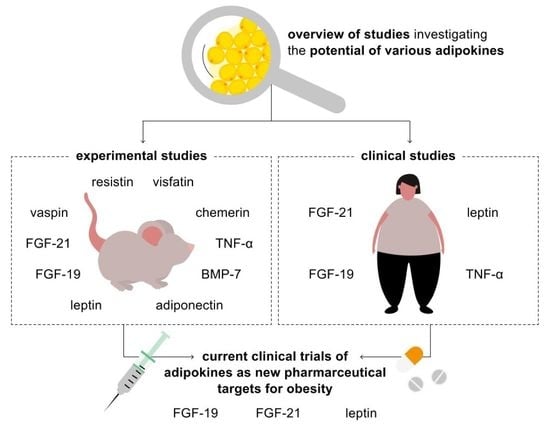
:
1. Introduction
2. Adipokines with Approval for Application in Humans
2.1. FGF-21 (Fibroblast Growth Factor-21)
2.2. FGF-19 (Fibroblast Growth Factor-19)
2.3. Leptin
3. Selected Adipokines with Potential Clinical Relevance
3.1. Adiponectin
3.2. Vaspin
3.3. Resistin
3.4. Chemerin
3.5. Visfatin
3.6. Bone Morphogenetic Protein 7 (BMP-7)
3.7. Tumor Necrosis Factor Alpha (TNF-α)
4. Conclusions and Outlook: The Evolutionary Path of Adipokines from Biomarker to Therapeutic Strategy

{kind=link}
{kind=link}
| Adipokine | Pharmacological Adipokine (Substance Class) | Target Tissue Mediating Metabolic Effects | Mode of Action | Metabolic Effect in Humans |
|---|---|---|---|---|
| Fibroblast growth factor-21 (FGF-21) | BFKB8488 (bispecific anti-FGFR1/ ß-Klotho agonist antibody) | - adipose tissue - CNS | - activation in hypothalamic glutaminergic neurons [37] | - ↓ body weight - ↓ caloric intake - ↓ LDL-C - ↓ triglycerides - ↓ fasting insulin - ↑ HDL-C - ↑ adiponectin |
| LLF580 (FGF21 analogue binding to FGFR1 and β-Klotho) | - liver - adipose tissue | - acting on a body-weight independent manner [51] - ↓ hepatic de novo lipogenesis [51] - ↑ fat oxidation [51] - ↓ of fatty acid flux from adipose tissue to the liver [51] - ↓ steatosis and lipotoxic damage [51] - action of LFF580 on triglyceride metabolism remains unclear [51] | - improved liver fat in patients suffering from NAFLD - ↓ triglycerides and hepatic fat - ↓ total cholesterol - ↓ LDL-C - ↓ insulin resistance - ↓ bone-specific alkaline phosphatase - ↓ procollagen type I N-terminal propeptide - ↓ osteocalcin - ↑ HDL-C - ↑ adiponectin | |
| Pegozafermin/BIO89-100 (FGF21 analogue binding to FGFR1 and β-Klotho) | - liver - adipose tissue | - under investigation [52] | - ↓ hepatic fat - ↓ body weight - ↓ LDL-C - ↓ non-HDL-C - ↓ serum triglycerides - ↑ HDL-C - ↑ adiponectin | |
| PF-05231023 (FGF-21 analogue binding to FGF R1 and β-Klotho) | - adipose tissue - CNS | - ↓ expression of adiponectin receptor (AdipoR) - ↓ expression of peroxisome proliferator activated receptor y (PPARy) - ↓ leptin - ↓ lipid synthesis - ↓ pro-inflammatory markers (IL1β, IFNy) - ↑ anti-inflammatory marker (IL10) [35] - regulation of food intake [35] | - ↓ body weight - ↓ total cholesterol - ↓ LDL-C - ↑ HDL-C - ↑ adiponectin | |
| LY2405319 (FGF-21 analogue FGFR1 and β-Klotho) | - liver - adipose tissue | - ↑ hepatic mitochondrial function - ↑ fatty acid oxidation - ↓ inflammatory signalling [172] | - ↓ triglycerides - ↓ body weight - ↑ HDL-C - ↑ adiponectin | |
| Pegbelfermin/ BMS-986036 (FGF-21 analogue FGFR1 and β-Klotho) | - liver - adipose tissue | - ↓ choloylglycine hydrolase gene expression - ↓ faecal secondary bile acid levels [47] | - improved metabolic parameters - ↓ absolute liver fat percentage in patients with non-alcoholic steatohepatitis - ↓ secondary bile acids - improved HDL-C - improved triglycerides - improved fibrosis biomarkers - ↑ adiponectin levels | |
| Efruxifermin/AKR-001 (FcFGF21 analogue FGFR1 and β-Klotho) | - liver - adipose tissue | - direct anti-fibrotic activity | - ↓ plasma triglycerides - ↓ LDL-C - ↑ HDL-C - ↑ adiponectin levels - ↓ hepatic steatosis - improved body weight - improved glycaemic control - improved liver fat | |
| Fibroblast growth factor-19 (FGF-19) | Aldafermin/NGM282 (non-mitogenic FGF-19 variant) | - liver | - ↓ serum concentration of 7α-hydroxy-4-cholesten-3-one (surrogate marker for enzymatic activity of CYP7A1) [63] | - ↓ liver fat content in patients with NASH - improved liver fibrosis |
| Leptin | r-metHuLeptin (recombinant methionyl human leptin) | - CNS | - ↑ centrally acting - ↑ leptin concentrations [78] | - weight and fat mass loss in patients with obesity and elevated endogenous serum leptin concentrations |
| metreleptin (recombinant human leptin analogue) | - CNS | - signalling via hypothalamus [67,89] | - ↑ satiety time and decreased ghrelin levels in patients with lipodystrophy - ↓ HbA1c - ↓ triglycerides - ↓ LDL-C - ↓ albuminuria |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kershaw, E.E.; Flier, J.S. Adipose Tissue as an Endocrine Organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Unamuno, X.; Gómez-Ambrosi, J.; Rodríguez, A.; Becerril, S.; Frühbeck, G.; Catalán, V. Adipokine dysregulation and adipose tissue inflammation in human obesity. Eur. J. Clin. Investig. 2018, 48, e12997. [Google Scholar] [CrossRef]
- Deng, Y.; Scherer, P.E. Adipokines as novel biomarkers and regulators of the metabolic syndrome. Ann. N. Y. Acad. Sci. 2010, 1212, E1–E19. [Google Scholar] [CrossRef] [PubMed]
- Vegiopoulos, A.; Rohm, M.; Herzig, S. Focus: Metabolism: Adipose tissue: Between the extremes. EMBO J. 2017, 36, 1999–2017. [Google Scholar] [CrossRef] [PubMed]
- Fasshauer, M.; Blüher, M. Adipokines in health and disease. Trends Pharmacol. Sci. 2015, 36, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Manjowk, G.-M.; Wagner, I.V.; Klöting, N.; Ebert, T.; Jessnitzer, B.; Lössner, U.; Stukenborg, J.-B.; Blüher, M.; Stumvoll, M.; et al. Leptin Within the Subphysiological to Physiological Range Dose Dependently Improves Male Reproductive Function in an Obesity Mouse Model. Endocrinology 2016, 157, 2461–2468. [Google Scholar] [CrossRef]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Crewe, C.; An, Y.A.; Scherer, P.E. The ominous triad of adipose tissue dysfunction: Inflammation, fibrosis, and impaired angiogenesis. J. Clin. Investig. 2017, 127, 74–82. [Google Scholar] [CrossRef]
- Hulthe, J.; Hultén, L.M.; Fagerberg, B. Low adipocyte-derived plasma protein adiponectin concentrations are associated with the metabolic syndrome and small dense low-density lipoprotein particles: Atherosclerosis and insulin resistance study. Metabolism 2003, 52, 1612–1614. [Google Scholar] [CrossRef]
- Arita, Y.; Kihara, S.; Ouchi, N.; Takahashi, M.; Maeda, K.; Miyagawa, J.; Hotta, K.; Shimomura, I.; Nakamura, T.; Miyaoka, K.; et al. Paradoxical Decrease of an Adipose-Specific Protein, Adiponectin, in Obesity. Biochem. Biophys. Res. Commun. 1999, 257, 79–83. [Google Scholar] [CrossRef]
- Bozaoglu, K.; Bolton, K.; McMillan, J.; Zimmet, P.; Jowett, J.; Collier, G.; Walder, K.; Segal, D. Chemerin Is a Novel Adipokine Associated with Obesity and Metabolic Syndrome. Endocrinology 2007, 148, 4687–4694. [Google Scholar] [CrossRef] [PubMed]
- Ebert, T.; Gebhardt, C.; Scholz, M.; Wohland, T.; Schleinitz, D.; Fasshauer, M.; Blüher, M.; Stumvoll, M.; Kovács, P.; Tönjes, A. Relationship Between 12 Adipocytokines and Distinct Components of the Metabolic Syndrome. J. Clin. Endocrinol. Metab. 2018, 103, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Yue, T.; Chen, Z.; Wu, W.; Xu, S.; Weng, J. Targeting FGF21 in cardiovascular and metabolic diseases: From mechanism to medicine. Int. J. Biol. Sci. 2023, 19, 66–88. [Google Scholar] [CrossRef]
- Yang, C.; Jin, C.; Li, X.; Wang, F.; McKeehan, W.L.; Luo, Y. Differential Specificity of Endocrine FGF19 and FGF21 to FGFR1 and FGFR4 in Complex with KLB. PLoS ONE 2012, 7, e33870. [Google Scholar] [CrossRef]
- Markan, K.R.; Naber, M.C.; Ameka, M.K.; Anderegg, M.D.; Mangelsdorf, D.J.; Kliewer, S.A.; Mohammadi, M.; Potthoff, M.J. Circulating FGF21 Is Liver Derived and Enhances Glucose Uptake During Refeeding and Overfeeding. Diabetes 2014, 63, 4057–4063. [Google Scholar] [CrossRef] [PubMed]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic Fibroblast Growth Factor 21 Is Regulated by PPARα and Is a Key Mediator of Hepatic Lipid Metabolism in Ketotic States. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef]
- De Sousa-Coelho, A.L.; Marrero, P.F.; Haro, D. Activating transcription factor 4-dependent induction of FGF21 during amino acid deprivation. Biochem. J. 2012, 443, 165–171. [Google Scholar] [CrossRef]
- Lundsgaard, A.-M.; Fritzen, A.M.; Sjøberg, K.A.; Myrmel, L.S.; Madsen, L.; Wojtaszewski, J.F.; Richter, E.A.; Kiens, B. Circulating FGF21 in humans is potently induced by short term overfeeding of carbohydrates. Mol. Metab. 2017, 6, 22–29. [Google Scholar] [CrossRef]
- Ying, L.; Li, N.; He, Z.; Zeng, X.; Nan, Y.; Chen, J.; Miao, P.; Ying, Y.; Lin, W.; Zhao, X.; et al. Fibroblast growth factor 21 Ameliorates diabetes-induced endothelial dysfunction in mouse aorta via activation of the CaMKK2/AMPKα signaling pathway. Cell Death Dis. 2019, 10, 655. [Google Scholar] [CrossRef]
- Zhang, N.; Liu, C.; Zhang, Y.; Xu, D.; Gui, L.; Lu, Y.; Zhang, Q. Liraglutide regulates lipid metabolism via FGF21- LKB1- AMPK- ACC1 pathway in white adipose tissues and macrophage of type 2 diabetic mice. Biochem. Biophys. Res. Commun. 2021, 548, 120–126. [Google Scholar] [CrossRef]
- Yang, H.; Feng, A.; Lin, S.; Yu, L.; Lin, X.; Yan, X.; Lu, X.; Zhang, C. Fibroblast growth factor-21 prevents diabetic cardiomyopathy via AMPK-mediated antioxidation and lipid-lowering effects in the heart. Cell Death Dis. 2018, 9, 227. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Qiang, L.; Farmer, S.R. Identification of a Domain within Peroxisome Proliferator-Activated Receptor γ Regulating Expression of a Group of Genes Containing Fibroblast Growth Factor 21 That Are Selectively Repressed by SIRT1 in Adipocytes. Mol. Cell. Biol. 2008, 28, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N. FGF21 as a Hepatokine, Adipokine, and Myokine in Metabolism and Diseases. Front. Endocrinol. 2014, 5, 107. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; Dunbar, J.D.; Bina, H.A.; Bright, S.; Moyers, J.S.; Zhang, C.; Ding, L.; Micanovic, R.; Mehrbod, S.F.; Knierman, M.D.; et al. FGF-21/FGF-21 receptor interaction and activation is determined by βKlotho. J. Cell. Physiol. 2008, 215, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Mangelsdorf, D.J. A Dozen Years of Discovery: Insights into the Physiology and Pharmacology of FGF21. Cell Metab. 2019, 29, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Lam, K.S.L.; Xu, A. The therapeutic potential of FGF21 in metabolic diseases: From bench to clinic. Nat. Rev. Endocrinol. 2020, 16, 654–667. [Google Scholar] [CrossRef]
- Sonoda, J.; Chen, M.Z.; Baruch, A. FGF21-receptor agonists: An emerging therapeutic class for obesity-related diseases. Horm. Mol. Biol. Clin. Investig. 2017, 30, 4. [Google Scholar] [CrossRef]
- Kolumam, G.; Chen, M.Z.; Tong, R.; Zavala-Solorio, J.; Kates, L.; van Bruggen, N.; Ross, J.; Wyatt, S.K.; Gandham, V.D.; Carano, R.A.; et al. Sustained Brown Fat Stimulation and Insulin Sensitization by a Humanized Bispecific Antibody Agonist for Fibroblast Growth Factor Receptor 1/βKlotho Complex. Ebiomedicine 2015, 2, 730–743. [Google Scholar] [CrossRef]
- Fisher, F.M.; Chui, P.C.; Antonellis, P.J.; Bina, H.A.; Kharitonenkov, A.; Flier, J.S.; Maratos-Flier, E. Obesity Is a Fibroblast Growth Factor 21 (FGF21)-Resistant State. Diabetes 2010, 59, 2781–2789. [Google Scholar] [CrossRef]
- Zhang, X.; Yeung, D.C.Y.; Karpisek, M.; Stejskal, D.; Zhou, Z.-G.; Liu, F.; Wong, R.L.C.; Chow, W.-S.; Tso, A.W.K.; Lam, K.S.L.; et al. Serum FGF21 Levels Are Increased in Obesity and Are Independently Associated with the Metabolic Syndrome in Humans. Diabetes 2008, 57, 1246–1253. [Google Scholar] [CrossRef]
- Martínez-Garza, Ú.; Torres-Oteros, D.; Yarritu-Gallego, A.; Marrero, P.F.; Haro, D.; Relat, J. Fibroblast Growth Factor 21 and the Adaptive Response to Nutritional Challenges. Int. J. Mol. Sci. 2019, 20, 4692. [Google Scholar] [CrossRef] [PubMed]
- Zhen, E.Y.; Jin, Z.; Ackermann, B.L.; Thomas, M.K.; Gutierrez, J.A. Circulating FGF21 proteolytic processing mediated by fibroblast activation protein. Biochem. J. 2016, 473, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Escuredo, J.M.; Gomez-Ambrosi, J.; Catalan, V.; Domingo, P.; Giralt, M.; Frühbeck, G.; Villarroya, F. Opposite alterations in FGF21 and FGF19 levels and disturbed expression of the receptor machinery for endocrine FGFs in obese patients. Int. J. Obes. 2015, 39, 121–129. [Google Scholar] [CrossRef]
- Gaich, G.; Chien, J.Y.; Fu, H.; Glass, L.C.; Deeg, M.A.; Holland, W.L.; Kharitonenkov, A.; Bumol, T.; Schilske, H.K.; Moller, D.E. The Effects of LY2405319, an FGF21 Analog, in Obese Human Subjects with Type 2 Diabetes. Cell Metab. 2013, 18, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, S.; Zhou, Y.; Li, D.; Rossulek, M.; Dong, J.; Somayaji, V.; Weng, Y.; Clark, R.; Lanba, A.; Owen, B.M.; et al. A Long-Acting FGF21 Molecule, PF-05231023, Decreases Body Weight and Improves Lipid Profile in Non-human Primates and Type 2 Diabetic Subjects. Cell Metab. 2016, 23, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; Wroblewski, V.J.; Koester, A.; Chen, Y.-F.; Clutinger, C.K.; Tigno, X.T.; Hansen, B.C.; Shanafelt, A.B.; Etgen, G.J. The Metabolic State of Diabetic Monkeys Is Regulated by Fibroblast Growth Factor-21. Endocrinology 2007, 148, 774–781. [Google Scholar] [CrossRef]
- Baruch, A.; Wong, C.; Chinn, L.W.; Vaze, A.; Sonoda, J.; Gelzleichter, T.; Chen, S.; Lewin-Koh, N.; Morrow, L.; Dheerendra, S.; et al. Antibody-mediated activation of the FGFR1/Klothoβ complex corrects metabolic dysfunction and alters food preference in obese humans. Proc. Natl. Acad. Sci. USA 2020, 117, 28992–29000. [Google Scholar] [CrossRef]
- Epperlein, S.; Gebhardt, C.; Rohde, K.; Chakaroun, R.; Patt, M.; Schamarek, I.; Kralisch, S.; Heiker, J.T.; Scholz, M.; Stumvoll, M.; et al. The Effect of FGF21 and Its Genetic Variants on Food and Drug Cravings, Adipokines and Metabolic Traits. Biomedicines 2021, 9, 345. [Google Scholar] [CrossRef]
- Jensen-Cody, S.O.; Flippo, K.H.; Claflin, K.E.; Yavuz, Y.; Sapouckey, S.A.; Walters, G.C.; Usachev, Y.M.; Atasoy, D.; Gillum, M.P.; Potthoff, M.J. FGF21 Signals to Glutamatergic Neurons in the Ventromedial Hypothalamus to Suppress Carbohydrate Intake. Cell Metab. 2020, 32, 273–286.e6. [Google Scholar] [CrossRef]
- Hill, C.M.; Qualls-Creekmore, E.; Berthoud, H.-R.; Soto, P.; Yu, S.; McDougal, D.H.; Münzberg, H.; Morrison, C.D. FGF21 and the Physiological Regulation of Macronutrient Preference. Endocrinology 2020, 161, bqaa019. [Google Scholar] [CrossRef]
- Solon-Biet, S.M.; Cogger, V.C.; Pulpitel, T.; Heblinski, M.; Wahl, D.; McMahon, A.C.; Warren, A.; Durrant-Whyte, J.; Walters, K.A.; Krycer, J.R.; et al. Defining the Nutritional and Metabolic Context of FGF21 Using the Geometric Framework. Cell Metab. 2016, 24, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Bookout, A.L.; De Groot, M.H.M.; Owen, B.; Lee, S.; Gautron, L.; Lawrence, H.L.; Ding, X.; Elmquist, J.K.; Takahashi, J.; Mangelsdorf, D.; et al. FGF21 regulates metabolism and circadian behavior by acting on the nervous system. Nat. Med. 2013, 19, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a novel metabolic regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Dunshee, D.R.; Bainbridge, T.W.; Kljavin, N.M.; Zavala-Solorio, J.; Schroeder, A.C.; Chan, R.; Corpuz, R.; Wong, M.; Zhou, W.; Deshmukh, G.; et al. Fibroblast Activation Protein Cleaves and Inactivates Fibroblast Growth Factor 21. J. Biol. Chem. 2016, 291, 5986–5996. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.; Charles, E.D.; Neuschwander-Tetri, B.A.; Loomba, R.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.J.; Halegoua-DeMarzio, D.; Kundu, S.; Noviello, S.; et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: A randomised, double-blind, placebo-controlled, phase 2a trial. Lancet 2019, 392, 2705–2717. [Google Scholar] [CrossRef]
- Luo, Y.; Decato, B.E.; Charles, E.D.; Shevell, D.E.; McNaney, C.; Shipkova, P.; Apfel, A.; Tirucherai, G.S.; Sanyal, A.J. Pegbelfermin selectively reduces secondary bile acid concentrations in patients with non-alcoholic steatohepatitis. JHEP Rep. Innov. Hepatol. 2022, 4, 100392. [Google Scholar] [CrossRef]
- Charles, E.D.; Neuschwander-Tetri, B.A.; Frias, J.P.; Kundu, S.; Luo, Y.; Tirucherai, G.S.; Christian, R. Pegbelfermin (BMS-986036), PEGylated FGF21, in Patients with Obesity and Type 2 Diabetes: Results from a Randomized Phase 2 Study. Obesity 2019, 27, 41–49. [Google Scholar] [CrossRef]
- Kaufman, A.; Abuqayyas, L.; Denney, W.S.; Tillman, E.J.; Rolph, T. AKR-001, an Fc-FGF21 Analog, Showed Sustained Pharmacodynamic Effects on Insulin Sensitivity and Lipid Metabolism in Type 2 Diabetes Patients. Cell Rep. Med. 2020, 1, 100057. [Google Scholar] [CrossRef]
- In Akero Therapeutics’ Phase 2b HARMONY Study, Both the 50 mg and 28 mg EFX Doses Achieved Statistical Significance on Primary and Secondary Histology Endpoints after 24 Weeks—Akero Therapeutics, Inc. Available online: https://ir.akerotx.com/news-releases/news-release-details/akero-therapeutics-phase-2b-harmony-study-both-50mg-and-28mg-efx/ (accessed on 11 January 2023).
- LLF580, an FGF21 Analog, Reduces Triglycerides and Hepatic Fat in Obese Adults with Modest Hypertriglyceridemia—PMC. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8914500/ (accessed on 13 April 2023).
- Loomba, R.; Lawitz, E.J.; Frias, J.P.; Ortiz-Lasanta, G.; Johansson, L.; Franey, B.B.; Morrow, L.; Rosenstock, M.; Hartsfield, C.L.; Chen, C.-Y.; et al. Safety, pharmacokinetics, and pharmacodynamics of pegozafermin in patients with non-alcoholic steatohepatitis: A randomised, double-blind, placebo-controlled, phase 1b/2a multiple-ascending-dose study. Lancet Gastroenterol. Hepatol. 2023, 8, 120–132. [Google Scholar] [CrossRef]
- Strack, A.M.; Myers, R.W. Modulation of Metabolic Syndrome by Fibroblast Growth Factor 19 (FGF19)? Endocrinology 2004, 145, 2591–2593. [Google Scholar] [CrossRef] [PubMed]
- Jones, S. Mini-Review: Endocrine Actions of Fibroblast Growth Factor 19. Mol. Pharm. 2008, 5, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Kliewer, S.A.; Mangelsdorf, D.J. Endocrine fibroblast growth factors 15/19 and 21: From feast to famine. Genes Dev. 2012, 26, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Barutcuoglu, B.; Basol, G.; Çakır, Y.; Cetinkalp, S.; Parildar, Z.; Kabaroglu, C.; Ozmen, D.; Mutaf, I.; Bayindir, O. Fibroblast growth factor-19 levels in type 2 diabetic patients with metabolic syndrome. Ann. Clin. Lab. Sci. 2011, 41, 390–396. [Google Scholar] [PubMed]
- Huang, X.; Yang, C.; Luo, Y.; Jin, C.; Wang, F.; McKeehan, W.L. FGFR4 Prevents Hyperlipidemia and Insulin Resistance but Underlies High-Fat Diet–Induced Fatty Liver. Diabetes 2007, 56, 2501–2510. [Google Scholar] [CrossRef]
- Fu, L.; John, L.M.; Adams, S.H.; Yu, X.X.; Tomlinson, E.; Renz, M.; Williams, P.M.; Soriano, R.; Corpuz, R.; Moffat, B.; et al. Fibroblast Growth Factor 19 Increases Metabolic Rate and Reverses Dietary and Leptin-Deficient Diabetes. Endocrinology 2004, 145, 2594–2603. [Google Scholar] [CrossRef]
- Ryan, K.K.; Kohli, R.; Gutierrez-Aguilar, R.; Gaitonde, S.G.; Woods, S.C.; Seeley, R.J. Fibroblast Growth Factor-19 Action in the Brain Reduces Food Intake and Body Weight and Improves Glucose Tolerance in Male Rats. Endocrinology 2013, 154, 9–15. [Google Scholar] [CrossRef]
- Attia, S.L.; Softic, S.; Mouzaki, M. Evolving Role for Pharmacotherapy in NAFLD/NASH. Clin. Transl. Sci. 2021, 14, 11–19. [Google Scholar] [CrossRef]
- Harrison, S.A.; Rinella, M.E.; Abdelmalek, M.F.; Trotter, J.F.; Paredes, A.H.; Arnold, H.L.; Kugelmas, M.; Bashir, M.R.; Jaros, M.J.; Ling, L.; et al. NGM282 for treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2018, 391, 1174–1185. [Google Scholar] [CrossRef]
- Harrison, S.A.; Abdelmalek, M.F.; Neff, G.; Gunn, N.; Guy, C.D.; Alkhouri, N.; Bashir, M.R.; Freilich, B.; Kohli, A.; Khazanchi, A.; et al. Aldafermin in patients with non-alcoholic steatohepatitis (ALPINE 2/3): A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Gastroenterol. Hepatol. 2022, 7, 603–616. [Google Scholar] [CrossRef]
- Mayo, M.J.; Wigg, A.J.; Leggett, B.A.; Arnold, H.; Thompson, A.J.; Weltman, M.; Carey, E.J.; Muir, A.J.; Ling, L.; Rossi, S.J.; et al. NGM282 for Treatment of Patients With Primary Biliary Cholangitis: A Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial. Hepatol. Commun. 2018, 2, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.; Cline, D.L.; Glavas, M.M.; Covey, S.D.; Kieffer, T.J. Tissue-Specific Effects of Leptin on Glucose and Lipid Metabolism. Endocr. Rev. 2020, 42, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.-Y.; Cheng, K.K.-Y.; Hoo, R.L.-C.; Siu, P.M.-F.; Yau, S.-Y. The Novel Perspectives of Adipokines on Brain Health. Int. J. Mol. Sci. 2019, 20, 5638. [Google Scholar] [CrossRef] [PubMed]
- Papathanasiou, A.E.; Nolen-Doerr, E.; Farr, O.M.; Mantzoros, C.S. GEOFFREY HARRIS PRIZE LECTURE 2018: Novel pathways regulating neuroendocrine function, energy homeostasis and metabolism in humans. Eur. J. Endocrinol. 2019, 180, R59–R71. [Google Scholar] [CrossRef]
- Schlögl, H.; Müller, K.; Horstmann, A.; Miehle, K.; Püschel, J.; Villringer, A.; Pleger, B.; Stumvoll, M.; Fasshauer, M. Leptin Substitution in Patients With Lipodystrophy: Neural Correlates for Long-term Success in the Normalization of Eating Behavior. Diabetes 2016, 65, 2179–2186. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Magkos, F.; Mantzoros, C.S.; Kang, E.S. Effects of leptin and adiponectin on pancreatic β-cell function. Metabolism 2011, 60, 1664–1672. [Google Scholar] [CrossRef]
- Montague, C.T.; Farooqi, I.S.; Whitehead, J.P.; Soos, M.A.; Rau, H.; Wareham, N.J.; Sewter, C.P.; Digby, J.E.; Mohammed, S.N.; Hurst, J.A.; et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997, 387, 903–908. [Google Scholar] [CrossRef]
- Wabitsch, M.; Funcke, J.-B.; Lennerz, B.; Kuhnle-Krahl, U.; Lahr, G.; Debatin, K.-M.; Vatter, P.; Gierschik, P.; Moepps, B.; Fischer-Posovszky, P. Biologically Inactive Leptin and Early-Onset Extreme Obesity. N. Engl. J. Med. 2015, 372, 48–54. [Google Scholar] [CrossRef]
- Gibson, W.T.; Farooqi, I.S.; Moreau, M.; DePaoli, A.M.; Lawrence, E.; O’rahilly, S.; Trussell, R.A. Congenital Leptin Deficiency Due to Homozygosity for the ?133G Mutation: Report of Another Case and Evaluation of Response to Four Years of Leptin Therapy. J. Clin. Endocrinol. Metab. 2004, 89, 4821–4826. [Google Scholar] [CrossRef]
- Paz-Filho, G.; Mastronardi, C.A.; Licinio, J. Leptin treatment: Facts and expectations. Metabolism 2015, 64, 146–156. [Google Scholar] [CrossRef]
- Lin, S.; Thomas, T.C.; Storlien, L.H.; Huang, X.F. Development of high fat diet-induced obesity and leptin resistance in C57Bl/6J mice. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 2000, 24, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.G.; Cowley, M.A.; Münzberg, H. Mechanisms of Leptin Action and Leptin Resistance. Annu. Rev. Physiol. 2008, 70, 537–556. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, A.G.; Crujeiras, A.B.; Casanueva, F.F.; Carreira, M.C. Leptin, Obesity, and Leptin Resistance: Where Are We 25 Years Later? Nutrients 2019, 11, 2704. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.G., Jr.; Heymsfield, S.B.; Haft, C.; Kahn, B.B.; Laughlin, M.; Leibel, R.L.; Tschöp, M.H.; Yanovski, J.A. Defining Clinical Leptin Resistance—Challenges and Opportunities. Cell Metab. 2012, 15, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Heymsfield, S.B.; Greenberg, A.S.; Fujioka, K.; Dixon, R.M.; Kushner, R.; Hunt, T.; Lubina, J.A.; Patane, J.; Self, B.; Hunt, P.; et al. Recombinant Leptin for Weight Loss in Obese and Lean Adults: A Randomized, Controlled, Dose-Escalation Trial. JAMA 1999, 282, 1568–1575. [Google Scholar] [CrossRef]
- Seeley, R.J.; van Dijk, G.; Campfield, L.A.; Smith, F.J.; Burn, P.; Nelligan, J.A.; Bell, S.M.; Baskin, D.G.; Woods, S.C.; Schwartz, M.W. Intraventricular Leptin Reduces Food Intake and Body Weight of Lean Rats but Not Obese Zucker Rats. Horm. Metab. Res. Horm. Stoffwechs. Horm. Metab. 1996, 28, 664–668. [Google Scholar] [CrossRef]
- Hukshorn, C.J.; Saris, W.H.M.; Westerterp-Plantenga, M.S.; Farid, A.R.; Smith, F.J.; Campfield, L.A. Weekly Subcutaneous Pegylated Recombinant Native Human Leptin (PEG-OB) Administration in Obese Men. J. Clin. Endocrinol. Metab. 2000, 85, 4003–4009. [Google Scholar] [CrossRef]
- Zelissen, P.M.J.; Stenlof, K.; Lean, M.E.J.; Fogteloo, J.; Keulen, E.T.P.; Wilding, J.; Finer, N.; Rossner, S.; Lawrence, E.; Fletcher, C.; et al. Effect of three treatment schedules of recombinant methionyl human leptin on body weight in obese adults: A randomized, placebo-controlled trial. Diabetes Obes. Metab. 2005, 7, 755–761. [Google Scholar] [CrossRef]
- Moon, H.-S.; Matarese, G.; Brennan, A.M.; Chamberland, J.P.; Liu, X.; Fiorenza, C.G.; Mylvaganam, G.H.; Abanni, L.; Carbone, F.; Williams, C.J.; et al. Efficacy of Metreleptin in Obese Patients With Type 2 Diabetes: Cellular and Molecular Pathways Underlying Leptin Tolerance. Diabetes 2011, 60, 1647–1656. [Google Scholar] [CrossRef]
- Chan, J.L.; Koda, J.; Heilig, J.S.; Cochran, E.K.; Gorden, P.; Oral, E.A.; Brown, R.J. Immunogenicity associated with metreleptin treatment in patients with obesity or lipodystrophy. Clin. Endocrinol. 2016, 85, 137–149. [Google Scholar] [CrossRef]
- Brown, R.J.; Araujo-Vilar, D.; Cheung, P.T.; Dunger, D.; Garg, A.; Jack, M.; Mungai, L.; Oral, E.A.; Patni, N.; Rother, K.I.; et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 4500–4511. [Google Scholar] [CrossRef] [PubMed]
- Diker-Cohen, T.; Cochran, E.; Gorden, P.; Brown, R.J. Partial and Generalized Lipodystrophy: Comparison of Baseline Characteristics and Response to Metreleptin. J. Clin. Endocrinol. Metab. 2015, 100, 1802–1810. [Google Scholar] [CrossRef] [PubMed]
- Haque, W.A.; Shimomura, I.; Matsuzawa, Y.; Garg, A. Serum Adiponectin and Leptin Levels in Patients with Lipodystrophies. J. Clin. Endocrinol. Metab. 2002, 87, 2395. [Google Scholar] [CrossRef] [PubMed]
- McDuffie, J.R.; Riggs, P.A.; Calis, K.A.; Freedman, R.J.; Oral, E.A.; DePaoli, A.M.; Yanovski, J.A. Effects of Exogenous Leptin on Satiety and Satiation in Patients with Lipodystrophy and Leptin Insufficiency. J. Clin. Endocrinol. Metab. 2004, 89, 4258–4263. [Google Scholar] [CrossRef] [PubMed]
- Ebihara, K.; Kusakabe, T.; Hirata, M.; Masuzaki, H.; Miyanaga, F.; Kobayashi, N.; Tanaka, T.; Chusho, H.; Miyazawa, T.; Hayashi, T.; et al. Efficacy and Safety of Leptin-Replacement Therapy and Possible Mechanisms of Leptin Actions in Patients with Generalized Lipodystrophy. J. Clin. Endocrinol. Metab. 2007, 92, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Oral, E.A.; Ruiz, E.; Andewelt, A.; Sebring, N.; Wagner, A.J.; DePaoli, A.M.; Gorden, P. Effect of Leptin Replacement on Pituitary Hormone Regulation in Patients with Severe Lipodystrophy. J. Clin. Endocrinol. Metab. 2002, 87, 3110–3117. [Google Scholar] [CrossRef]
- Püschel, J.; Miehle, K.; Müller, K.; Villringer, A.; Stumvoll, M.; Fasshauer, M.; Schlögl, H. Beneficial effects of leptin substitution on impaired eating behavior in lipodystrophy are sustained beyond 150 weeks of treatment. Cytokine 2019, 113, 400–404. [Google Scholar] [CrossRef]
- Würfel, M.; Breitfeld, J.; Gebhard, C.; Scholz, M.; Baber, R.; Riedel-Heller, S.G.; Blüher, M.; Stumvoll, M.; Kovacs, P.; Tönjes, A. Interplay between adipose tissue secreted proteins, eating behavior and obesity. Eur. J. Nutr. 2021, 61, 885–899. [Google Scholar] [CrossRef]
- Kaszubska, W.; Falls, H.; Schaefer, V.G.; Haasch, D.; Frost, L.; Hessler, P.; Kroeger, P.E.; White, D.W.; Jirousek, M.R.; Trevillyan, J.M. Protein tyrosine phosphatase 1B negatively regulates leptin signaling in a hypothalamic cell line. Mol. Cell. Endocrinol. 2002, 195, 109–118. [Google Scholar] [CrossRef]
- Reed, A.S.; Unger, E.K.; Olofsson, L.E.; Piper, M.L., Jr.; Myers, M.G.; Xu, A.W. Functional Role of Suppressor of Cytokine Signaling 3 Upregulation in Hypothalamic Leptin Resistance and Long-Term Energy Homeostasis. Diabetes 2010, 59, 894–906. [Google Scholar] [CrossRef]
- Lee, J.; Liu, J.; Feng, X.; Hernández, M.A.S.; Mucka, P.; Ibi, D.; Choi, J.W.; Ozcan, U. Withaferin A is a leptin sensitizer with strong antidiabetic properties in mice. Nat. Med. 2016, 22, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lee, J.; Hernandez, M.A.S.; Mazitschek, R.; Ozcan, U. Treatment of Obesity with Celastrol. Cell 2015, 161, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhu, Y.; Schultz, R.D.; Li, N.; He, Z.; Zhang, Z.; Caron, A.; Zhu, Q.; Sun, K.; Xiong, W.; et al. Partial Leptin Reduction as an Insulin Sensitization and Weight Loss Strategy. Cell Metab. 2019, 30, 706–719.e6. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Kihara, S.; Ouchi, N.; Nishida, M.; Arita, Y.; Kumada, M.; Ohashi, K.; Sakai, N.; Shimomura, I.; Kobayashi, H.; et al. Adiponectin Reduces Atherosclerosis in Apolipoprotein E-Deficient Mice. Circulation 2002, 106, 2767–2770. [Google Scholar] [CrossRef]
- Matsuda, M.; Shimomura, I.; Sata, M.; Arita, Y.; Nishida, M.; Maeda, N.; Kumada, M.; Okamoto, Y.; Nagaretani, H.; Nishizawa, H.; et al. Role of Adiponectin in Preventing Vascular Stenosis: The Missing Link of Adipo-Vascular Axis. J. Biol. Chem. 2002, 277, 37487–37491. [Google Scholar] [CrossRef]
- Achari, A.E.; Jain, S.K. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int. J. Mol. Sci. 2017, 18, 1321. [Google Scholar] [CrossRef]
- Turer, A.T.; Scherer, P.E. Adiponectin: Mechanistic insights and clinical implications. Diabetologia 2012, 55, 2319–2326. [Google Scholar] [CrossRef]
- Li, X.; Zhang, D.; Vatner, D.F.; Goedeke, L.; Hirabara, S.M.; Zhang, Y.; Perry, R.J.; Shulman, G.I. Mechanisms by which adiponectin reverses high fat diet-induced insulin resistance in mice. Proc. Natl. Acad. Sci. USA 2020, 117, 32584–32593. [Google Scholar] [CrossRef]
- Padmalayam, I.; Suto, M. Role of Adiponectin in the Metabolic Syndrome: Current Perspectives on Its Modulation as a Treatment Strategy. Curr. Pharm. Des. 2013, 19, 5755–5763. [Google Scholar] [CrossRef]
- Pita, J.; Panadero, A.; Soriano-Guillén, L.; Rodríguez, E.; Rovira, A. The insulin sensitizing effects of PPAR-γ agonist are associated to changes in adiponectin index and adiponectin receptors in Zucker fatty rats. Regul. Pept. 2012, 174, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Hoehn, K.; Lawrence, R.T.; Sawbridge, L.; Talbot, N.A.; Tomsig, J.L.; Turner, N.; Cooney, G.J.; Whitehead, J.; Kraegen, E.W.; et al. Overexpression of the Adiponectin Receptor AdipoR1 in Rat Skeletal Muscle Amplifies Local Insulin Sensitivity. Endocrinology 2012, 153, 5231–5246. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Li, H.; Li, W.; Wu, X.; Ding, X. Pioglitazone prevents hyperglycemia induced decrease of AdipoR1 and AdipoR2 in coronary arteries and coronary VSMCs. Mol. Cell. Endocrinol. 2012, 363, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Okada-Iwabu, M.; Yamauchi, T.; Iwabu, M.; Honma, T.; Hamagami, K.-I.; Matsuda, K.; Yamaguchi, M.; Tanabe, H.; Kimura-Someya, T.; Shirouzu, M.; et al. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature 2013, 503, 493–499. [Google Scholar] [CrossRef]
- Okada-Iwabu, M.; Iwabu, M.; Yamauchi, T.; Kadowaki, T. Drug development research for novel adiponectin receptor-targeted antidiabetic drugs contributing to healthy longevity. Diabetol. Int. 2019, 10, 237–244. [Google Scholar] [CrossRef]
- Kim, Y.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Yoon, H.E.; Shin, S.J.; Choi, B.S.; Kim, Y.-S.; Chang, Y.S.; Park, C.W. The Adiponectin Receptor Agonist AdipoRon Ameliorates Diabetic Nephropathy in a Model of Type 2 Diabetes. J. Am. Soc. Nephrol. JASN 2018, 29, 1108–1127. [Google Scholar] [CrossRef]
- Tumminia, A.; Vinciguerra, F.; Parisi, M.; Graziano, M.; Sciacca, L.; Baratta, R.; Frittitta, L. Adipose Tissue, Obesity and Adiponectin: Role in Endocrine Cancer Risk. Int. J. Mol. Sci. 2019, 20, 2863. [Google Scholar] [CrossRef]
- Liao, Q.; Long, C.; Deng, Z.; Bi, X.; Hu, J. The role of circulating adiponectin in prostate cancer: A meta-analysis. Int. J. Biol. Markers 2015, 30, e22–e31. [Google Scholar] [CrossRef]
- Stevens, V.L.; Jacobs, E.J.; Sun, J.; Gapstur, S.M. No Association of Plasma Levels of Adiponectin and c-peptide with Risk of Aggressive Prostate Cancer in the Cancer Prevention Study II Nutrition Cohort. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2014, 23, 890–892. [Google Scholar] [CrossRef]
- Heiker, J.T. Vaspin (serpinA12) in obesity, insulin resistance, and inflammation. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2014, 20, 299–306. [Google Scholar] [CrossRef]
- Blüher, M. Vaspin in obesity and diabetes: Pathophysiological and clinical significance. Endocrine 2012, 41, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Klöting, N.; Kovacs, P.; Kern, M.; Heiker, J.T.; Fasshauer, M.; Schön, M.R.; Stumvoll, M.; Beck-Sickinger, A.G.; Blüher, M. Central vaspin administration acutely reduces food intake and has sustained blood glucose-lowering effects. Diabetologia 2011, 54, 1819–1823. [Google Scholar] [CrossRef] [PubMed]
- Hida, K.; Wada, J.; Eguchi, J.; Zhang, H.; Baba, M.; Seida, A.; Hashimoto, I.; Okada, T.; Yasuhara, A.; Nakatsuka, A.; et al. Visceral adipose tissue-derived serine protease inhibitor: A unique insulin-sensitizing adipocytokine in obesity. Proc. Natl. Acad. Sci. USA 2005, 102, 10610–10615. [Google Scholar] [CrossRef]
- Breitfeld, J.; Tonjes, A.; Gast, M.-T.; Schleinitz, R.; Blüher, M.; Stumvoll, M.; Kovacs, P.; Böttcher, Y. Role of Vaspin in Human Eating Behaviour. PLoS ONE 2013, 8, e54140. [Google Scholar] [CrossRef] [PubMed]
- Heiker, J.T.; Klöting, N.; Kovacs, P.; Kuettner, E.B.; Sträter, N.; Schultz, S.; Kern, M.; Stumvoll, M.; Blüher, M.; Beck-Sickinger, A.G. Vaspin inhibits kallikrein 7 by serpin mechanism. Cell. Mol. Life Sci. 2013, 70, 2569–2583. [Google Scholar] [CrossRef]
- Chang, H.M.; Lee, H.J.; Park, H.S.; Kang, J.H.; Kim, K.S.; Song, Y.S.; Jang, Y.J. Effects of Weight Reduction on Serum Vaspin Concentrations in Obese Subjects: Modification by Insulin Resistance. Obesity 2010, 18, 2105–2110. [Google Scholar] [CrossRef]
- Martos-Moreno, G.; Kratzsch, J.; Körner, A.; Barrios, V.; Hawkins, F.; Kiess, W.; Argente, J. Serum visfatin and vaspin levels in prepubertal children: Effect of obesity and weight loss after behavior modifications on their secretion and relationship with glucose metabolism. Int. J. Obes. 2011, 35, 1355–1362. [Google Scholar] [CrossRef]
- Koiou, E.; Tziomalos, K.; Dinas, K.; Katsikis, I.; Kalaitzakis, E.; Delkos, D.; Kandaraki, E.A.; Panidis, D. The effect of weight loss and treatment with metformin on serum vaspin levels in women with polycystic ovary syndrome. Endocr. J. 2011, 58, 237–246. [Google Scholar] [CrossRef]
- Steppan, C.M.; Lazar, M.A. Resistin and obesity-associated insulin resistance. Trends Endocrinol. Metab. 2002, 13, 18–23. [Google Scholar] [CrossRef]
- Rajala, M.W.; Qi, Y.; Patel, H.R.; Takahashi, N.; Banerjee, R.; Pajvani, U.B.; Sinha, M.K.; Gingerich, R.L.; Scherer, P.E.; Ahima, R.S. Regulation of Resistin Expression and Circulating Levels in Obesity, Diabetes, and Fasting. Diabetes 2004, 53, 1671–1679. [Google Scholar] [CrossRef]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.I.Z.; Shirwany, T.A.K. Relationship of serum resistin with insulin resistance and obesity. J. Ayub Med. Coll. Abbottabad JAMC 2015, 27, 552–555. [Google Scholar] [PubMed]
- Lazar, M.A. Resistin- and Obesity-associated Metabolic Diseases. Horm. Metab. Res. Horm. Stoffwechs. Horm. Metab. 2007, 39, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Su, K.-Z.; Li, Y.-R.; Zhang, D.; Yuan, J.-H.; Zhang, C.-S.; Liu, Y.; Song, L.-M.; Lin, Q.; Li, M.-W.; Dong, J. Relation of Circulating Resistin to Insulin Resistance in Type 2 Diabetes and Obesity: A Systematic Review and Meta-Analysis. Front. Physiol. 2019, 10, 1399. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.H.; Di, J.; Jin, Y.; Zhang, Y.C.; Wu, M.; Sun, Y.; Zhang, G.Z. Resistin is expressed in human hepatocytes and induces insulin resistance. Endocrine 2008, 33, 135–143. [Google Scholar] [CrossRef]
- Silswal, N.; Singh, A.K.; Aruna, B.; Mukhopadhyay, S.; Ghosh, S.; Ehtesham, N.Z. Human resistin stimulates the pro-inflammatory cytokines TNF-α and IL-12 in macrophages by NF-κB-dependent pathway. Biochem. Biophys. Res. Commun. 2005, 334, 1092–1101. [Google Scholar] [CrossRef]
- Qatanani, M.; Szwergold, N.R.; Greaves, D.R.; Ahima, R.S.; Lazar, M.A. Macrophage-derived human resistin exacerbates adipose tissue inflammation and insulin resistance in mice. J. Clin. Investig. 2009, 119, 531–539. [Google Scholar] [CrossRef]
- Amato, M.C.; Pizzolanti, G.; Torregrossa, V.; Misiano, G.; Milano, S.; Giordano, C. Visceral Adiposity Index (VAI) Is Predictive of an Altered Adipokine Profile in Patients with Type 2 Diabetes. PLoS ONE 2014, 9, e91969. [Google Scholar] [CrossRef]
- Monzillo, L.U.; Hamdy, O.; Horton, E.S.; Ledbury, S.; Mullooly, C.; Jarema, C.; Porter, S.; Ovalle, K.; Moussa, A.; Mantzoros, C.S. Effect of Lifestyle Modification on Adipokine Levels in Obese Subjects with Insulin Resistance. Obes. Res. 2003, 11, 1048–1054. [Google Scholar] [CrossRef]
- Wolfe, B.E.; Jimerson, D.C.; Orlova, C.; Mantzoros, C.S. Effect of dieting on plasma leptin, soluble leptin receptor, adiponectin and resistin levels in healthy volunteers. Clin. Endocrinol. 2004, 61, 332–338. [Google Scholar] [CrossRef]
- Wittamer, V.; Franssen, J.D.; Vulcano, M.; Mirjolet, J.F.; Le Poul, E.; Migeotte, I.; Brézillon, S.; Tyldesley, R.; Blanpain, C.; Detheux, M.; et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J. Exp. Med. 2003, 198, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, S.; Patel, S.; Jacobe, H.; DiSepio, D.; Ghosn, C.; Malhotra, M.; Teng, M.; Duvic, M.; Chandraratna, R.A. Tazarotene-induced Gene 2 (TIG2), a Novel Retinoid-Responsive Gene in Skin. J. Investig. Dermatol. 1997, 109, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Bozaoglu, K.; Segal, D.; Shields, K.A.; Cummings, N.; Curran, J.E.; Comuzzie, A.G.; Mahaney, M.C.; Rainwater, D.L.; VandeBerg, J.L.; Maccluer, J.W.; et al. Chemerin Is Associated with Metabolic Syndrome Phenotypes in a Mexican-American Population. J. Clin. Endocrinol. Metab. 2009, 94, 3085–3088. [Google Scholar] [CrossRef]
- Chakaroun, R.; Raschpichler, M.; Klöting, N.; Oberbach, A.; Flehmig, G.; Kern, M.; Schön, M.R.; Shang, E.; Lohmann, T.; Dreßler, M.; et al. Effects of weight loss and exercise on chemerin serum concentrations and adipose tissue expression in human obesity. Metabolism 2012, 61, 706–714. [Google Scholar] [CrossRef]
- Ba, H.-J.; Xu, L.-L.; Qin, Y.-Z.; Chen, H.-S. Serum Chemerin Levels Correlate With Determinants of Metabolic Syndrome in Obese Children and Adolescents. Clin. Med. Insights Pediatr. 2019, 13, 1179556519853780. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Liu, L. Role of Chemerin/ChemR23 axis as an emerging therapeutic perspective on obesity-related vascular dysfunction. J. Transl. Med. 2022, 20, 141. [Google Scholar] [CrossRef]
- Sell, H.; Laurencikiene, J.; Taube, A.; Eckardt, K.; Cramer, A.; Horrighs, A.; Arner, P.; Eckel, J. Chemerin Is a Novel Adipocyte-Derived Factor Inducing Insulin Resistance in Primary Human Skeletal Muscle Cells. Diabetes 2009, 58, 2731–2740. [Google Scholar] [CrossRef]
- Carracedo, M.; Witasp, A.; Qureshi, A.R.; Laguna-Fernandez, A.; Brismar, T.; Stenvinkel, P.; Bäck, M. Chemerin inhibits vascular calcification through ChemR23 and is associated with lower coronary calcium in chronic kidney disease. J. Intern. Med. 2019, 286, 449–457. [Google Scholar] [CrossRef]
- Esteghamati, A.; Ghasemiesfe, M.; Mousavizadeh, M.; Noshad, S.; Nakhjavani, M. Pioglitazone and metformin are equally effective in reduction of chemerin in patients with type 2 diabetes. J. Diabetes Investig. 2014, 5, 327–332. [Google Scholar] [CrossRef]
- Helfer, G.; Ross, A.W.; Thomson, L.M.; Mayer, C.D.; Stoney, P.N.; McCaffery, P.J.; Morgan, P.J. A neuroendocrine role for chemerin in hypothalamic remodelling and photoperiodic control of energy balance. Sci. Rep. 2016, 6, 26830. [Google Scholar] [CrossRef]
- Adeghate, E. Visfatin: Structure, Function and Relation to Diabetes Mellitus and Other Dysfunctions. Curr. Med. Chem. 2008, 15, 1851–1862. [Google Scholar] [CrossRef] [PubMed]
- Sethi, J.K.; Vidal-Puig, A. Visfatin: The missing link between intra-abdominal obesity and diabetes? Trends Mol. Med. 2005, 11, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, M.M.I. Role of visfatin in obesity-induced insulin resistance. World J. Clin. Cases 2022, 10, 10840–10851. [Google Scholar] [CrossRef] [PubMed]
- Haider, D.G.; Schaller, G.; Kapiotis, S.; Maier, C.; Luger, A.; Wolzt, M. The release of the adipocytokine visfatin is regulated by glucose and insulin. Diabetologia 2006, 49, 1909–1914. [Google Scholar] [CrossRef] [PubMed]
- Bala, M.; Martin, J.; Kopp, A.; Hanses, F.; Buechler, C.; Schäffler, A. In Vivo Suppression of Visfatin by Oral Glucose Uptake: Evidence for a Novel Incretin-Like Effect by Glucagon-Like Peptide-1 (GLP-1). J. Clin. Endocrinol. Metab. 2011, 96, 2493–2501. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-P.; Chung, F.-M.; Chang, D.-M.; Tsai, J.C.-R.; Huang, H.-F.; Shin, S.-J.; Lee, Y.-J. Elevated Plasma Level of Visfatin/Pre-B Cell Colony-Enhancing Factor in Patients with Type 2 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2006, 91, 295–299. [Google Scholar] [CrossRef]
- Filippatos, T.D.; Derdemezis, C.S.; Gazi, I.F.; Lagos, K.; Kiortsis, D.N.; Tselepis, A.D.; Elisaf, M.S. Increased plasma visfatin levels in subjects with the metabolic syndrome. Eur. J. Clin. Investig. 2008, 38, 71–72. [Google Scholar] [CrossRef]
- Laudes, M.; Oberhauser, F.; Schulte, D.M.; Freude, S.; Bilkovski, R.; Mauer, J.; Rappl, G.; Abken, H.; Hahn, M.; Schulz, O.; et al. Visfatin/PBEF/Nampt and Resistin Expressions in Circulating Blood Monocytes are Differentially Related to Obesity and Type 2 Diabetes in Humans. Horm. Metab. Res. Horm. Stoffwechs. Horm. Metab. 2010, 42, 268–273. [Google Scholar] [CrossRef]
- Moschen, A.R.; Kaser, A.; Enrich, B.; Mosheimer, B.; Theurl, M.; Niederegger, H.; Tilg, H. Visfatin, an Adipocytokine with Proinflammatory and Immunomodulating Properties. J. Immunol. 2007, 178, 1748–1758. [Google Scholar] [CrossRef]
- Ezzati-Mobaser, S.; Malekpour-Dehkordi, Z.; Nourbakhsh, M.; Tavakoli-Yaraki, M.; Ahmadpour, F.; Golpour, P.; Nourbakhsh, M. The up-regulation of markers of adipose tissue fibrosis by visfatin in pre-adipocytes as well as obese children and adolescents. Cytokine 2020, 134, 155193. [Google Scholar] [CrossRef]
- Chen, D.; Zhao, M.; Mundy, G.R. Bone Morphogenetic Proteins. Growth Factors 2004, 22, 233–241. [Google Scholar] [CrossRef]
- Bragdon, B.; Moseychuk, O.; Saldanha, S.; King, D.; Julian, J.; Nohe, A. Bone Morphogenetic Proteins: A critical review. Cell. Signal. 2011, 23, 609–620. [Google Scholar] [CrossRef]
- Tseng, Y.-H.; Kokkotou, E.; Schulz, T.J.; Huang, T.L.; Winnay, J.N.; Taniguchi, C.M.; Tran, T.T.; Suzuki, R.; Espinoza, D.O.; Yamamoto, Y.; et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 2008, 454, 1000–1004. [Google Scholar] [CrossRef]
- Townsend, K.L.; Suzuki, R.; Huang, T.L.; Jing, E.; Schulz, T.J.; Lee, K.; Taniguchi, C.M.; Espinoza, D.O.; McDougall, L.E.; Zhang, H.; et al. Bone morphogenetic protein 7 (BMP7) reverses obesity and regulates appetite through a central mTOR pathway. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 2187–2196. [Google Scholar] [CrossRef]
- Ohyama, K.; Das, R.; Placzek, M. Temporal progression of hypothalamic patterning by a dual action of BMP. Development 2008, 135, 3325–3331. [Google Scholar] [CrossRef]
- Beutler, B.; Cerami, A. The Biology of Cachectin/TNF -- A Primary Mediator of the Host Response. Annu. Rev. Immunol. 1989, 7, 625–655. [Google Scholar] [CrossRef]
- Pennica, D.; Nedwin, G.E.; Hayflick, J.S.; Seeburg, P.H.; Derynck, R.; Palladino, M.A.; Kohr, W.J.; Aggarwal, B.B.; Goeddel, D.V. Human tumour necrosis factor: Precursor structure, expression and homology to lymphotoxin. Nature 1984, 312, 724–729. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Spiegelman, B.M. Tumor Necrosis Factor α: A Key Component of the Obesity-Diabetes Link. Diabetes 1994, 43, 1271–1278. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose Expression of Tumor Necrosis Factor-α: Direct Role in Obesity-Linked Insulin Resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Grunfeld, C.; Feingold, K.R. The metabolic effects of tumor necrosis factor and other cytokines. Biotherapy 1991, 3, 143–158. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef] [PubMed]
- Hamann, A.; Benecke, H.; Le Marchand-Brustel, Y.; Susulic, V.S.; Lowell, B.B.; Flier, J.S. Characterization of Insulin Resistance and NIDDM in Transgenic Mice With Reduced Brown Fat. Diabetes 1995, 44, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef] [PubMed]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-α function. Nature 1997, 389, 610–614. [Google Scholar] [CrossRef]
- Ofei, F.; Hurel, S.; Newkirk, J.; Sopwith, M.; Taylor, R. Effects of an Engineered Human Anti–TNF-α Antibody (CDP571) on Insulin Sensitivity and Glycemic Control in Patients With NIDDM. Diabetes 1996, 45, 881–885. [Google Scholar] [CrossRef]
- Wascher, T.C.; Lindeman, J.H.N.; Sourij, H.; Kooistra, T.; Pacini, G.; Roden, M. Chronic TNF-α Neutralization Does Not Improve Insulin Resistance or Endothelial Function in “Healthy” Men with Metabolic Syndrome. Mol. Med. Camb. Mass 2011, 17, 189–193. [Google Scholar] [CrossRef]
- Di Rocco, P.; Manco, M.; Rosa, G.; Greco, A.V.; Mingrone, G. Lowered Tumor Necrosis Factor Receptors, but Not Increased Insulin Sensitivity, with Infliximab. Obes. Res. 2004, 12, 734–739. [Google Scholar] [CrossRef]
- Dominguez, H.; Storgaard, H.; Rask-Madsen, C.; Hermann, T.S.; Ihlemann, N.; Nielsen, D.B.; Spohr, C.; Kober, L.; Vaag, A.; Torp-Pedersen, C. Metabolic and Vascular Effects of Tumor Necrosis Factor-α Blockade with Etanercept in Obese Patients with Type 2 Diabetes. J. Vasc. Res. 2005, 42, 517–525. [Google Scholar] [CrossRef]
- Bernstein, L.E.; Berry, J.; Kim, S.; Canavan, B.; Grinspoon, S.K. Effects of Etanercept in Patients with the Metabolic Syndrome. Arch. Intern. Med. 2006, 166, 902–908. [Google Scholar] [CrossRef]
- Lee, J.H.; Kang, Y.E.; Chang, J.Y.; Park, K.C.; Kim, H.-W.; Kim, J.T.; Kim, H.J.; Yi, H.-S.; Shong, M.; Chung, H.K.; et al. An engineered FGF21 variant, LY2405319, can prevent non-alcoholic steatohepatitis by enhancing hepatic mitochondrial function. Am. J. Transl. Res. 2016, 8, 4750–4763. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Würfel, M.; Blüher, M.; Stumvoll, M.; Ebert, T.; Kovacs, P.; Tönjes, A.; Breitfeld, J. Adipokines as Clinically Relevant Therapeutic Targets in Obesity. Biomedicines 2023, 11, 1427. https://doi.org/10.3390/biomedicines11051427
Würfel M, Blüher M, Stumvoll M, Ebert T, Kovacs P, Tönjes A, Breitfeld J. Adipokines as Clinically Relevant Therapeutic Targets in Obesity. Biomedicines. 2023; 11(5):1427. https://doi.org/10.3390/biomedicines11051427
Chicago/Turabian StyleWürfel, Marleen, Matthias Blüher, Michael Stumvoll, Thomas Ebert, Peter Kovacs, Anke Tönjes, and Jana Breitfeld. 2023. "Adipokines as Clinically Relevant Therapeutic Targets in Obesity" Biomedicines 11, no. 5: 1427. https://doi.org/10.3390/biomedicines11051427