
Combined Pulmonary Fibrosis and Emphysema: Comparative Evidence on a Complex Condition





Abstract
:1. Introduction
2. Definition
3. Epidemiology
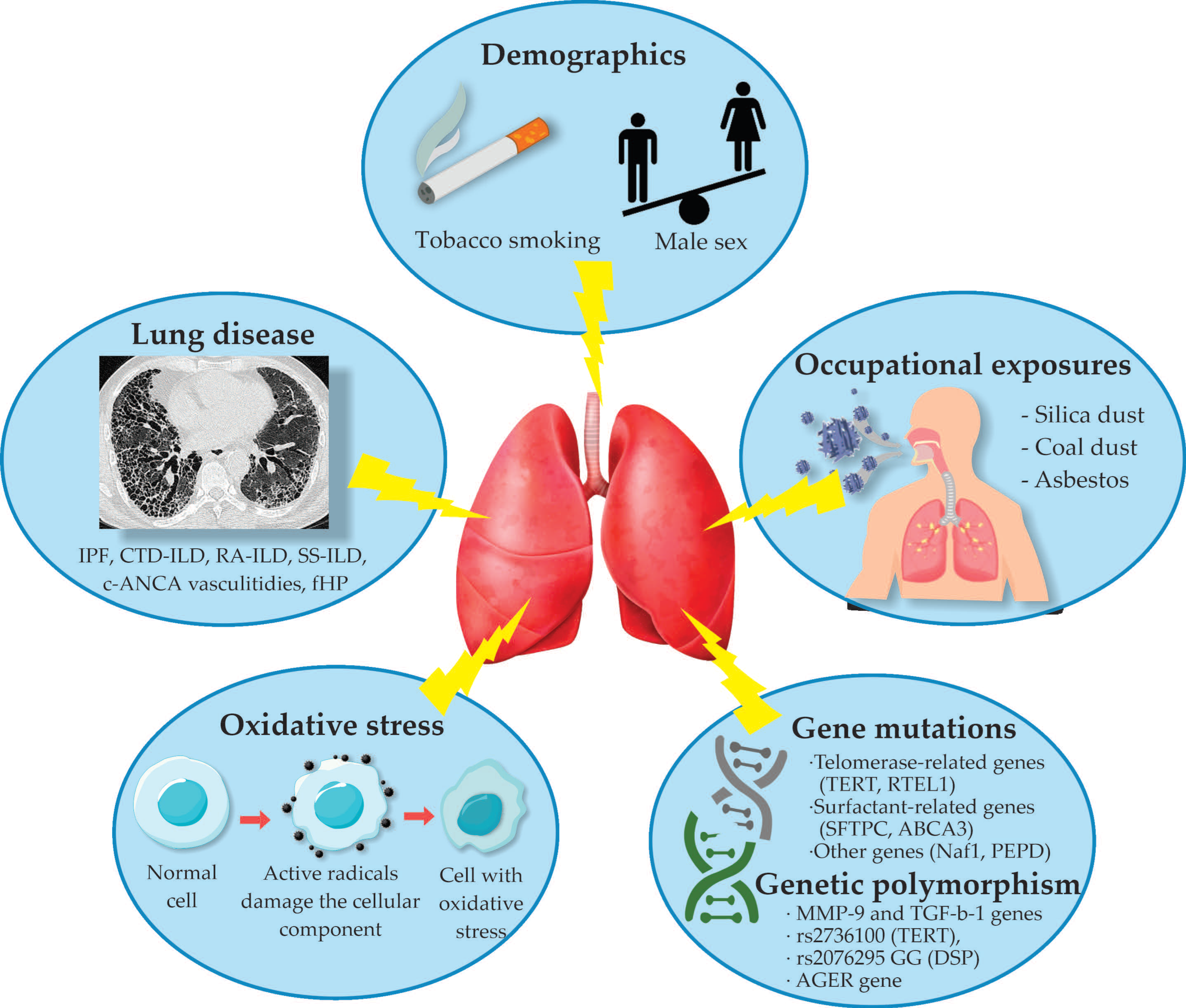
4. Etiology and Pathogenesis
5. Main Pathological Features
6. Clinical Features
7. Lung Function
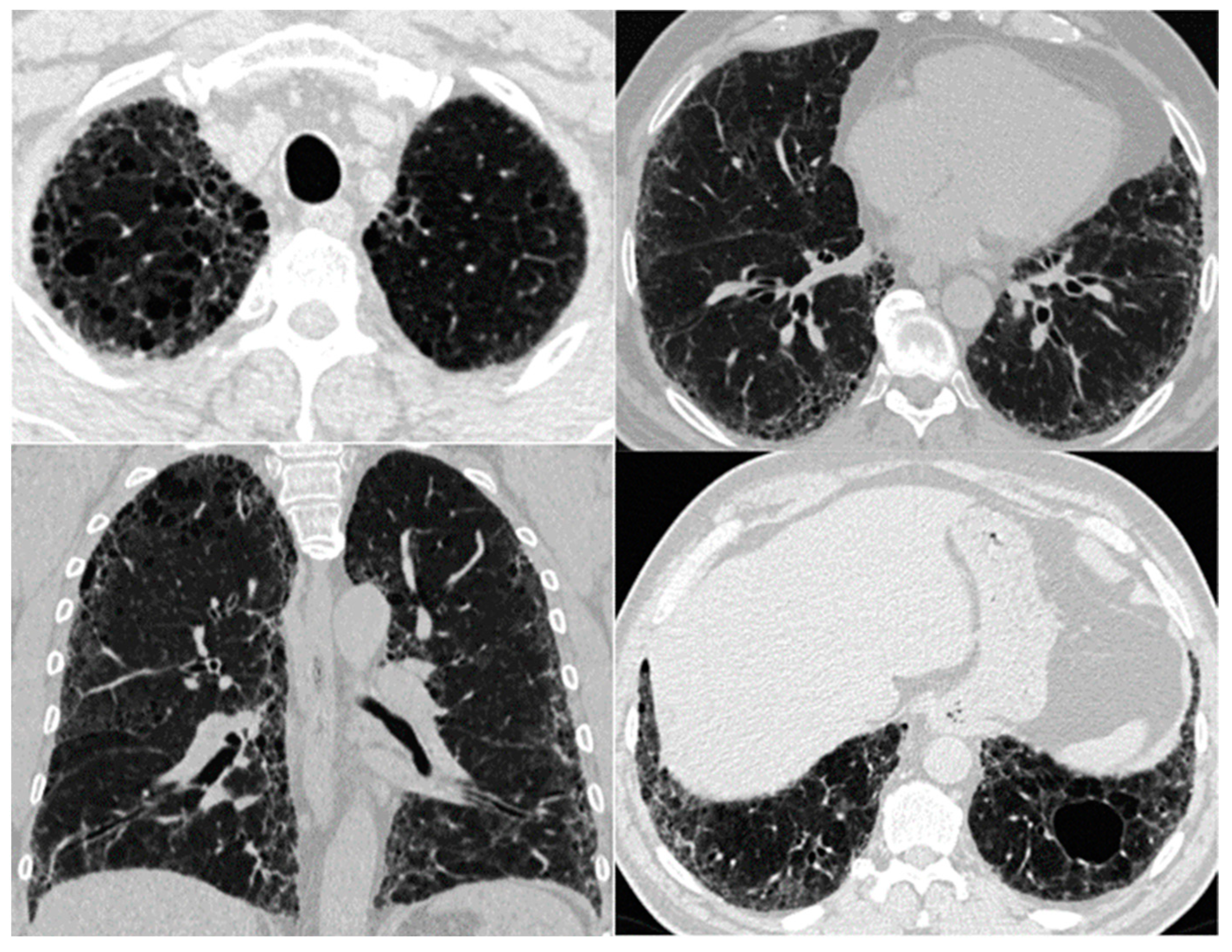
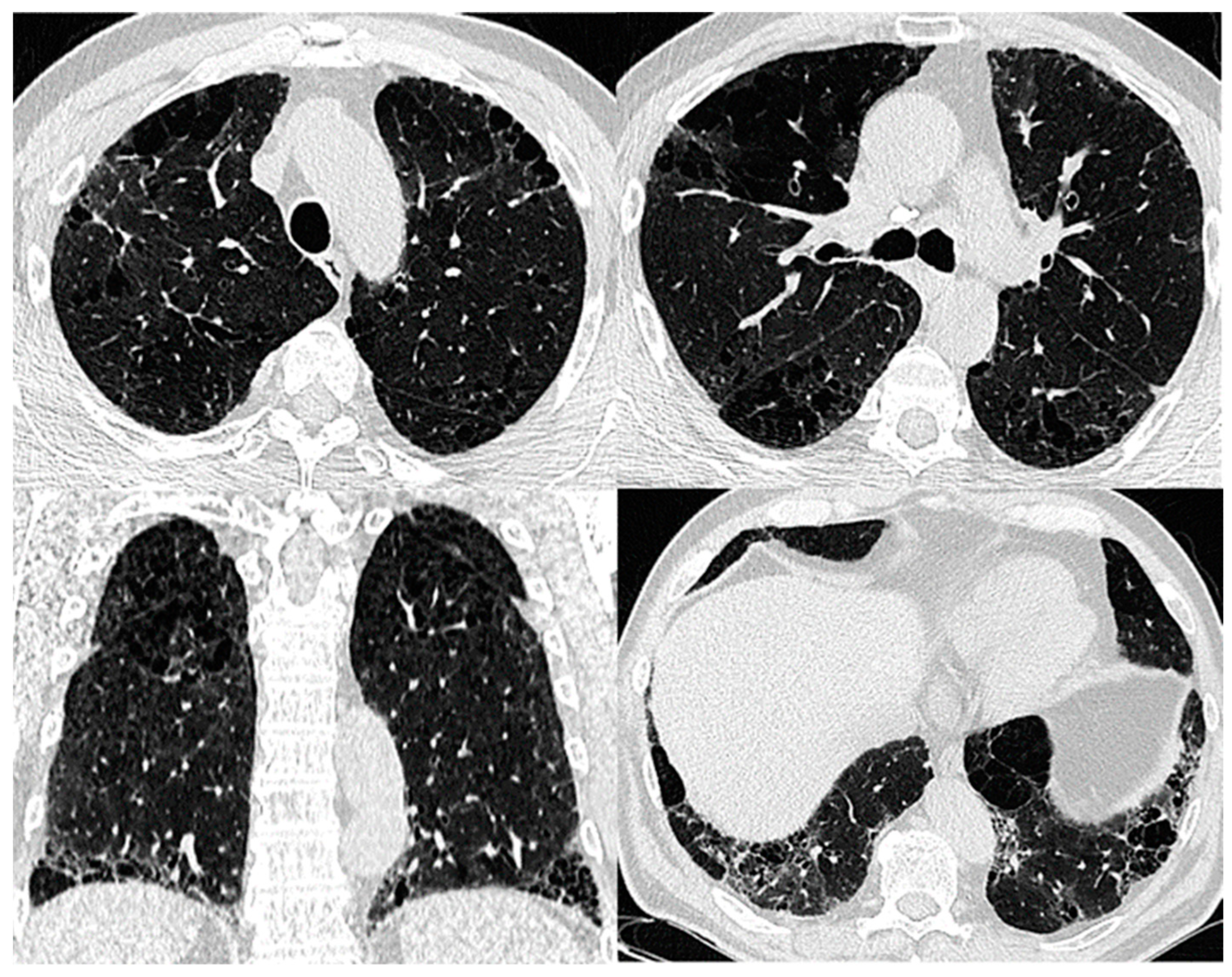
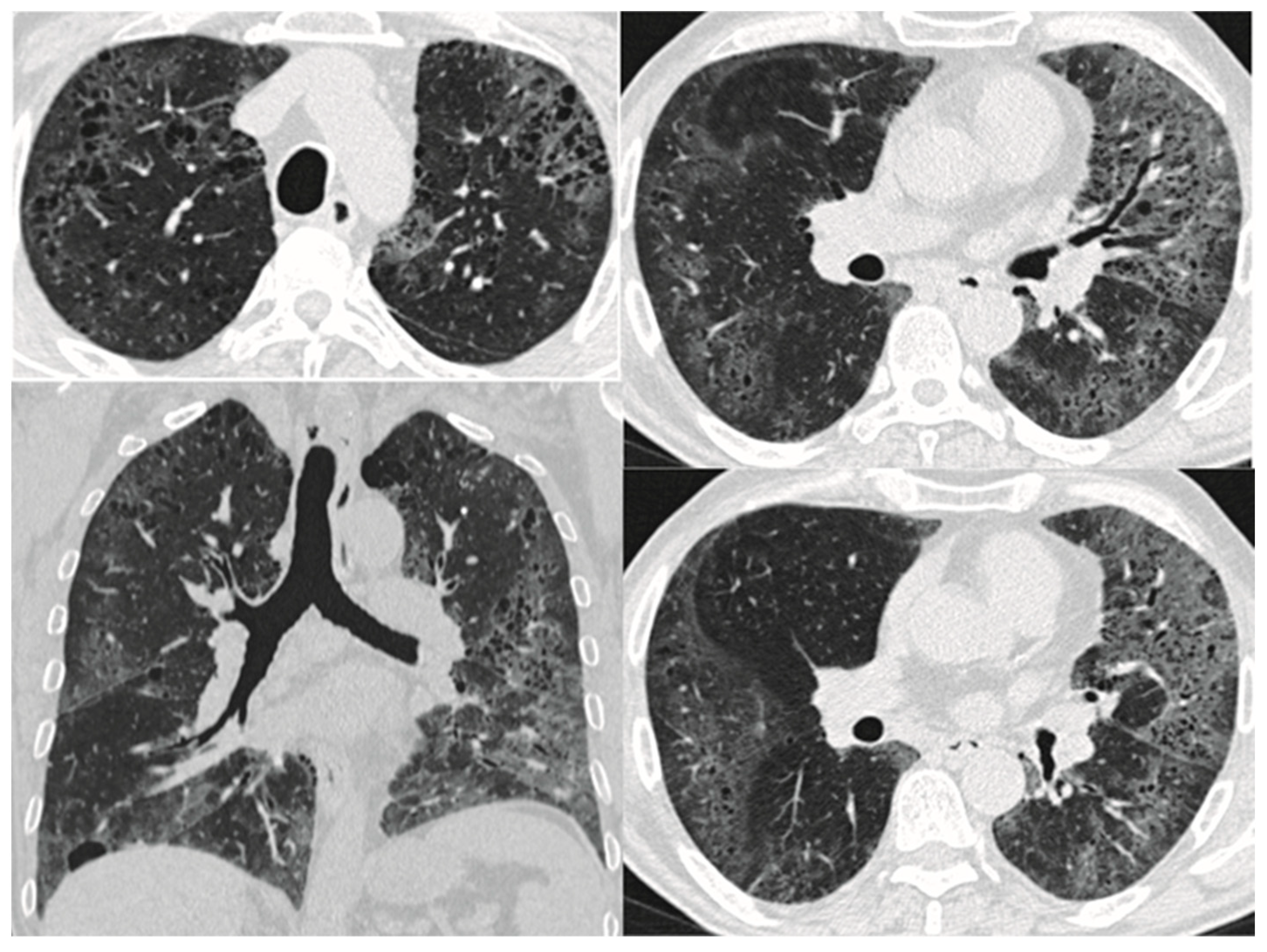
8. Imaging
9. Comorbidities
9.1. Lung Cancer
9.2. Pulmonary Hypertension
10. Treatment
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Auerbach, O.; Garfinkel, L.; Hammond, E.C. Relation of Smoking and Age to Findings in Lung Parenchyma: A Microscopic Study. Chest 1974, 65, 29–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottin, V.; Nunes, H.; Brillet, P.-Y.; Delaval, P.; Devouassaoux, G.; Tillie-Leblond, I.; Israel-Biet, D.; Court-Fortune, I.; Valeyre, D.; Cordier, J.-F. Combined pulmonary fibrosis and emphysema: A distinct underrecognised entity. Eur. Respir. J. 2005, 26, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Safiri, S.; Carson-Chahhoud, K.; Noori, M.; Nejadghaderi, S.A.; Sullman, M.J.M.; Heris, J.A.; Ansarin, K.; Mansournia, M.A.; Collins, G.S.; Kolahi, A.-A.; et al. Burden of chronic obstructive pulmonary disease and its attributable risk factors in 204 countries and territories, 1990-2019: Results from the Global Burden of Disease Study 2019. BMJ 2022, 378, e069679. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Bendstrup, E.; Dron, L.; Langley, J.; Smith, G.; Khalid, J.M.; Patel, H.; Kreuter, M. Global incidence and prevalence of idiopathic pulmonary fibrosis. Respir. Res. 2021, 22, 197. [Google Scholar] [CrossRef]
- Cottin, V.; Hansell, D.M.; Sverzellati, N.; Weycker, D.; Antoniou, K.M.; Atwood, M.; Oster, G.; Kirchgaessler, K.-U.; Collard, H.R.; Wells, A.U. Effect of Emphysema Extent on Serial Lung Function in Patients with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 1162–1171. [Google Scholar] [CrossRef]
- Cottin, V.; Selman, M.; Inoue, Y.; Wong, A.W.; Corte, T.J.; Flaherty, K.R.; Han, M.K.; Jacob, J.; Johannson, K.A.; Kitaichi, M.; et al. Syndrome of Combined Pulmonary Fibrosis and Emphysema: An Official ATS/ERS/JRS/ALAT Research Statement. Am. J. Respir. Crit. Care Med. 2022, 206, e7–e41. [Google Scholar] [CrossRef]
- Sakai, F.; Tominaga, J.; Kaga, A.; Usui, Y.; Kanazawa, M.; Ogura, T.; Yanagawa, N.; Takemura, T. Imaging Diagnosis of Interstitial Pneumonia with Emphysema (Combined Pulmonary Fibrosis and Emphysema). Pulm. Med. 2012, 2012, 816541. [Google Scholar] [CrossRef] [Green Version]
- Sangani, R.; Ghio, A.; Culp, S.; Patel, Z.; Sharma, S. Combined Pulmonary Fibrosis Emphysema: Role of Cigarette Smoking and Pulmonary Hypertension in a Rural Cohort. Int. J. Chronic Obstr. Pulm. Dis. 2021, 16, 1873–1885. [Google Scholar] [CrossRef]
- Lin, H.; Jiang, S. Combined pulmonary fibrosis and emphysema (CPFE): An entity different from emphysema or pulmonary fibrosis alone. J. Thorac. Dis. 2015, 7, 767–779. [Google Scholar]
- Kitaguchi, Y.; Fujimoto, K.; Hayashi, R.; Hanaoka, M.; Honda, T.; Kubo, K. Annual changes in pulmonary function in combined pulmonary fibrosis and emphysema: Over a 5-year follow-up. Respir. Med. 2013, 107, 1986–1992. [Google Scholar] [CrossRef] [Green Version]
- Antoniou, K.M.; Walsh, S.L.; Hansell, D.M.; Rubens, M.R.; Marten, K.; Tennant, R.; Hansel, T.; Desai, S.R.; Siafakas, N.M.; du Bois, R.M.; et al. Smoking-related emphysema is associated with idiopathic pulmonary fibrosis and rheumatoid lung. Respirology 2013, 18, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Jankowich, M.D.; Rounds, S.I. Combined Pulmonary Fibrosis and Emphysema Syndrome: A Review. Chest 2012, 141, 222–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kärkkäinen, M.; Kettunen, H.-P.; Nurmi, H.; Selander, T.; Purokivi, M.; Kaarteenaho, R. Effect of smoking and comorbidities on survival in idiopathic pulmonary fibrosis. Respir. Res. 2017, 18, 160. [Google Scholar] [CrossRef] [Green Version]
- Grubstein, A.; Bendayan, D.; Schactman, I.; Cohen, M.; Shitrit, D.; Kramer, M.R. Concomitant upper-lobe bullous emphysema, lower-lobe interstitial fibrosis and pulmonary hypertension in heavy smokers: Report of eight cases and review of the literature. Respir. Med. 2005, 99, 948–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottin, V. The impact of emphysema in pulmonary fibrosis. Eur. Respir. Rev. 2013, 22, 153–157. [Google Scholar] [CrossRef] [Green Version]
- Cottin, V.; Cordier, J.-F. Combined pulmonary fibrosis and emphysema in connective tissue disease. Curr. Opin. Pulm. Med. 2012, 18, 418–427. [Google Scholar] [CrossRef]
- Morse, D.; Rosas, I.O. Tobacco Smoke–Induced Lung Fibrosis and Emphysema. Annu. Rev. Physiol. 2014, 76, 493–513. [Google Scholar] [CrossRef]
- Ye, Q.; Huang, K.; Ding, Y.; Lou, B.; Hou, Z.; Dai, H.; Wang, C. Cigarette smoking contributes to idiopathic pulmonary fibrosis associated with emphysema. Chin. Med. J. 2014, 127, 469–474. [Google Scholar]
- Chae, K.J.; Jin, G.Y.; Jung, H.N.; Kwon, K.S.; Choi, H.; Lee, Y.C.; Chung, M.J.; Park, H.S. Differentiating Smoking-Related Interstitial Fibrosis (SRIF) from Usual Interstitial Pneumonia (UIP) with Emphysema Using CT Features Based on Pathologically Proven Cases. PLoS ONE 2016, 11, e0162231. [Google Scholar] [CrossRef] [Green Version]
- Kitaguchi, Y.; Fujimoto, K.; Hanaoka, M.; Kawakami, S.; Honda, T.; Kubo, K. Clinical characteristics of combined pulmonary fibrosis and emphysema. Respirology 2010, 15, 265–271. [Google Scholar] [CrossRef]
- Joshi, J.M.; Karkhanis, V.S. Combined pulmonary fibrosis and emphysema in a tyre industry worker. Lung India 2012, 29, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Roshan, R.; Gupta, M.; Kulshrestha, R.; Menon, B.; Chhabra, S. Combined Pulmonary Fibrosis and Emphysema in a welder. Monaldi Arch. Chest Dis. 2012, 77, 26–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cormier, Y.; Brown, M.; Worthy, S.; Racine, G.; Muller, N.L. High-resolution computed tomographic characteristics in acute farmer’s lung and in its follow-up. Eur. Respir. J. 2000, 16, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.; Song, J.W.; Yoon, H.-Y.; Cross, G.; Barnett, J.; Woo, W.L.; Adams, F.; Kokosi, M.; Devaraj, A.; Renzoni, E.; et al. Prevalence and Effects of Emphysema in Never-Smokers with Rheumatoid Arthritis Interstitial Lung Disease. Ebiomedicine 2018, 28, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Antoniou, K.M.; Margaritopoulos, G.A.; Goh, N.S.; Karagiannis, K.; Desai, S.R.; Nicholson, A.G.; Siafakas, N.M.; Coghlan, J.G.; Denton, C.P.; Hansell, D.M.; et al. Combined Pulmonary Fibrosis and Emphysema in Scleroderma-Related Lung Disease Has a Major Confounding Effect on Lung Physiology and Screening for Pulmonary Hypertension. Arthritis Rheumatol. 2016, 68, 1004–1012. [Google Scholar] [CrossRef]
- Gocho, K.; Sugino, K.; Sato, K.; Hasegawa, C.; Uekusa, T.; Homma, S. Microscopic polyangiitis preceded by combined pulmonary fibrosis and emphysema. Respir. Med. Case Rep. 2015, 15, 128–132. [Google Scholar] [CrossRef] [Green Version]
- Tzouvelekis, A.; Zacharis, G.; Oikonomou, A.; Mikroulis, D.; Margaritopoulos, G.; Koutsopoulos, A.; Antoniadis, A.; Koulelidis, A.; Steiropoulos, P.; Boglou, P.; et al. Increased incidence of autoimmune markers in patients with combined pulmonary fibrosis and emphysema. BMC Pulm. Med. 2013, 13, 31. [Google Scholar] [CrossRef] [Green Version]
- Manni, M.; Oury, T. Oxidative Stress and Pulmonary Fibrosis. In Systems Biology of Free Radicals and Antioxidants; Laher, I., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 1611–1631. [Google Scholar]
- Marginean, C.; Popescu, M.S.; Vladaia, M.; Tudorascu, D.; Pirvu, D.C.; Petrescu, F. Involvement of Oxidative Stress in COPD. Curr. Health Sci. J. 2018, 44, 48–55. [Google Scholar]
- Finicelli, M.; Digilio, F.A.; Galderisi, U.; Peluso, G. The Emerging Role of Macrophages in Chronic Obstructive Pulmonary Disease: The Potential Impact of Oxidative Stress and Extracellular Vesicle on Macrophage Polarization and Function. Antioxidants 2022, 11, 464. [Google Scholar] [CrossRef]
- Estornut, C.; Milara, J.; Bayarri, M.A.; Belhadj, N.; Cortijo, J. Targeting Oxidative Stress as a Therapeutic Approach for Idiopathic Pulmonary Fibrosis. Front. Pharmacol. 2022, 12, 794997. [Google Scholar] [CrossRef]
- Duckworth, A.; Gibbons, M.A.; Allen, R.J.; Almond, H.; Beaumont, R.N.; Wood, A.R.; Lunnon, K.; Lindsay, M.A.; Wain, L.V.; Tyrrell, J.; et al. Telomere length and risk of idiopathic pulmonary fibrosis and chronic obstructive pulmonary disease: A mendelian randomisation study. Lancet Respir. Med. 2021, 9, 285–294. [Google Scholar] [CrossRef] [PubMed]
- van Moorsel, C.H.; van Oosterhout, M.F.; Barlo, N.P.; de Jong, P.A.; van der Vis, J.J.; Ruven, H.J.; van Es, H.W.; van den Bosch, J.M.; Grutters, J.C. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am. J. Respir. Crit. Care Med. 2010, 182, 1419–1425. [Google Scholar] [CrossRef]
- Cronkhite, J.T.; Xing, C.; Raghu, G.; Chin, K.M.; Torres, F.; Rosenblatt, R.L.; Garcia, C.K. Faculty Opinions recommendation of Telomere shortening in familial and sporadic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 729–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanaoka, M.; Ito, M.; Droma, Y.; Ushiki, A.; Kitaguchi, Y.; Yasuo, M.; Kubo, K. Comparison of gene expression profiling between lung fibrotic and emphysematous tissues sampled from patients with combined pulmonary fibrosis and emphysema. Fibrogenesis Tissue Repair 2012, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, O.M.; Baldi, B.G.; Costa, A.N.; Carvalho, C.R.R. Combined pulmonary fibrosis and emphysema: An increasingly recognized condition. J. Bras. Pneumol. 2014, 40, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Liang, B.; Yang, J.; Xiao, J.; Ma, C.; Xu, S.; Lei, J.; Xu, X.; Liao, Z.; Liu, H.; et al. Association of FAM13A polymorphisms with COPD and COPD-related phenotypes in Han Chinese. Clin. Biochem. 2013, 46, 1683–1688. [Google Scholar] [CrossRef]
- Fingerlin, T.E.; Murphy, E.; Zhang, W.; Peljto, A.L.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; Cosgrove, G.P.; Lynch, D.; Groshong, S.; et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 2013, 45, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, B.D.; de Jong, K.; Lamontagne, M.; Bossé, Y.; Shrine, N.; Artigas, M.S.; Wain, L.V.; Hall, I.P.; Jackson, V.E.; COPDGene Investigators; et al. Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat. Genet. 2017, 49, 426–432. [Google Scholar] [CrossRef] [Green Version]
- Kinjo, T.; Kitaguchi, Y.; Droma, Y.; Yasuo, M.; Wada, Y.; Ueno, F.; Ota, M.; Hanaoka, M. The Gly82Ser mutation in AGER contributes to pathogenesis of pulmonary fibrosis in combined pulmonary fibrosis and emphysema (CPFE) in Japanese patients. Sci. Rep. 2020, 10, 12811. [Google Scholar] [CrossRef]
- Xu, L.; Bian, W.; Gu, X.-H.; Shen, C. Genetic polymorphism in matrix metalloproteinase-9 and transforming growth factor-β1 and susceptibility to combined pulmonary fibrosis and emphysema in a Chinese population. Kaohsiung J. Med. Sci. 2017, 33, 124–129. [Google Scholar] [CrossRef]
- Guzmán-Vargas, J.; Ambrocio-Ortiz, E.; Pérez-Rubio, G.; Ponce-Gallegos, M.A.; Hernández-Zenteno, R.D.J.; Mejía, M.; Ramírez-Venegas, A.; Buendia-Roldan, I.; Falfán-Valencia, R. Differential Genomic Profile in TERT, DSP, and FAM13A Between COPD Patients With Emphysema, IPF, and CPFE Syndrome. Front. Med. 2021, 8, 725144. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.J.; Hobbs, B.D.; Yun, J.H.; Saferali, A.; Moll, M.; Xu, Z.; Chase, R.P.; Morrow, J.; Ziniti, J.; Sciurba, F.; et al. Lung tissue shows divergent gene expression between chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Respir. Res. 2022, 23, 97. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, P.; Semenzato, U. Revealing the pathogenic and ageing-related mechanisms of the enigmatic idiopathic pulmonary fibrosis (and chronic obstructive pulmonary disease). Curr. Opin. Pulm. Med. 2022, 28, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.A. Commonalities between the pro-fibrotic mechanisms in COPD and IPF. Pulm. Pharmacol. Ther. 2012, 25, 276–280. [Google Scholar] [CrossRef]
- Beghé, B.; Cerri, S.; Fabbri, L.M.; Marchioni, A. COPD, Pulmonary Fibrosis and ILAs in Aging Smokers: The Paradox of Striking Different Responses to the Major Risk Factors. Int. J. Mol. Sci. 2021, 22, 9292. [Google Scholar] [CrossRef]
- Miravitlles, M.; Ribera, A. Understanding the impact of symptoms on the burden of COPD. Respir. Res. 2017, 18, 67. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.S.; Mira-Avendano, I.; Ryu, J.H.; Daniels, C.E. The burden of idiopathic pulmonary fibrosis: An unmet public health need. Respir. Med. 2014, 108, 955–967. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.; Goh, N.S.L.; Holland, A.E.; Khor, Y.H. Cough in Idiopathic Pulmonary Fibrosis. Front. Rehabil. Sci. 2021, 2, 751798. [Google Scholar] [CrossRef]
- Costa, C.M.; Neder, J.A.; Verrastro, C.G.; Paula-Ribeiro, M.; Ramos, R.; Ferreira, E.M.; Nery, L.E.; O’Donnell, D.E.; Pereira, C.A.; Ota-Arakaki, J. Uncovering the mechanisms of exertional dyspnoea in combined pulmonary fibrosis and emphysema. Eur. Respir. J. 2020, 55, 1901319. [Google Scholar] [CrossRef]
- Cottin, V.; Le Pavec, J.; Prévot, G.; Mal, H.; Humbert, M.; Simonneau, G.; Cordier, J.F. Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur. Respir. J. 2010, 35, 105–111. [Google Scholar] [CrossRef]
- Kusaka, K.; Morio, Y.; Kimura, Y.; Takeda, K.; Kawashima, M.; Masuda, K.; Matsui, H. Improvement of pulmonary arterial compliance by pulmonary vasodilator in pulmonary hypertension from combined pulmonary fibrosis and emphysema. Respir. Med. Case Rep. 2019, 28, 100940. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.Y.; Lee, Y.S.; Min, K.H.; Hur, G.Y.; Lee, S.Y.; Kang, K.H.; Shim, J.J. Impact and prognosis of lung cancer in patients with combined pulmonary fibrosis and emphysema. Sarcoidosis Vasc. Diffus. Lung Dis. 2020, 37, e2020020. [Google Scholar]
- Çiftci, F.; Gülpınar, B.; Atasoy, Ç.; Kayacan, O.; Saryal, S. Combined pulmonary fibrosis and emphysema: How does cohabitation affect respiratory functions? Adv. Med. Sci. 2019, 64, 285–291. [Google Scholar] [CrossRef]
- Brillet, P.; Cottin, V.; Letoumelin, P.; Landino, F.; Brauner, M.; Valeyre, D.; Cordier, J.; Nunes, H. Combined apical emphysema and basal fibrosis syndrome [emphysema/fibrosis syndrome]: CT imaging features and pulmonary function tests. Clin. Imaging 2009, 33, 332. (In French) [Google Scholar] [CrossRef]
- Sekine, Y.; Katsura, H.; Koh, E.; Hiroshima, K.; Fujisawa, T. Early detection of COPD is important for lung cancer surveillance. Eur. Respir. J. 2011, 39, 1230–1240. [Google Scholar] [CrossRef] [Green Version]
- Mimae, T.; Suzuki, K.; Tsuboi, M.; Ikeda, N.; Takamochi, K.; Aokage, K.; Shimada, Y.; Miyata, Y.; Okada, M. Severity of lung fibrosis affects early surgical outcomes of lung cancer among patients with combined pulmonary fibrosis and emphysema. Medicine 2016, 95, e4314. [Google Scholar] [CrossRef] [PubMed]
- IInomata, M.; Ikushima, S.; Awano, N.; Kondoh, K.; Satake, K.; Masuo, M.; Kusunoki, Y.; Moriya, A.; Kamiya, H.; Ando, T.; et al. An autopsy study of combined pulmonary fibrosis and emphysema: Correlations among clinical, radiological, and pathological features. BMC Pulm. Med. 2014, 14, 104. [Google Scholar] [CrossRef] [Green Version]
- Ando, K.; Sekiya, M.; Tobino, K.; Takahashi, K. Relationship Between Quantitative CT Metrics and Pulmonary Function in Combined Pulmonary Fibrosis and Emphysema. Lung 2013, 191, 585–591. [Google Scholar] [CrossRef]
- Matsuoka, S.; Yamashiro, T.; Matsushita, S.; Fujikawa, A.; Kotoku, A.; Yagihashi, K.; Kurihara, Y.; Nakajima, Y. Morphological disease progression of combined pulmonary fibrosis and emphysema: Comparison with emphysema alone and pulmonary fibrosis alone. J. Comput. Assist. Tomogr. 2015, 39, 153–159. [Google Scholar] [CrossRef]
- Wilson, D.O.; Weissfeld, J.L.; Balkan, A.; Schragin, J.G.; Fuhrman, C.R.; Fisher, S.N.; Wilson, J.; Leader, J.K.; Siegfried, J.M.; Shapiro, S.D.; et al. Association of radiographic emphysema and airflow obstruction with lung cancer. Am. J. Respir. Crit. Care Med. 2008, 178, 738–744. [Google Scholar] [CrossRef] [Green Version]
- Ballester, B.; Milara, J.; Cortijo, J. Idiopathic Pulmonary Fibrosis and Lung Cancer: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2019, 20, 593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, C.; Sun, S.-W.; Xiong, X.-Z. From COPD to Lung Cancer: Mechanisms Linking, Diagnosis, Treatment, and Prognosis. Int. J. Chronic Obstr. Pulm. Dis. 2022, 17, 2603–2621. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; Jeong, B.-H.; Chung, M.J.; Lee, K.S.; Kwon, O.J.; Chung, M.P. Risk factors and clinical characteristics of lung cancer in idiopathic pulmonary fibrosis: A retrospective cohort study. BMC Pulm. Med. 2019, 19, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, H.J.; Do, K.-H.; Lee, J.B.; Alblushi, S.; Lee, S.M. Lung Cancer in Combined Pulmonary Fibrosis and Emphysema: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0161437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Wu, W.; Chen, N.; Song, H.; Lu, T.; Yang, Z.; Wang, Z.; Zhou, J.; Liu, L. Clinical characteristics and outcomes of lung cancer patients with combined pulmonary fibrosis and emphysema: A systematic review and meta-analysis of 13 studies. J. Thorac. Dis. 2017, 9, 5322–5334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, S.W.; Park, M.S.; Kim, Y.S.; Jang, J.; Lee, J.H.; Lee, C.-T.; Chung, J.-H.; Shim, H.S.; Lee, K.W.; Kim, S.-S.; et al. Combined pulmonary fibrosis and emphysema and idiopathic pulmonary fibrosis in non-small cell lung cancer: Impact on survival and acute exacerbation. BMC Pulm. Med. 2019, 19, 177. [Google Scholar] [CrossRef] [Green Version]
- Seeger, W.; Adir, Y.; Barberà, J.A.; Champion, H.; Coghlan, J.G.; Cottin, V.; De Marco, T.; Galiè, N.; Ghio, S.; Gibbs, S.; et al. Pulmonary hypertension in chronic lung diseases. J. Am. Coll. Cardiol. 2013, 62, D109–D116. [Google Scholar] [CrossRef]
- Caminati, A.; Cassandro, R.; Harari, S.; Wener, R.R.; Bel, E.H. Pulmonary hypertension in chronic interstitial lung diseases. Eur. Respir. Rev. 2013, 22, 292–301. [Google Scholar] [CrossRef]
- Sato, T.; Tsujino, I.; Tanino, M.; Ohira, H.; Nishimura, M. Broad and heterogeneous vasculopathy in pulmonary fibrosis and emphysema with pulmonary hypertension. Respirol. Case Rep. 2013, 1, 10–13. [Google Scholar] [CrossRef]
- Mejía, M.; Carrillo, G.; Rojas-Serrano, J.; Estrada, A.; Suárez, T.; Alonso, D.; Barrientos, E.; Gaxiola, M.; Navarro, C.; Selman, M. Idiopathic pulmonary fibrosis and emphysema: Decreased survival associated with severe pulmonary arterial hypertension. Chest 2009, 136, 10–15. [Google Scholar] [CrossRef]
- 2023 Gold Report. Global Strategy for Prevention, Diagnosis and Management of COPD: 2023 Report. Available online: https://goldcopd.org/2023-gold-report-2/ (accessed on 26 May 2023).
- Dong, F.; Zhang, Y.; Chi, F.; Song, Q.; Zhang, L.; Wang, Y.; Che, C. Clinical efficacy and safety of ICS/LABA in patients with combined idiopathic pulmonary fibrosis and emphysema. Int. J. Clin. Exp. Med. 2015, 8, 8617–8625. [Google Scholar] [PubMed]
- Zhang, L.; Zhang, C.; Dong, F.; Song, Q.; Chi, F.; Liu, L.; Wang, Y.; Che, C. Combined pulmonary fibrosis and emphysema: A retrospective analysis of clinical characteristics, treatment and prognosis. BMC Pulm. Med. 2016, 16, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richeldi, L.; Du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef] [Green Version]
- Wijsenbeek, M.; Cottin, V. Spectrum of Fibrotic Lung Diseases. N. Engl. J. Med. 2020, 383, 958–968. [Google Scholar] [CrossRef]
- Nathan, S.D.; Barbera, J.A.; Gaine, S.P.; Harari, S.; Martinez, F.J.; Olschewski, H.; Olsson, K.M.; Peacock, A.J.; Pepke-Zaba, J.; Provencher, S.; et al. Pulmonary hypertension in chronic lung disease and hypoxia. Eur. Respir. J. 2019, 53, 1801914. [Google Scholar] [CrossRef] [Green Version]
- Kolb, M.; Raghu, G.; Wells, A.U.; Behr, J.; Richeldi, L.; Schinzel, B.; Quaresma, M.; Stowasser, S.; Martinez, F.J. Nintedanib plus Sildenafil in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 379, 1722–1731. [Google Scholar] [CrossRef]
- Zimmermann, G.S.; Von Wulffen, W.; Huppmann, P.; Meis, T.; Ihle, F.; Geiseler, J.; Leuchte, H.H.; Tufman, A.; Behr, J.; Neurohr, C. Haemodynamic changes in pulmonary hypertension in patients with interstitial lung disease treated with PDE-5 inhibitors. Respirology 2014, 19, 700–706. [Google Scholar] [CrossRef]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 2022, 43, 3618–3731. [Google Scholar] [CrossRef]
- Girard, N.; Marchand-Adam, S.; Naccache, J.-M.; Borie, R.; Urban, T.; Jouneau, S.; Marchand, E.; Ravel, A.-C.; Kiakouama, L.; Etienne-Mastroianni, B.; et al. Lung Cancer in Combined Pulmonary Fibrosis and Emphysema: A Series of 47 Western Patients. J. Thorac. Oncol. 2014, 9, 1162–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pulmonary Function Test Variable | CPFE | Fibrotic ILD | Emphysema |
|---|---|---|---|
| FVC | N/↓ | ↓ | N/↓ |
| FEV1 | N/↓ | ↓ | N/↓ |
| FEV1/FVC | N/↓/↑ | N/↑ | N/↓ |
| TLC | N/↓/↑ | N/↓ | ↑ |
| FRC | N/↓/↑ | N/↓ | ↑ |
| RV | N/↓/↑ | N/↓ | ↑ |
| DLCO | ↓ disproportionately | ↓ | ↓ |
| KCO | ↓↓ | N/↓ | ↓ |
| SaO2 following 6 min walk test | ↓↓ | ↓ | ↓ |
| COPD | CPFE | IPF | |
|---|---|---|---|
| Prevalence | ↑↑↑ | ↓↓ | ↓ |
| Smoking history | ↑↑↑ | ↑↑↑/− | ↑ |
| Gender predilection | male predominance | male predominance | male predominance |
| Age of clinical manifestations | 5th–6th decade | 6th–7th decade/earlier in CTD-ILD | 5th–6th decade |
| Spectrum of environmental exposures | Tobacco, air pollution | Tobacco, noxious particles, or gases other than tobacco, asbestos, silica dust, and agrochemical chemicals, organic dust | Organic dust, metal and mineral dust, wood dust, asbestos, and ambient particulate matter |
| Autoimmunity | − | −/+ | − |
| Response to oxidative stress | cellular injury and death and ECM ↓ | cellular injury and death and ECM ↓ in the upper lobes, Activation of fibroblasts, ECM ↑ in the lower lobes | Activation of fibroblasts, ECM ↑ |
| Role of epigenetic factors | |||
| ↓ | ↑ | ↑ |
| ↑ | ↑ | ↓ |
| ↓ | ↑ | ↑ |
| − | ↓ | ↓ |
| Pathology | Small airways remodeling, fibrosis, and destruction of the lung parenchyma with an upper lobe predominance | Emphysema in the upper lobes and patchy fibrosis, fibroblast foci, and honeycombing—UIP/NSIP/DIP pattern—in the lower lobes. Presence of thick-walled cysts on a UIP background | Lung parenchyma and interstitium, with a lower lobe predilection |
| Clinical features | |||
| Cough with sputum production, dyspnea later in the disease course | Dyspnea, dry cough later in the disease course | Dyspnea, dry cough later in the disease course |
| − | + | + |
| Not a typical sign | 50% | 70% |
| Lung function | Obstruction +/− Low DLCO | Normal Low DLCO | Restriction Low DLCO |
| Imaging | Centrilobular and/or bullous emphysema | Coexistence of paraseptal emphysema in the upper zones of the lungs and the UIP/NSIP/DIP in the lower zones, thick-walled cysts in the area of fibrosis | UIP pattern |
| Comorbidities | |||
| ↑ | ↑↑ | ↑ |
| ↑ | ↑↑ | ↑ |
| Treatment options | Bronchodilators +/− ICS +/− Oxygen therapy | Bronchodilators +/− ICS Antifibrotics (pirfenidone, nintedanib) Oxygen therapy | Antifibrotics (pirfenidone, nintedanib) Oxygen therapy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calaras, D.; Mathioudakis, A.G.; Lazar, Z.; Corlateanu, A. Combined Pulmonary Fibrosis and Emphysema: Comparative Evidence on a Complex Condition. Biomedicines 2023, 11, 1636. https://doi.org/10.3390/biomedicines11061636
Calaras D, Mathioudakis AG, Lazar Z, Corlateanu A. Combined Pulmonary Fibrosis and Emphysema: Comparative Evidence on a Complex Condition. Biomedicines. 2023; 11(6):1636. https://doi.org/10.3390/biomedicines11061636
Chicago/Turabian StyleCalaras, Diana, Alexander G. Mathioudakis, Zsofia Lazar, and Alexandru Corlateanu. 2023. "Combined Pulmonary Fibrosis and Emphysema: Comparative Evidence on a Complex Condition" Biomedicines 11, no. 6: 1636. https://doi.org/10.3390/biomedicines11061636