
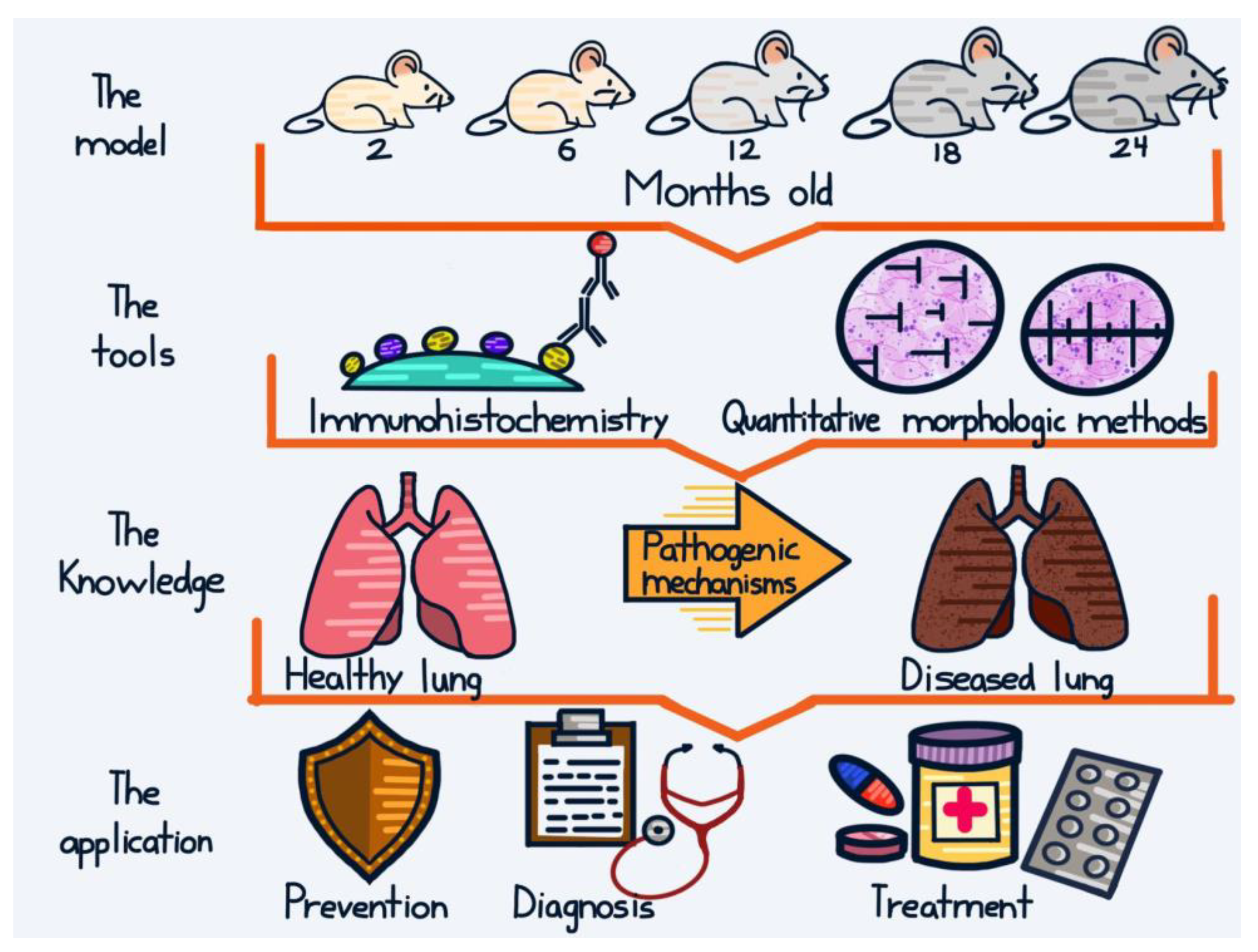
Back to the Basics: Usefulness of Naturally Aged Mouse Models and Immunohistochemical and Quantitative Morphologic Methods in Studying Mechanisms of Lung Aging and Associated Diseases

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Studies in Mechanisms of Lung Aging
3. Studies in Lung Cancer
4. Studies in COPD
5. Studies in Infectious Diseases
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 4.1B | Band 4.1 proteins |
| 8OHdG | 8-hydroxy-2-deoxyguanosine |
| ADMD | Age-dependent macrophage dysfunction |
| AECII | Alveolar epithelial type II cells |
| BALF | Bronchoalveolar lavage fluid |
| BPD | Bronchopulmonary dysplasia |
| CAP | Community-acquired pneumonia |
| CC-10 | Clara cell 10 protein |
| COPD | Chronic obstructive pulmonary disease |
| CS | Cigarette smoke |
| CSCs | Cancer stem cells |
| CSE | Cigarette smoke extract |
| CXCL1 | C-X-C motif chemokine ligand 1 |
| DAB | Diaminobenzidine |
| DI | Destructive index |
| DLBCL | Diffuse large B cell lymphoma |
| ECM | Extracellular matrix |
| ELF | Epithelial lining fluid |
| ELISA | Enzyme-linked immunosorbent assay |
| GCLM | Glutamate-cysteineligase modifier subunit |
| GEFs | Guanine nucleotide exchange factors |
| GSH | Glutathione |
| GSR | Glutathione reductase |
| HA | Hemagglutinin |
| hK14 | Human keratin 14 |
| HO-1 | Heme oxygenase-1 |
| HP1β | Heterochromatin protein 1 beta |
| HR | Homologous recombination |
| hVEGF-D | Human vascular endothelial growth factor D |
| ICR | Imprinting control region |
| IFU | Inclusion forming units |
| IGF | Insulin growth factor |
| IHC | Immunohistochemistry |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-6 |
| IPF | Idiopathic pulmonary fibrosis |
| ISA | Internal surface area |
| K10 | Keratin 10 |
| K14 | Keratin 14 |
| KC | Keratinocyte-derived chemokine |
| LR | Laminin receptor |
| LRI | Lower respiratory infections |
| MCh | Methacholine |
| MDA | Malondialdehyde |
| mH2A | Histone macro H2A |
| MIP | Macrophage inflammatory protein |
| MLI | Mean linear intercept |
| NAD | Nicotinamide adenine dinucleotide |
| NFκB | Nuclear factor kappa B |
| NOS | Nitric oxide synthase |
| Nrf2 | NF-E2-related factor 2 |
| p16 | p16INK4a |
| p38 MAPK | p38 mitogen-activated protein kinase |
| PAFr | Platelet-activating factor receptor |
| PCNA | Proliferating cell nuclear antigen |
| PD50 | Median pneumonia doses |
| PNX | Pneumonectomy |
| QM | Quantitative morphologic |
| RAC | Radial alveolar count |
| RaDR | Rosa26 direct repeat |
| RAGE | Receptor for advanced glycation end products |
| ROS | Reactive oxygen species |
| RSV | Respiratory syncytial virus |
| RT-PCR | Reverse transcription polymerase chain reaction |
| SAHF | Senescence-associated heterochromatin foci |
| SCGB | Secretoglobin |
| SIRT | Sirtuin |
| SNPs | Single nucleotide polymorphisms |
| SOD | Superoxide dismutase |
| SP-C | Surfactant protein C |
| SQCA | Squamous cell carcinoma |
| SV | Sendai virus |
| TGF | Transforming growth factor |
| TNF-α | Tumor necrosis factor-α |
| TUNEL | Terminal Transferase dUTP Nick End Labeling |
| VEGFs | Vascular endothelial growth factors |
| VLPs | Virus-like particles |
| αSMA | Alpha smooth muscle actin |
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Keshavarz, M.; Xie, K.; Schaaf, K.; Bano, D.; Ehninger, D. Targeting the “hallmarks of aging” to slow aging and treat age-related disease: Fact or fiction? Mol. Psychiatry 2023, 28, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; de Wit, S.; Učambarlić, E.; Markousis-Mavrogenis, G.; Screever, E.M.; Meijers, W.C.; de Boer, R.A.; Aboumsallem, J.P. Multifactorial diseases of the heart, kidneys, lungs, and liver and incident cancer: Epidemiology and shared mechanisms. Cancers 2023, 15, 729. [Google Scholar] [CrossRef]
- Miller, M.R. Structural and physiological age-associated changes in aging lungs. Semin. Respir. Crit. Care Med. 2010, 31, 521–527. [Google Scholar] [CrossRef]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- Corren, J. Small airways disease in asthma. Curr. Allergy Asthma Rep. 2008, 8, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Soma, T.; Nagata, M. Immunosenescence, inflammaging, and lung senescence in asthma in the elderly. Biomolecules 2022, 12, 1456. [Google Scholar] [CrossRef]
- Torrelles, J.B.; Restrepo, B.I.; Bai, Y.; Ross, C.; Schlesinger, L.S.; Turner, J. The impact of aging on the lung alveolar environment, predetermining susceptibility to respiratory infections. Front. Aging 2022, 3, 818700. [Google Scholar] [CrossRef] [PubMed]
- Meiners, S.; Eickelberg, O.; Königshoff, M. Hallmarks of the ageing lung. Eur. Respir. J. 2015, 45, 807–827. [Google Scholar] [CrossRef] [Green Version]
- Ishii, M.; Yamaguchi, Y.; Yamamoto, H.; Hanaoka, Y.; Ouchi, Y. Airspace enlargement with airway cell apoptosis in klotho mice: A model of aging lung. J. Gerontol. A Biol. Sci. Med. Sci. 2008, 63, 1289–1298. [Google Scholar] [CrossRef] [Green Version]
- Glaab, T.; Braun, A. Noninvasive measurement of pulmonary function in experimental mouse models of airway disease. Lung 2021, 199, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Cai, N.; Wu, Y.; Huang, Y. Induction of accelerated aging in a mouse model. Cells 2022, 11, 1418. [Google Scholar] [CrossRef] [PubMed]
- Kõks, S.; Dogan, S.; Tuna, B.G.; González-Navarro, H.; Potter, P.; Vandenbroucke, R.E. Mouse models of ageing and their relevance to disease. Mech. Ageing Dev. 2016, 160, 41–53. [Google Scholar] [CrossRef]
- Dutta, S.; Sengupta, P. Men and mice: Relating their ages. Life Sci. 2016, 152, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Burke, W.; Psaty, B.M. Personalized medicine in the era of genomics. JAMA 2007, 298, 1682–1684. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine (IOM). Evolution of Translational Omics: Lessons Learned and the Path Forward; The National Academies Press: Washington, DC, USA, 2012; pp. 17–18. [Google Scholar]
- D’Adamo, G.L.; Widdop, J.T.; Giles, E.M. The future is now? Clinical and translational aspects of “Omics” technologies. Immunol. Cell Biol. 2021, 99, 168–176. [Google Scholar] [CrossRef]
- Cui, M.; Cheng, C.; Zhang, L. High-throughput proteomics: A methodological mini-review. Lab. Investig. 2022, 102, 1170–1181. [Google Scholar] [CrossRef]
- Ramos-Vara, J.A. Technical aspects of immunohistochemistry. Vet. Pathol. 2005, 42, 405–426. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Vara, J.A.; Miller, M.A. When tissue antigens and antibodies get along: Revisiting the technical aspects of immunohistochemistry—The red, brown, and blue technique. Vet. Pathol. 2014, 51, 42–87. [Google Scholar] [CrossRef] [Green Version]
- Mandarim-de-Lacerda, C.A.; Del Sol, M. Tips for studies with quantitative morphology (morphometry and stereology). Int. J. Morphol. 2017, 35, 1482–1494. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.L. Practical stereology applications for the pathologist. Vet. Pathol. 2017, 54, 358–368. [Google Scholar] [CrossRef]
- Weibel, E.R.; Hsia, C.C.; Ochs, M. How much is there really? Why stereology is essential in lung morphometry. J. Appl. Physiol. (1985) 2007, 102, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Brandenberger, C.; Ochs, M.; Mühlfeld, C. Assessing particle and fiber toxicology in the respiratory system: The stereology toolbox. Part. Fibre Toxicol. 2015, 12, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoshiba, K.; Nagai, A. Chronic lung inflammation in aging mice. FEBS Lett. 2007, 581, 3512–3516. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, J.; Bu, X.; Liu, X.; Tankersley, C.G.; Wang, C.; Huang, K. Age-induced augmentation of p38 MAPK phosphorylation in mouse lung. Exp. Gerontol. 2011, 46, 694–702. [Google Scholar] [CrossRef]
- Paidi, R.K.; Jana, M.; Mishra, R.K.; Dutta, D.; Raha, S.; Pahan, K. ACE-2-interacting domain of SARS-CoV-2 (AIDS) peptide suppresses inflammation to reduce fever and protect lungs and heart in mice: Implications for COVID-19 therapy. J. Neuroimmune Pharmacol. 2021, 16, 59–70. [Google Scholar] [CrossRef]
- Paidi, R.K.; Jana, M.; Raha, S.; Mishra, R.K.; Jeong, B.; Sheinin, M.; Pahan, K. Prenol, but not vitamin C, of fruit binds to SARS-CoV-2 spike S1 to inhibit viral entry: Implications for COVID-19. J. Immunol. 2023, ji2200279. [Google Scholar] [CrossRef]
- Gomez, C.R.; Hirano, S.; Cutro, B.T.; Birjandi, S.; Baila, H.; Nomellini, V.; Kovacs, E.J. Advanced age exacerbates the pulmonary inflammatory response after lipopolysaccharide exposure. Crit. Care Med. 2007, 35, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Vegeto, E.; Cuzzocrea, S.; Crisafulli, C.; Mazzon, E.; Sala, A.; Krust, A.; Maggi, A. Estrogen receptor-alpha as a drug target candidate for preventing lung inflammation. Endocrinology 2010, 151, 174–184. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Dugan, B.; Conway, J.; Duggal, N.A. Inflammaging as a target for healthy ageing. Age Ageing 2023, 52, afac328. [Google Scholar] [CrossRef] [PubMed]
- Faniyi, A.A.; Hughes, M.J.; Scott, A.; Belchamber, K.B.R.; Sapey, E. Inflammation, ageing and diseases of the lung: Potential therapeutic strategies from shared biological pathways. Br. J. Pharmacol. 2022, 179, 1790–1807. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D. Healing and hurting: Molecular mechanisms, functions, and pathologies of cellular senescence. Mol. Cell 2009, 36, 2–14. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Narita, M.; Muñoz-Cánoves, P. Cellular senescence: Beneficial, harmful, and highly complex. FEBS J. 2023, 290, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef]
- Wang, C.; Jurk, D.; Maddick, M.; Nelson, G.; Martin-Ruiz, C.; von Zglinicki, T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 2009, 8, 311–323. [Google Scholar] [CrossRef]
- Narita, M.; Nũnez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Kreiling, J.A.; Tamamori-Adachi, M.; Sexton, A.N.; Jeyapalan, J.C.; Munoz-Najar, U.; Peterson, A.L.; Manivannan, J.; Rogers, E.S.; Pchelintsev, N.A.; Adams, P.D.; et al. Age-associated increase in heterochromatic marks in murine and primate tissues. Aging Cell 2011, 10, 292–304. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.; Kim, J.; Lee, C.Y.; Kim, H. Expression of SIRT1 and SIRT3 varies according to age in mice. Anat. Cell Biol. 2015, 48, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Mahur, P.; Muthukumaran, J.; Singh, A.K.; Jain, M. Shedding light on structure, function and regulation of human sirtuins: A comprehensive review. 3 Biotech 2023, 13, 29. [Google Scholar] [CrossRef]
- Calvi, C.L.; Podowski, M.; D’Alessio, F.R.; Metzger, S.L.; Misono, K.; Poonyagariyagorn, H.; Lopez-Mercado, A.; Ku, T.; Lauer, T.; Cheadle, C.; et al. Critical transition in tissue homeostasis accompanies murine lung senescence. PLoS ONE 2011, 6, e20712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, J.E.; Mantilla, C.B.; Pabelick, C.M.; Roden, A.C.; Sieck, G.C. Aging-related changes in respiratory system mechanics and morphometry in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L167–L176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, V.P.; Aryal, P.; Darkwah, E.K. Advanced glycation end products in health and disease. Microorganisms 2022, 10, 1848. [Google Scholar] [CrossRef] [PubMed]
- Fineschi, S.; De Cunto, G.; Facchinetti, F.; Civelli, M.; Imbimbo, B.P.; Carnini, C.; Villetti, G.; Lunghi, B.; Stochino, S.; Gibbons, D.L.; et al. Receptor for advanced glycation end products contributes to postnatal pulmonary development and adult lung maintenance program in mice. Am. J. Respir. Cell Mol. Biol. 2013, 48, 164–171. [Google Scholar] [CrossRef]
- Weibel, E.R.; Gomez, D.M. Architecture of the human lung. Use of quantitative methods establishes fundamental relations between size and number of lung structures. Science 1962, 137, 577–585. [Google Scholar] [CrossRef]
- Herring, M.J.; Avdalovic, M.V.; Quesenberry, C.L.; Putney, L.F.; Tyler, N.K.; Ventimiglia, F.F.; St George, J.A.; Hyde, D.M. Accelerated structural decrements in the aging female rhesus macaque lung compared with males. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L125–L134. [Google Scholar] [CrossRef] [Green Version]
- Hyde, D.M.; Robinson, N.E.; Gillespie, J.R.; Tyler, W.S. Morphometry of the distal air spaces in lungs of aging dogs. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1977, 43, 86–91. [Google Scholar] [CrossRef]
- Kerr, J.S.; Yu, S.Y.; Riley, D.J. Strain specific respiratory air space enlargement in aged rats. Exp. Gerontol. 1990, 25, 563–574. [Google Scholar] [CrossRef]
- Kawakami, M.; Paul, J.L.; Thurlbeck, W.M. The effect of age on lung structure in male BALB/cNNia inbred mice. Am. J. Anat. 1984, 170, 1–21. [Google Scholar] [CrossRef]
- Jaramillo-Rangel, G.; Gutiérrez-Arenas, E.; Ancer-Arellano, A.; Chávez-Briones, M.L.; Cerda-Flores, R.M.; Ortega-Martínez, M. Determination of the area and number of pulmonary alveoli through the normal aging process in CD1 mouse. In Research Aspects in Biological Science; Ozcan, G., Ed.; Book Publisher International: London, UK, 2022; Volume 3, pp. 103–109. [Google Scholar]
- Massa, C.B.; Groves, A.M.; Jaggernauth, S.U.; Laskin, D.L.; Gow, A.J. Histologic and biochemical alterations predict pulmonary mechanical dysfunction in aging mice with chronic lung inflammation. PLoS Comput. Biol. 2017, 13, e1005570. [Google Scholar] [CrossRef] [Green Version]
- Emery, J.L.; Mithal, A. The number of alveoli in the terminal respiratory unit of man during late intrauterine life and childhood. Arch. Dis. Child. 1960, 35, 544–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooney, T.P.; Thurlbeck, W.M. The radial alveolar count method of Emery and Mithal: A reappraisal 1—Postnatal lung growth. Thorax 1982, 37, 572–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooney, T.P.; Thurlbeck, W.M. The radial alveolar count method of Emery and Mithal: A reappraisal 2—Intrauterine and early postnatal lung growth. Thorax 1982, 37, 580–583. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Martínez, M.; Gutiérrez-Marín, A.; Coronado-Hernández, I.; Cerda-Flores, R.M.; Ancer-Arellano, A.; de la Garza-González, C.; Rodríguez-Flores, L.E.; Ancer-Rodríguez, J.; Jaramillo-Rangel, G. Radial alveolar count assessment in the aging. In Microscopy: Advances in Scientific Research and Education; Méndez-Vilas, A., Ed.; Formatex Research Center: Badajoz, Spain, 2014; Volume 1, pp. 344–347. [Google Scholar]
- Ortega-Martínez, M.; Gutiérrez-Dávila, V.; Niderhauser-García, A.; Cerda-Flores, R.M.; García-Juárez, J.; de-la-Garza-González, C.; Jaramillo-Rangel, G. Morphometric analysis of the non-epithelial areas of mouse bronchioles through the normal aging process. Am. J. Transl. Res. 2019, 11, 3637–3644. [Google Scholar] [PubMed]
- Pearson, M. Is the primary mechanism underlying COPD: Inflammation or ischaemia? COPD 2013, 10, 536–541. [Google Scholar] [CrossRef]
- Ortega-Martínez, M.; Gopar-Cuevas, Y.; Cerda-Flores, R.M.; Ancer-Arellano, A.; Chávez-Briones, M.L.; de la Garza-González, C.; Rodríguez-Flores, L.E.; Ancer-Rodríguez, J.; Jaramillo-Rangel, G. Morphometric analysis of the bronchiolar arterioles through the normal aging process. In Microscopy and Imagen Science; Méndez-Vilas, A., Ed.; Formatex Research Center: Badajoz, Spain, 2017; Volume 1, pp. 289–292. [Google Scholar]
- Laros, C.D.; Westermann, C.J. Dilatation, compensatory growth, or both after pneumonectomy during childhood and adolescence. A thirty-year follow-up study. J. Thorac. Cardiovasc. Surg. 1987, 93, 570–576. [Google Scholar] [CrossRef]
- Voswinckel, R.; Motejl, V.; Fehrenbach, A.; Wegmann, M.; Mehling, T.; Fehrenbach, H.; Seeger, W. Characterisation of post-pneumonectomy lung growth in adult mice. Eur. Respir. J. 2004, 24, 524–532. [Google Scholar] [CrossRef] [Green Version]
- Fehrenbach, H.; Voswinckel, R.; Michl, V.; Mehling, T.; Fehrenbach, A.; Seeger, W.; Nyengaard, J.R. Neoalveolarisation contributes to compensatory lung growth following pneumonectomy in mice. Eur. Respir. J. 2008, 31, 515–522. [Google Scholar] [CrossRef]
- Paxson, J.A.; Gruntman, A.; Parkin, C.D.; Mazan, M.R.; Davis, A.; Ingenito, E.P.; Hoffman, A.M. Age-dependent decline in mouse lung regeneration with loss of lung fibroblast clonogenicity and increased myofibroblastic differentiation. PLoS ONE 2011, 6, e23232. [Google Scholar] [CrossRef] [Green Version]
- Rawlins, E.L.; Okubo, T.; Xue, Y.; Brass, D.M.; Auten, R.L.; Hasegawa, H.; Wang, F.; Hogan, B.L. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell 2009, 4, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Watson, J.K.; Sanders, P.; Dunmore, R.; Rosignoli, G.; Julé, Y.; Rawlins, E.L.; Mustelin, T.; May, R.; Clarke, D.; Finch, D.K. Distal lung epithelial progenitor cell function declines with age. Sci. Rep. 2020, 10, 10490. [Google Scholar] [CrossRef]
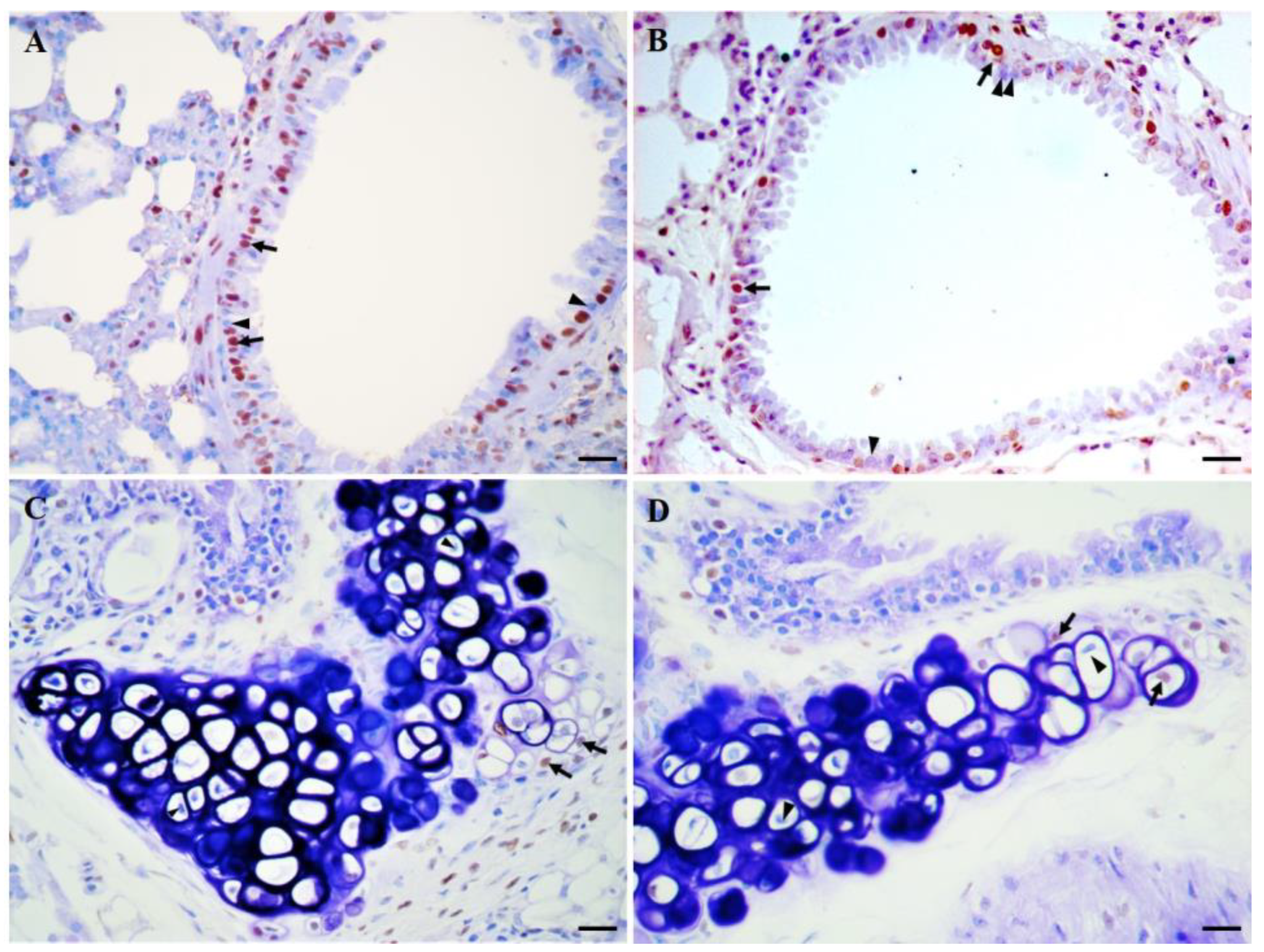
- Ortega-Martínez, M.; Rodríguez-Flores, L.E.; Ancer-Arellano, A.; Cerda-Flores, R.M.; de-la-Garza-González, C.; Ancer-Rodríguez, J.; Jaramillo-Rangel, G. Analysis of cell turnover in the bronchiolar epithelium through the normal aging process. Lung 2016, 194, 581–587. [Google Scholar] [CrossRef]
- Bernal, A.; Arranz, L. Nestin-expressing progenitor cells: Function, identity and therapeutic implications. Cell. Mol. Life Sci. 2018, 75, 2177–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muir, H. The chondrocyte, architect of cartilage. Biomechanics, structure, function and molecular biology of cartilage matrix macromolecules. Bioessays 1995, 17, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Marcu, K.B. Cartilage homeostasis in health and rheumatic diseases. Arthritis Res. Ther. 2009, 11, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umlauf, D.; Frank, S.; Pap, T.; Bertrand, J. Cartilage biology, pathology, and repair. Cell. Mol. Life Sci. 2010, 67, 4197–4211. [Google Scholar] [CrossRef]
- Henrikson, R.C.; Kaye, G.I.; Mazurkiewicz, J.E. Respiratory system. In Histology; Nieginski, E.A., Ed.; Lippincott Williams & Wilkins: New York, NY, USA, 1997; pp. 311–321. [Google Scholar]
- Ortega-Martínez, M.; Romero-Núñez, E.; Niderhauser-García, A.; de-la-Garza-González, C.; Ancer-Rodríguez, J.; Jaramillo-Rangel, G. Evidence of chondrocyte turnover in lung cartilage, with the probable participation of nestin-positive cells. Cell Biol. Int. 2013, 37, 239–241. [Google Scholar] [CrossRef]
- Ortega-Martínez, M.; de-la-Garza-González, C.; Ancer-Rodríguez, J.; Jaramillo-Rangel, G. Nestin-positive stem cells participate in chondrocyte renewal in healthy adult lung cartilage. Int. J. Morphol. 2014, 32, 151–153. [Google Scholar] [CrossRef] [Green Version]
- Gopar-Cuevas, Y.; Niderhauser-García, A.; Ancer-Arellano, A.; Miranda-Maldonado, I.C.; Chávez-Briones, M.L.; Rodríguez-Flores, L.E.; Ortega-Martínez, M.; Jaramillo-Rangel, G. Chondrocyte turnover in lung cartilage. In Cartilage Repair and Regeneration; Zorzi, A.R., Batista de Miranda, J., Eds.; InTech Publisher: Rijeka, Croacia, 2018; pp. 25–42. [Google Scholar]
- Ortega-Martínez, M.; Rodríguez-Flores, L.E.; de-la-Garza-González, C.; Ancer-Rodríguez, J.; Jaramillo-Rangel, G. Detection of a novel stem cell probably involved in normal turnover of the lung airway epithelium. J. Cell. Mol. Med. 2015, 19, 2679–2681. [Google Scholar] [CrossRef]
- Jaramillo-Rangel, G.; Chávez-Briones, M.D.; Ancer-Arellano, A.; Ortega-Martínez, M. Nestin-expressing cells in the lung: The bad and the good parts. Cells 2021, 10, 3413. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Coll, P.P.; Korc-Grodzicki, B.; Ristau, B.T.; Shahrokni, A.; Koshy, A.; Filippova, O.T.; Ali, I. Cancer prevention and screening for older adults: Part 1. Lung, colorectal, bladder, and kidney cancer. J. Am. Geriatr. Soc. 2020, 68, 2399–2406. [Google Scholar] [CrossRef]
- Szymanska, H.; Lechowska-Piskorowska, J.; Krysiak, E.; Strzalkowska, A.; Unrug-Bielawska, K.; Grygalewicz, B.; Skurzak, H.M.; Pienkowska-Grela, B.; Gajewska, M. Neoplastic and nonneoplastic lesions in aging mice of unique and common inbred strains contribution to modeling of human neoplastic diseases. Vet. Pathol. 2014, 51, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Haines, D.C.; Chattopadhyay, S.; Ward, J.M. Pathology of aging B6;129 mice. Toxicol. Pathol. 2001, 29, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.X.; Robb, V.A.; Gutmann, D.H. Protein 4.1 tumor suppressors: Getting a FERM grip on growth regulation. J. Cell Sci. 2002, 115, 3991–4000. [Google Scholar] [CrossRef] [PubMed]
- Tran, Y.K.; Bögler, O.; Gorse, K.M.; Wieland, I.; Green, M.R.; Newsham, I.F. A novel member of the NF2/ERM/4.1 superfamily with growth suppressing properties in lung cancer. Cancer Res. 1999, 59, 35–43. [Google Scholar]
- Yi, C.; McCarty, J.H.; Troutman, S.A.; Eckman, M.S.; Bronson, R.T.; Kissil, J.L. Loss of the putative tumor suppressor band 4.1B/Dal1 gene is dispensable for normal development and does not predispose to cancer. Mol. Cell. Biol. 2005, 25, 10052–10059. [Google Scholar] [CrossRef] [Green Version]
- Chu, P.G.; Weiss, L.M. Keratin expression in human tissues and neoplasms. Histopathology 2002, 40, 403–439. [Google Scholar] [CrossRef]
- Jetten, A.M.; Nervi, C.; Vollberg, T.M. Control of squamous differentiation in tracheobronchial and epidermal epithelial cells: Role of retinoids. J. Natl. Cancer Inst. Monogr. 1992, 13, 93–100. [Google Scholar]
- Dakir, E.L.H.; Feigenbaum, L.; Linnoila, R.I. Constitutive expression of human keratin 14 gene in mouse lung induces premalignant lesions and squamous differentiation. Carcinogenesis 2008, 29, 2377–2384. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.; Gao, L.; Wu, X.; Hade, E.M.; Gao, J.X.; Ding, H.; Barsky, S.H.; Otterson, G.A.; Villalona-Calero, M.A. Expression of a mutant p53 results in an age-related demographic shift in spontaneous lung tumor formation in transgenic mice. PLoS ONE 2009, 4, e5563. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Kärkkäinen, A.M.; Kotimaa, A.; Huusko, J.; Kholova, I.; Heinonen, S.E.; Stefanska, A.; Dijkstra, M.H.; Purhonen, H.; Hämäläinen, E.; Mäkinen, P.I.; et al. Vascular endothelial growth factor-D transgenic mice show enhanced blood capillary density, improved postischemic muscle regeneration, and increased susceptibility to tumor formation. Blood 2009, 113, 4468–4475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, D.R.; Rossman, K.L.; Der, C.J. Rho guanine nucleotide exchange factors: Regulators of Rho GTPase activity in development and disease. Oncogene 2014, 33, 4021–4035. [Google Scholar] [CrossRef] [Green Version]
- Ognibene, M.; Barbieri, O.; Vanni, C.; Mastracci, L.; Astigiano, S.; Emionite, L.; Salani, B.; Fedele, M.; Resaz, R.; Tenca, C.; et al. High frequency of development of B cell lymphoproliferation and diffuse large B cell lymphoma in Dbl knock-in mice. J. Mol. Med. 2011, 89, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Zhao, X.; Tang, L.; Chen, J.; Zhao, J.; Guo, M.; Chen, C.; Zhou, Y.; Xu, L. Thyroid Transcription Factor-1: Structure, expression, function and its relationship with disease. Biomed Res. Int. 2021, 2021, 9957209. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Yokoyama, S.; Pilon, A.L.; Kurotani, R. Emerging role of an immunomodulatory protein secretoglobin 3A2 in human diseases. Pharmacol. Ther. 2022, 236, 108112. [Google Scholar] [CrossRef]
- Tachihara-Yoshikawa, M.; Ishida, T.; Watanabe, K.; Sugawara, A.; Kanazawa, K.; Kanno, R.; Suzuki, T.; Niimi, T.; Kimura, S.; Munakata, M. Expression of secretoglobin3A2 (SCGB3A2) in primary pulmonary carcinomas. Fukushima J. Med. Sci. 2008, 54, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Kurotani, R.; Kumaki, N.; Naizhen, X.; Ward, J.M.; Linnoila, R.I.; Kimura, S. Secretoglobin 3A2/uteroglobin-related protein 1 is a novel marker for pulmonary carcinoma in mice and humans. Lung Cancer 2011, 71, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Doig, K.D.; Fellowes, A.P.; Fox, S.B. Homologous recombination repair deficiency: An overview for pathologists. Mod. Pathol. 2023, 36, 100049. [Google Scholar] [CrossRef]
- Sukup-Jackson, M.R.; Kiraly, O.; Kay, J.E.; Na, L.; Rowland, E.A.; Winther, K.E.; Chow, D.N.; Kimoto, T.; Matsuguchi, T.; Jonnalagadda, V.S.; et al. Rosa26-GFP direct repeat (RaDR-GFP) mice reveal tissue- and age-dependence of homologous recombination in mammals in vivo. PLoS Genet. 2014, 10, e1004299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimoto, T.; Kay, J.E.; Li, N.; Engelward, B.P. Recombinant cells in the lung increase with age via de novo recombination events and clonal expansion. Environ. Mol. Mutagen. 2017, 58, 135–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agustí, A.; Celli, B.R.; Criner, G.J.; Halpin, D.; Anzueto, A.; Barnes, P.; Bourbeau, J.; Han, M.K.; Martinez, F.J.; Montes de Oca, M.; et al. Global Initiative for Chronic Obstructive Lung Disease 2023 Report: GOLD Executive Summary. Eur. Respir. J. 2023, 61, 2300239. [Google Scholar] [CrossRef]
- Gomes, F.; Cheng, S.L. Pathophysiology, therapeutic targets, and future therapeutic alternatives in COPD: Focus on the importance of the cholinergic system. Biomolecules 2023, 13, 476. [Google Scholar] [CrossRef]
- Safiri, S.; Carson-Chahhoud, K.; Noori, M.; Nejadghaderi, S.A.; Sullman, M.J.M.; Ahmadian Heris, J.; Ansarin, K.; Mansournia, M.A.; Collins, G.S.; Kolahi, A.A.; et al. Burden of chronic obstructive pulmonary disease and its attributable risk factors in 204 countries and territories, 1990-2019: Results from the Global Burden of Disease Study 2019. BMJ 2022, 378, e069679. [Google Scholar] [CrossRef]
- Wasswa-Kintu, S.; Gan, W.Q.; Man, S.F.; Pare, P.D.; Sin, D.D. Relationship between reduced forced expiratory volume in one second and the risk of lung cancer: A systematic review and meta-analysis. Thorax 2005, 60, 570–575. [Google Scholar] [CrossRef] [Green Version]
- Forder, A.; Zhuang, R.; Souza, V.G.P.; Brockley, L.J.; Pewarchuk, M.E.; Telkar, N.; Stewart, G.L.; Benard, K.; Marshall, E.A.; Reis, P.P.; et al. Mechanisms contributing to the comorbidity of COPD and lung cancer. Int. J. Mol. Sci. 2023, 24, 2859. [Google Scholar] [CrossRef] [PubMed]
- Palmer, L.J.; Celedón, J.C.; Chapman, H.A.; Speizer, F.E.; Weiss, S.T.; Silverman, E.K. Genome-wide linkage analysis of bronchodilator responsiveness and post-bronchodilator spirometric phenotypes in chronic obstructive pulmonary disease. Hum. Mol. Genet. 2003, 12, 1199–1210. [Google Scholar] [CrossRef] [Green Version]
- Demeo, D.L.; Mariani, T.J.; Lange, C.; Srisuma, S.; Litonjua, A.A.; Celedon, J.C.; Lake, S.L.; Reilly, J.J.; Chapman, H.A.; Mecham, B.H.; et al. The SERPINE2 gene is associated with chronic obstructive pulmonary disease. Am. J. Hum. Genet. 2006, 78, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J. Oxidative stress in chronic obstructive pulmonary disease. Antioxidants 2022, 11, 965. [Google Scholar] [CrossRef]
- Suzuki, M.; Betsuyaku, T.; Ito, Y.; Nagai, K.; Nasuhara, Y.; Kaga, K.; Kondo, S.; Nishimura, M. Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2008, 39, 673–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, B.M.; Voynow, J.A.; Ghio, A.J. COPD: Balancing oxidants and antioxidants. Int. J. Chronic Obstr. Pulm. Dis. 2015, 10, 261–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, N.S.; Min, E.; Gauthier, S.; Chu, H.W.; Martin, R.; Day, B.J. Aging adversely affects the cigarette smoke-induced glutathione adaptive response in the lung. Am. J. Respir. Crit. Care Med. 2010, 182, 1114–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriyama, C.; Betsuyaku, T.; Ito, Y.; Hamamura, I.; Hata, J.; Takahashi, H.; Nasuhara, Y.; Nishimura, M. Aging enhances susceptibility to cigarette smoke-induced inflammation through bronchiolar chemokines. Am. J. Respir. Cell Mol. Biol. 2010, 42, 304–311. [Google Scholar] [CrossRef]
- Brasier, A.R. Therapeutic targets for inflammation-mediated airway remodeling in chronic lung disease. Expert Rev. Respir. Med. 2018, 12, 931–939. [Google Scholar] [CrossRef]
- Zhou, S.; Wright, J.L.; Liu, J.; Sin, D.D.; Churg, A. Aging does not enhance experimental cigarette smoke-induced COPD in the mouse. PLoS ONE 2013, 8, e71410. [Google Scholar] [CrossRef]
- Ray, D.; Yung, R. Immune senescence, epigenetics and autoimmunity. Clin. Immunol. 2018, 196, 59–63. [Google Scholar] [CrossRef]
- Murray, M.A.; Chotirmall, S.H. The impact of immunosenescence on pulmonary disease. Mediat. Inflamm. 2015, 2015, 692546. [Google Scholar] [CrossRef]
- Esme, M.; Topeli, A.; Yavuz, B.B.; Akova, M. Infections in the elderly critically-ill patients. Front. Med. 2019, 6, 118. [Google Scholar] [CrossRef] [Green Version]
- Castle, S.C.; Uyemura, K.; Fulop, T.; Makinodan, T. Host resistance and immune responses in advanced age. Clin. Geriatr. Med. 2007, 23, 463–479. [Google Scholar] [CrossRef]
- Knight, A.C.; Montgomery, S.A.; Fletcher, C.A.; Baxter, V.K. Mouse models for the study of SARS-CoV-2 infection. Comp. Med. 2021, 71, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.S.; Chen, R.E.; Alam, F.; Yildiz, S.; Case, J.B.; Uccellini, M.B.; Holtzman, M.J.; Garcia-Sastre, A.; Schotsaert, M.; Diamond, M.S. SARS-CoV-2 causes lung infection without severe disease in human ACE2 knock-in mice. J. Virol. 2022, 96, e0151121. [Google Scholar] [CrossRef] [PubMed]
- Kolopp-Sarda, M.N.; Bene, M.C.; Massin, N.; Moulin, J.J.; Faure, G.C. Immunohistological analysis of macrophages, B-cells, and T-cells in the mouse lung. Anat. Rec. 1994, 239, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Yarbro, J.R.; Emmons, R.S.; Pence, B.D. Macrophage immunometabolism and inflammaging: Roles of mitochondrial dysfunction, cellular senescence, CD38, and NAD. Immunometabolism 2020, 2, e200026. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.H.; Sun, S.C. Tumor necrosis factor receptor-associated factor regulation of nuclear factor κB and mitogen-activated protein kinase pathways. Front. Immunol. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Hinojosa, C.A.; Akula Suresh Babu, R.; Rahman, M.M.; Fernandes, G.; Boyd, A.R.; Orihuela, C.J. Elevated A20 contributes to age-dependent macrophage dysfunction in the lungs. Exp. Gerontol. 2014, 54, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Shivshankar, P.; Boyd, A.R.; Le Saux, C.J.; Yeh, I.T.; Orihuela, C.J. Cellular senescence increases expression of bacterial ligands in the lungs and is positively correlated with increased susceptibility to pneumococcal pneumonia. Aging Cell 2011, 10, 798–806. [Google Scholar] [CrossRef] [Green Version]
- Cheok, Y.Y.; Lee, C.Y.Q.; Cheong, H.C.; Looi, C.Y.; Wong, W.F. Chronic inflammatory diseases at secondary sites ensuing urogenital or pulmonary Chlamydia infections. Microorganisms 2020, 8, 127. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Sun, J.; Che, G.; Cheng, H. Systematic infection of Chlamydia pneumoniae. Clin. Lab. 2022, 68. [Google Scholar] [CrossRef]
- Gnarpe, J.; Gnarpe, H.; Gause-Nilsson, I.; Lundorg, P.; Steen, B. Seroprevalence of antibodies to Chlamydia pneumoniae in elderly people: A two-decade longitudinal and cohort difference study. Scand. J. Infect. Dis. 2000, 32, 177–179. [Google Scholar] [CrossRef]
- Little, C.S.; Bowe, A.; Lin, R.; Litsky, J.; Fogel, R.M.; Balin, B.J.; Fresa-Dillon, K.L. Age alterations in extent and severity of experimental intranasal infection with Chlamydophila pneumoniae in BALB/c mice. Infect. Immun. 2005, 73, 1723–1734. [Google Scholar] [CrossRef] [Green Version]
- Mohd-Qawiem, F.; Nawal-Amani, A.R.; Faranieyza-Afiqah, F.; Yasmin, A.R.; Arshad, S.S.; Norfitriah, M.S.; Nur-Fazila, S.H. Paramyxoviruses in rodents: A review. Open Vet. J. 2022, 12, 868–876. [Google Scholar] [CrossRef]
- Jacoby, R.O.; Bhatt, P.N.; Barthold, S.W.; Brownstein, D.G. Sendai viral pneumonia in aged BALB/c mice. Exp. Gerontol. 1994, 29, 89–100. [Google Scholar] [CrossRef]
- Binns, E.; Tuckerman, J.; Licciardi, P.V.; Wurzel, D. Respiratory syncytial virus, recurrent wheeze and asthma: A narrative review of pathophysiology, prevention and future directions. J. Paediatr. Child Health 2022, 58, 1741–1746. [Google Scholar] [CrossRef]
- Collins, R.A.; Gualano, R.C.; Zosky, G.R.; Chiappetta, C.L.; Turner, D.J.; Colasurdo, G.N.; Hantos, Z.; Sly, P.D. Lack of long-term effects of respiratory syncytial virus infection on airway function in mice. Respir. Physiol. Neurobiol. 2007, 156, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Giles, B.M.; Bissel, S.J.; Craigo, J.K.; Dealmeida, D.R.; Wiley, C.A.; Tumpey, T.M.; Ross, T.M. Elicitation of anti-1918 influenza virus immunity early in life prevents morbidity and lower levels of lung infection by 2009 pandemic H1N1 influenza virus in aged mice. J. Virol. 2012, 86, 1500–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheinin, M.; Jeong, B.; Paidi, R.K.; Pahan, K. Regression of lung cancer in mice by intranasal administration of SARS-CoV-2 Spike S1. Cancers 2022, 14, 5648. [Google Scholar] [CrossRef] [PubMed]
- Rydell-Törmänen, K.; Johnson, J.R. The applicability of mouse models to the study of human disease. Methods Mol. Biol. 2019, 1940, 3–22. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaramillo-Rangel, G.; Chávez-Briones, M.-d.-L.; Ancer-Arellano, A.; Miranda-Maldonado, I.; Ortega-Martínez, M. Back to the Basics: Usefulness of Naturally Aged Mouse Models and Immunohistochemical and Quantitative Morphologic Methods in Studying Mechanisms of Lung Aging and Associated Diseases. Biomedicines 2023, 11, 2075. https://doi.org/10.3390/biomedicines11072075
Jaramillo-Rangel G, Chávez-Briones M-d-L, Ancer-Arellano A, Miranda-Maldonado I, Ortega-Martínez M. Back to the Basics: Usefulness of Naturally Aged Mouse Models and Immunohistochemical and Quantitative Morphologic Methods in Studying Mechanisms of Lung Aging and Associated Diseases. Biomedicines. 2023; 11(7):2075. https://doi.org/10.3390/biomedicines11072075
Chicago/Turabian StyleJaramillo-Rangel, Gilberto, María-de-Lourdes Chávez-Briones, Adriana Ancer-Arellano, Ivett Miranda-Maldonado, and Marta Ortega-Martínez. 2023. "Back to the Basics: Usefulness of Naturally Aged Mouse Models and Immunohistochemical and Quantitative Morphologic Methods in Studying Mechanisms of Lung Aging and Associated Diseases" Biomedicines 11, no. 7: 2075. https://doi.org/10.3390/biomedicines11072075