
The Impact of Muscarinic Antagonism on Psychosis-Relevant Behaviors and Striatal [11C] Raclopride Binding in Tau Mouse Models of Alzheimer’s Disease
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Pharmacology
2.3. Locomotor Activity/Open Field
2.4. Acoustic Startle and Sensory Motor Gating
2.5. Bulk RNA-Seq
2.6. MicroPET
2.6.1. Image Acquisition
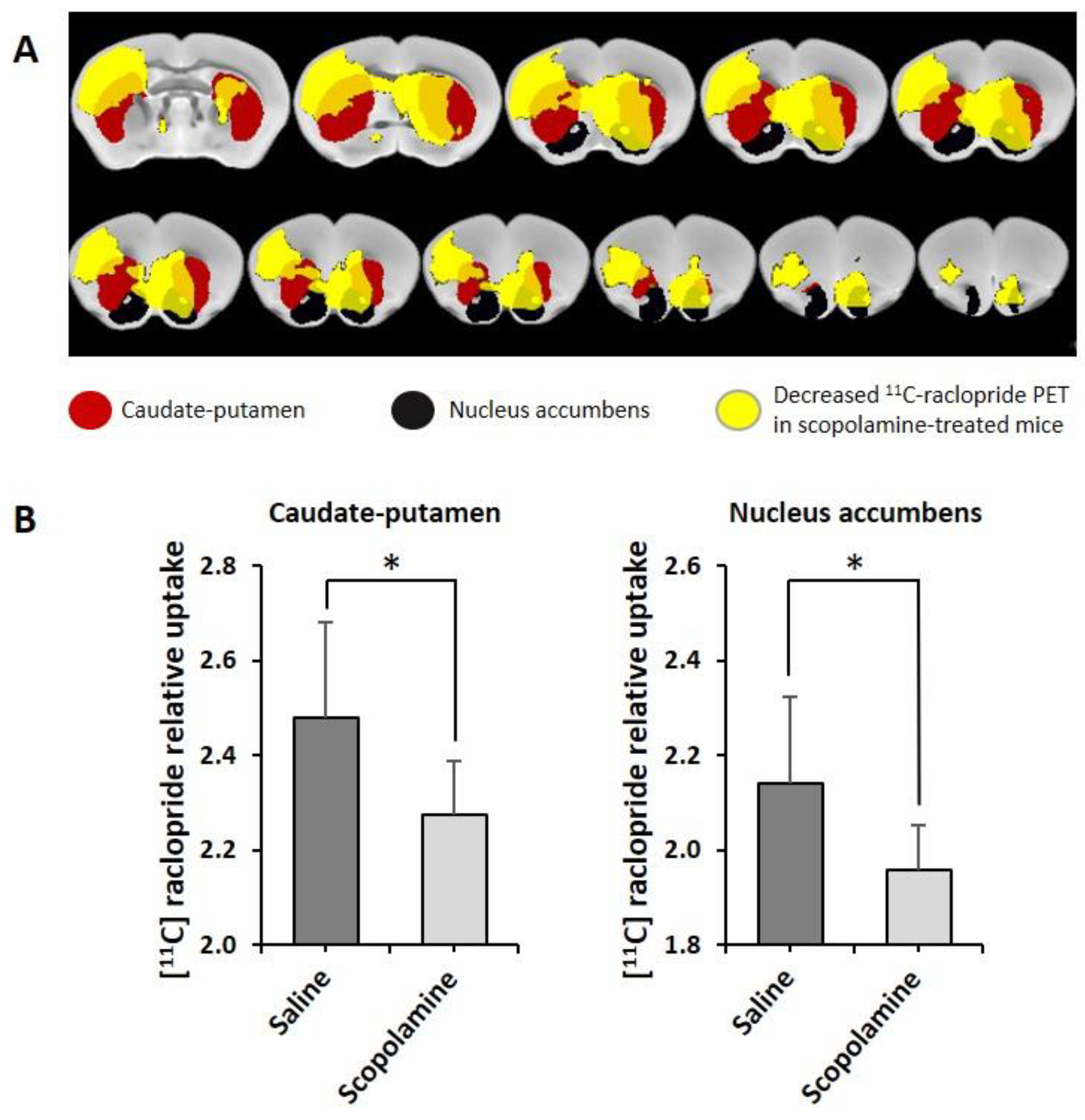
2.6.2. Image Analysis
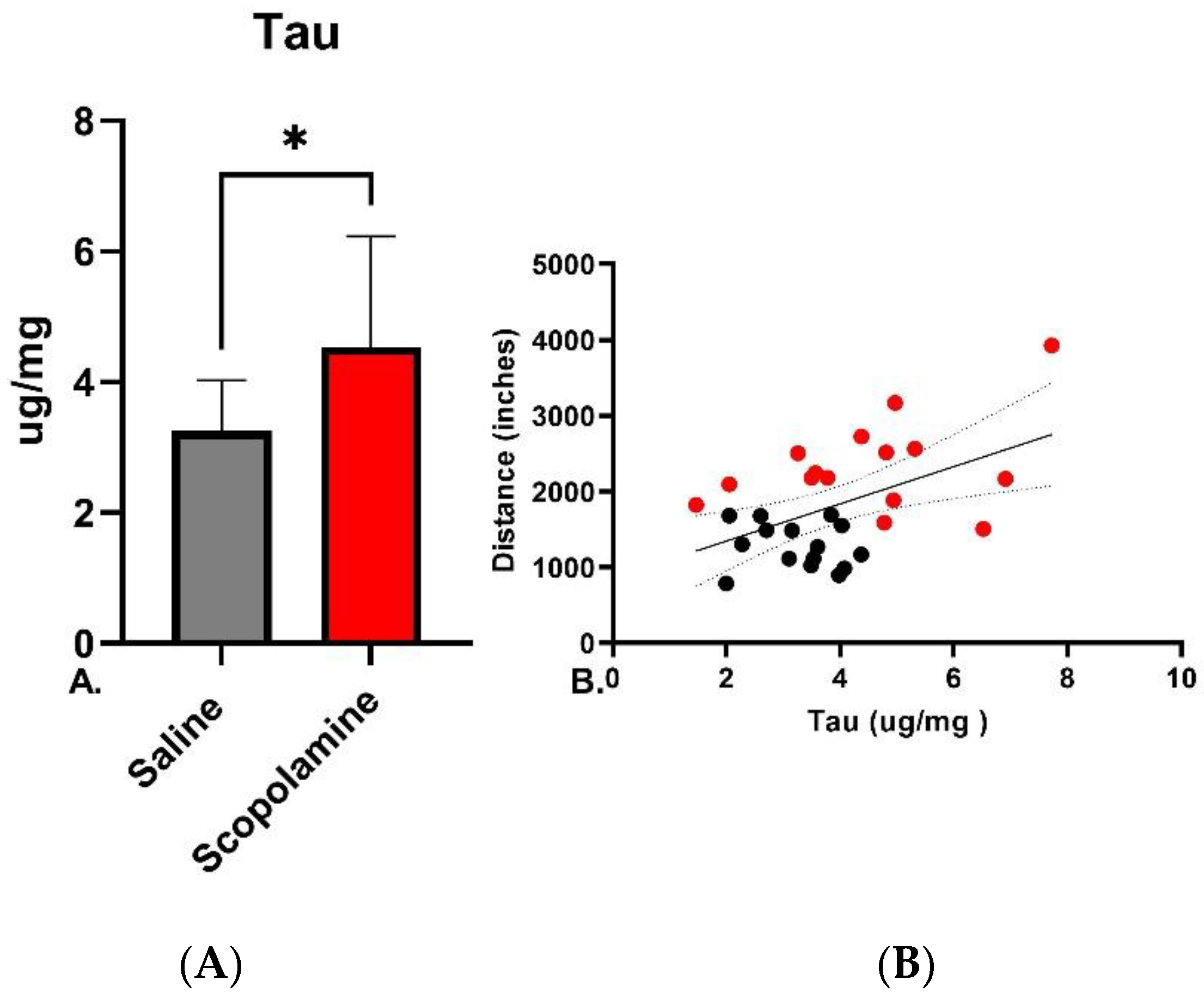
2.6.3. Tau ELISA
2.7. Statistics
3. Results
3.1. Behavior
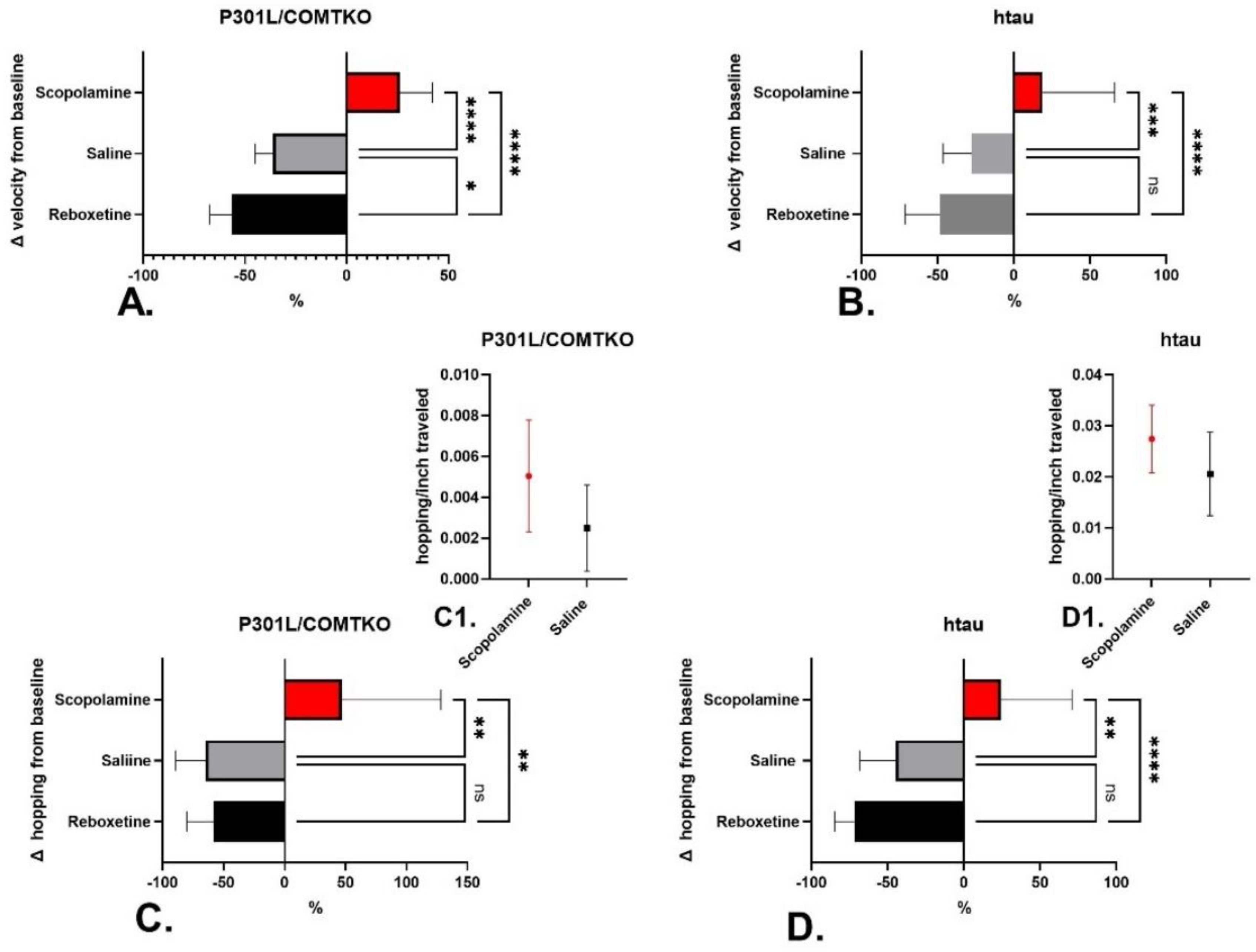
3.1.1. Locomotion
3.1.2. Acoustic Startle/PPI
3.2. Neurobiology
3.2.1. RNA-Seq
3.2.2. Tau and Locomotion
3.2.3. MicroPET
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ballard, C.; Kales, H.C.; Lyketsos, C.; Aarsland, D.; Creese, B.; Mills, R.; Williams, H.; Sweet, R.A. Psychosis in Alzheimer’s Disease. Curr. Neurol. Neurosci. Rep. 2020, 20, 57. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, L.H.; Bylsma, F.W.; Rovner, B.W.; Steele, C.; Folstein, M.F. Psychosis and physical aggression in probable Alzheimer’s disease. Am. J. Psychiatry 1991, 148, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, D.; Cummings, J.L.; Yenner, G.; Miller, B. Relationship of aggressive behavior to other neuropsychiatric symptoms in patients with Alzheimer’s disease. Am. J. Psychiatry 1996, 153, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Koppel, J.; Greenwald, B.S. Optimal treatment of Alzheimer’s disease psychosis: Challenges and solutions. Neuropsychiatr. Dis. Treat. 2014, 10, 2253–2262. [Google Scholar] [CrossRef] [Green Version]
- Koppel, J.; Sunday, S.; Buthorn, J.; Goldberg, T.; Davies, P.; Greenwald, B.; Alzheimer’s Disease Neuroimaging, I. Elevated CSF Tau is associated with psychosis in Alzheimer’s disease. Am. J. Psychiatry 2013, 170, 1212–1213. [Google Scholar] [CrossRef]
- Murray, P.S.; Kirkwood, C.M.; Gray, M.C.; Fish, K.N.; Ikonomovic, M.D.; Hamilton, R.L.; Kofler, J.K.; Klunk, W.E.; Lopez, O.L.; Sweet, R.A. Hyperphosphorylated tau is elevated in Alzheimer’s disease with psychosis. J. Alzheimers Dis. 2014, 39, 759–773. [Google Scholar] [CrossRef]
- Koppel, J.; Acker, C.; Davies, P.; Lopez, O.L.; Jimenez, H.; Azose, M.; Greenwald, B.S.; Murray, P.S.; Kirkwood, C.M.; Kofler, J.; et al. Psychotic Alzheimer’s disease is associated with gender-specific tau phosphorylation abnormalities. Neurobiol. Aging 2014, 35, 2021–2028. [Google Scholar] [CrossRef] [Green Version]
- Farber, N.B.; Rubin, E.H.; Newcomer, J.W.; Kinscherf, D.A.; Miller, J.P.; Morris, J.C.; Olney, J.W.; McKeel, D.W., Jr. Increased neocortical neurofibrillary tangle density in subjects with Alzheimer disease and psychosis. Arch. Gen. Psychiatry 2000, 57, 1165–1173. [Google Scholar] [CrossRef]
- Gomar, J.J.; Tan, G.; Halpern, J.; Gordon, M.L.; Greenwald, B.; Koppel, J. Increased retention of tau PET ligand [(18)F]-AV1451 in Alzheimer’s Disease Psychosis. Transl. Psychiatry 2022, 12, 82. [Google Scholar] [CrossRef]
- New warning on antipsychotic drugs used to treat older people. FDA Consum. 2005, 39, 2–3.
- Schneider, L.S.; Dagerman, K.S.; Insel, P. Risk of death with atypical antipsychotic drug treatment for dementia: Meta-analysis of randomized placebo-controlled trials. JAMA 2005, 294, 1934–1943. [Google Scholar] [CrossRef]
- Krivinko, J.M.; Koppel, J.; Savonenko, A.; Sweet, R.A. Animal Models of Psychosis in Alzheimer Disease. Am. J. Geriatr. Psychiatry 2020, 28, 1–19. [Google Scholar] [CrossRef]
- McKean, N.E.; Handley, R.R.; Snell, R.G. A Review of the Current Mammalian Models of Alzheimer’s Disease and Challenges That Need to Be Overcome. Int. J. Mol. Sci. 2021, 22, 13168. [Google Scholar] [CrossRef]
- Gogos, A.; van den Buuse, M. Sex Differences in Psychosis: Focus on Animal Models. Curr. Top. Behav. Neurosci. 2022, 2022, 133–163. [Google Scholar] [CrossRef]
- Price, D.L.; Bonhaus, D.W.; McFarland, K. Pimavanserin, a 5-HT2A receptor inverse agonist, reverses psychosis-like behaviors in a rodent model of Alzheimer’s disease. Behav. Pharmacol. 2012, 23, 426–433. [Google Scholar] [CrossRef]
- Krivinko, J.M.; Erickson, S.L.; MacDonald, M.L.; Garver, M.E.; Sweet, R.A. Fingolimod mitigates synaptic deficits and psychosis-like behavior in APP/PSEN1 mice. Alzheimers Dement. 2022, 8, e12324. [Google Scholar] [CrossRef]
- Yeomans, J.S.; Frankland, P.W. The acoustic startle reflex: Neurons and connections. Brain Res. Brain Res. Rev. 1995, 21, 301–314. [Google Scholar] [CrossRef]
- Braff, D.; Stone, C.; Callaway, E.; Geyer, M.; Glick, I.; Bali, L. Prestimulus effects on human startle reflex in normals and schizophrenics. Psychophysiology 1978, 15, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, N.R.; Braff, D.L.; Geyer, M.A. Sensorimotor gating of the startle reflex: What we said 25 years ago, what has happened since then, and what comes next. J. Psychopharmacol. 2016, 30, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, M.; Wei, B.; Shi, J.; Yu, T. Research Progress in the Study of Startle Reflex to Disease States. Neuropsychiatr. Dis. Treat. 2022, 18, 427–435. [Google Scholar] [CrossRef]
- Gomez-Nieto, R.; Hormigo, S.; Lopez, D.E. Prepulse Inhibition of the Auditory Startle Reflex Assessment as a Hallmark of Brainstem Sensorimotor Gating Mechanisms. Brain Sci. 2020, 10, 639. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Hranilovic, D.; Fetsko, L.A.; Bucan, M.; Wang, Y. Dopamine D2S and D2L receptors may differentially contribute to the actions of antipsychotic and psychotic agents in mice. Mol. Psychiatry 2002, 7, 1075–1082. [Google Scholar] [CrossRef] [Green Version]
- Perriol, M.P.; Dujardin, K.; Derambure, P.; Marcq, A.; Bourriez, J.L.; Laureau, E.; Pasquier, F.; Defebvre, L.; Destee, A. Disturbance of sensory filtering in dementia with Lewy bodies: Comparison with Parkinson’s disease dementia and Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2005, 76, 106–108. [Google Scholar] [CrossRef] [Green Version]
- Jafari, Z.; Kolb, B.E.; Mohajerani, M.H. Prepulse inhibition of the acoustic startle reflex and P50 gating in aging and alzheimer’s disease. Ageing Res. Rev. 2020, 59, 101028. [Google Scholar] [CrossRef] [PubMed]
- Koda, K.; Ago, Y.; Yano, K.; Nishimura, M.; Kobayashi, H.; Fukada, A.; Takuma, K.; Matsuda, T. Involvement of decreased muscarinic receptor function in prepulse inhibition deficits in mice reared in social isolation. Br. J. Pharmacol. 2011, 162, 763–772. [Google Scholar] [CrossRef] [Green Version]
- Singer, P.; Yee, B.K. Reversal of scopolamine-induced disruption of prepulse inhibition by clozapine in mice. Pharmacol. Biochem. Behav. 2012, 101, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Fiorentini, A.; Cantu, F.; Crisanti, C.; Cereda, G.; Oldani, L.; Brambilla, P. Substance-Induced Psychoses: An Updated Literature Review. Front. Psychiatry 2021, 12, 694863. [Google Scholar] [CrossRef] [PubMed]
- Barak, S. Modeling cholinergic aspects of schizophrenia: Focus on the antimuscarinic syndrome. Behav. Brain Res. 2009, 204, 335–351. [Google Scholar] [CrossRef]
- Carruthers, S.P.; Gurvich, C.T.; Rossell, S.L. The muscarinic system, cognition and schizophrenia. Neurosci. Biobehav. Rev. 2015, 55, 393–402. [Google Scholar] [CrossRef]
- Davis, M. Cocaine: Excitatory effects on sensorimotor reactivity measured with acoustic startle. Psychopharmacology 1985, 86, 31–36. [Google Scholar] [CrossRef]
- Swerdlow, N.R.; Eastvold, A.; Karban, B.; Ploum, Y.; Stephany, N.; Geyer, M.A.; Cadenhead, K.; Auerbach, P.P. Dopamine agonist effects on startle and sensorimotor gating in normal male subjects: Time course studies. Psychopharmacology 2002, 161, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Sweet, R.A.; Hamilton, R.L.; Healy, M.T.; Wisniewski, S.R.; Henteleff, R.; Pollock, B.G.; Lewis, D.A.; DeKosky, S.T. Alterations of striatal dopamine receptor binding in Alzheimer disease are associated with Lewy body pathology and antemortem psychosis. Arch. Neurol. 2001, 58, 466–472. [Google Scholar] [CrossRef] [Green Version]
- Reeves, S.; McLachlan, E.; Bertrand, J.; D’Antonio, F.; Brownings, S.; Nair, A.; Greaves, S.; Smith, A.; Taylor, D.; Dunn, J.; et al. Therapeutic window of dopamine D2/3 receptor occupancy to treat psychosis in Alzheimer’s disease. Brain 2017, 140, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Grant, K.M.; LeVan, T.D.; Wells, S.M.; Li, M.; Stoltenberg, S.F.; Gendelman, H.E.; Carlo, G.; Bevins, R.A. Methamphetamine-associated psychosis. J. Neuroimmune Pharmacol. 2012, 7, 113–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Martin, N.L.; Cotes, R.O. Cocaine-induced psychotic disorders: Presentation, mechanism, and management. J. Dual Diagn. 2014, 10, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Dave, S.; Weintraub, D.; Aarsland, D.; Ffytche, D.H. Drug and Disease Effects in Parkinson’s Psychosis: Revisiting the Role of Dopamine. Mov. Disord. Clin. Pract. 2020, 7, 32–36. [Google Scholar] [CrossRef] [Green Version]
- Kishi, T.; Sakuma, K.; Iwata, N. Efficacy and Safety of Psychostimulants for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Pharmacopsychiatry 2020, 53, 109–114. [Google Scholar] [CrossRef]
- Mintzer, J.; Lanctot, K.L.; Scherer, R.W.; Rosenberg, P.B.; Herrmann, N.; van Dyck, C.H.; Padala, P.R.; Brawman-Mintzer, O.; Porsteinsson, A.P.; Lerner, A.J.; et al. Effect of Methylphenidate on Apathy in Patients with Alzheimer Disease: The ADMET 2 Randomized Clinical Trial. JAMA Neurol. 2021, 78, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Back, C. The cholinergic hypothesis of neuropsychiatric symptoms in Alzheimer’s disease. Am. J. Geriatr. Psychiatry 1998, 6 (Suppl. S1), S64–S78. [Google Scholar] [CrossRef]
- Lai, M.K.; Lai, O.F.; Keene, J.; Esiri, M.M.; Francis, P.T.; Hope, T.; Chen, C.P. Psychosis of Alzheimer’s disease is associated with elevated muscarinic M2 binding in the cortex. Neurology 2001, 57, 805–811. [Google Scholar] [CrossRef]
- Cancelli, I.; Beltrame, M.; D’Anna, L.; Gigli, G.L.; Valente, M. Drugs with anticholinergic properties: A potential risk factor for psychosis onset in Alzheimer’s disease? Expert. Opin. Drug Saf. 2009, 8, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Cancelli, I.; Valentinis, L.; Merlino, G.; Valente, M.; Gigli, G.L. Drugs with anticholinergic properties as a risk factor for psychosis in patients affected by Alzheimer’s disease. Clin. Pharmacol. Ther. 2008, 84, 63–68. [Google Scholar] [CrossRef]
- Wynn, Z.J.; Cummings, J.L. Cholinesterase inhibitor therapies and neuropsychiatric manifestations of Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2004, 17, 100–108. [Google Scholar] [CrossRef]
- Figiel, G.; Sadowsky, C. A systematic review of the effectiveness of rivastigmine for the treatment of behavioral disturbances in dementia and other neurological disorders. Curr. Med. Res. Opin. 2008, 24, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Mackell, J.; Kaufer, D. Behavioral effects of current Alzheimer’s disease treatments: A descriptive review. Alzheimers Dement. 2008, 4, 49–60. [Google Scholar] [CrossRef]
- Dall’Igna, O.P.; Hoffmann, A.; da Silva, A.L.; Souza, D.O.; Lara, D.R. Beta-amyloid treatment sensitizes mice to amphetamine-induced locomotion but reduces response to caffeine. Neurodegener. Dis. 2004, 1, 38–43. [Google Scholar] [CrossRef]
- Kruk, M.; Tendera, K.; Biala, G. Memory-related effects of cholinergic receptor ligands in mice as measured by the elevated plus maze test. Pharmacol. Rep. 2011, 63, 1372–1382. [Google Scholar] [CrossRef]
- Lindner, M.D.; Hogan, J.B.; Hodges, D.B., Jr.; Orie, A.F.; Chen, P.; Corsa, J.A.; Leet, J.E.; Gillman, K.W.; Rose, G.M.; Jones, K.M.; et al. Donepezil primarily attenuates scopolamine-induced deficits in psychomotor function, with moderate effects on simple conditioning and attention, and small effects on working memory and spatial mapping. Psychopharmacology 2006, 188, 629–640. [Google Scholar] [CrossRef]
- Sunderland, T.; Tariot, P.N.; Weingartner, H.; Murphy, D.L.; Newhouse, P.A.; Mueller, E.A.; Cohen, R.M. Pharmacologic modelling of Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 1986, 10, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Takahata, K.; Kimura, Y.; Sahara, N.; Koga, S.; Shimada, H.; Ichise, M.; Saito, F.; Moriguchi, S.; Kitamura, S.; Kubota, M.; et al. PET-detectable tau pathology correlates with long-term neuropsychiatric outcomes in patients with traumatic brain injury. Brain 2019, 142, 3265–3279. [Google Scholar] [CrossRef]
- Moriguchi, S.; Takahata, K.; Shimada, H.; Kubota, M.; Kitamura, S.; Kimura, Y.; Tagai, K.; Tarumi, R.; Tabuchi, H.; Meyer, J.H.; et al. Excess tau PET ligand retention in elderly patients with major depressive disorder. Mol. Psychiatry 2020, 26, 5856–5863. [Google Scholar] [CrossRef] [PubMed]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, M.; Kobayashi, H.; Tatsumi, L.; Tomita, T. Mouse Models of Alzheimer’s Disease. Front. Mol. Neurosci. 2022, 15, 912995. [Google Scholar] [CrossRef]
- Koppel, J.; Jimenez, H.; Azose, M.; D’Abramo, C.; Acker, C.; Buthorn, J.; Greenwald, B.S.; Lewis, J.; Lesser, M.; Liu, Z.; et al. Pathogenic tau species drive a psychosis-like phenotype in a mouse model of Alzheimer’s disease. Behav. Brain Res. 2014, 275, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, H.; Adrien, L.; Wolin, A.; Eun, J.; Chang, E.H.; Burstein, E.S.; Gomar, J.; Davies, P.; Koppel, J. The impact of pimavanserin on psychotic phenotypes and tau phosphorylation in the P301L/COMT- and rTg(P301L)4510 mouse models of Alzheimer’s disease. Alzheimers Dement 2022, 8, e12247. [Google Scholar] [CrossRef]
- Lewis, J.; McGowan, E.; Rockwood, J.; Melrose, H.; Nacharaju, P.; Van Slegtenhorst, M.; Gwinn-Hardy, K.; Paul Murphy, M.; Baker, M.; Yu, X.; et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000, 25, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Gogos, J.A.; Morgan, M.; Luine, V.; Santha, M.; Ogawa, S.; Pfaff, D.; Karayiorgou, M. Catechol-O-methyltransferase-deficient mice exhibit sexually dimorphic changes in catecholamine levels and behavior. Proc. Natl. Acad. Sci. USA 1998, 95, 9991–9996. [Google Scholar] [CrossRef]
- Koppel, J.; Jimenez, H.; Adrien, L.E.H.C.; Malhotra, A.K.; Davies, P. Increased tau phosphorylation follows impeded dopamine clearance in a P301L and novel P301L/COMT-deleted (DM) tau mouse model. J. Neurochem. 2019, 148, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Andorfer, C.; Kress, Y.; Espinoza, M.; de Silva, R.; Tucker, K.L.; Barde, Y.A.; Duff, K.; Davies, P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem. 2003, 86, 582–590. [Google Scholar] [CrossRef]
- Wysocka, A.; Palasz, E.; Steczkowska, M.; Niewiadomska, G. Dangerous Liaisons: Tau Interaction with Muscarinic Receptors. Curr. Alzheimer Res. 2020, 17, 224–237. [Google Scholar] [CrossRef]
- Ramsden, M.; Kotilinek, L.; Forster, C.; Paulson, J.; McGowan, E.; SantaCruz, K.; Guimaraes, A.; Yue, M.; Lewis, J.; Carlson, G.; et al. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J. Neurosci. 2005, 25, 10637–10647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buccarello, L.; Grignaschi, G.; Castaldo, A.M.; Di Giancamillo, A.; Domeneghini, C.; Melcangi, R.C.; Borsello, T. Sex. Impact on Tau-Aggregation and Postsynaptic Protein Levels in the P301L Mouse Model of Tauopathy. J. Alzheimers Dis. 2017, 56, 1279–1292. [Google Scholar] [CrossRef] [PubMed]
- Asante, C.O.; Chu, A.; Fisher, M.; Benson, L.; Beg, A.; Scheiffele, P.; Martin, J. Cortical control of adaptive locomotion in wild-type mice and mutant mice lacking the ephrin-Eph effector protein alpha2-chimaerin. J. Neurophysiol. 2010, 104, 3189–3202. [Google Scholar] [CrossRef] [Green Version]
- Bellardita, C.; Kiehn, O. Phenotypic characterization of speed-associated gait changes in mice reveals modular organization of locomotor networks. Curr. Biol. 2015, 25, 1426–1436. [Google Scholar] [CrossRef] [Green Version]
- Eun, J.D.; Jimenez, H.; Adrien, L.; Wolin, A.; Marambaud, P.; Davies, P.; Koppel, J.L. Anesthesia promotes acute expression of genes related to Alzheimer’s disease and latent tau aggregation in transgenic mouse models of tauopathy. Mol. Med. 2022, 28, 83. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.D.; Wang, G.J.; Fowler, J.S.; Logan, J.; Schlyer, D.; Hitzemann, R.; Lieberman, J.; Angrist, B.; Pappas, N.; MacGregor, R.; et al. Imaging endogenous dopamine competition with [11C]raclopride in the human brain. Synapse 1994, 16, 255–262. [Google Scholar] [CrossRef]
- Ma, Y.; Hof, P.R.; Grant, S.C.; Blackband, S.J.; Bennett, R.; Slatest, L.; McGuigan, M.D.; Benveniste, H. A three-dimensional digital atlas database of the adult C57BL/6J mouse brain by magnetic resonance microscopy. Neuroscience 2005, 135, 1203–1215. [Google Scholar] [CrossRef]
- Johnson, G.A.; Badea, A.; Brandenburg, J.; Cofer, G.; Fubara, B.; Liu, S.; Nissanov, J. Waxholm space: An image-based reference for coordinating mouse brain research. Neuroimage 2010, 53, 365–372. [Google Scholar] [CrossRef] [Green Version]
- Sawiak, S.J.; Wood, N.I.; Williams, G.B.; Morton, A.J.; Carpenter, T.A. Voxel-based morphometry in the R6/2 transgenic mouse reveals differences between genotypes not seen with manual 2D morphometry. Neurobiol. Dis. 2009, 33, 20–27. [Google Scholar] [CrossRef]
- Acker, C.M.; Forest, S.K.; Zinkowski, R.; Davies, P.; d’Abramo, C. Sensitive quantitative assays for tau and phospho-tau in transgenic mouse models. Neurobiol. Aging 2013, 34, 338–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppel, J.; Jimenez, H.; Adrien, L.; Greenwald, B.S.; Marambaud, P.; Cinamon, E.; Davies, P. Haloperidol inactivates AMPK and reduces tau phosphorylation in a tau mouse model of Alzheimer’s disease. Alzheimers Dement. 2016, 2, 121–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oral, S.; Goktalay, G. Prepulse inhibition based grouping of rats and assessing differences in response to pharmacological agents. Neurosci. Lett. 2021, 755, 135913. [Google Scholar] [CrossRef] [PubMed]
- Baik, J.H.; Picetti, R.; Saiardi, A.; Thiriet, G.; Dierich, A.; Depaulis, A.; Le Meur, M.; Borrelli, E. Parkinsonian-like locomotor impairment in mice lacking dopamine D2 receptors. Nature 1995, 377, 424–428. [Google Scholar] [CrossRef]
- Gomeza, J.; Zhang, L.; Kostenis, E.; Felder, C.; Bymaster, F.; Brodkin, J.; Shannon, H.; Xia, B.; Deng, C.; Wess, J. Enhancement of D1 dopamine receptor-mediated locomotor stimulation in M(4) muscarinic acetylcholine receptor knockout mice. Proc. Natl. Acad. Sci. USA 1999, 96, 10483–10488. [Google Scholar] [CrossRef]
- Ferreira, J.A. The Benjamini-Hochberg method in the case of discrete test statistics. Int. J. Biostat. 2007, 3, 11. [Google Scholar] [CrossRef] [Green Version]
- Morozova, V.; Cohen, L.S.; Makki, A.E.; Shur, A.; Pilar, G.; El Idrissi, A.; Alonso, A.D. Normal and Pathological Tau Uptake Mediated by M1/M3 Muscarinic Receptors Promotes Opposite Neuronal Changes. Front. Cell Neurosci. 2019, 13, 403. [Google Scholar] [CrossRef] [Green Version]
- Yohn, S.E.; Weiden, P.J.; Felder, C.C.; Stahl, S.M. Muscarinic acetylcholine receptors for psychotic disorders: Bench-side to clinic. Trends Pharmacol. Sci. 2022, 43, 1098–1112. [Google Scholar] [CrossRef]
- Asai, M.; Fujikawa, A.; Noda, A.; Miyoshi, S.; Matsuoka, N.; Nishimura, S. Donepezil- and scopolamine-induced rCMRglu changes assessed by PET in conscious rhesus monkeys. Ann. Nucl. Med. 2009, 23, 877–882. [Google Scholar] [CrossRef]
- Graff-Guerrero, A.; Willeit, M.; Ginovart, N.; Mamo, D.; Mizrahi, R.; Rusjan, P.; Vitcu, I.; Seeman, P.; Wilson, A.A.; Kapur, S. Brain region binding of the D2/3 agonist [11C]-(+)-PHNO and the D2/3 antagonist [11C]raclopride in healthy humans. Hum. Brain Mapp. 2008, 29, 400–410. [Google Scholar] [CrossRef]
- Falsafi, S.K.; Deli, A.; Hoger, H.; Pollak, A.; Lubec, G. Scopolamine administration modulates muscarinic, nicotinic and NMDA receptor systems. PLoS ONE 2012, 7, e32082. [Google Scholar] [CrossRef]
- Threlfell, S.; Cragg, S.J. Dopamine signaling in dorsal versus ventral striatum: The dynamic role of cholinergic interneurons. Front. Syst. Neurosci. 2011, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Dewey, S.L.; Smith, G.S.; Logan, J.; Brodie, J.D.; Simkowitz, P.; MacGregor, R.R.; Fowler, J.S.; Volkow, N.D.; Wolf, A.P. Effects of central cholinergic blockade on striatal dopamine release measured with positron emission tomography in normal human subjects. Proc. Natl. Acad. Sci. USA 1993, 90, 11816–11820. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, H.; Harada, N.; Nishiyama, S.; Ohba, H.; Kakiuchi, T. Cholinergic neuronal modulation alters dopamine D2 receptor availability in vivo by regulating receptor affinity induced by facilitated synaptic dopamine turnover: Positron emission tomography studies with microdialysis in the conscious monkey brain. J. Neurosci. 2000, 20, 7067–7073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, D.J.; Sotnikova, T.D.; Gainetdinov, R.R.; Huang, S.Y.; Caron, M.G.; Tonegawa, S. Hyperactivity, elevated dopaminergic transmission, and response to amphetamine in M1 muscarinic acetylcholine receptor-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 15312–15317. [Google Scholar] [CrossRef]
- Bradley, S.J.; Molloy, C.; Valuskova, P.; Dwomoh, L.; Scarpa, M.; Rossi, M.; Finlayson, L.; Svensson, K.A.; Chernet, E.; Barth, V.N.; et al. Biased M1-muscarinic-receptor-mutant mice inform the design of next-generation drugs. Nat. Chem. Biol. 2020, 16, 240–249. [Google Scholar] [CrossRef]
- Koshimizu, H.; Leiter, L.M.; Miyakawa, T. M4 muscarinic receptor knockout mice display abnormal social behavior and decreased prepulse inhibition. Mol. Brain 2012, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Moya, N.A.; Yun, S.; Fleps, S.W.; Martin, M.M.; Nadel, J.A.; Beutler, L.R.; Zweifel, L.S.; Parker, J.G. The effect of selective nigrostriatal dopamine excess on behaviors linked to the cognitive and negative symptoms of schizophrenia. Neuropsychopharmacology 2023, 48, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Aoki, K. Development of a strain of spontaneously hypertensive rats. Jpn. Circ. J. 1963, 27, 282–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viggiano, D.; Ruocco, L.A.; Arcieri, S.; Sadile, A.G. Involvement of norepinephrine in the control of activity and attentive processes in animal models of attention deficit hyperactivity disorder. Neural Plast. 2004, 11, 133–149. [Google Scholar] [CrossRef] [Green Version]
- Hajos, M.; Fleishaker, J.C.; Filipiak-Reisner, J.K.; Brown, M.T.; Wong, E.H. The selective norepinephrine reuptake inhibitor antidepressant reboxetine: Pharmacological and clinical profile. CNS Drug Rev. 2004, 10, 23–44. [Google Scholar] [CrossRef]
- Kaenmaki, M.; Tammimaki, A.; Myohanen, T.; Pakarinen, K.; Amberg, C.; Karayiorgou, M.; Gogos, J.A.; Mannisto, P.T. Quantitative role of COMT in dopamine clearance in the prefrontal cortex of freely moving mice. J. Neurochem. 2010, 114, 1745–1755. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.H.; Sonders, M.S.; Amara, S.G.; Tinholt, P.M.; Piercey, M.F.; Hoffmann, W.P.; Hyslop, D.K.; Franklin, S.; Porsolt, R.D.; Bonsignori, A.; et al. Reboxetine: A pharmacologically potent, selective, and specific norepinephrine reuptake inhibitor. Biol. Psychiatry 2000, 47, 818–829. [Google Scholar] [CrossRef]
- Kuczenski, R.; Segal, D.S.; Cho, A.K.; Melega, W. Hippocampus norepinephrine, caudate dopamine and serotonin, and behavioral responses to the stereoisomers of amphetamine and methamphetamine. J. Neurosci. 1995, 15, 1308–1317. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Moratalla, R.; Gold, L.H.; Hiroi, N.; Koob, G.F.; Graybiel, A.M.; Tonegawa, S. Dopamine D1 receptor mutant mice are deficient in striatal expression of dynorphin and in dopamine-mediated behavioral responses. Cell 1994, 79, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Centonze, D.; Grande, C.; Saulle, E.; Martin, A.B.; Gubellini, P.; Pavon, N.; Pisani, A.; Bernardi, G.; Moratalla, R.; Calabresi, P. Distinct roles of D1 and D5 dopamine receptors in motor activity and striatal synaptic plasticity. J. Neurosci. 2003, 23, 8506–8512. [Google Scholar] [CrossRef] [Green Version]
- Granado, N.; Ortiz, O.; Suarez, L.M.; Martin, E.D.; Cena, V.; Solis, J.M.; Moratalla, R. D1 but not D5 dopamine receptors are critical for LTP, spatial learning, and LTP-Induced arc and zif268 expression in the hippocampus. Cereb. Cortex 2008, 18, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNamara, F.N.; Clifford, J.J.; Tighe, O.; Kinsella, A.; Drago, J.; Croke, D.T.; Waddington, J.L. Congenic D1A dopamine receptor mutants: Ethologically based resolution of behavioural topography indicates genetic background as a determinant of knockout phenotype. Neuropsychopharmacology 2003, 28, 86–99. [Google Scholar] [CrossRef] [Green Version]
- Ariano, M.A.; Sibley, D.R. Dopamine receptor distribution in the rat CNS: Elucidation using anti-peptide antisera directed against D1A and D3 subtypes. Brain Res. 1994, 649, 95–110. [Google Scholar] [CrossRef]
- Meador-Woodruff, J.H.; Damask, S.P.; Wang, J.; Haroutunian, V.; Davis, K.L.; Watson, S.J. Dopamine receptor mRNA expression in human striatum and neocortex. Neuropsychopharmacology 1996, 15, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Hurd, Y.L.; Suzuki, M.; Sedvall, G.C. D1 and D2 dopamine receptor mRNA expression in whole hemisphere sections of the human brain. J. Chem. Neuroanat. 2001, 22, 127–137. [Google Scholar] [CrossRef]
- Ernst, M.; Zametkin, A.J.; Matochik, J.A.; Jons, P.H.; Cohen, R.M. DOPA decarboxylase activity in attention deficit hyperactivity disorder adults. A [fluorine-18]fluorodopa positron emission tomographic study. J. Neurosci. 1998, 18, 5901–5907. [Google Scholar] [CrossRef] [Green Version]
- Greenhill, L.L.; Pliszka, S.; Dulcan, M.K.; Bernet, W.; Arnold, V.; Beitchman, J.; Benson, R.S.; Bukstein, O.; Kinlan, J.; McClellan, J.; et al. Practice Parameter for the use of stimulant medications in the treatment of children, adolescents, and adults. J. Am. Acad. Child. Adolesc. Psychiatry 2002, 41 (Suppl. S2), 26S–49S. [Google Scholar] [CrossRef]
- Takamatsu, Y.; Hagino, Y.; Sato, A.; Takahashi, T.; Nagasawa, S.Y.; Kubo, Y.; Mizuguchi, M.; Uhl, G.R.; Sora, I.; Ikeda, K. Improvement of learning and increase in dopamine level in the frontal cortex by methylphenidate in mice lacking dopamine transporter. Curr. Mol. Med. 2015, 15, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svensson, L. The role of the dopaminergic system in the modulation of the acoustic startle response in the rat. Eur. J. Pharmacol. 1990, 175, 107–111. [Google Scholar] [CrossRef]
- Halberstadt, A.L.; Geyer, M.A. Habituation and sensitization of acoustic startle: Opposite influences of dopamine D1 and D2-family receptors. Neurobiol. Learn. Mem. 2009, 92, 243–248. [Google Scholar] [CrossRef] [Green Version]
- Koch, M. The neurobiology of startle. Prog. Neurobiol. 1999, 59, 107–128. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Nagai, T.; Kamei, H.; Maeda, K.; Matsuya, T.; Arai, S.; Mizoguchi, H.; Yoneda, Y.; Nabeshima, T.; Takuma, K.; et al. Neural circuits containing pallidotegmental GABAergic neurons are involved in the prepulse inhibition of the startle reflex in mice. Biol. Psychiatry 2007, 62, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Fendt, M.; Koch, M. Cholinergic modulation of the acoustic startle response in the caudal pontine reticular nucleus of the rat. Eur. J. Pharmacol. 1999, 370, 101–107. [Google Scholar] [CrossRef]
- Philippens, I.H.; Olivier, B.; Melchers, B.P. Effects of physostigmine on the startle in guinea pigs: Two mechanisms involved. Pharmacol. Biochem. Behav. 1997, 58, 909–913. [Google Scholar] [CrossRef]
- Wu, M.F.; Jenden, D.J.; Fairchild, M.D.; Siegel, J.M. Cholinergic mechanisms in startle and prepulse inhibition: Effects of the false cholinergic precursor N-aminodeanol. Behav. Neurosci. 1993, 107, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.K.; Shannon, H.E. Muscarinic cholinergic modulation of prepulse inhibition of the acoustic startle reflex. J. Pharmacol. Exp. Ther. 2000, 294, 1017–1023. [Google Scholar] [PubMed]
- Zhang, J.; Forkstam, C.; Engel, J.A.; Svensson, L. Role of dopamine in prepulse inhibition of acoustic startle. Psychopharmacology 2000, 149, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–278. [Google Scholar] [CrossRef]
- Takeda, S. Progression of Alzheimer’s disease, tau propagation, and its modifiable risk factors. Neurosci. Res. 2019, 141, 36–42. [Google Scholar] [CrossRef]
- Jul, P.; Volbracht, C.; de Jong, I.E.; Helboe, L.; Elvang, A.B.; Pedersen, J.T. Hyperactivity with Agitative-Like Behavior in a Mouse Tauopathy Model. J. Alzheimers Dis. 2016, 49, 783–795. [Google Scholar] [CrossRef]
- Gilley, J.; Seereeram, A.; Ando, K.; Mosely, S.; Andrews, S.; Kerschensteiner, M.; Misgeld, T.; Brion, J.P.; Anderton, B.; Hanger, D.P.; et al. Age-dependent axonal transport and locomotor changes and tau hypophosphorylation in a “P301L” tau knockin mouse. Neurobiol. Aging 2012, 33, 621.e1–621.e15. [Google Scholar] [CrossRef]
- Boxer, A.L.; Yu, J.T.; Golbe, L.I.; Litvan, I.; Lang, A.E.; Hoglinger, G.U. Advances in progressive supranuclear palsy: New diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol. 2017, 16, 552–563. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, E.C.; Graybiel, A.M.; Duyckaerts, C.; Javoy-Agid, F. Neuronal loss in the pedunculopontine tegmental nucleus in Parkinson disease and in progressive supranuclear palsy. Proc. Natl. Acad. Sci. USA 1987, 84, 5976–5980. [Google Scholar] [CrossRef]
- King, G.; Veros, K.M.; MacLaren, D.A.A.; Leigh, M.P.K.; Spernyak, J.A.; Clark, S.D. Human wildtype tau expression in cholinergic pedunculopontine tegmental neurons is sufficient to produce PSP-like behavioural deficits and neuropathology. Eur. J. Neurosci. 2021, 54, 7688–7709. [Google Scholar] [CrossRef]
- Robert, A.; Scholl, M.; Vogels, T. Tau Seeding Mouse Models with Patient Brain-Derived Aggregates. Int. J. Mol. Sci. 2021, 22, 6132. [Google Scholar] [CrossRef]
- Ellison, G.D.; Eison, M.S. Continuous amphetamine intoxication: An animal model of the acute psychotic episode. Psychol. Med. 1983, 13, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.C.; Baskaran, R.; Tsao, C.Y.; Tuan, L.H.; Siow, P.F.; Palani, M.; Lee, L.J.; Liu, C.M.; Hwu, H.G.; Lee, L.J. Chronic N-Acetylcysteine Treatment Prevents Amphetamine-Induced Hyperactivity in Heterozygous Disc1 Mutant Mice, a Putative Prodromal Schizophrenia Animal Model. Int. J. Mol. Sci. 2022, 23, 9419. [Google Scholar] [CrossRef]
- Petty, A.; Cui, X.; Ali, A.; Du, Z.; Srivastav, S.; Kesby, J.P.; Kirik, D.; Howes, O.; Eyles, D. Positive symptom phenotypes appear progressively in “EDiPS”, a new animal model of the schizophrenia prodrome. Sci. Rep. 2021, 11, 4294. [Google Scholar] [CrossRef] [PubMed]
- Correia, B.S.B.; Nani, J.V.; Waladares Ricardo, R.; Stanisic, D.; Costa, T.; Hayashi, M.A.F.; Tasic, L. Effects of Psychostimulants and Antipsychotics on Serum Lipids in an Animal Model for Schizophrenia. Biomedicines 2021, 9, 235. [Google Scholar] [CrossRef]
- Pfeiffer, C.C.; Jenney, E.H. The inhibition of the conditioned response and the counteraction of schizophrenia by muscarinic stimulation of the brain. Ann. N. Y. Acad. Sci. 1957, 66, 753–764. [Google Scholar] [CrossRef]
- Shekhar, A.; Potter, W.Z.; Lightfoot, J.; Lienemann, J.; Dube, S.; Mallinckrodt, C.; Bymaster, F.P.; McKinzie, D.L.; Felder, C.C. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am. J. Psychiatry 2008, 165, 1033–1039. [Google Scholar] [CrossRef]
- Bodick, N.C.; Offen, W.W.; Levey, A.I.; Cutler, N.R.; Gauthier, S.G.; Satlin, A.; Shannon, H.E.; Tollefson, G.D.; Rasmussen, K.; Bymaster, F.P.; et al. Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch. Neurol. 1997, 54, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Breier, A.; Brannan, S.K.; Paul, S.M.; Miller, A.C. Evidence of trospium’s ability to mitigate cholinergic adverse events related to xanomeline: Phase 1 study results. Psychopharmacology 2023, 240, 1191–1198. [Google Scholar] [CrossRef]
- Solmi, M.; Murru, A.; Pacchiarotti, I.; Undurraga, J.; Veronese, N.; Fornaro, M.; Stubbs, B.; Monaco, F.; Vieta, E.; Seeman, M.V.; et al. Safety, tolerability, and risks associated with first- and second-generation antipsychotics: A state-of-the-art clinical review. Ther. Clin. Risk Manag. 2017, 13, 757–777. [Google Scholar] [CrossRef] [Green Version]
- Giros, B.; Jaber, M.; Jones, S.R.; Wightman, R.M.; Caron, M.G. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 1996, 379, 606–612. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jimenez, H.; Carrion, J.; Adrien, L.; Wolin, A.; Eun, J.; Cinamon, E.; Chang, E.H.; Davies, P.; Vo, A.; Koppel, J. The Impact of Muscarinic Antagonism on Psychosis-Relevant Behaviors and Striatal [11C] Raclopride Binding in Tau Mouse Models of Alzheimer’s Disease. Biomedicines 2023, 11, 2091. https://doi.org/10.3390/biomedicines11082091
Jimenez H, Carrion J, Adrien L, Wolin A, Eun J, Cinamon E, Chang EH, Davies P, Vo A, Koppel J. The Impact of Muscarinic Antagonism on Psychosis-Relevant Behaviors and Striatal [11C] Raclopride Binding in Tau Mouse Models of Alzheimer’s Disease. Biomedicines. 2023; 11(8):2091. https://doi.org/10.3390/biomedicines11082091
Chicago/Turabian StyleJimenez, Heidy, Joseph Carrion, Leslie Adrien, Adam Wolin, John Eun, Ezra Cinamon, Eric H. Chang, Peter Davies, An Vo, and Jeremy Koppel. 2023. "The Impact of Muscarinic Antagonism on Psychosis-Relevant Behaviors and Striatal [11C] Raclopride Binding in Tau Mouse Models of Alzheimer’s Disease" Biomedicines 11, no. 8: 2091. https://doi.org/10.3390/biomedicines11082091