
Puzzling Out the Genetic Architecture of Endometriosis: Whole-Exome Sequencing and Novel Candidate Gene Identification in a Deeply Clinically Characterised Cohort
 , , , , , , , , , , , and
, , , , , , , , , , , and
Abstract
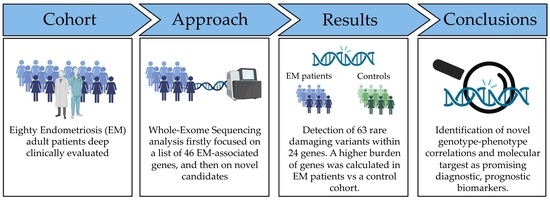
:
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Participants’ Recruitment and Clinical Evaluation
2.3. DNA Extraction and Quality Control
2.4. Whole-Exome Sequencing (WES)
2.5. WES Data Analysis and Variant Selection
- Variants with a quality score < 20, Variant Allele Frequency < 30, or called in off-target regions were excluded;
- A Minor Allele Frequency (MAF) cut-off of 0.1% was considered. The variant frequency was verified both in NCBI dbSNP (https://www.ncbi.nlm.nih.gov/snp/, accessed on 30 April 2023) and gnomAD (https://gnomad.broadinstitute.org/, accessed on 30 April 2023);
- The effect of the genetic variants was evaluated with in silico prediction tools, such as PolyPhen-2 (tolerated for scores < 0.5, damaging for scores ≥ 0.5) [22], SIFT [23], PaPI (tolerated for scores ≤ 0.5, damaging for scores > 0.5) [24], DANN (tolerated for scores ≤ 0.9, damaging for scores > 0.9) [25], the dbscSNV score (tolerated for scores ≤ 0.9, damaging for scores > 0.9) [26], and SpliceAI (tolerated for scores < 0.5, probably damaging for scores ranging from 0.5 to 0.8, damaging for scores ≥ 0.8) [27];
- SNVs leading to synonymous aminoacidic substitutions not predicted as damaging, not affecting splicing, or highly conserved residues were excluded.
2.6. Control Cohort
2.7. Statistical Analysis
3. Results
3.1. Demographic Data and Clinical Features of EM Patients
3.2. WES Analysis and Results Classification
- A total of 34/80 (43%) patients carried different rare, predicted, and damaging variants within 13 recurrent genes (FCRL3, LAMA5, SYNE1, SYNE2, GREB1, MAP3K4, C3, MMP3, MMP9, TYK2, VEGFA, VEZT, RHOJ);
- A total of 7/80 (8.8%) patients (i.e., patients 3, 9, 19, 50, 67, 14, 19, 13) carried different private, rare, predicted, and damaging variants within eight single genes (KAZN, IL18, WT1, CYP19A1, IL1A, IL2RB, LILRB2, ZNF366);
- A total of 19/80 (24%) patients carried different rare, predicted, and damaging variants within three novel candidate genes (ABCA13, NEB, CSMD1).
3.3. Rare Variants within Recurrent Genes
3.4. Rare, Private Variants within Specific Genes in Single Patients
3.5. Identification of Novel Candidate Genes
3.6. Burden of Genes Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smolarz, B.; Szyłło, K.; Romanowicz, H. Endometriosis: Epidemiology, Classification, Pathogenesis, Treatment and Genetics (Review of Literature). Int. J. Mol. Sci. 2021, 22, 10554. [Google Scholar] [CrossRef] [PubMed]
- Monnin, N.; Fattet, A.J.; Koscinski, I. Endometriosis: Update of Pathophysiology, (Epi) Genetic and Environmental Involvement. Biomedicines 2023, 11, 978. [Google Scholar] [CrossRef]
- Leyendecker, G.; Wildt, L.; Mall, G. The Pathophysiology of Endometriosis and Adenomyosis: Tissue Injury and Repair. Arch. Gynecol. Obstet. 2009, 280, 529–538. [Google Scholar] [CrossRef] [Green Version]
- Da Broi, M.G.; Ferriani, R.A.; Navarro, P.A. Ethiopathogenic Mechanisms of Endometriosis-Related Infertility. J. Bras. Reprod. Assist. 2019, 23, 273–280. [Google Scholar]
- Cousins, F.L.; McKinnon, B.D.; Mortlock, S.; Fitzgerald, H.C.; Zhang, C.; Montgomery, G.W.; Gargett, C.E. New Concepts on the Etiology of Endometriosis. J. Obstet. Gynaecol. Res. 2023, 49, 1090–1105. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T. A Revised Stem Cell Theory for the Pathogenesis of Endometriosis. J. Pers. Med. 2022, 12, 216. [Google Scholar] [CrossRef]
- Savone, R.; Salvatore, G.; Di Francesco, A. Endometriosis in a Patient with Mayer-Rokitansky-Küister-Hauser Syndrome. Gazz. Medica Ital. Arch. Per Le Sci. Mediche 2014, 173, 141–143. [Google Scholar] [CrossRef] [Green Version]
- Rei, C.; Williams, T.; Feloney, M. Endometriosis in a Man as a Rare Source of Abdominal Pain: A Case Report and Review of the Literature. Case Rep. Obstet. Gynecol. 2018, 2018, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, G.W.; Mortlock, S.; Giudice, L.C. Should Genetics Now Be Considered the Pre-Eminent Etiologic Factor in Endometriosis? J. Minim. Invasive Gynecol. 2020, 27, 280–286. [Google Scholar] [CrossRef]
- Bellelis, P.; Podgaec, S.; Abrão, M.S. Environmental Factors and Endometriosis. Rev. Da Assoc. Médica Bras. 2011, 57, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Birnbaum, L.S.; Cummings, A.M. Dioxins and Endometriosis: A Plausible Hypothesis. Environ. Health Perspect. 2002, 110, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parazzini, F.; Viganò, P.; Candiani, M.; Fedele, L. Diet and Endometriosis Risk: A Literature Review. Reprod. Biomed. Online 2013, 26, 323–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, J.L.; Elias, S.; Malinak, L.R.; Buttram, V.C. Heritable Aspects of Endometriosis. I. Genetic Studies. Am. J. Obstet. Gynecol. 1980, 137, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.; Pettersson, H.J.; Svedberg, P.; Olovsson, M.; Bergqvist, A.; Marions, L.; Tornvall, P.; Kuja-Halkola, R. Heritability of Endometriosis. Fertil. Steril. 2015, 104, 947–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapkota, Y.; Steinthorsdottir, V.; Morris, A.P.; Fassbender, A.; Rahmioglu, N.; De Vivo, I.; Buring, J.E.; Zhang, F.; Edwards, T.L.; Jones, S.; et al. Meta-Analysis Identifies Five Novel Loci Associated with Endometriosis Highlighting Key Genes Involved in Hormone Metabolism. Nat. Commun. 2017, 8, 15539. [Google Scholar] [CrossRef] [Green Version]
- Rahmioglu, N.; Mortlock, S.; Ghiasi, M.; Møller, P.L.; Stefansdottir, L.; Galarneau, G.; Turman, C.; Danning, R.; Law, M.H.; Sapkota, Y.; et al. The Genetic Basis of Endometriosis and Comorbidity with Other Pain and Inflammatory Conditions. Nat. Genet. 2023, 55, 423–436. [Google Scholar] [CrossRef]
- Guerriero, S.; Condous, G.; van den Bosch, T.; Valentin, L.; Leone, F.P.G.; Van Schoubroeck, D.; Exacoustos, C.; Installé, A.J.F.; Martins, W.P.; Abrao, M.S.; et al. Systematic Approach to Sonographic Evaluation of the Pelvis in Women with Suspected Endometriosis, Including Terms, Definitions and Measurements: A Consensus Opinion from the International Deep Endometriosis Analysis (IDEA) Group. Ultrasound Obstet. Gynecol. 2016, 48, 318–332. [Google Scholar] [CrossRef]
- Bazot, M.; Bharwani, N.; Huchon, C.; Kinkel, K.; Cunha, T.M.; Guerra, A.; Manganaro, L.; Buñesch, L.; Kido, A.; Togashi, K.; et al. European Society of Urogenital Radiology (ESUR) Guidelines: MR Imaging of Pelvic Endometriosis. Eur. Radiol. 2017, 27, 2765–2775. [Google Scholar] [CrossRef] [Green Version]
- American Society for Reproductive Medicine. Revised American Society for Reproductive Medicine Classification of Endometriosis: 1996. Fertil Steril. 1997, 67, 817–821. [Google Scholar] [CrossRef]
- Haefeli, M.; Elfering, A. Pain Assessment. Eur. Spine J. 2006, 15, S17–S24. [Google Scholar] [CrossRef]
- Spedicati, B.; Santin, A.; Nardone, G.G.; Rubinato, E.; Lenarduzzi, S.; Graziano, C.; Garavelli, L.; Miccoli, S.; Bigoni, S.; Morgan, A.; et al. The Enigmatic Genetic Landscape of Hereditary Hearing Loss: A Multistep Diagnostic Strategy in the Italian Population. Biomedicines 2023, 11, 703. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013. [CrossRef] [PubMed] [Green Version]
- Ng, P.C.; Henikoff, S. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic. Acids. Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limongelli, I.; Marini, S.; Bellazzi, R. PaPI: Pseudo Amino Acid Composition to Score Human Protein-Coding Variants. BMC Bioinform. 2015, 16, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quang, D.; Chen, Y.; Xie, X. DANN: A Deep Learning Approach for Annotating the Pathogenicity of Genetic Variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [Green Version]
- Jian, X.; Boerwinkle, E.; Liu, X. In Silico Prediction of Splice-Altering Single Nucleotide Variants in the Human Genome. Nucleic. Acids. Res. 2014, 42, 13534–13544. [Google Scholar] [CrossRef] [Green Version]
- de Sainte Agathe, J.M.; Filser, M.; Isidor, B.; Besnard, T.; Gueguen, P.; Perrin, A.; Van Goethem, C.; Verebi, C.; Masingue, M.; Rendu, J.; et al. SpliceAI-Visual: A Free Online Tool to Improve SpliceAI Splicing Variant Interpretation. Hum. Genomics. 2023, 17, 1–16. [Google Scholar] [CrossRef]
- Wiel, L.; Baakman, C.; Gilissen, D.; Veltman, J.A.; Vriend, G.; Gilissen, C. MetaDome: Pathogenicity Analysis of Genetic Variants through Aggregation of Homologous Human Protein Domains. Hum. Mutat. 2019, 40, 1030–1038. [Google Scholar] [CrossRef] [Green Version]
- Signorile, P.G.; Viceconte, R.; Baldi, A. New Insights in Pathogenesis of Endometriosis. Front. Med. 2022, 9, 879015. [Google Scholar] [CrossRef]
- Culley, L.; Law, C.; Hudson, N.; Denny, E.; Mitchell, H.; Baumgarten, M.; Raine-Fenning, N. The Social and Psychological impact of Endometriosis on Women’s Lives: A Critical Narrative Review. Hum. Reprod. Update 2013, 19, 625–639. [Google Scholar] [CrossRef] [Green Version]
- Hung, S.W.; Zhang, R.; Tan, Z.; Chung, J.P.W.; Zhang, T.; Wang, C.C. Pharmaceuticals Targeting Signaling Pathways of Endometriosis as Potential New Medical Treatment: A Review. Med. Res. Rev. 2021, 41, 2489–2564. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Zhao, L.; Wang, L.; Wu, Z.; Mei, Q.; Nie, J.; Li, X.; Li, Y.; Fu, X.; et al. Whole-Exome Sequencing of Endometriosis Identifies Frequent Alterations in Genes Involved in Cell Adhesion and Chromatin-Remodeling Complexes. Hum. Mol. Genet. 2014, 23, 6008–6021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianco, B.; André, G.M.; Vilarino, F.L.; Peluso, C.; Mafra, F.A.; Christofolini, D.M.; Barbosa, C.P. The Possible Role of Genetic Variants in Autoimmune-Related Genes in the Development of Endometriosis. Hum. Immunol. 2012, 73, 306–315. [Google Scholar] [CrossRef]
- Bianco, B.; Teles, J.S.; Lerner, T.G.; Vilarino, F.L.; Christofolini, D.M.; Barbosa, C.P. Association of FCRL3-169T/C Polymorphism with Endometriosis and Identification of a Protective Haplotype against the Development of the Disease in Brazilian Population. Hum. Immunol. 2011, 72, 774–778. [Google Scholar] [CrossRef]
- Berbic, M.; Hey-Cunningham, A.J.; Ng, C.; Tokushige, N.; Ganewatta, S.; Markham, R.; Russell, P.; Fraser, I.S. The Role of Foxp3+ Regulatory T-Cells in Endometriosis: A Potential Controlling Mechanism for a Complex, Chronic Immunological Condition. Hum. Reprod. 2010, 25, 900–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zhang, Z.; Li, G.; Wang, S.; Zhang, S.; Xie, B. Association of FCRL3 Genetic Polymorphisms with Endometriosis-Related Infertility Risk. Medicine 2015, 94, e1168. [Google Scholar] [CrossRef]
- Christofolini, D.M.; Mafra, F.A.; Catto, M.C.; Bianco, B.; Barbosa, C.P. New Candidate Genes Associated to Endometriosis. Gynecol. Endocrinol. 2019, 35, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Laudanski, P.; Charkiewicz, R.; Kuzmicki, M.; Szamatowicz, J.; Świa̧tecka, J.; Mroczko, B.; Niklinski, J. Profiling of Selected Angiogenesis-Related Genes in Proliferative Eutopic Endometrium of Women with Endometriosis. Eur. J. Obstet. Gynecol. Reprod. Biol. 2014, 172, 85–92. [Google Scholar] [CrossRef]
- Tamaresis, J.S.; Irwin, J.C.; Goldfien, G.A.; Rabban, J.T.; Burney, R.O.; Nezhat, C.; DePaolo, L.V.; Giudice, L.C. Molecular Classification of Endometriosis and Disease Stage Using High-Dimensional Genomic Data. Endocrinology 2014, 155, 4986–4999. [Google Scholar] [CrossRef] [Green Version]
- Luo, Q.; Ning, W.; Wu, Y.; Zhu, X.; Jin, F.; Sheng, J.; Huang, H. Altered Expression of Interleukin-18 in the Ectopic and Eutopic Endometrium of Women with Endometriosis. J. Reprod. Immunol. 2006, 72, 108–117. [Google Scholar] [CrossRef]
- Arici, A.; Matalliotakis, I.; Goumenou, A.; Koumantakis, G.; Vassiliadis, S.; Mahutte, N.G. Altered Expression of Interleukin-18 in the Peritoneal Fluid of Women with Endometriosis. Fertil. Steril. 2003, 80, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Lédée-Bataille, N.; Olivennes, F.; Kadoch, J.; Dubanchet, S.; Frydman, N.; Chaouat, G.; Frydman, R. Detectable Levels of Interleukin-18 in Uterine Luminal Secretions at Oocyte Retrieval Predict Failure of the Embryo Transfer. Hum. Reprod. 2004, 19, 1968–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbitoff, Y.A.; Tsarev, A.A.; Vashukova, E.S.; Maksiutenko, E.M.; Kovalenko, L.V.; Belotserkovtseva, L.D.; Glotov, A.S. A Data-Driven Review of the Genetic Factors of Pregnancy Complications. Int. J. Mol. Sci. 2020, 21, 3384. [Google Scholar] [CrossRef] [PubMed]
- Nathan, A.; Reinhardt, P.; Kruspe, D.; Jörß, T.; Groth, M.; Nolte, H.; Habenicht, A.; Herrmann, J.; Holschbach, V.; Toth, B.; et al. The Wilms Tumor Protein Wt1 Contributes to Female Fertility by Regulating Oviductal Proteostasis. Hum. Mol. Genet. 2017, 26, 1694–1705. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, S.; Canis, M.; Darcha, C.; Déchelotte, P.J.; Pouly, J.L.; Mage, G. Expression of WT1 Is Down-Regulated in Eutopic Endometrium Obtained during the Midsecretory Phase from Patients with Endometriosis. Fertil. Steril. 2006, 86, 554–558. [Google Scholar] [CrossRef]
- Nakato, M.; Shiranaga, N.; Tomioka, M.; Watanabe, H.; Kurisu, J.; Kengaku, M.; Komura, N.; Ando, H.; Kimura, Y.; Kioka, N.; et al. ABCA13 Dysfunction Associated with Psychiatric Disorders Causes Impaired Cholesterol Trafficking. J. Biol. Chem. 2021, 296, 100166. [Google Scholar] [CrossRef]
- Sántha, P.; Dobos, I.; Kis, G.; Jancsó, G. Role of Gangliosides in Peripheral Pain Mechanisms. Int. J. Mol. Sci. 2020, 21, 1005. [Google Scholar] [CrossRef] [Green Version]
- Pappas, C.T.; Bliss, K.T.; Zieseniss, A.; Gregorio, C.C. The Nebulin Family: An Actin Support Group. Trends Cell Biol. 2011, 21, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Althubiti, M. Mutation Frequencies in Endometrial Cancer Patients of Different Ethnicities and Tumor Grades: An Analytical Study. Saudi. J. Med. Med. Sci. 2019, 7, 16. [Google Scholar] [CrossRef]
- Lee, A.S.; Rusch, J.; Lima, A.C.; Usmani, A.; Huang, N.; Lepamets, M.; Vigh-Conrad, K.A.; Worthington, R.E.; Mägi, R.; Wu, X.; et al. Rare Mutations in the Complement Regulatory Gene CSMD1 Are Associated with Male and Female Infertility. Nat. Commun. 2019, 10, 4626. [Google Scholar] [CrossRef] [Green Version]
- Uimari, O.; Rahmioglu, N.; Nyholt, D.R.; Vincent, K.; Missmer, S.A.; Becker, C.; Morris, A.P.; Montgomery, G.W.; Zondervan, K.T. Genome-Wide Genetic Analyses Highlight Mitogen-Activated Protein Kinase (MAPK) Signaling in the Pathogenesis of Endometriosis. Hum. Reprod. 2017, 32, 780–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamal, M.; Holliday, D.L.; Morrison, E.E.; Speirs, V.; Toomes, C.; Bell, S.M. Loss of CSMD1 Expression Disrupts Mammary Duct Formation While Enhancing Proliferation, Migration and Invasion. Oncol. Rep. 2017, 38, 283–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Demographic and Clinical Data (n = 80) | Mean ± sd or N (%) |
|---|---|
| Age (years) | 37.6 ± 9.2 |
| Body-mass index (kg/m2) | 22.9 ± 4.0 |
| Age at menarche (years) | 12.1 ± 1.5 |
| EM diagnosis | |
| Surgery and histopathological exam | 73 (91.3%) |
| Transvaginal and transrectal ultrasound and/or magnetic resonance imaging (MRI) | 7 (8.75%) |
| rASRM classification | |
| Stage I | 8 (10.0%) |
| Surgical confirmation | 4 (5.00%) |
| Stage II | 13 (16.3%) |
| Surgical confirmation | 13 (16.3%) |
| Stage III–IV | 59 (73.8%) |
| Surgical confirmation | 56 (70.0%) |
| Gynaecological data | |
| Pregnancies | 42 (52.5%) |
| Parity | 38 (47.5%) |
| Term Labour | 38 (47.5%) |
| Abortion | 14 (17.5%) |
| VTP | 4 (5.00%) |
| MAP | 2 (2.50%) |
| Infertility diagnosis * | 14 (20.6%) |
| EM Medical therapy | 50 (62.5%) |
| Ongoing medical therapy | 36 (45.0%) |
| Progestins ** | 16 (51.6%) |
| Combined oestrogen–progestins ** | 11 (35.5%) |
| Hormone-releasing intrauterine devices (IUDs) ** | 4 (5.00%) |
| Anti-inflammatory drugs only ** | 13 (16.3%) |
| Pain evaluation and intensity assessment *** | |
| Ovulation pain | 28 (40.6%) |
| Intensity | 6.9 ± 2.1 |
| Pre-menstrual pain | 27 (39.1%) |
| Intensity | 6.3 ± 2.4 |
| Post-menstrual pain | 14 (20.3%) |
| Intensity | 8.1 ± 2.0 |
| Dysmenorrhea | 44 (63.8%) |
| Intensity | 6.8 ± 1.8 |
| Dyspareunia | 30 (43.5%) |
| Intensity | 6.0 ± 2.5 |
| Dyschezia | 24 (34.8%) |
| Intensity | 5.4 ± 2.0 |
| Dysuria | 4 (5.8%) |
| Intensity | 5.0 ± 3.5 |
| Gene Name (Isoform), Size | HGVS Coding, Protein | AF | PaPI | PolyPhen | SIFT | DANN | dbscSNV | SpliceAI | MetaDome Analysis | Patient ID |
|---|---|---|---|---|---|---|---|---|---|---|
| LAMA5 (NM_005560.6) 59 kb | c.7375G>A, p.(Ala2459Thr) | 0.00003 | D | D | T | D | NA | NA | Slightly intolerant | 5 |
| c.2185G>A, p.(Gly729Ser) | 2.2 × 10−5 | D | D | D | D | NA | NA | Slightly intolerant | 12 | |
| c.1043C>T, p.(Ala348Val) | 0.00004 | D | D | D | D | NA | NA | Intolerant | 23 | |
| c.2248G>A, p.(Val750Met) | 0.00011 | D | D | D | D | NA | NA | Intolerant | 55 | |
| c.8269G>A, p.(Ala2757Thr) | 4 × 10−6 | D | D | D | D | NA | NA | Intolerant | 77 | |
| SYNE2 (NM_182914.3) 373 kb | c.12856A>C, p.(Ile4286Leu) | 2.9 × 10−5 | D | D | T | D | NA | NA | Neutral | 16 |
| c.18001G>A, p.(Asp6001Asn) | 0.00012 | D | D | T | D | NA | NA | Intolerant | 23 | |
| c.15757G>T, p.(Glu5253 *) | NA | D | NA | NA | D | NA | NA | Tolerant | 26 | |
| c.16018G>T, p.(Val5340Phe) | 2.8 × 10−5 | D | D | D | D | NA | NA | Slightly tolerant | 31 | |
| c.18565C>T, p.(Arg6189Trp) | 4 × 10−6 | D | D | D | D | NA | NA | Slightly tolerant | 45 | |
| MAP3K4 (NM_005922.4) 126 kb | c.2566C>A, p.(Pro856Thr) | NA | D | D | T | D | NA | NA | Intolerant | 4 |
| c.2659G>A, p.(Asp887Asn) | 1.4 × 10−5 | D | D | T | D | NA | NA | Intolerant | 28, 63, 64 ¶ | |
| c.3590_3598dupCTGCTGCTG, p.(Ala1197_Ala1199dup) | 0.00014 | D | NA | NA | NA | NA | NA | NA | 68 | |
| FCRL3 (NM_052939.4) 27 kb | c.1776_1783dupTCTGCTGC, p.(His595fs) | NA | D | NA | NA | NA | NA | NA | Neutral | 2 |
| c.1643A>G, p.(Asn548Ser) | 5.7 × 10−5 | D | D | D | D | NA | NA | Intolerant | 15 | |
| c.958T>A, p.(Phe320Ile) | 1.2 × 10−5 | D | D | D | D | NA | NA | Tolerant | 34, 35 ¶ | |
| GREB1 (NM_014668.4) 109 kb | c.5780G>A, p.(Arg1927His) | 3.2 × 10−5 | D | D | D | D | NA | NA | Neutral | 3 |
| c.5782G>A, p.(Asp1928Asn) | NA | D | D | D | D | NA | NA | Neutral | 8 | |
| c.1241C>T, p.(Ser414Phe) | 2.8 × 10−5 | D | D | T | D | NA | NA | Intolerant | 54 | |
| VEZT (NM_017599.4) 85 kb | c.514T>C, p.(Trp172Arg) | 0.00004 | D | D | T | D | NA | NA | Intolerant | 24 |
| c.1428G>T, p.(Lys476Asn) | 0.00019 | D | T | D | D | NA | NA | Intolerant | 42, 61 | |
| SYNE1 (NM_182961.4) 516 kb | c.21095A>G, p.(Gln7032Arg) | 4.4 × 10−5 | D | D | D | D | NA | NA | Intolerant | 40 |
| c.16111C>T, p.(Arg5371 *) | 1.6 × 10−5 | D | NA | NA | D | NA | NA | Tolerant | 47 | |
| C3 (NM_000064.4) 53 kb | c.2951-5_2951-3delTGC | 0.00074 | NA | NA | NA | NA | NA | D | NA | 24 |
| NM_000064.4:c.3431C>T | 6.7 × 10−5 | D | D | D | D | NA | NA | Intolerant | 80 | |
| MMP3 (NM_002422.5) 8.0 kb | c.1153G>A, p.(Val385Met) | 0.00039 | D | D | D | D | NA | NA | Intolerant | 22 |
| c.484T>C, p.(Ser162Pro) | 4 × 10−6 | D | D | D | D | NA | NA | Neutral | 32 | |
| MMP9 (NM_004994.3) 7.7 kb | c.1420dupA, p.(Thr474fs) | 0.00011 | D | NA | NA | NA | NA | NA | Neutral | 22 |
| c.1127C>T, p.(Thr376Ile) | 4 × 10−6 | D | D | T | D | NA | NA | Intolerant | 36 | |
| TYK2 (NM_003331.5) 30 kb | c.3475C>T, p.(Arg1159Cys) | 1.2 × 10−5 | D | D | D | D | NA | NA | Intolerant | 15 |
| c.256C>A, p.(Pro86Thr) | 5.3 × 10−5 | D | D | D | D | NA | NA | Intolerant | 59 | |
| VEGFA (NM_003376.6) 16 kb | c.337G>C, p.(Ala113Pro) | 8 × 10−6 | D | D | D | D | NA | NA | NA | 67 |
| c.1184G>C, p.(Arg395Pro) | NA | D | D | D | D | NA | NA | Intolerant | 70 | |
| RHOJ (NM_020663.4) 89 kb | c.554C>T, p.(Ala185Val) | 2.5 × 10−5 | D | T | D | D | NA | NA | Slightly tolerant | 4, 69 |
| Gene Name (Isoform), Size | HGVS Coding, Protein | AF | PaPI | PolyPhen | SIFT | DANN | dbscSNV | SpliceAI | MetaDome Analysis | Patient ID |
|---|---|---|---|---|---|---|---|---|---|---|
| KAZN (NM_201628.2) 519 kb | c.236G>A, p.(Arg79Gln) | 0.000049 | D | D | T | D | NA | NA | Tolerant | 3 |
| IL18 (NM_001562.3) 21 kb | c.113T>C, p.(Phe38Ser) | NA | D | D | D | D | NA | NA | Highly intolerant | 9 |
| WT1 (NM_024426.6) 48 kb | c.475G>A, p.(Glu159Lys) | 0.000034 | D | T | D | D | NA | NA | NA | 19 |
| CYP19A1 (NM_000103.4) 130 kb | c.1327G>A, p.(Ala443Thr) | 0.000012 | D | D | D | D | NA | NA | Neutral | 50 |
| IL1A (NM_000575.5) 11 kb | c.526G>C, p.(Asp176His) | 0.000064 | D | D | T | D | NA | NA | Slightly intolerant | 67 |
| IL2RB (NM_000878.3) 49 kb | c.1640C>G, p.(Pro547Arg) | NA | D | D | D | D | NA | NA | Slightly tolerant | 14 |
| LILRB2 (NM_001278406.1) 7.4 kb | c.964C>T, p.(Arg322Cys) | 0.000004 | T | D | T | D | NA | NA | Slightly tolerant | 14 |
| ZNF366 (NM_152625.1) 65 kb | c.1402G>A, p.(Val468Met) | NA | D | D | T | D | NA | NA | Intolerant | 13 |
| Gene Name (Isoform), Size | HGVS Coding, Protein | AF | PaPI | PolyPhen | SIFT | DANN | dbscSNV | SpliceAI | MetaDome Analysis | Patient ID |
|---|---|---|---|---|---|---|---|---|---|---|
| ABCA13 (NM_152701.5) 476 kb | c.14579G>A, p.(Gly4860Glu) | 0.000004 | D | D | D | D | NA | NA | Intolerant | 6 |
| c.2039A>G, p.(Asn680Ser) | 0.000004 | T | T | D | D | NA | NA | Slightly intolerant | 9 | |
| c.410_421delGACTTTGGGTAG, p.(Arg137_Glu141delinsLys) | 0.00009 | D | NA | NA | NA | NA | NA | NA | 12 | |
| c.13246A>G, p.(lle4416Val) | 0.000007 | T | D | T | D | NA | NA | Intolerant | 14 | |
| c.13243delC, p.(Ile4416fs) | 0.000024 | D | NA | NA | NA | NA | NA | Intolerant | 27 | |
| c.3248T>A, p.(Met1083Lys) | 0.000032 | D | T | D | D | NA | NA | Intolerant | 31 | |
| c.8030T>C, p.(Ile2677Thr) | NA | D | D | D | D | NA | NA | Highly tolerant | 31 | |
| c.11981C>T, p.(Ser3994Leu) | 0.000024 | D | D | D | D | NA | NA | Tolerant | 56 | |
| c.14185C>T, p.(Arg4729Cys) | 0.000336 | D | D | T | D | NA | NA | Slightly intolerant | 75 | |
| NEB (NM_001164508.2) 249 kb | c.16817A>G, p.(Tyr5606Cys) | 0.000098 | D | D | D | D | NA | NA | Neutral | 7 |
| c.8674C>T, p.(Leu2892Phe) | NA | D | D | T | D | NA | NA | Slightly intolerant | 8 | |
| c.4105G>A, p.(Glu1369Lys) | 0.00002 | D | D | T | D | NA | NA | Intolerant | 13 | |
| c.4558C>A, p.(Pro1520Thr) | NA | D | D | T | D | NA | NA | Intolerant | 17 | |
| c.4289T>A, p.(Ile1430Asn) | 0.000004 | D | D | D | D | NA | NA | Intolerant | 32 | |
| c.8317C>T, p.(Arg2773Trp) | 0.000036 | D | D | D | D | NA | NA | Neutral | 33 | |
| c.2771A>C, p.(Tyr924Ser) | 0.000247 | D | D | D | D | NA | NA | Intolerant | 60 | |
| c.18862G>A, p.(Val6288Ile) | 0.000385 | D | T | D | D | NA | NA | Tolerant | 61 | |
| CSMD1 (NM_033225.6) 2.1 Mb | c.2783C>T, p.(Ala928Val) | 0.000004 | D | D | T | D | D | NA | NA | 10 |
| c.3023T>A, p.(Ile1008Asn) | NA | D | D | T | D | NA | NA | NA | 17 | |
| c.4553T>C, p.(Ile1518Thr) | 0.000005 | D | D | T | D | NA | NA | NA | 58 | |
| c.3333T>A, p.(Asn1111Lys) | 0.000049 | D | D | T | D | NA | NA | NA | 62 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santin, A.; Spedicati, B.; Morgan, A.; Lenarduzzi, S.; Tesolin, P.; Nardone, G.G.; Mazzà, D.; Di Lorenzo, G.; Romano, F.; Buonomo, F.; et al. Puzzling Out the Genetic Architecture of Endometriosis: Whole-Exome Sequencing and Novel Candidate Gene Identification in a Deeply Clinically Characterised Cohort. Biomedicines 2023, 11, 2122. https://doi.org/10.3390/biomedicines11082122
Santin A, Spedicati B, Morgan A, Lenarduzzi S, Tesolin P, Nardone GG, Mazzà D, Di Lorenzo G, Romano F, Buonomo F, et al. Puzzling Out the Genetic Architecture of Endometriosis: Whole-Exome Sequencing and Novel Candidate Gene Identification in a Deeply Clinically Characterised Cohort. Biomedicines. 2023; 11(8):2122. https://doi.org/10.3390/biomedicines11082122
Chicago/Turabian StyleSantin, Aurora, Beatrice Spedicati, Anna Morgan, Stefania Lenarduzzi, Paola Tesolin, Giuseppe Giovanni Nardone, Daniela Mazzà, Giovanni Di Lorenzo, Federico Romano, Francesca Buonomo, and et al. 2023. "Puzzling Out the Genetic Architecture of Endometriosis: Whole-Exome Sequencing and Novel Candidate Gene Identification in a Deeply Clinically Characterised Cohort" Biomedicines 11, no. 8: 2122. https://doi.org/10.3390/biomedicines11082122