
Recent Advancements in AAV-Vectored Immunoprophylaxis in the Nonhuman Primate Model
 , , and
, , and
Abstract
:1. Introduction
2. AAV VIP in the NHP Model
2.1. AAV-Mediated Expression of Immunoadhesins
2.2. AAV-Mediated Expression of Antibody-like Molecules
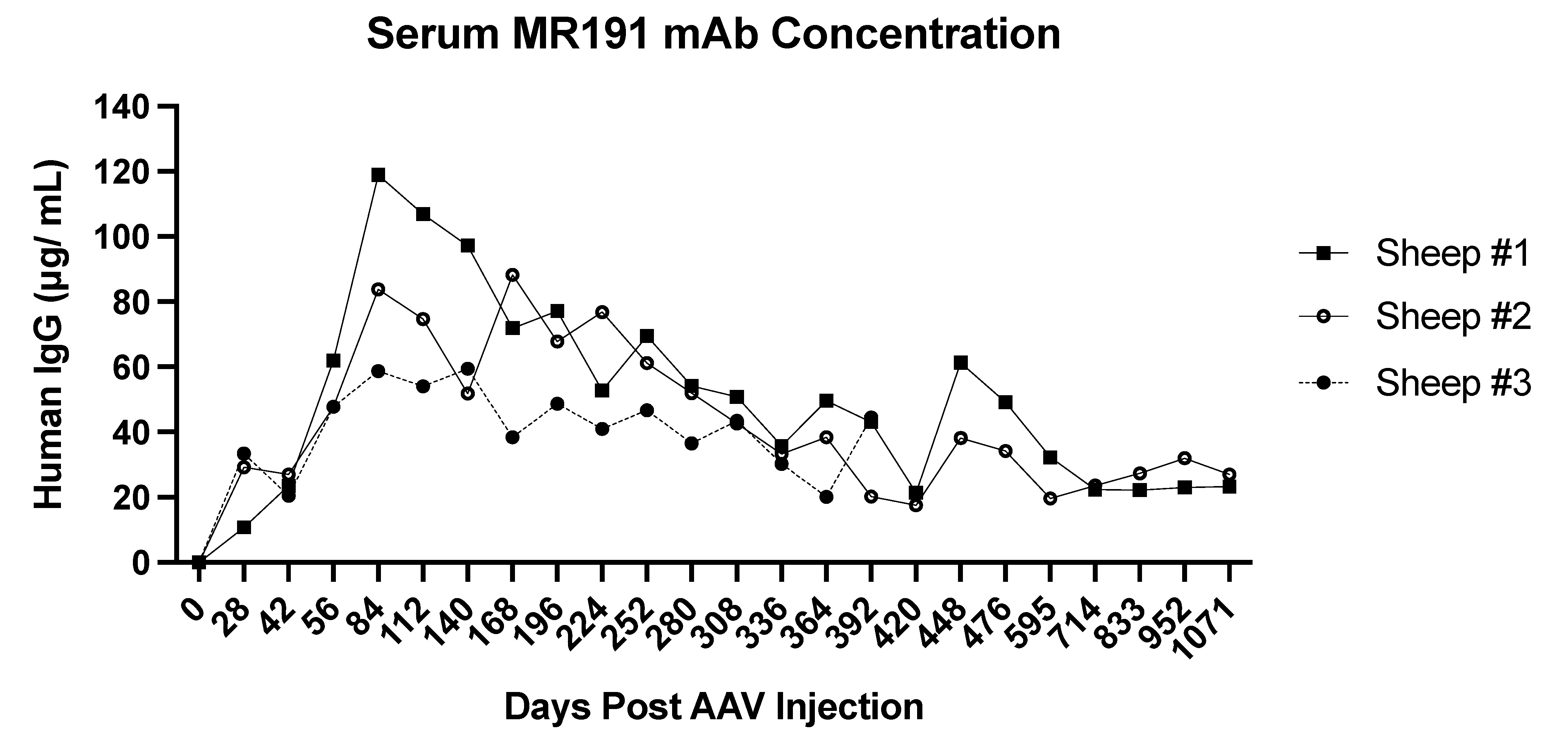
2.3. AAV-Mediated Expression of Non-Native Full-Length Antibodies
2.4. AAV-Mediated Expression of Native Full-Length Antibodies
3. AAV VIP in Other Large-Animal Models
4. AAV VIP in Human Clinical Trials
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amanna, I.J.; Slifka, M.K. Successful Vaccines. In Vaccination Strategies Against Highly Variable Pathogens; Hangartner, L., Burton, D.R., Eds.; Current Topics in Microbiology and Immunology; Springer International Publishing: Cham, Switzerland, 2018; Volume 428, pp. 1–30. ISBN 978-3-030-58003-2. [Google Scholar]
- Welles, H.C.; Jennewein, M.F.; Mason, R.D.; Narpala, S.; Wang, L.; Cheng, C.; Zhang, Y.; Todd, J.-P.; Lifson, J.D.; Balazs, A.B.; et al. Vectored Delivery of Anti-SIV Envelope Targeting MAb via AAV8 Protects Rhesus Macaques from Repeated Limiting Dose Intrarectal Swarm SIVsmE660 Challenge. PLoS Pathog. 2018, 14, e1007395. [Google Scholar] [CrossRef]
- Marcotte, H.; Hammarström, L. Passive Immunization. In Mucosal Immunology; Elsevier: Amsterdam, The Netherlands, 2015; pp. 1403–1434. ISBN 978-0-12-415847-4. [Google Scholar]
- Balazs, A.B.; Chen, J.; Hong, C.M.; Rao, D.S.; Yang, L.; Baltimore, D. Antibody-Based Protection against HIV Infection by Vectored Immunoprophylaxis. Nature 2012, 481, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Chng, J.; Wang, T.; Nian, R.; Lau, A.; Hoi, K.M.; Ho, S.C.; Gagnon, P.; Bi, X.; Yang, Y. Cleavage Efficient 2A Peptides for High Level Monoclonal Antibody Expression in CHO Cells. mAbs 2015, 7, 403–412. [Google Scholar] [CrossRef] [Green Version]
- Lopes, J.A.; Rghei, A.D.; Thompson, B.; Susta, L.; Khursigara, C.M.; Wootton, S.K. Overcoming Barriers to Preventing and Treating P. Aeruginosa Infections Using AAV Vectored Immunoprophylaxis. Biomedicines 2022, 10, 3162. [Google Scholar] [CrossRef]
- Verdera, H.C.; Kuranda, K.; Mingozzi, F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. 2020, 28, 723–746. [Google Scholar] [CrossRef] [PubMed]
- Daya, S.; Berns, K.I. Gene Therapy Using Adeno-Associated Virus Vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.R.; Al-Zaidy, S.A.; Rodino-Klapac, L.R.; Goodspeed, K.; Gray, S.J.; Kay, C.N.; Boye, S.L.; Boye, S.E.; George, L.A.; Salabarria, S.; et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2021, 29, 464–488. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Anselmo, A.C.; Mitragotri, S. Viral VECTOR-BASED Gene Therapies in the Clinic. Bioeng. Transl. Med. 2022, 7, e10258. [Google Scholar] [CrossRef] [PubMed]
- Penaud-Budloo, M.; Le Guiner, C.; Nowrouzi, A.; Toromanoff, A.; Chérel, Y.; Chenuaud, P.; Schmidt, M.; Von Kalle, C.; Rolling, F.; Moullier, P.; et al. Adeno-Associated Virus Vector Genomes Persist as Episomal Chromatin in Primate Muscle. J. Virol. 2008, 82, 7875–7885. [Google Scholar] [CrossRef] [Green Version]
- Toromanoff, A.; Chérel, Y.; Guilbaud, M.; Penaud-Budloo, M.; Snyder, R.O.; Haskins, M.E.; Deschamps, J.-Y.; Guigand, L.; Podevin, G.; Arruda, V.R.; et al. Safety and Efficacy of Regional Intravenous (RI) Versus Intramuscular (IM) Delivery of RAAV1 and RAAV8 to Nonhuman Primate Skeletal Muscle. Mol. Ther. 2008, 16, 1291–1299. [Google Scholar] [CrossRef]
- Balazs, A.B.; Ouyang, Y.; Hong, C.M.; Chen, J.; Nguyen, S.M.; Rao, D.S.; An, D.S.; Baltimore, D. Vectored Immunoprophylaxis Protects Humanized Mice from Mucosal HIV Transmission. Nat. Med. 2014, 20, 296–300. [Google Scholar] [CrossRef] [Green Version]
- Johnson, P.R.; Schnepp, B.C.; Zhang, J.; Connell, M.J.; Greene, S.M.; Yuste, E.; Desrosiers, R.C.; Reed Clark, K. Vector-Mediated Gene Transfer Engenders Long-Lived Neutralizing Activity and Protection against SIV Infection in Monkeys. Nat. Med. 2009, 15, 901–906. [Google Scholar] [CrossRef] [Green Version]
- Saunders, K.O.; Wang, L.; Joyce, M.G.; Yang, Z.-Y.; Balazs, A.B.; Cheng, C.; Ko, S.-Y.; Kong, W.-P.; Rudicell, R.S.; Georgiev, I.S.; et al. Broadly Neutralizing Human Immunodeficiency Virus Type 1 Antibody Gene Transfer Protects Nonhuman Primates from Mucosal Simian-Human Immunodeficiency Virus Infection. J. Virol. 2015, 89, 8334–8345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, S.P.; Martinez-Navio, J.M.; Piatak, M.; Lifson, J.D.; Gao, G.; Desrosiers, R.C. AAV-Delivered Antibody Mediates Significant Protective Effects against SIVmac239 Challenge in the Absence of Neutralizing Activity. PLoS Pathog. 2015, 11, e1005090. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.R.; Fellinger, C.H.; Kattenhorn, L.M.; Davis-Gardner, M.E.; Weber, J.A.; Alfant, B.; Zhou, A.S.; Prasad, N.R.; Kondur, H.R.; Newton, W.A.; et al. AAV-Delivered ECD4-Ig Protects Rhesus Macaques from High-Dose SIVmac239 Challenges. Sci. Transl. Med. 2019, 11, eaau5409. [Google Scholar] [CrossRef] [PubMed]
- Priddy, F.H.; Lewis, D.J.M.; Gelderblom, H.C.; Hassanin, H.; Streatfield, C.; LaBranche, C.; Hare, J.; Cox, J.H.; Dally, L.; Bendel, D.; et al. Adeno-Associated Virus Vectored Immunoprophylaxis to Prevent HIV in Healthy Adults: A Phase 1 Randomised Controlled Trial. Lancet HIV 2019, 6, e230–e239. [Google Scholar] [CrossRef] [Green Version]
- Casazza, J.P.; Cale, E.M.; Narpala, S.; Yamshchikov, G.V.; Coates, E.E.; Hendel, C.S.; Novik, L.; Holman, L.A.; Widge, A.T.; Apte, P.; et al. Safety and Tolerability of AAV8 Delivery of a Broadly Neutralizing Antibody in Adults Living with HIV: A Phase 1, Dose-Escalation Trial. Nat. Med. 2022, 28, 1022–1030. [Google Scholar] [CrossRef]
- Deal, C.; Balazs, A.B.; Espinosa, D.A.; Zavala, F.; Baltimore, D.; Ketner, G. Vectored Antibody Gene Delivery Protects against Plasmodium Falciparum Sporozoite Challenge in Mice. Proc. Natl. Acad. Sci. USA 2014, 111, 12528–12532. [Google Scholar] [CrossRef]
- Skaricic, D.; Traube, C.; De, B.; Joh, J.; Boyer, J.; Crystal, R.G.; Worgall, S. Genetic Delivery of an Anti-RSV Antibody to Protect against Pulmonary Infection with RSV. Virology 2008, 378, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Balazs, A.B.; Bloom, J.D.; Hong, C.M.; Rao, D.S.; Baltimore, D. Broad Protection against Influenza Infection by Vectored Immunoprophylaxis in Mice. Nat. Biotechnol. 2013, 31, 647–652. [Google Scholar] [CrossRef]
- Van Lieshout, L.P.; Soule, G.; Sorensen, D.; Frost, K.L.; He, S.; Tierney, K.; Safronetz, D.; Booth, S.A.; Kobinger, G.P.; Qiu, X.; et al. Intramuscular Adeno-Associated Virus–Mediated Expression of Monoclonal Antibodies Provides 100% Protection Against Ebola Virus Infection in Mice. J. Infect. Dis. 2018, 217, 916–925. [Google Scholar] [CrossRef]
- Rghei, A.D.; Van Lieshout, L.P.; Cao, W.; He, S.; Tierney, K.; Lopes, J.A.; Zielinska, N.; Baracuhy, E.M.; Campbell, E.S.B.; Minott, J.A.; et al. Adeno-Associated Virus Mediated Expression of Monoclonal Antibody MR191 Protects Mice against Marburg Virus and Provides Long-Term Expression in Sheep. Gene Ther. 2022. [Google Scholar] [CrossRef] [PubMed]
- Backes, I.M.; Byrd, B.K.; Slein, M.D.; Patel, C.D.; Taylor, S.A.; Garland, C.R.; MacDonald, S.W.; Balazs, A.B.; Davis, S.C.; Ackerman, M.E.; et al. Maternally Transferred MAbs Protect Neonatal Mice from HSV-Induced Mortality and Morbidity. J. Exp. Med. 2022, 219, e20220110. [Google Scholar] [CrossRef] [PubMed]
- Limberis, M.P.; Adam, V.S.; Wong, G.; Gren, J.; Kobasa, D.; Ross, T.M.; Kobinger, G.P.; Tretiakova, A.; Wilson, J.M. Intranasal Antibody Gene Transfer in Mice and Ferrets Elicits Broad Protection Against Pandemic Influenza. Sci. Transl. Med. 2013, 5, 187ra72. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Hernandez, T.; Carnathan, D.G.; Moyle, P.M.; Toth, I. The Contribution of Non-Human Primate Models to the Development of Human Vaccines. Discov. Med. 2015, 18, 313. [Google Scholar]
- Sundling, C.; Li, Y.; Huynh, N.; Poulsen, C.; Wilson, R.; O’Dell, S.; Feng, Y.; Mascola, J.R.; Wyatt, R.T.; Karlsson Hedestam, G.B. High-Resolution Definition of Vaccine-Elicited B Cell Responses Against the HIV Primary Receptor Binding Site. Sci. Transl. Med. 2012, 4, 142ra96. [Google Scholar] [CrossRef] [Green Version]
- Estes, J.D.; Wong, S.W.; Brenchley, J.M. Nonhuman Primate Models of Human Viral Infections. Nat. Rev. Immunol. 2018, 18, 390–404. [Google Scholar] [CrossRef]
- Gardner, M.R.; Kattenhorn, L.M.; Kondur, H.R.; Von Schaewen, M.; Dorfman, T.; Chiang, J.J.; Haworth, K.G.; Decker, J.M.; Alpert, M.D.; Bailey, C.C.; et al. AAV-Expressed ECD4-Ig Provides Durable Protection from Multiple SHIV Challenges. Nature 2015, 519, 87–91. [Google Scholar] [CrossRef] [Green Version]
- Gardner, M.R.; Fetzer, I.; Kattenhorn, L.M.; Davis-Gardner, M.E.; Zhou, A.S.; Alfant, B.; Weber, J.A.; Kondur, H.R.; Martinez-Navio, J.M.; Fuchs, S.P.; et al. Anti-Drug Antibody Responses Impair Prophylaxis Mediated by AAV-Delivered HIV-1 Broadly Neutralizing Antibodies. Mol. Ther. 2019, 27, 650–660. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Navio, J.M.; Fuchs, S.P.; Pantry, S.N.; Lauer, W.A.; Duggan, N.N.; Keele, B.F.; Rakasz, E.G.; Gao, G.; Lifson, J.D.; Desrosiers, R.C. Adeno-Associated Virus Delivery of Anti-HIV Monoclonal Antibodies Can Drive Long-Term Virologic Suppression. Immunity 2019, 50, 567–575.e5. [Google Scholar] [CrossRef] [Green Version]
- Davis-Gardner, M.E.; Weber, J.A.; Xie, J.; Pekrun, K.; Alexander, E.A.; Weisgrau, K.L.; Furlott, J.R.; Rakasz, E.G.; Kay, M.A.; Gao, G.; et al. A Strategy for High Antibody Expression with Low Anti-Drug Antibodies Using AAV9 Vectors. Front. Immunol. 2023, 14, 1105617. [Google Scholar] [CrossRef]
- Lewis, A.D.; Chen, R.; Montefiori, D.C.; Johnson, P.R.; Clark, K.R. Generation of Neutralizing Activity against Human Immunodeficiency Virus Type 1 in Serum by Antibody Gene Transfer. J. Virol. 2002, 76, 8769–8775. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A.; Chamow, S. Immunoadhesins as Research Tools and Therapeutic Agents. Curr. Opin. Immunol. 1997, 9, 195–200. [Google Scholar] [CrossRef]
- Rizzuto, C.D.; Wyatt, R.; Hernández-Ramos, N.; Sun, Y.; Kwong, P.D.; Hendrickson, W.A.; Sodroski, J. A Conserved HIV Gp120 Glycoprotein Structure Involved in Chemokine Receptor Binding. Science 1998, 280, 1949–1953. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Lam, S.N.; Acharya, P.; Tang, M.; Xiang, S.-H.; Hussan, S.S.; Stanfield, R.L.; Robinson, J.; Sodroski, J.; Wilson, I.A.; et al. Structures of the CCR5 N Terminus and of a Tyrosine-Sulfated Antibody with HIV-1 Gp120 and CD4. Science 2007, 317, 1930–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagenaur, L.A.; Villarroel, V.A.; Bundoc, V.; Dey, B.; Berger, E.A. SCD4-17b Bifunctional Protein: Extremely Broad and Potent Neutralization of HIV-1 Env Pseudotyped Viruses from Genetically Diverse Primary Isolates. Retrovirology 2010, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Humes, D.; Emery, S.; Laws, E.; Overbaugh, J. A Species-Specific Amino Acid Difference in the Macaque CD4 Receptor Restricts Replication by Global Circulating HIV-1 Variants Representing Viruses from Recent Infection. J. Virol. 2012, 86, 12472–12483. [Google Scholar] [CrossRef] [Green Version]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [Green Version]
- Rghei, A.D.; Van Lieshout, L.P.; Santry, L.A.; Guilleman, M.M.; Thomas, S.P.; Susta, L.; Karimi, K.; Bridle, B.W.; Wootton, S.K. AAV Vectored Immunoprophylaxis for Filovirus Infections. Trop. Med. Infect. Dis. 2020, 5, 169. [Google Scholar] [CrossRef]
- De, B.P.; Hackett, N.R.; Crystal, R.G.; Boyer, J.L. Rapid/Sustained Anti-Anthrax Passive Immunity Mediated by Co-Administration of Ad/AAV. Mol. Ther. 2008, 16, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Salazar, G.; Zhang, N.; Fu, T.-M.; An, Z. Antibody Therapies for the Prevention and Treatment of Viral Infections. npj Vaccines 2017, 2, 19. [Google Scholar] [CrossRef] [Green Version]
- West, A.P.; Galimidi, R.P.; Gnanapragasam, P.N.P.; Bjorkman, P.J. Single-Chain Fv-Based Anti-HIV Proteins: Potential and Limitations. J. Virol. 2012, 86, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mingozzi, F.; Chen, Y.; Murphy, S.L.; Edmonson, S.C.; Tai, A.; Price, S.D.; Metzger, M.E.; Zhou, S.; Wright, J.F.; Donahue, R.E.; et al. Pharmacological Modulation of Humoral Immunity in a Nonhuman Primate Model of AAV Gene Transfer for Hemophilia B. Mol. Ther. 2012, 20, 1410–1416. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Navio, J.M.; Fuchs, S.P.; Mendes, D.E.; Rakasz, E.G.; Gao, G.; Lifson, J.D.; Desrosiers, R.C. Long-Term Delivery of an Anti-SIV Monoclonal Antibody With AAV. Front. Immunol. 2020, 11, 449. [Google Scholar] [CrossRef] [Green Version]
- Ko, S.-Y.; Pegu, A.; Rudicell, R.S.; Yang, Z.; Joyce, M.G.; Chen, X.; Wang, K.; Bao, S.; Kraemer, T.D.; Rath, T.; et al. Enhanced Neonatal Fc Receptor Function Improves Protection against Primate SHIV Infection. Nature 2014, 514, 642–645. [Google Scholar] [CrossRef] [Green Version]
- Zalevsky, J.; Chamberlain, A.K.; Horton, H.M.; Karki, S.; Leung, I.W.L.; Sproule, T.J.; Lazar, G.A.; Roopenian, D.C.; Desjarlais, J.R. Enhanced Antibody Half-Life Improves in Vivo Activity. Nat. Biotechnol. 2010, 28, 157–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boisgerault, F.; Gross, D.-A.; Ferrand, M.; Poupiot, J.; Darocha, S.; Richard, I.; Galy, A. Prolonged Gene Expression in Muscle Is Achieved Without Active Immune Tolerance Using MicrorRNA 142.3p-Regulated RAAV Gene Transfer. Hum. Gene Ther. 2013, 24, 393–405. [Google Scholar] [CrossRef]
- Majowicz, A.; Maczuga, P.; Kwikkers, K.L.; Van Der Marel, S.; Van Logtenstein, R.; Petry, H.; Van Deventer, S.J.; Konstantinova, P.; Ferreira, V. Mir-142-3p Target Sequences Reduce Transgene-Directed Immunogenicity Following Intramuscular Adeno-Associated Virus 1 Vector-Mediated Gene Delivery: Reduction of Transgene Directed Immune Responses in AAV1 Gene Delivery. J. Gene Med. 2013, 15, 219–232. [Google Scholar] [CrossRef]
- Agudo, J.; Ruzo, A.; Tung, N.; Salmon, H.; Leboeuf, M.; Hashimoto, D.; Becker, C.; Garrett-Sinha, L.-A.; Baccarini, A.; Merad, M.; et al. The MiR-126–VEGFR2 Axis Controls the Innate Response to Pathogen-Associated Nucleic Acids. Nat. Immunol. 2014, 15, 54–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shingai, M.; Donau, O.K.; Schmidt, S.D.; Gautam, R.; Plishka, R.J.; Buckler-White, A.; Sadjadpour, R.; Lee, W.R.; LaBranche, C.C.; Montefiori, D.C.; et al. Most Rhesus Macaques Infected with the CCR5-Tropic SHIVAD8 Generate Cross-Reactive Antibodies That Neutralize Multiple HIV-1 Strains. Proc. Natl. Acad. Sci. USA 2012, 109, 19769–19774. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, S.-R.; Li, L.-H.; Park, H.-J.; Park, J.-H.; Lee, K.Y.; Kim, M.-K.; Shin, B.A.; Choi, S.-Y. High Cleavage Efficiency of a 2A Peptide Derived from Porcine Teschovirus-1 in Human Cell Lines, Zebrafish and Mice. PLoS ONE 2011, 6, e18556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pekrun, K.; De Alencastro, G.; Luo, Q.-J.; Liu, J.; Kim, Y.; Nygaard, S.; Galivo, F.; Zhang, F.; Song, R.; Tiffany, M.R.; et al. Using a Barcoded AAV Capsid Library to Select for Clinically Relevant Gene Therapy Vectors. JCI Insight 2019, 4, e131610. [Google Scholar] [CrossRef] [Green Version]
- Paulk, N.K.; Pekrun, K.; Charville, G.W.; Maguire-Nguyen, K.; Wosczyna, M.N.; Xu, J.; Zhang, Y.; Lisowski, L.; Yoo, B.; Vilches-Moure, J.G.; et al. Bioengineered Viral Platform for Intramuscular Passive Vaccine Delivery to Human Skeletal Muscle. Mol. Ther. Methods Clin. Dev. 2018, 10, 144–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, L.M.; Phogat, S.K.; Chan-Hui, P.-Y.; Wagner, D.; Phung, P.; Goss, J.L.; Wrin, T.; Simek, M.D.; Fling, S.; Mitcham, J.L.; et al. Broad and Potent Neutralizing Antibodies from an African Donor Reveal a New HIV-1 Vaccine Target. Science 2009, 326, 285–289. [Google Scholar] [CrossRef] [Green Version]
- Euler, Z.; Bunnik, E.M.; Burger, J.A.; Boeser-Nunnink, B.D.M.; Grijsen, M.L.; Prins, J.M.; Schuitemaker, H. Activity of Broadly Neutralizing Antibodies, Including PG9, PG16, and VRC01, against Recently Transmitted Subtype B HIV-1 Variants from Early and Late in the Epidemic. J. Virol. 2011, 85, 7236–7245. [Google Scholar] [CrossRef] [Green Version]
- Phelps, M.; Balazs, A.B. Contribution to HIV Prevention and Treatment by Antibody-Mediated Effector Function and Advances in Broadly Neutralizing Antibody Delivery by Vectored Immunoprophylaxis. Front. Immunol. 2021, 12, 734304. [Google Scholar] [CrossRef]
- Xiang, Z.; Kurupati, R.K.; Li, Y.; Kuranda, K.; Zhou, X.; Mingozzi, F.; High, K.A.; Ertl, H.C.J. The Effect of CpG Sequences on Capsid-Specific CD8+ T Cell Responses to AAV Vector Gene Transfer. Mol. Ther. 2020, 28, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.K.; Wang, S.K.; Chu, C.J.; Copland, D.A.; Letizia, A.J.; Costa Verdera, H.; Chiang, J.J.; Sethi, M.; Wang, M.K.; Neidermyer, W.J.; et al. Engineering Adeno-Associated Viral Vectors to Evade Innate Immune and Inflammatory Responses. Sci. Transl. Med. 2021, 13, eabd3438. [Google Scholar] [CrossRef] [PubMed]
- Meliani, A.; Boisgerault, F.; Hardet, R.; Marmier, S.; Collaud, F.; Ronzitti, G.; Leborgne, C.; Costa Verdera, H.; Simon Sola, M.; Charles, S.; et al. Antigen-Selective Modulation of AAV Immunogenicity with Tolerogenic Rapamycin Nanoparticles Enables Successful Vector Re-Administration. Nat. Commun. 2018, 9, 4098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Z.; Kuranda, K.; Quinn, W.; Chekaoui, A.; Ambrose, R.; Hasanpourghai, M.; Novikov, M.; Newman, D.; Cole, C.; Zhou, X.; et al. The Effect of Rapamycin and Ibrutinib on Antibody Responses to Adeno-Associated Virus Vector-Mediated Gene Transfer. Hum. Gene Ther. 2022, 33, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Yuan, N.; Shen, Z.; Tang, C.; Shipp, S.; Qian, L.; Lu, Y.; Andolina, I.M.; Zhang, S.; Wu, J.; et al. Transduction Catalysis: Doxorubicin Amplifies RAAV-Mediated Gene Expression in the Cortex of Higher-Order Vertebrates. iScience 2021, 24, 102685. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Zak, R.; Zhang, Y.; Ding, W.; Godwin, S.; Munson, K.; Peluso, R.; Engelhardt, J.F. Distinct Classes of Proteasome-Modulating Agents Cooperatively Augment Recombinant Adeno-Associated Virus Type 2 and Type 5-Mediated Transduction from the Apical Surfaces of Human Airway Epithelia. J. Virol. 2004, 78, 2863–2874. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Transgene Class | Transgene Species Origin | Transgene Name | Transgene Function | Antibody Isotype | AAV Capsid Serotype | Vector Modification | AAV Vector Dose | Serum Concentration of Transgene * | ADA Response | Challenge Virus | Pre-Screening of NHPs ** | Immune Modulation | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Immunoadhesin | Simian | 4L6 | binds gp120 | rhesus IgG2 Fc | scAAV1 | NA | 2 × 1013 vg | 100–190 μg/mL time of challenge | 0 of 3 macaques | SIVmac316 IV | SIV negative | NA | [14] |
| 5L7 | 0–175 μg/mL time of challenge | 2 of 3 macaque | |||||||||||
| N4 | binds CD4 receptor on Env | ssAAV1 | 3–10 μg/mL time of challenge | 1 of 3 macaque | |||||||||
| Fc-Fusion Protein | Simian-Human chimera | rh-eCD4-IgG2I39N,mim2 | binds CD4 receptor on Env | rhesus CD4 domain 1 & 2 and IgG2 Fc & hinge | ssAAV1 | Ile39 mutation in CD4 domain | 2 × 1013 gc | 13–44 μg/mL | all 4 macque | SIVmac239 IV | AAV1 negative SIV negative | Co-administered 0.5 × 1013 gc AAV1-TPST2 | [17] |
| Simian-Human chimera | rh-eCD4-IgG2I39N,mim2 | binds CD4 receptor on Env | rhesus CD4 domain 1 & 2 and IgG2 Fc & hinge | ssAAV1 | Ile39 mutation in CD4 domain | 2.5 × 1013 gc | plateau 17–77 μg/mL for 40 weeks | 2 of 4 macaque | SHIV-AD8-EO IV | AAV1 negative | Co-administered 0.5 × 1013 gc AAV1-TPST2 | [30] | |
| Non-native full length antibodies | Human | VRC07 | CD4 receptor on Env | simianized IgG1 | AAV8 | somatic mutation of framework CDR region from human to macaque | 1 × 1013 vg | 2.5–7.7 μg/mL | all 4 macaque | NA | AAV8 negative | NA | [15] |
| 38.12 μg/mL | 3 of 6 macaque | CCR5-tropic SHIV-BaLP4 REC | CsA infusion 5 mg/kg; oral 15–30 mg/kg | ||||||||||
| Simian-Simian chimera | 5L7 | Env gp140 | rhesus IgG1 CH1 and CL domains | scAAV1 | NA | 2× 8 × 1012 vg | 20–55 μg/mL | 1 of 3 macaque | NA | AAV1 negative SIV negative | NA | [16] | |
| ssAAV1 | 1.6 × 1013 vg | 53–270 μg/mL | 2 of 3 macaque | SIVmac239 IV | |||||||||
| 4L6 | ssAAV1 | 2.5 × 1013 vg | 45–150 μg/mL | all 6 macaque | |||||||||
| Human | 3BNC117 | CD4 receptor on Env | rhesus IgG1 or IgG2 constant regions | AAV1 | NA | 1 × 1013 gc | 3–69 μg/mL | all 12 macaque | SHIV-AD8 IV | AAV1 negative SIV negative | NA | [31] | |
| NIH45–46 | |||||||||||||
| 10-1074 | N332 V3-glycan site on Env | ||||||||||||
| PGT121 | |||||||||||||
| Human | 10E8 | epitope in gp41 | rhesus IgG1 constant heavy and light chains | AAV1 | “LS” mutation in Fc & mir-142-3p sequence in vector | 2 × 1012 vg/kg | 2–10 μg/mL | all 4 macaque | SHIV-AD8e0 IV | AAV1 negative HIV negative SIV negative | NA | [32] | |
| 3BNC117 | CD4 receptor on Env gp120 | 0–150 μg/mL | |||||||||||
| 10-1074 | C2 V3 region of gp120 | 20–200 μg/mL | |||||||||||
| 3BNC117 | CD4 receptor on Env gp120 | AAV8 prime AAV1 boost | 2 × 1012 vg/kg prime 1 × 1012 vg/kg boost | 0.9–5.8 μg/mL | all 12 macaques | AAV1 negative AAV8 negative HIV negative | Gammagard 10% liquid (IVIG) 1 g/kg | ||||||
| 10-1074 | C2 V3 region of gp120 | 0.2–75 μg/mL | |||||||||||
| N6 | CD4 receptor on Env | 0–13 μg/mL | |||||||||||
| 35O22 | novel Env epitope | 0–8.9 μg/mL | |||||||||||
| PGT128 | C2 V3 region of gp120 | 0–60 μg/mL | |||||||||||
| PGT145 | oligomannose glycans | 0.2–53 μg/mL | |||||||||||
| Native full-length antibodies | Simian | ITS01 | binds CD4 receptor on Env | fully native rhesus IgG1 | AAV8 | NA | 1 × 1013 gc/animal | 21.2 μg/mL | ~20% of macaques | SIVsmE660 REC | AAV8 negative | NA | [2] |
| ITS06.02 | binds variable loop 1 | 8.6 μg/mL | |||||||||||
| ITS01 | binds CD4 receptor on Env | 5 × 1012 gc/animal | 20–100 μg/mL | ||||||||||
| ITS06.02 | binds variable loop 1 | ||||||||||||
| Simian | ITS01 | binds CD4 receptor on Env | fully native rhesus IgG1 | AAV1 | NA | 2.5 × 1012 vg/kg | 216–243 μg/mL | 2 of 3 macaque | not a challenge model | screened for pre-existing AAV neutralizing antibodies and grouped accordingly | NA | [33] | |
| AAV8 | >100 μg/mL | 3 of 5 macaque | |||||||||||
| AAV9 | 224–302 μg/mL | 0 of 5 macaque | |||||||||||
| AAV-NP22 | >100 μg/mL | ||||||||||||
| AAV-KP1 | 1 of 3 macaque |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campbell, E.S.B.; Goens, M.M.; Cao, W.; Thompson, B.; Susta, L.; Banadyga, L.; Wootton, S.K. Recent Advancements in AAV-Vectored Immunoprophylaxis in the Nonhuman Primate Model. Biomedicines 2023, 11, 2223. https://doi.org/10.3390/biomedicines11082223
Campbell ESB, Goens MM, Cao W, Thompson B, Susta L, Banadyga L, Wootton SK. Recent Advancements in AAV-Vectored Immunoprophylaxis in the Nonhuman Primate Model. Biomedicines. 2023; 11(8):2223. https://doi.org/10.3390/biomedicines11082223
Chicago/Turabian StyleCampbell, Elena S. B., Melanie M. Goens, Wenguang Cao, Brad Thompson, Leonardo Susta, Logan Banadyga, and Sarah K. Wootton. 2023. "Recent Advancements in AAV-Vectored Immunoprophylaxis in the Nonhuman Primate Model" Biomedicines 11, no. 8: 2223. https://doi.org/10.3390/biomedicines11082223