
A Protective Effect of Pirfenidone in Lung Fibroblast–Endothelial Cell Network via Inhibition of Rho-Kinase Activity
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Co-Culture Endothelial Network Formation
2.3. Flow Cytometry Analysis
2.4. Immunostaining
2.5. Evaluation of Endothelial Network
2.6. Rho-Kinase Activity Assay
2.7. Collagen Assay
2.8. Apoptosis Evaluation
2.9. Statistical Analysis
3. Results
3.1. Co-Culture Endothelial Network Formation
3.2. Evaluation of Endothelial Network Length and Branch Number in the Co-Culture Model
3.3. Assessment of Cell Proportions in Cell Sheet of Co-Culture Model
3.4. Rho-Kinase Activity in Co-Culture Model
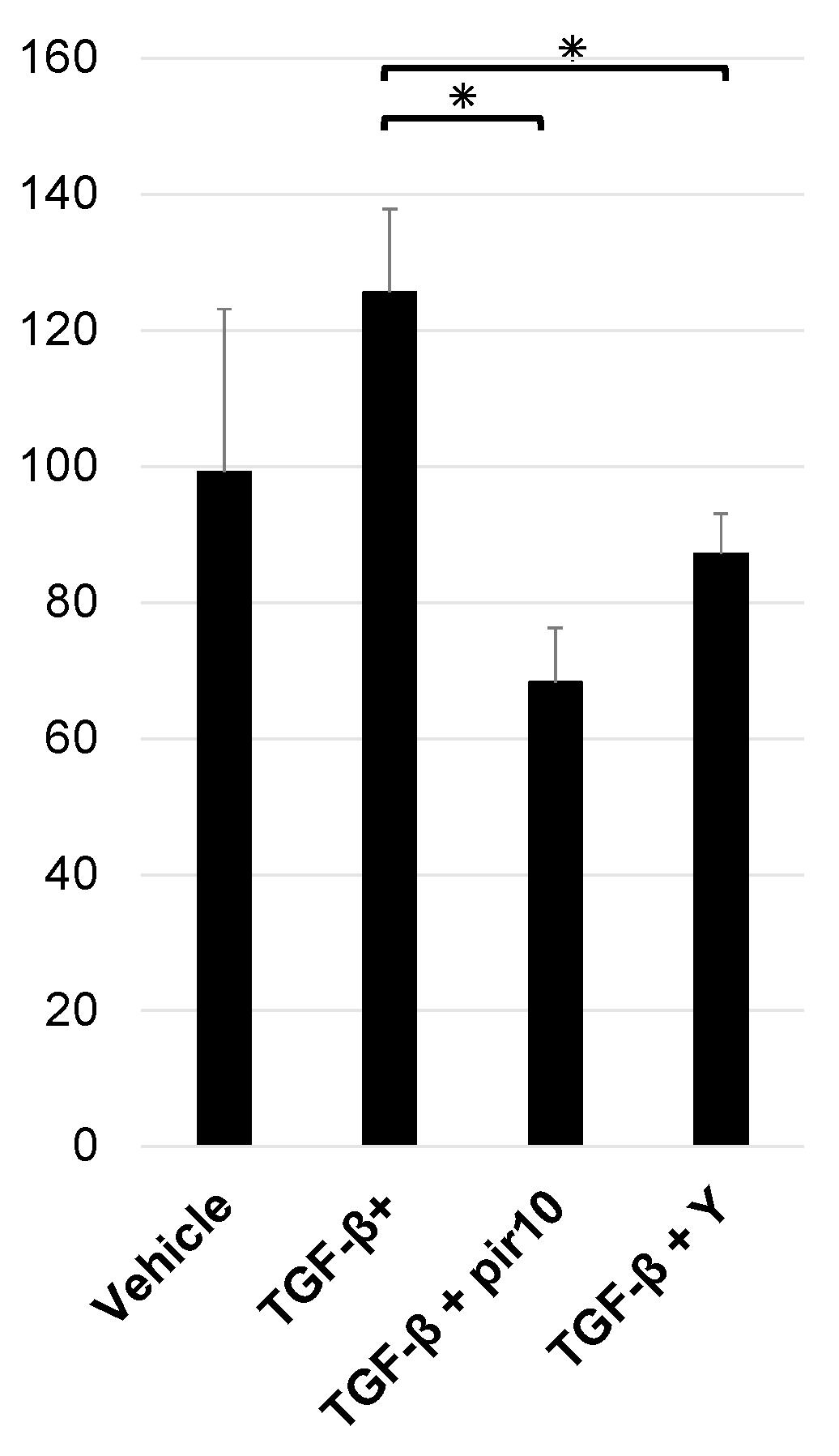
3.5. Collagen Production in the Co-Culture Model
3.6. Measurement of Apoptosis in the Co-Culture Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wijsenbeek, M.; Kreuter, M.; Olson, A.; Fischer, A.; Bendstrup, E.; Wells, C.D.; Denton, C.P.; Mounir, B.; Zouad-Lejour, L.; Quaresma, M.; et al. Progressive fibrosing interstitial lung diseases: Current practice in diagnosis and management. Curr. Med. Res. Opin. 2019, 35, 2015–2024. [Google Scholar] [PubMed] [Green Version]
- Wells, A.U.; Flaherty, K.R.; Brown, K.K.; Inoue, Y.; Devaraj, A.; Richeldi, L.; Moua, T.; Crestani, B.; Wuyts, W.A.; Stowasser, S.; et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases-subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: A randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir. Med. 2020, 8, 453–460. [Google Scholar] [CrossRef]
- Richeldi, L.; Davies, H.R.; Ferrara, G.; Franco, F. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst. Rev. 2003, 2003, Cd002880. [Google Scholar]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.F.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef] [Green Version]
- Behr, J.; Prasse, A.; Kreuter, M.; Johow, J.; Rabe, K.F.; Bonella, F.; Bonnet, R.; Grohe, C.; Held, M.; Wilkens, H.; et al. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): A double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2021, 9, 476–486. [Google Scholar]
- Maher, T.M.; Corte, T.J.; Fischer, A.; Kreuter, M.; Lederer, D.J.; Molina-Molina, M.; Axmann, J.; Kirchgaessler, K.U.; Samara, K.; Gilberg, F.; et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir. Med. 2020, 8, 147–157. [Google Scholar] [CrossRef]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar]
- Natsuizaka, M.; Chiba, H.; Kuronuma, K.; Otsuka, M.; Kudo, K.; Mori, M.; Bando, M.; Sugiyama, Y.; Takahashi, H. Epidemiologic survey of Japanese patients with idiopathic pulmonary fibrosis and investigation of ethnic differences. Am. J. Respir. Crit. Care Med. 2014, 190, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.K.; Martinez, F.J.; Walsh, S.L.F.; Thannickal, V.J.; Prasse, A.; Schlenker-Herceg, R.; Goeldner, R.G.; Clerisme-Beaty, E.; Tetzlaff, K.; Cottin, V.; et al. The natural history of progressive fibrosing interstitial lung diseases. Eur. Respir. J. 2020, 55, 2000085. [Google Scholar] [CrossRef] [PubMed]
- Wijsenbeek, M.; Cottin, V. Spectrum of Fibrotic Lung Diseases. N. Engl. J. Med. 2020, 383, 958–968. [Google Scholar] [CrossRef]
- Distler, J.H.W.; Györfi, A.H.; Ramanujam, M.; Whitfield, M.L.; Königshoff, M.; Lafyatis, R. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730. [Google Scholar] [PubMed]
- Winters, N.I.; Burman, A.; Kropski, J.A.; Blackwell, T.S. Epithelial Injury and Dysfunction in the Pathogenesis of Idiopathic PulmonaryFibrosis. Am. J. Med. Sci. 2019, 357, 374–378. [Google Scholar] [CrossRef] [Green Version]
- Wells, R.G. Tissue mechanics and fibrosis. Biochim. Biophys. Acta 2013, 1832, 884–890. [Google Scholar]
- Lokmic, Z.; Musyoka, J.; Hewitson, T.D.; Darby, I.A. Hypoxia and hypoxia signaling in tissue repair and fibrosis. Int. Rev. Cell Mol. Biol. 2012, 296, 139–185. [Google Scholar] [PubMed]
- Parker, M.W.; Rossi, D.; Peterson, M.; Smith, K.; Sikström, K.; White, E.S.; Connett, J.E.; Henke, C.A.; Larsson, O.; Bitterman, P.B. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J. Clin. Invest. 2014, 124, 1622–1635. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-β on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef]
- Nakamura, Y.; Shimizu, Y. Cellular and Molecular Control of Lipid Metabolism in Idiopathic Pulmonary Fibrosis: Clinical Application of the Lysophosphatidic Acid Pathway. Cells 2023, 12, 548. [Google Scholar]
- Hanumegowda, C.; Farkas, L.; Kolb, M. Angiogenesis in pulmonary fibrosis: Too much or not enough? Chest 2012, 142, 200–207. [Google Scholar] [PubMed]
- Ebina, M.; Taniguchi, H.; Miyasho, T.; Yamada, S.; Shibata, N.; Ohta, H.; Hisata, S.; Ohkouchi, S.; Tamada, T.; Nishimura, H.; et al. Gradual increase of high mobility group protein b1 in the lungs after the onset of acute exacerbation of idiopathic pulmonary fibrosis. Pulm. Med. 2011, 2011, 916486. [Google Scholar]
- Cao, Z.; Lis, R.; Ginsberg, M.; Chavez, D.; Shido, K.; Rabbany, S.Y.; Fong, G.H.; Sakmar, T.P.; Rafii, S.; Ding, B.S. Targeting of the pulmonary capillary vascular niche promotes lung alveolar repair and ameliorates fibrosis. Nat. Med. 2016, 22, 154–162. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Yang, Y.; Guo, X.; Liu, L.; Wu, K.; Yu, M. The Antiangiogenesis Effect of Pirfenidone in Wound Healing In Vitro. J. Ocul. Pharmacol. Ther. 2017, 33, 693–703. [Google Scholar] [CrossRef]
- Gan, D.; Cheng, W.; Ke, L.; Sun, A.R.; Jia, Q.; Chen, J.; Xu, Z.; Xu, J.; Zhang, P. Biphasic Effect of Pirfenidone on Angiogenesis. Front. Pharmacol. 2021, 12, 804327. [Google Scholar] [CrossRef]
- Ackermann, M.; Kim, Y.O.; Wagner, W.L.; Schuppan, D.; Valenzuela, C.D.; Mentzer, S.J.; Kreuz, S.; Stiller, D.; Wollin, L.; Konerding, M.A. Effects of nintedanib on the microvascular architecture in a lung fibrosis model. Angiogenesis 2017, 20, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Shimizu, Y.; Horibata, Y.; Tei, R.; Koike, R.; Masawa, M.; Watanabe, T.; Shiobara, T.; Arai, R.; Chibana, K.; et al. Changes of plasmalogen phospholipid levels during differentiation of induced pluripotent stem cells 409B2 to endothelial phenotype cells. Sci. Rep. 2017, 7, 9377. [Google Scholar] [CrossRef] [PubMed]
- Caporarello, N.; Ligresti, G. Vascular Contribution to Lung Repair and Fibrosis. Am. J. Respir Cell Mol. Biol. 2023, 69, 135–146. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Mumford, J.A.; Murray, S.; Kazerooni, E.A.; Gross, B.H.; Colby, T.V.; Travis, W.D.; Flint, A.; Toews, G.B.; Lynch, J.P., 3rd; et al. Prognostic implications of physiologic and radiographic changes in idiopathic interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2003, 168, 543–548. [Google Scholar] [CrossRef]
- Ma, Z.; Zhao, C.; Chen, Q.; Yu, C.; Zhang, H.; Zhang, Z.; Huang, W.; Shen, Z. Antifibrotic effects of a novel pirfenidone derivative in vitro and in vivo. Pulm. Pharmacol. Ther. 2018, 53, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Ruwanpura, S.M.; Thomas, B.J.; Bardin, P.G. Pirfenidone: Molecular Mechanisms and Potential Clinical Applications in Lung Disease. Am. J. Respir. Cell Mol. Biol. 2020, 62, 413–422. [Google Scholar] [CrossRef]
- Pourgholamhossein, F.; Rasooli, R.; Pournamdari, M.; Pourgholi, L.; Samareh-Fekri, M.; Ghazi-Khansari, M.; Iranpour, M.; Poursalehi, H.R.; Heidari, M.R.; Mandegary, A. Pirfenidone protects against paraquat-induced lung injury and fibrosis in mice by modulation of inflammation, oxidative stress, and gene expression. Food Chem. Toxicol. 2018, 112, 39–46. [Google Scholar] [CrossRef]
- Conte, E.; Gili, E.; Fagone, E.; Fruciano, M.; Iemmolo, M.; Vancheri, C. Effect of pirfenidone on proliferation, TGF-β-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur. J. Pharm. Sci. 2014, 58, 13–19. [Google Scholar] [CrossRef]
- Molina-Molina, M.; Machahua-Huamani, C.; Vicens-Zygmunt, V.; Llatjós, R.; Escobar, I.; Sala-Llinas, E.; Luburich-Hernaiz, P.; Dorca, J.; Montes-Worboys, A. Anti-fibrotic effects of pirfenidone and rapamycin in primary IPF fibroblasts and human alveolar epithelial cells. BMC Pulm. Med. 2018, 18, 63. [Google Scholar] [CrossRef] [Green Version]
- Gurujeyalakshmi, G.; Hollinger, M.A.; Giri, S.N. Pirfenidone inhibits PDGF isoforms in bleomycin hamster model of lung fibrosis at the translational level. Am. J. Physiol. 1999, 276, L311–L318. [Google Scholar] [CrossRef]
- Oku, H.; Shimizu, T.; Kawabata, T.; Nagira, M.; Hikita, I.; Ueyama, A.; Matsushima, S.; Torii, M.; Arimura, A. Antifibrotic action of pirfenidone and prednisolone: Different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur. J. Pharmacol. 2008, 590, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Hamidzadeh, K.; Christensen, S.M.; Dalby, E.; Chandrasekaran, P.; Mosser, D.M. Macrophages and the Recovery from Acute and Chronic Inflammation. Annu. Rev. Physiol. 2017, 79, 567–592. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.N.; Gurujeyalakshmi, G.; Giri, S.N. Effects of pirfenidone on transforming growth factor-beta gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J. Pharmacol. Exp. Ther. 1999, 291, 367–373. [Google Scholar] [PubMed]
- Rouhani, F.N.; Brantly, M.L.; Markello, T.C.; Helip-Wooley, A.; O’Brien, K.; Hess, R.; Huizing, M.; Gahl, W.A.; Gochuico, B.R. Alveolar macrophage dysregulation in Hermansky-Pudlak syndrome type 1. Am. J. Respir. Crit. Care Med. 2009, 180, 1114–1121. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Zhang, M.; Leng, C.; Blokzijl, T.; Jansen, B.H.; Dijkstra, G.; Faber, K.N. Pirfenidone Inhibits Cell Proliferation and Collagen I Production of Primary Human Intestinal Fibroblasts. Cells 2020, 9, 775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannotta, M.; Trani, M.; Dejana, E. VE-cadherin and endothelial adherens junctions: Active guardians of vascular integrity. Dev. Cell 2013, 26, 441–454. [Google Scholar] [CrossRef] [Green Version]
- Leach, H.G.; Chrobak, I.; Han, R.; Trojanowska, M. Endothelial cells recruit macrophages and contribute to a fibrotic milieu in bleomycin lung injury. Am. J. Respir. Cell Mol. Biol. 2013, 49, 1093–1101. [Google Scholar] [CrossRef] [Green Version]
- Knipe, R.S.; Probst, C.K.; Lagares, D.; Franklin, A.; Spinney, J.J.; Brazee, P.L.; Grasberger, P.; Zhang, L.; Black, K.E.; Sakai, N.; et al. The Rho Kinase Isoforms ROCK1 and ROCK2 Each Contribute to the Development of Experimental Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Dobashi, K.; Iizuka, K.; Horie, T.; Suzuki, K.; Tukagoshi, H.; Nakazawa, T.; Nakazato, Y.; Mori, M. Contribution of small GTPase Rho and its target protein rock in a murine model of lung fibrosis. Am. J. Respir. Crit. Care Med. 2001, 163, 210–217. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, Y.; Dobashi, K.; Sano, T.; Yamada, M. ROCK activation in lung of idiopathic pulmonary fibrosis with oxidative stress. Int. J. Immunopathol. Pharmacol. 2014, 27, 37–44. [Google Scholar] [CrossRef]
- Wang, Y.C.; Chen, Q.; Luo, J.M.; Nie, J.; Meng, Q.H.; Shuai, W.; Xie, H.; Xia, J.M.; Wang, H. Notch1 promotes the pericyte-myofibroblast transition in idiopathic pulmonary fibrosis through the PDGFR/ROCK1 signal pathway. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Meekins, L.C.; Rosado-Adames, N.; Maddala, R.; Zhao, J.J.; Rao, P.V.; Afshari, N.A. Corneal Endothelial Cell Migration and Proliferation Enhanced by Rho Kinase (ROCK) Inhibitors in In Vitro and In Vivo Models. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6731–6738. [Google Scholar] [CrossRef] [PubMed]
- Clements, R.T.; Minnear, F.L.; Singer, H.A.; Keller, R.S.; Vincent, P.A. RhoA and Rho-kinase dependent and independent signals mediate TGF-beta-induced pulmonary endothelial cytoskeletal reorganization and permeability. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L294–L306. [Google Scholar] [CrossRef]
- Fukata, Y.; Amano, M.; Kaibuchi, K. Rho-Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol. Sci. 2001, 22, 32–39. [Google Scholar] [CrossRef]
- Zhang, J.; Tang, L.; Dai, F.; Qi, Y.; Yang, L.; Liu, Z.; Deng, L.; Yao, W. ROCK inhibitors alleviate myofibroblast transdifferentiation and vascular remodeling via decreasing TGFβ1-mediated RhoGDI expression. Gen. Physiol. Biophys. 2019, 38, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Pertz, O.; Hodgson, L.; Klemke, R.L.; Hahn, K.M. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature 2006, 440, 1069–1072. [Google Scholar] [CrossRef]
- Oshiro, N.; Fukata, Y.; Kaibuchi, K. Phosphorylation of moesin by rho-associated kinase (Rho-kinase) plays a crucial role in the formation of microvilli-like structures. J. Biol. Chem. 1998, 273, 34663–34666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebina, M.; Shimizukawa, M.; Shibata, N.; Kimura, Y.; Suzuki, T.; Endo, M.; Sasano, H.; Kondo, T.; Nukiwa, T. Heterogeneous increase in CD34-positive alveolar capillaries in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2004, 169, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Van Nieuw Amerongen, G.P.; Vermeer, M.A.; van Hinsbergh, V.W. Role of RhoA and Rho kinase in lysophosphatidic acid-induced endothelial barrier dysfunction. Arterioscler. Thromb. Vasc. Biol. 2000, 20, E127–E133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.Y.; Porte, J.; Knox, A.J.; Weinreb, P.H.; Maher, T.M.; Violette, S.M.; McAnulty, R.J.; Sheppard, D.; Jenkins, G. Lysophosphatidic acid induces alphavbeta6 integrin-mediated TGF-beta activation via the LPA2 receptor and the small G protein G alpha(q). Am. J. Pathol. 2009, 174, 1264–1279. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.S.; Fu, P.; Patel, P.; Harijith, A.; Sun, T.; Zhao, Y.; Garcia, J.G.; Chun, J.; Natarajan, V. Lysophosphatidic acid receptor-2 deficiency confers protection against bleomycin-induced lung injury and fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 2013, 49, 912–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikonomou, N.; Mouratis, M.A.; Tzouvelekis, A.; Kaffe, E.; Valavanis, C.; Vilaras, G.; Karameris, A.; Prestwich, G.D.; Bouros, D.; Aidinis, V. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 566–574. [Google Scholar] [CrossRef]
- Shimizu, Y.; Nakamura, Y.; Horibata, Y.; Fujimaki, M.; Hayashi, K.; Uchida, N.; Morita, H.; Arai, R.; Chibana, K.; Takemasa, A.; et al. Imaging of lysophosphatidylcholine in an induced pluripotent stem cell-derived endothelial cell network. Regen. Ther. 2020, 14, 299–305. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura, Y.; Shimizu, Y.; Fujimaki-Shiraishi, M.; Uchida, N.; Takemasa, A.; Niho, S. A Protective Effect of Pirfenidone in Lung Fibroblast–Endothelial Cell Network via Inhibition of Rho-Kinase Activity. Biomedicines 2023, 11, 2259. https://doi.org/10.3390/biomedicines11082259
Nakamura Y, Shimizu Y, Fujimaki-Shiraishi M, Uchida N, Takemasa A, Niho S. A Protective Effect of Pirfenidone in Lung Fibroblast–Endothelial Cell Network via Inhibition of Rho-Kinase Activity. Biomedicines. 2023; 11(8):2259. https://doi.org/10.3390/biomedicines11082259
Chicago/Turabian StyleNakamura, Yusuke, Yasuo Shimizu, Mio Fujimaki-Shiraishi, Nobuhiko Uchida, Akihiro Takemasa, and Seiji Niho. 2023. "A Protective Effect of Pirfenidone in Lung Fibroblast–Endothelial Cell Network via Inhibition of Rho-Kinase Activity" Biomedicines 11, no. 8: 2259. https://doi.org/10.3390/biomedicines11082259