
Assessment of Substrate Status of Drugs Metabolized by Polymorphic Cytochrome P450 (CYP) 2 Enzymes: An Analysis of a Large-Scale Dataset

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Preparation
2.2. Assessments of Substrate Status
2.2.1. Manual Assessment of Substrate Status
2.2.2. Automatic Assessment of Substrate Status
2.3. Statistical Analysis
3. Results
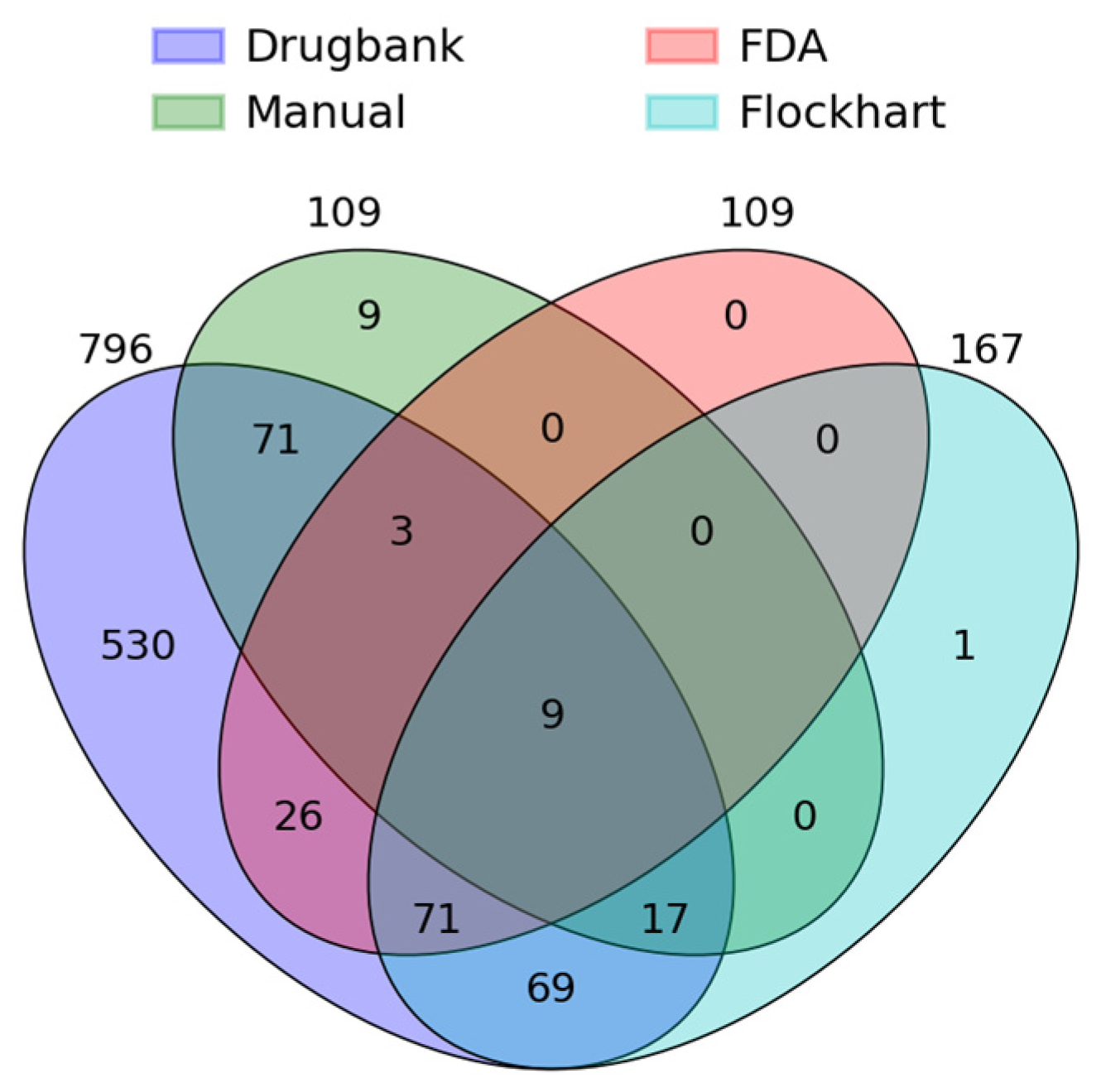
3.1. Completeness of Assessments
3.2. CYP Classification
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Relling, M.V.; Evans, W.E. Pharmacogenomics in the clinic. Nature 2015, 526, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Hamburg, M.A.; Collins, F.S. The path to personalized medicine. N. Engl. J. Med. 2010, 363, 301–304. [Google Scholar] [CrossRef]
- Stingl, J.; Brockmöller, J.; Viviani, R. Genetic variability of drug-metabolizing enzymes: The dual impact on psychiatric therapy and regulation of brain function. Mol. Psychiatry 2013, 18, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.E.; Relling, M.V. Pharmacogenomics: Translating functional genomics into rational therapeutics. Science 1999, 286, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Whirl-Carrillo, M.; Huddart, R.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Whaley, R.; Klein, T.E. An Evidence-Based Framework for Evaluating Pharmacogenomics Knowledge for Personalized Medicine. Clin. Pharmacol. Ther. 2021, 110, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Osanlou, O.; Pirmohamed, M.; Daly, A.K. Pharmacogenetics of Adverse Drug Reactions. Adv. Pharmacol. 2018, 83, 155–190. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U.A. Pharmacogenetics and adverse drug reactions. Lancet 2000, 356, 1667–1671. [Google Scholar] [CrossRef]
- Swen, J.J.; van der Wouden, C.H.; Manson, L.E.N.; Abdullah-Koolmees, H.; Blagec, K.; Blagus, T.; Böhringer, S.; Cambon-Thomsen, A.; Cecchin, E.; Cheung, K.-C.; et al. A 12-gene pharmacogenetic panel to prevent adverse drug reactions: An open-label, multicentre, controlled, cluster-randomised crossover implementation study. Lancet 2023, 401, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Just, K.S.; Dormann, H.; Schurig, M.; Böhme, M.; Fracowiak, J.; Steffens, M.; Scholl, C.; Seufferlein, T.; Gräff, I.; Schwab, M.; et al. Adverse Drug Reactions in the Emergency Department: Is There a Role for Pharmacogenomic Profiles at Risk?—Results from the ADRED Study. J. Clin. Med. 2020, 9, 1801. [Google Scholar] [CrossRef]
- Shah, R.R.; Smith, R.L. Addressing phenoconversion: The Achilles’ heel of personalized medicine. Br. J. Clin. Pharmacol. 2015, 79, 222–240. [Google Scholar] [CrossRef]
- Klomp, S.D.; Manson, M.L.; Guchelaar, H.J.; Swen, J.J. Phenoconversion of Cytochrome P450 Metabolism: A Systematic Review. J. Clin. Med. 2020, 9, 2890. [Google Scholar] [CrossRef]
- Cicali, E.J.; Elchynski, A.L.; Cook, K.J.; Houder, J.T.; Thomas, C.D.; Smith, D.M.; Elsey, A.; Johnson, J.A.; Cavallari, L.H.; Wiisanen, K. How to Integrate CYP2D6 Phenoconversion Into Clinical Pharmacogenetics: A Tutorial. Clin. Pharmacol. Ther. 2021, 110, 677–687. [Google Scholar] [CrossRef]
- Kheshti, R.; Aalipour, M.; Namazi, S. A comparison of five common drug-drug interaction software programs regarding accuracy and comprehensiveness. J. Res. Pharm. Pract. 2016, 5, 257–263. [Google Scholar] [CrossRef]
- Kuperman, G.J.; Bobb, A.; Payne, T.H.; Avery, A.J.; Gandhi, T.K.; Burns, G.; Classen, D.C.; Bates, D.W. Medication-related clinical decision support in computerized provider order entry systems: A review. J. Am. Med. Inform. Assoc. 2007, 14, 29–40. [Google Scholar] [CrossRef] [PubMed]
- FDA. Drug Development and Drug Interactions: Table of Substrates, Inhibitors, and Inducers. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers (accessed on 3 June 2023).
- Flockhart, D.A. Drug Interactions Flockhart Table™. Available online: https://drug-interactions.medicine.iu.edu/MainTable.aspx (accessed on 18 September 2023).
- Schurig, A.M.; Bohme, M.; Just, K.S.; Scholl, C.; Dormann, H.; Plank-Kiegele, B.; Seufferlein, T.; Graff, I.; Schwab, M.; Stingl, J.C. Adverse Drug Reactions (ADR) and Emergencies. Dtsch. Arztebl. Int. 2018, 115, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Just, K.S.; Dormann, H.; Böhme, M.; Schurig, M.; Schneider, K.L.; Steffens, M.; Dunow, S.; Plank-Kiegele, B.; Ettrich, K.; Seufferlein, T.; et al. Personalising drug safety—Results from the multi-centre prospective observational study on Adverse Drug Reactions in Emergency Departments (ADRED). Eur. J. Clin. Pharmacol. 2020, 76, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Landis, J.R.; Koch, G.G. The measurement of observer agreement for categorical data. Biometrics 1977, 33, 159–174. [Google Scholar] [CrossRef]
- Zanger, U.M.; Turpeinen, M.; Klein, K.; Schwab, M. Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal. Bioanal. Chem. 2008, 392, 1093–1108. [Google Scholar] [CrossRef]
- Castaño-Amores, C.; Díaz-Villamarín, X.; Pérez-Gutiérrez, A.M.; Antúnez-Rodríguez, A.; Pozo-Agundo, A.; Moreno-Escobar, E.; Sánchez-Ramos, J.G.; Martínez-González, L.J.; Dávila-Fajardo, C.L. Pharmacogenetic polymorphisms affecting bisoprolol response. Biomed. Pharmacother. 2021, 142, 112069. [Google Scholar] [CrossRef] [PubMed]
- KNMP. CYP2D6: Bisoprolol. Available online: https://www.g-standaard.nl/risicoanalyse/B0002457.PDF (accessed on 31 October 2023).
- Chang, S.Y.; Li, W.; Traeger, S.C.; Wang, B.; Cui, D.; Zhang, H.; Wen, B.; Rodrigues, A.D. Confirmation that cytochrome P450 2C8 (CYP2C8) plays a minor role in (S)-(+)- and (R)-(−)-ibuprofen hydroxylation in vitro. Drug Metab. Dispos. 2008, 36, 2513–2522. [Google Scholar] [CrossRef]
- Saiz-Rodríguez, M.; Valdez-Acosta, S.; Borobia, A.M.; Burgueño, M.; Gálvez-Múgica, M.; Acero, J.; Cabaleiro, T.; Muñoz-Guerra, M.F.; Puerro, M.; Llanos, L.; et al. Influence of Genetic Polymorphisms on the Response to Tramadol, Ibuprofen, and the Combination in Patients with Moderate to Severe Pain after Dental Surgery. Clin. Ther. 2021, 43, e86–e102. [Google Scholar] [CrossRef]
- Kitzmiller, J.P.; Mikulik, E.B.; Dauki, A.M.; Murkherjee, C.; Luzum, J.A. Pharmacogenomics of statins: Understanding susceptibility to adverse effects. Pharmacogenom. Pers. Med. 2016, 9, 97–106. [Google Scholar] [CrossRef]
- Choi, H.Y.; Bae, K.S.; Cho, S.H.; Ghim, J.L.; Choe, S.; Jung, J.A.; Jin, S.J.; Kim, H.S.; Lim, H.S. Impact of CYP2D6, CYP3A5, CYP2C19, CYP2A6, SLCO1B1, ABCB1, and ABCG2 gene polymorphisms on the pharmacokinetics of simvastatin and simvastatin acid. Pharmacogenet. Genom. 2015, 25, 595–608. [Google Scholar] [CrossRef]
- Nakashima, A.; Kawashita, H.; Masuda, N.; Saxer, C.; Niina, M.; Nagae, Y.; Iwasaki, K. Identification of cytochrome P450 forms involved in the 4-hydroxylation of valsartan, a potent and specific angiotensin II receptor antagonist, in human liver microsomes. Xenobiotica 2005, 35, 589–602. [Google Scholar] [CrossRef]
- Hallberg, P.; Karlsson, J.; Kurland, L.; Lind, L.; Kahan, T.; Malmqvist, K.; Ohman, K.P.; Nyström, F.; Melhus, H. The CYP2C9 genotype predicts the blood pressure response to irbesartan: Results from the Swedish Irbesartan Left Ventricular Hypertrophy Investigation vs Atenolol (SILVHIA) trial. J. Hypertens. 2002, 20, 2089–2093. [Google Scholar] [CrossRef]
- Joy, M.S.; Dornbrook-Lavender, K.; Blaisdell, J.; Hilliard, T.; Boyette, T.; Hu, Y.; Hogan, S.L.; Candiani, C.; Falk, R.J.; Goldstein, J.A. CYP2C9 genotype and pharmacodynamic responses to losartan in patients with primary and secondary kidney diseases. Eur. J. Clin. Pharmacol. 2009, 65, 947–953. [Google Scholar] [CrossRef]
- Grün, B.; Krautter, S.; Riedel, K.D.; Mikus, G. Inhibition of the active principle of the weak opioid tilidine by the triazole antifungal voriconazole. Br. J. Clin. Pharmacol. 2009, 68, 712–720. [Google Scholar] [CrossRef]
- Budnitz, D.S.; Shehab, N.; Kegler, S.R.; Richards, C.L. Medication use leading to emergency department visits for adverse drug events in older adults. Ann. Intern. Med. 2007, 147, 755–765. [Google Scholar] [CrossRef]
- Shehab, N.; Lovegrove, M.C.; Geller, A.I.; Rose, K.O.; Weidle, N.J.; Budnitz, D.S. US emergency department visits for outpatient adverse drug events, 2013–2014. JAMA 2016, 316, 2115–2125. [Google Scholar] [CrossRef]
- Pirmohamed, M.; James, S.; Meakin, S.; Green, C.; Scott, A.K.; Walley, T.J.; Farrar, K.; Park, B.K.; Breckenridge, A.M. Adverse drug reactions as cause of admission to hospital: Prospective analysis of 18,820 patients. BMJ 2004, 329, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Just, K.S.; Dormann, H.; Freitag, M.; Schurig, M.; Böhme, M.; Steffens, M.; Scholl, C.; Seufferlein, T.; Graeff, I.; Schwab, M.; et al. CYP2D6 in the Brain: Potential Impact on Adverse Drug Reactions in the Central Nervous System-Results From the ADRED Study. Front. Pharmacol. 2021, 12, 624104. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Drug | Manual Assessment Method | Automatic Assessment Method | Entries, n (%) | ||
|---|---|---|---|---|---|
| - | Drugbank | FDA | Flockhart TableTM | ||
| pantoprazole | - | - | x | - | 1147 (4.8) |
| acetylsalicylic acid | - | - | x | x | 952 (4.0) |
| torasemide | - | - | x | x | 725 (3.0) |
| ramipril | - | - | x | x | 725 (3.0) |
| levothyroxine | - | - | x | x | 619 (2.6) |
| metamizole | - | - | x | x | 567 (2.4) |
| hydrochlorothiazide | - | - | x | x | 454 (1.9) |
| amlodipine | - | - | x | - | 450 (1.9) |
| potassium | x | - | x | x | 419 (1.8) |
| cholecalciferol | - | - | x | x | 388 (1.6) |
| allopurinol | - | - | - | x | 327 (1.4) |
| phenprocoumon | - | - | - | x | 325 (1.4) |
| magnesium | x | - | - | - | 153 (0.6) |
| formoterol | x | - | - | - | 152 (0.6) |
| tiotropium bromide | - | x | - | - | 147 (0.6) |
| salbutamol | x | - | - | - | 122 (0.5) |
| levodopa | x | - | - | - | 120 (0.5) |
| ipratropium bromide | x | x | - | - | 113 (0.5) |
| oxycodone | x | - | - | - | 103 (0.4) |
| budesonide | x | - | - | - | 96 (0.4) |
| lorazepam | x | - | - | - | 85 (0.4) |
| nitrendipine | x | - | - | - | 85 (0.4) |
| aciclovir | - | x | - | - | 53 (0.2) |
| beclomethasone | - | x | - | - | 52 (0.2) |
| glycerol | - | x | - | - | 47 (0.2) |
| insulin-isophane | - | x | - | - | 43 (0.2) |
| glycopyrronium bromide | - | x | - | - | 36 (0.2) |
| amphotericin b | - | x | - | - | 33 (0.1) |
| ascorbic acid | - | x | - | - | 31 (0.1) |
| potassium iodide | - | x | - | - | 29 (0.1) |
| Manual Assessment Method, n (%) | Automatic Assessment Method, n (%) | Cohen’s Kappa (κ) | |||
|---|---|---|---|---|---|
| - | Drugbank 1 | FDA 2 | Flockhart TableTM 3 | ||
| CYP2D6 | 9 (9.0) | 12 (12.0) | - | - | 0.84 |
| CYP2C19 | 8 (8.0) | 11 (11.0) | - | - | 0.71 |
| CYP2C9 | 6 (6.0) | 17 (17.0) | - | - | 0.48 |
| CYP2D6 | 3 (25.0) | - | 1 (8.3) | - | 0.43 |
| CYP2C19 | 3 (25.0) | - | 1 (8.3) | - | 0.43 |
| CYP2C9 | 0 (0.0) | - | 0 (0.0) | - | - |
| CYP2D6 | 7 (26.9) | - | - | 6 (23.1) | 0.90 |
| CYP2C19 | 7 (26.9) | - | - | 6 (23.1) | 0.90 |
| CYP2C9 | 2 (7.7) | - | - | 5 (19.2) | 0.20 |
| Enzyme | Drug | Manual Assessment Method | Automatic Assessment Method | Entries, n (%) | ||
|---|---|---|---|---|---|---|
| - | Drugbank | FDA | Flockhart TableTM | |||
| CYP2D6 | simvastatin | x | x | - | - | 628 (2.6) |
| acetaminophen | - | x | NA | - | 56 (0.2) | |
| escitalopram | x | x | - | - | 31 (0.1) | |
| caffeine | - | x | - | - | 11 (0.1) | |
| dapagliflozin | - | x | NA | NA | 10 (<0.1) | |
| CYP2C19 | simvastatin | - | x | - | - | 628 (2.6) |
| (dex)ibuprofen | - | x | NA | - | 268 (1.1) | |
| clopidogrel | x | x | - | - | 257 (1.1) | |
| apixaban | x | x | NA | - | 218 (0.9) | |
| naloxone | - | x | NA | NA | 137 (0.6) | |
| tilidine | x | - | NA | NA | 110 (0.5) | |
| escitalopram | x | x | - | - | 31 (0.1) | |
| CYP2C9 | acetylsalicylic acid | - | x | NA | NA | 952 (4.0) |
| valsartan | - | x | NA | NA | 259 (1.1) | |
| clopidogrel | - | x | - | x | 257 (1.1) | |
| apixaban | x | x | NA | - | 218 (0.9) | |
| omeprazole | - | x | - | - | 121 (0.5) | |
| carvedilol | - | x | NA | - | 97 (0.4) | |
| ondansetron | - | x | NA | - | 87 (0.4) | |
| losartan | - | x | NA | x | 31 (0.1) | |
| irbesartan | - | x | NA | x | 25 (0.1) | |
| olodaterol | - | x | NA | x | 17 (0.1) | |
| caffeine | - | x | - | - | 11 (0.1) | |
| dapagliflozin | - | x | NA | - | 10 (<0.1) | |
| Manual Assessment Method | Automatic Assessment Method with Matching Approach to… | |||
|---|---|---|---|---|
| - | …Drugbank [19] | …FDA [15] | …Flockhart TableTM [16] | |
| Velocity of assessment process | Time consuming | Quick | Quick | Quick |
| Underlying data | Literature search, standardized approach is recommended for reproducibility of results | In vitro and in vivo data are summarized. CYP substrate classification also based on in vitro data | Data for drugs showing changes in AUC by administration of an inhibitor | Published evidence that a drug is at least in parts metabolized by a CYP enzyme |
| Dealing with conflicting evidence | Manpower needed for dealing with disagreements. An independent review approach is recommended | No independent reviewers needed, as one clear classification is given | No independent reviewers needed, as one clear classification is given | No independent reviewers needed, as one clear classification is given |
| Completeness of assessments | Often limited due to time-consuming process needing manpower | Almost complete | Only small number of drugs available | Moderate number of drugs available |
| Potential clinical relevance of assessments | High with focus on in vivo data | Unclear, as often in vitro data leads to classification of CYP substrates | Very high, as only clear substrates are classified | High with focus on data with evidence in vivo |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sommer, J.; Wozniak, J.; Schmitt, J.; Koch, J.; Stingl, J.C.; Just, K.S. Assessment of Substrate Status of Drugs Metabolized by Polymorphic Cytochrome P450 (CYP) 2 Enzymes: An Analysis of a Large-Scale Dataset. Biomedicines 2024, 12, 161. https://doi.org/10.3390/biomedicines12010161
Sommer J, Wozniak J, Schmitt J, Koch J, Stingl JC, Just KS. Assessment of Substrate Status of Drugs Metabolized by Polymorphic Cytochrome P450 (CYP) 2 Enzymes: An Analysis of a Large-Scale Dataset. Biomedicines. 2024; 12(1):161. https://doi.org/10.3390/biomedicines12010161
Chicago/Turabian StyleSommer, Jakob, Justyna Wozniak, Judith Schmitt, Jana Koch, Julia C. Stingl, and Katja S. Just. 2024. "Assessment of Substrate Status of Drugs Metabolized by Polymorphic Cytochrome P450 (CYP) 2 Enzymes: An Analysis of a Large-Scale Dataset" Biomedicines 12, no. 1: 161. https://doi.org/10.3390/biomedicines12010161