
Current Technologies and Future Perspectives in Immunotherapy towards a Clinical Oncology Approach

 ,
,  , , ,
, , ,  , , and
, , and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Modulation of Tumor Immunogenicity
2.1. Direct Modulation of Tumor Immunogenicity
2.2. Indirect Modulation of Tumor Immunogenicity
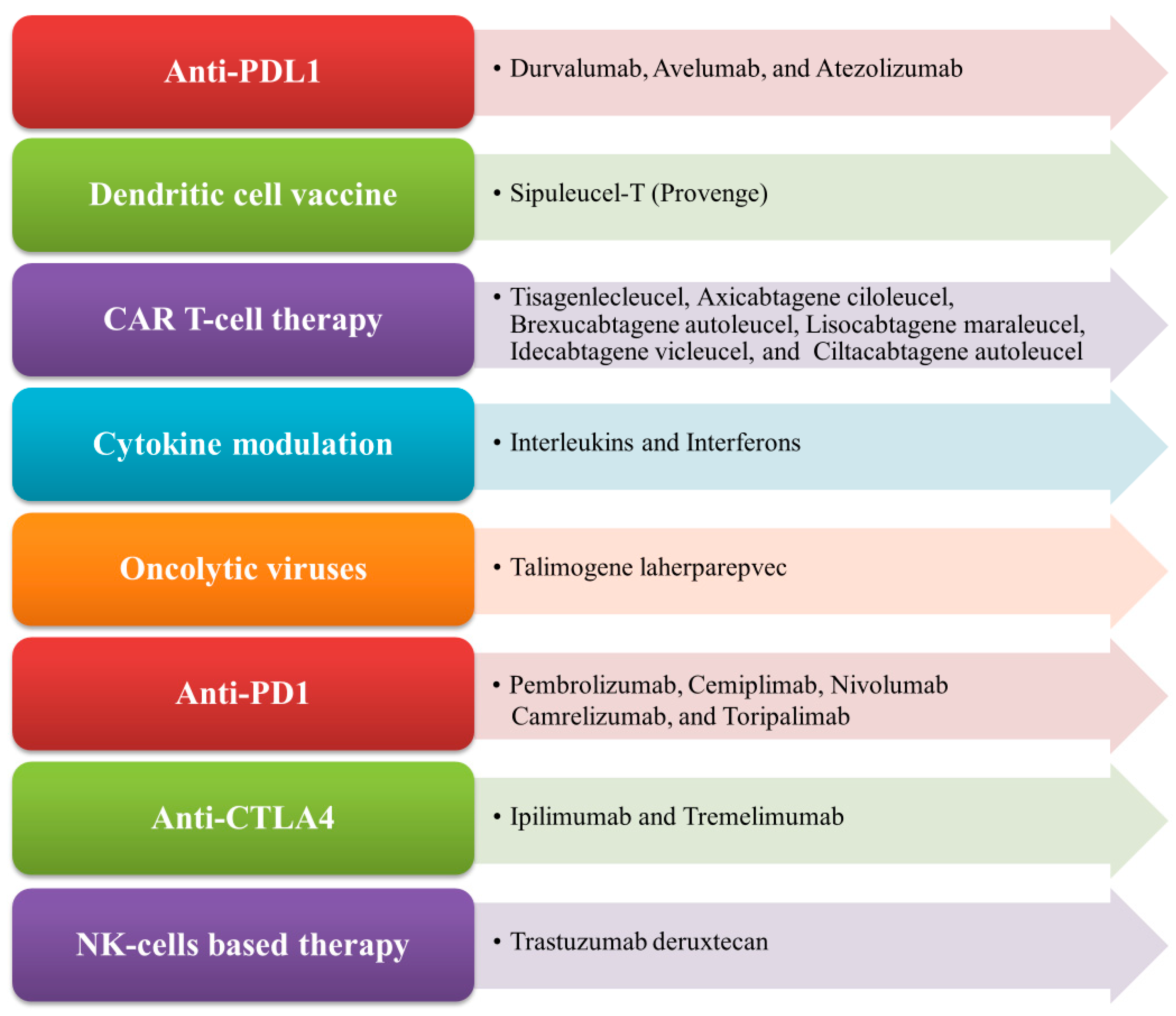
2.3. Existing Immunotherapies for the Indirect Modulation of Immunogenicity
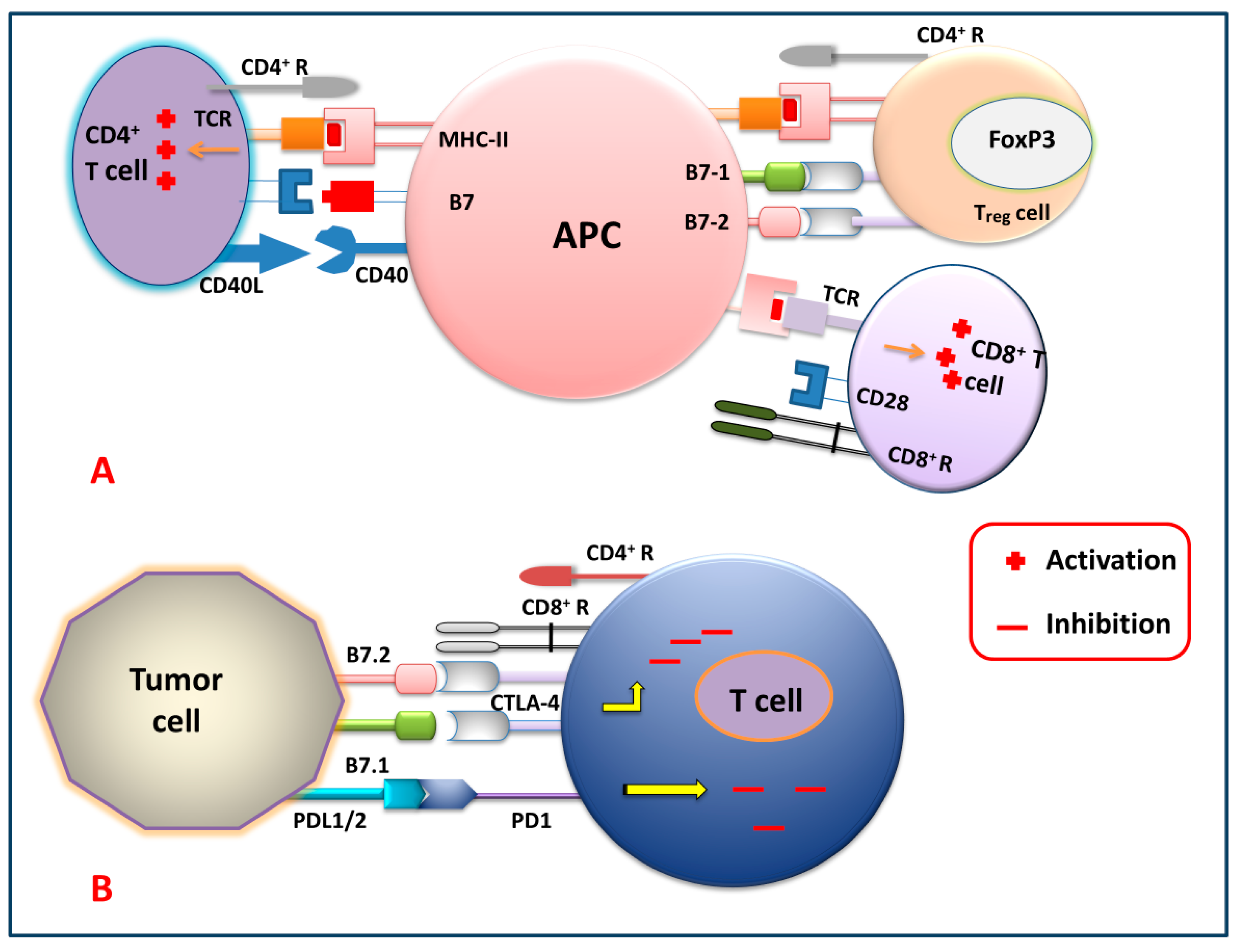
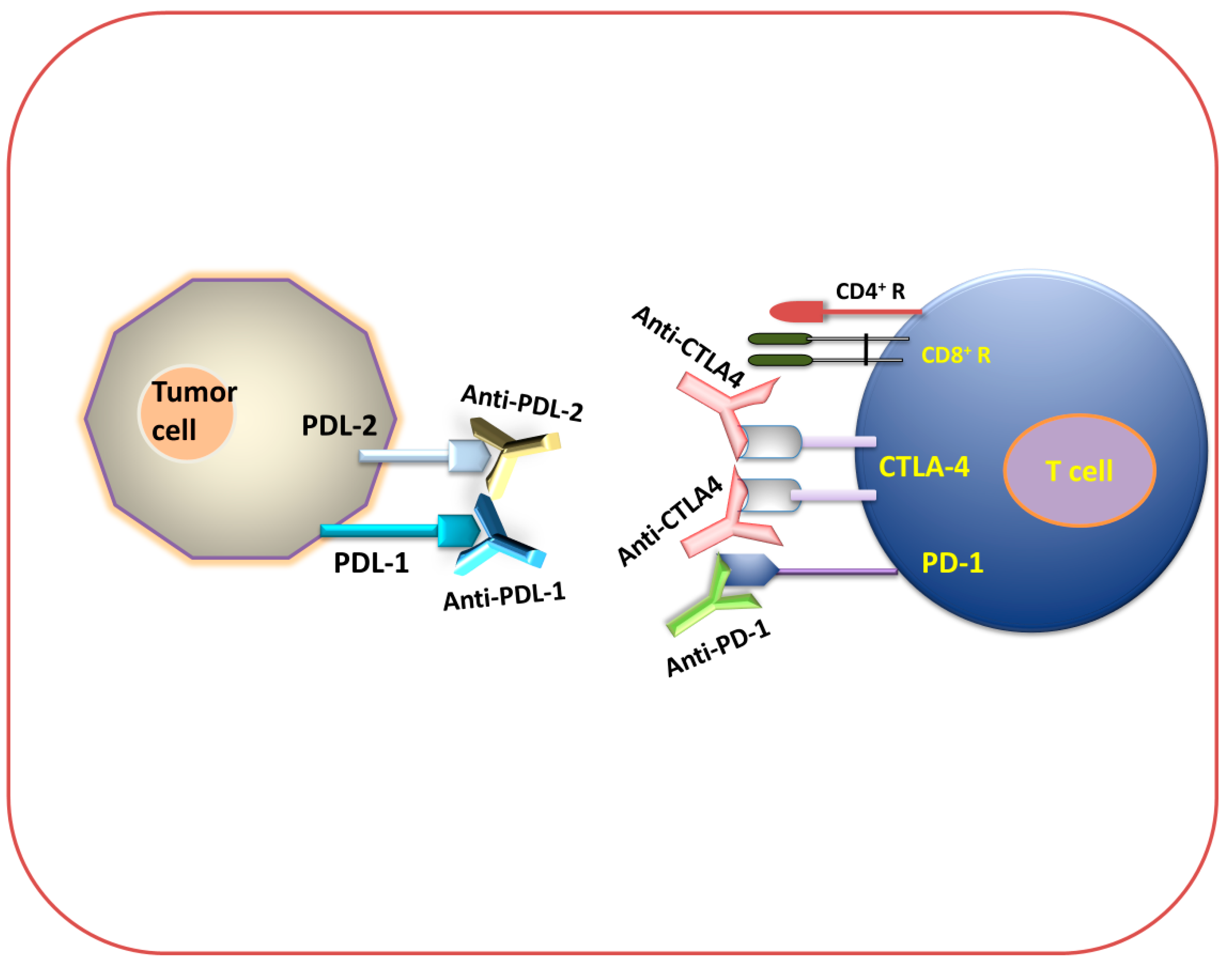
2.3.1. Checkpoint Blockade Therapy
Anti-PD-1 Therapy
Anti-PD-L1 and PD-L2 Therapy
Anti-CTLA-4 Therapy
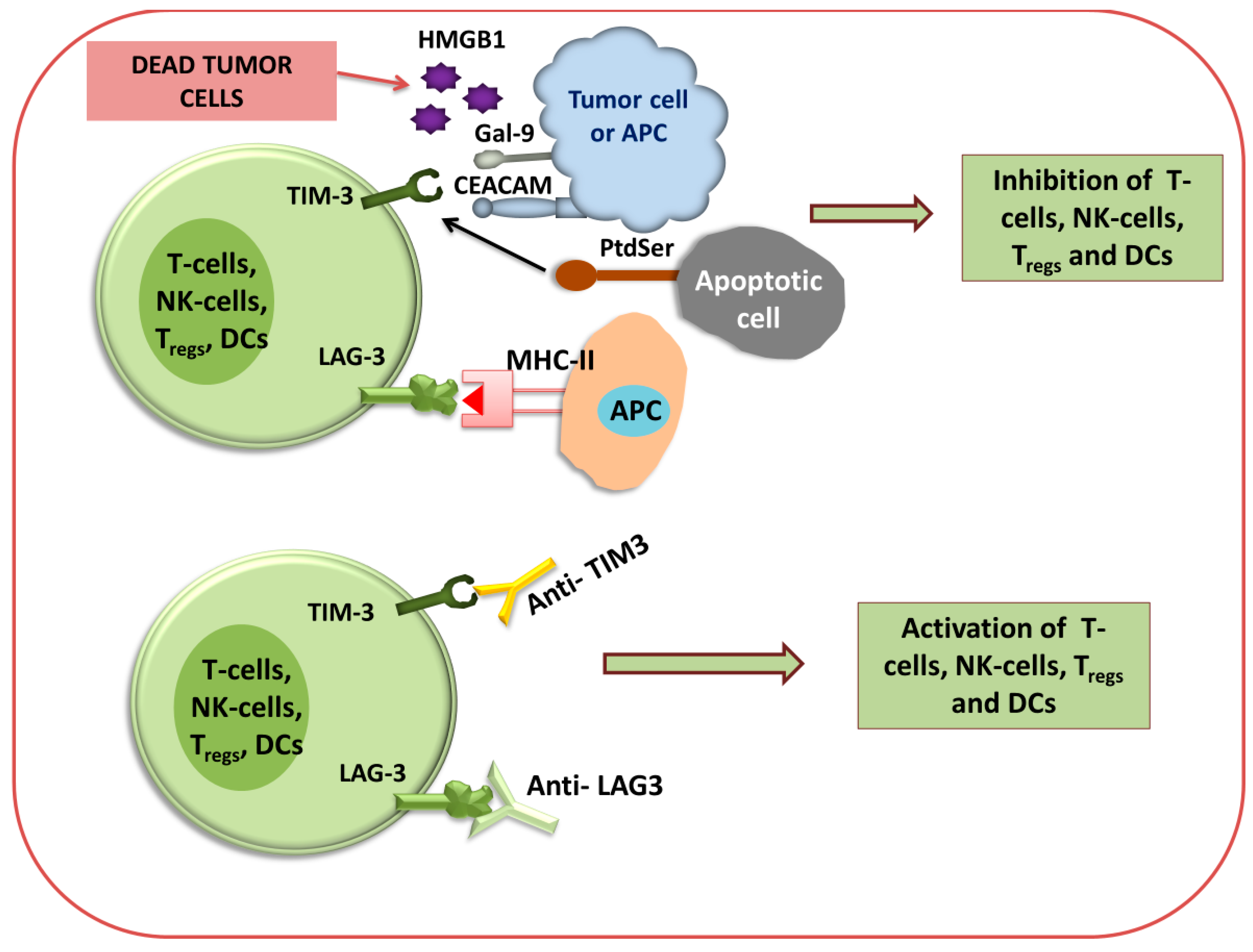
LAG3 and TIM-3 Immunotherapy
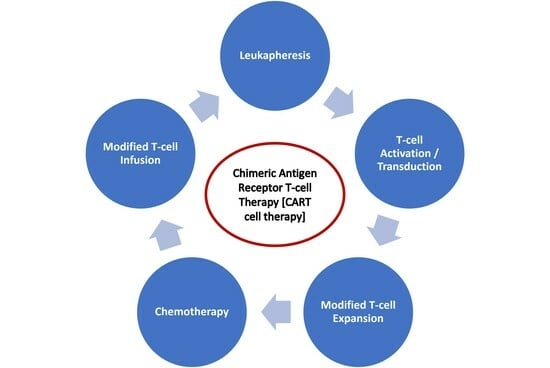
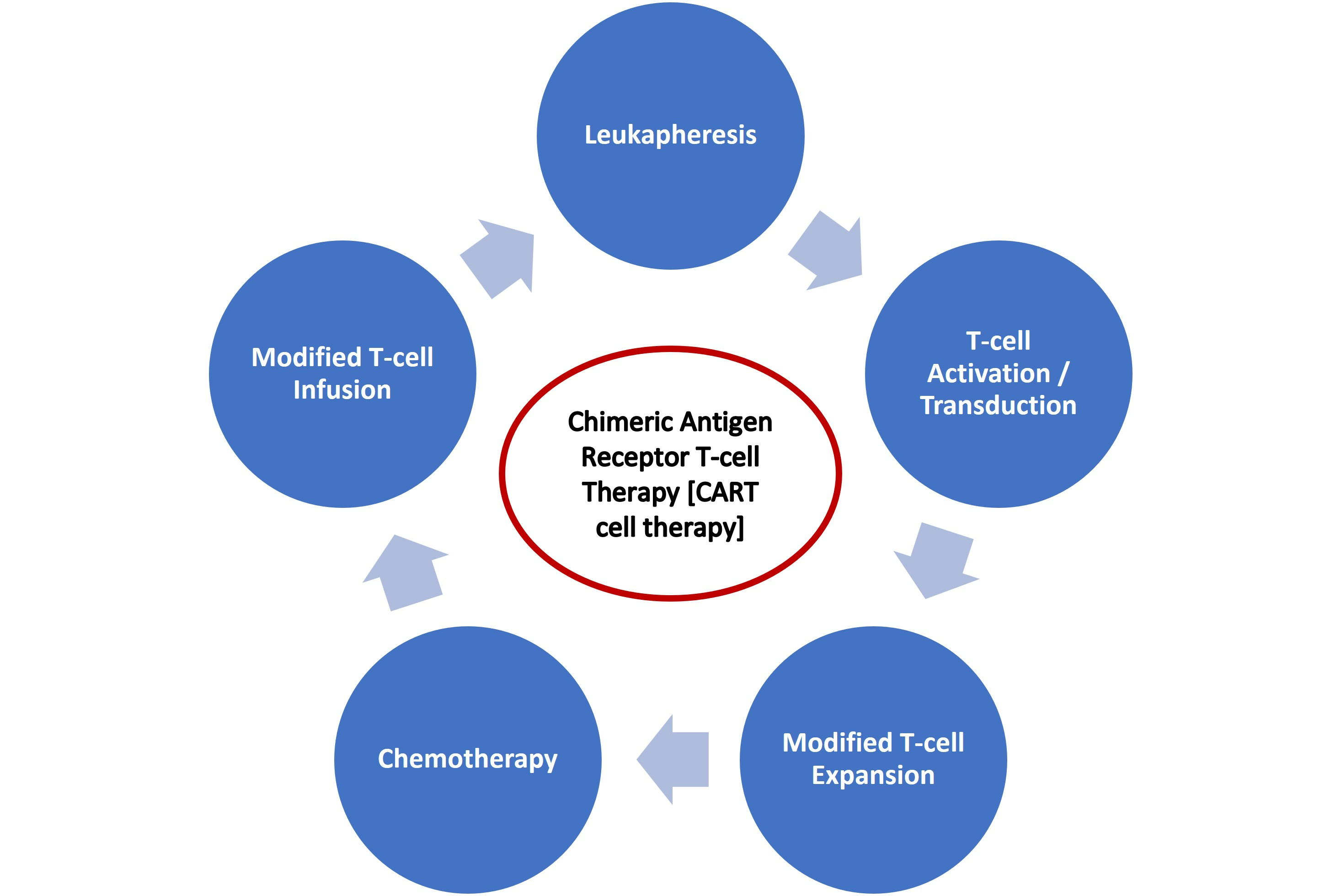
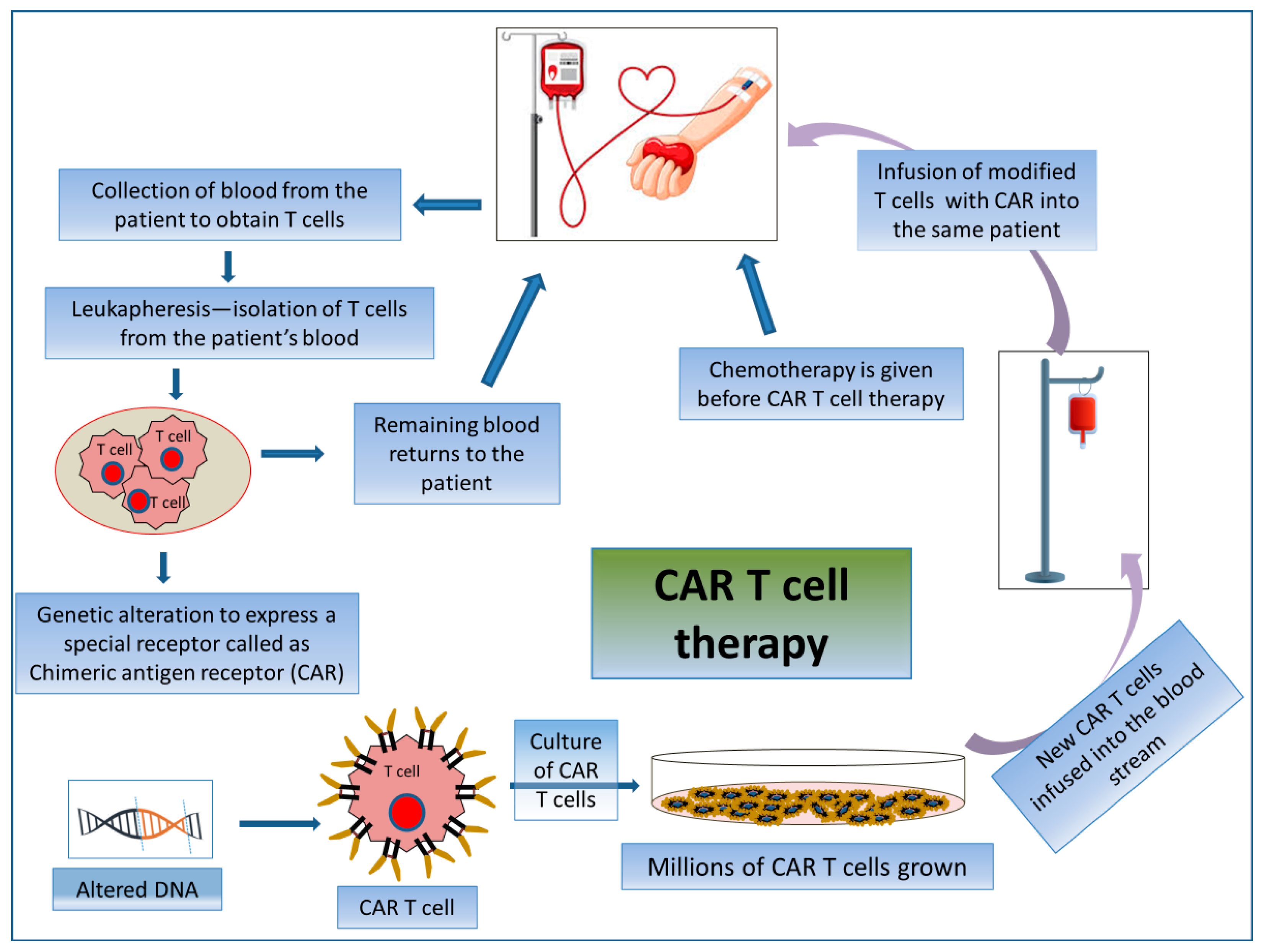
2.3.2. CAR T Cell Therapy
2.3.3. Natural Killer (NK) Cell-Based Immunotherapy (Alternative to CAR T Cell Therapy)
2.3.4. Dendritic Cell Vaccine Therapy (a Cross between a Vaccine and a Cell Therapy)
2.3.5. CRISPR-Cas9-Based Immunotherapy
3. COVID-19 and Immunotherapy
4. Resistance to Immunotherapy
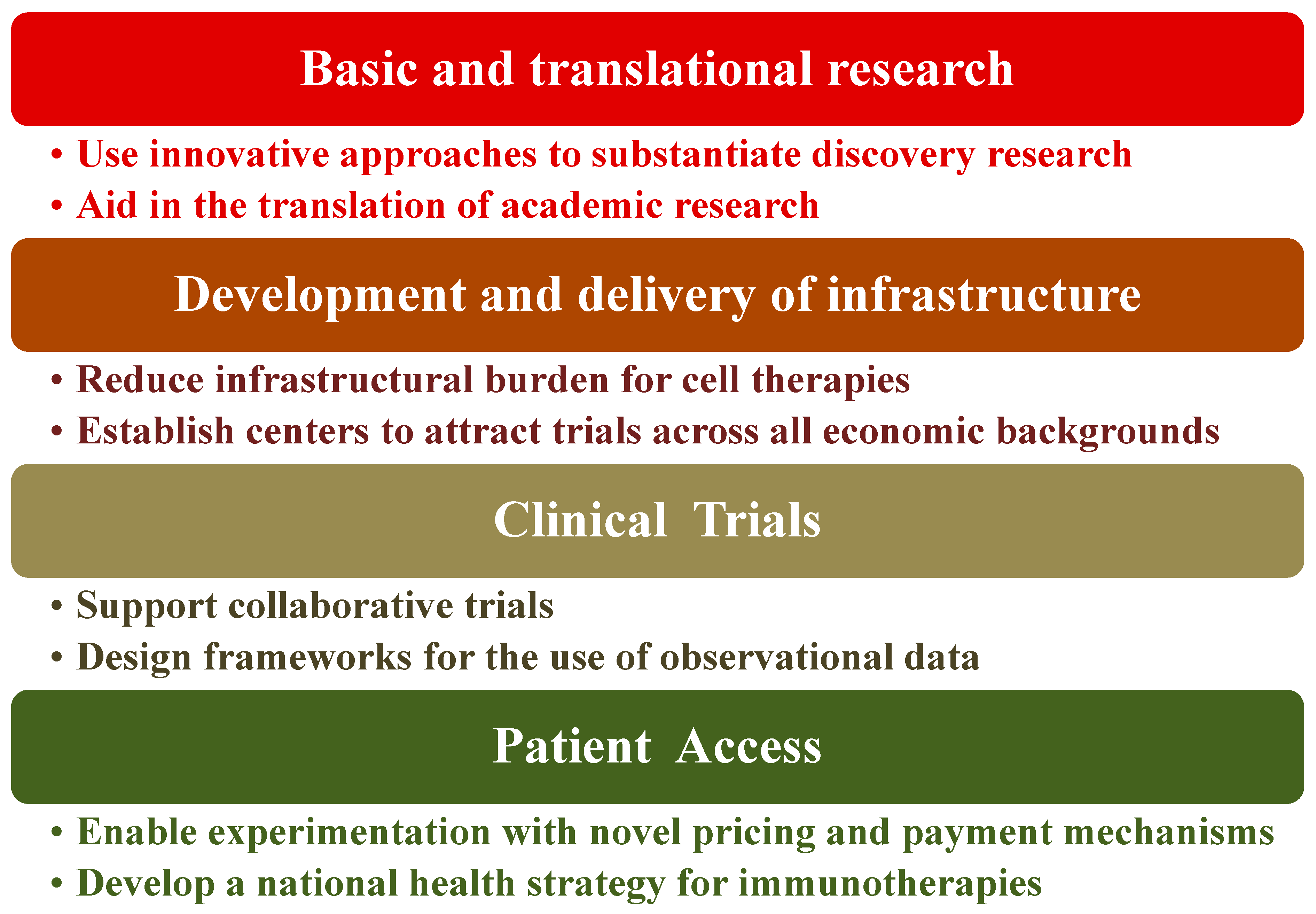
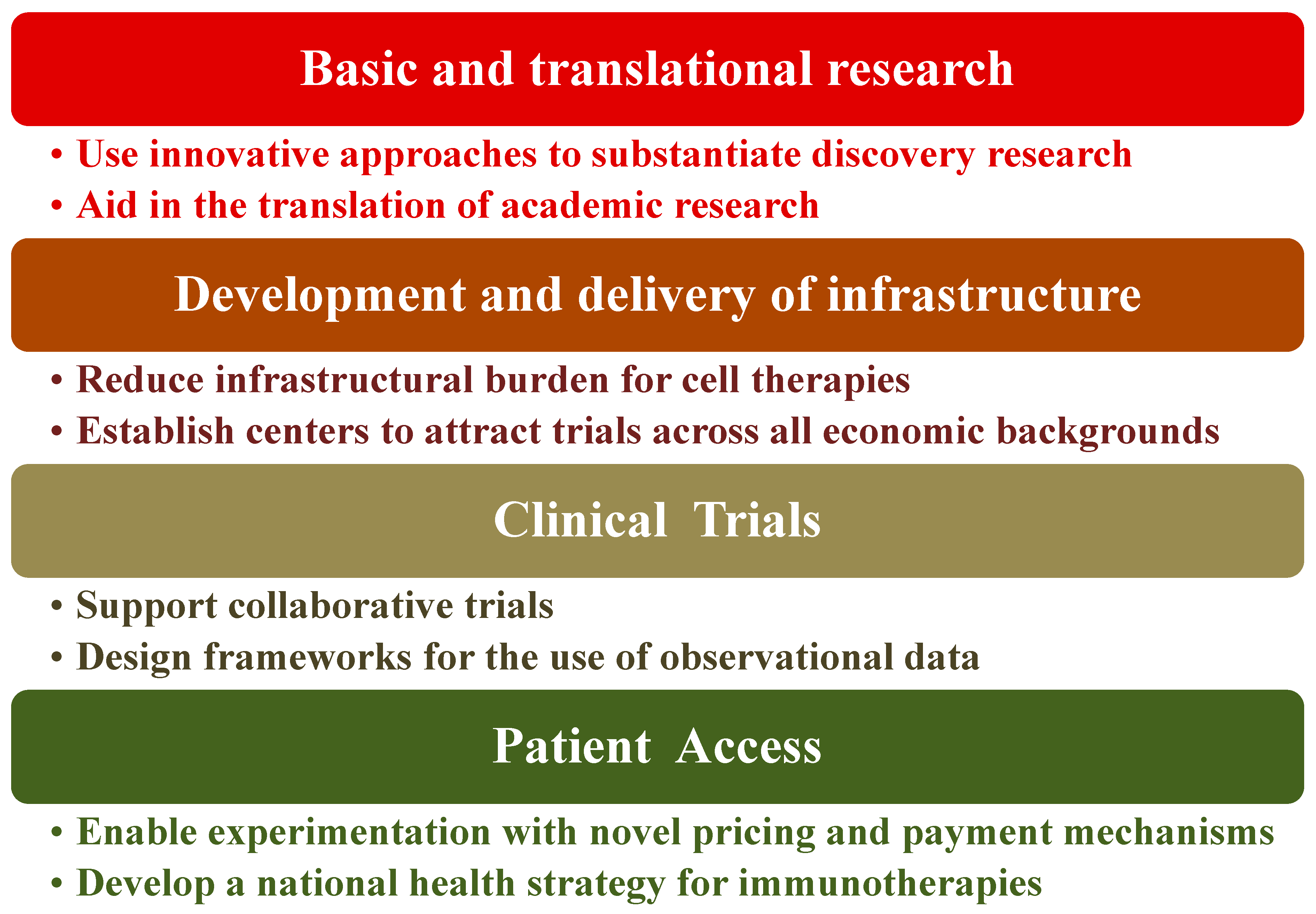
5. Policy Recommendations Regarding Immunotherapy
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Esfahani, K.; Elkrief, A.; Calabrese, C.; Lapointe, R.; Hudson, M.; Routy, B.; Miller, W.H., Jr.; Calabrese, L. Moving towards personalized treatments of immune-related adverse events. Nat. Rev. Clin. Oncol. 2020, 17, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, M.; Wu, H.-X.; Xu, R.-H. Advancing to the era of cancer immunotherapy. Cancer Commun. 2021, 41, 803–829. [Google Scholar] [CrossRef]
- Sharmiladevi, P.; Girigoswami, K.; Haribabu, V.; Girigoswami, A. Nano-enabled theranostics for cancer. Mater. Adv. 2021, 2, 2876–2891. [Google Scholar] [CrossRef]
- Yang, K.; Halima, A.; Chan, T.A. Antigen presentation in cancer—Mechanisms and clinical implications for immunotherapy. Nat. Rev. Clin. Oncol. 2023, 20, 604–623. [Google Scholar] [CrossRef]
- Kruger, S.; Ilmer, M.; Kobold, S.; Cadilha, B.L.; Endres, S.; Ormanns, S.; Schuebbe, G.; Renz, B.W.; D’Haese, J.G.; Schloesser, H.; et al. Advances in cancer immunotherapy 2019—Latest trends. J. Exp. Clin. Cancer Res. 2019, 38, 268. [Google Scholar] [CrossRef] [PubMed]
- Alatrash, G.; Jakher, H.; Stafford, P.D.; Mittendorf, E.A. Cancer immunotherapies, their safety and toxicity. Expert Opin. Drug Saf. 2013, 12, 631–645. [Google Scholar] [CrossRef]
- Pilard, C.; Ancion, M.; Delvenne, P.; Jerusalem, G.; Hubert, P.; Herfs, M. Cancer immunotherapy: It’s time to better predict patients’ response. Br. J. Cancer 2021, 125, 927–938. [Google Scholar] [CrossRef]
- Sahin, U.; Türeci, Ö. Personalized vaccines for cancer immunotherapy. Science 2018, 359, 1355–1360. [Google Scholar] [CrossRef]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Eno, J. Immunotherapy through the years. J. Adv. Pract. Oncol. 2017, 8, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.; Vincelette, N.D.; Green, M.R.; Wahner Hendrickson, A.E.; Abraham, I. Targeting immune checkpoints in unresectable metastatic cutaneous melanoma: A systematic review and meta-analysis of anti-CTLA-4 and anti-PD-1 agents trials. Cancer Med. 2016, 5, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Bachanova, V.; Miller, J.S. NK cells in therapy of cancer. Crit. Rev. Oncog. 2014, 19, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, K.; Janowicz, K.; Dyszkiewicz-Konwińska, M.; Hutchings, G.; Dompe, C.; Moncrieff, L.; Jankowski, M.; Machnik, M.; Oleksiewicz, U.; Kocherova, I.; et al. CRISPR/Cas9 in Cancer Immunotherapy: Animal Models and Human Clinical Trials. Genes 2020, 11, 921. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi Rad, H.; Monkman, J.; Warkiani, M.E.; Ladwa, R.; O’Byrne, K.; Rezaei, N.; Kulasinghe, A. Understanding the tumor microenvironment for effective immunotherapy. Med. Res. Rev. 2021, 41, 1474–1498. [Google Scholar] [CrossRef]
- Yalcin, G.D.; Danisik, N.; Baygin, R.C.; Acar, A. Systems Biology and Experimental Model Systems of Cancer. J. Pers. Med. 2020, 10, 180. [Google Scholar] [CrossRef]
- Zhang, J.; Tao, A. Antigenicity, immunogenicity, allergenicity. In Allergy Bioinformatics; Springer Nature B.V.: Dordrecht, The Netherlands, 2015; pp. 175–186. [Google Scholar] [CrossRef]
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef]
- Brode, S.; Cooke, A. Immune-potentiating effects of the chemotherapeutic drug cyclophosphamide. Crit. Rev. Immunol. 2008, 28, 109–126. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus chemotherapy in metastatic non–small-cell lung cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar]
- Campbell, A.M.; Decker, R.H. Harnessing the Immunomodulatory Effects of Radiation Therapy. Oncology 2018, 32, 370–374. [Google Scholar] [PubMed]
- Atkins, M.B.; Plimack, E.R.; Puzanov, I.; Fishman, M.N.; McDermott, D.F.; Cho, D.C.; Vaishampayan, U.; George, S.; Olencki, T.E.; Tarazi, J.C.; et al. Axitinib in combination with pembrolizumab in patients with advanced renal cell cancer: A non-randomised, open-label, dose-finding, and dose-expansion phase 1b trial. Lancet Oncol. 2018, 19, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Stanculeanu, D.L.; Daniela, Z.; Lazescu, A.; Bunghez, R.; Anghel, R. Development of new immunotherapy treatments in different cancer types. J. Med. Life 2016, 9, 240–248. [Google Scholar] [PubMed]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef]
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.; Kussie, P.; Ferguson, K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005, 7, 301–311. [Google Scholar] [CrossRef]
- Patel, D.; Bassi, R.; Hooper, A.; Prewett, M.; Hicklin, D.J.; Kang, X. Anti-epidermal growth factor receptor monoclonal antibody cetuximab inhibits EGFR/HER-2 heterodimerization and activation. Int. J. Oncol. 2009, 34, 25–32. [Google Scholar]
- Pelster, M.S.; Amaria, R.N. Combined targeted therapy and immunotherapy in melanoma: A review of the impact on the tumor microenvironment and outcomes of early clinical trials. Ther. Adv. Med. Oncol. 2019, 11, 1758835919830826. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Makker, V.; Rasco, D.; Vogelzang, N.J.; Brose, M.S.; Cohn, A.L.; Mier, J.; Di Simone, C.; Hyman, D.M.; Stepan, D.E.; Dutcus, C.E.; et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: An interim analysis of a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 711–718. [Google Scholar] [CrossRef]
- Makker, V.; Colombo, N.; Casado Herráez, A.; Santin, A.D.; Colomba, E.; Miller, D.S.; Fujiwara, K.; Pignata, S.; Baron-Hay, S.; Ray-Coquard, I.; et al. Lenvatinib plus Pembrolizumab for Advanced Endometrial Cancer. N. Engl. J. Med. 2022, 386, 437–448. [Google Scholar] [CrossRef]
- García Rubiño, M.E.; Carrillo, E.; Ruiz Alcalá, G.; Domínguez-Martín, A.; Marchal, J.A.; Boulaiz, H. Phenformin as an anticancer agent: Challenges and prospects. Int. J. Mol. Sci. 2019, 20, 3316. [Google Scholar] [CrossRef]
- Orecchioni, S.; Reggiani, F.; Talarico, G.; Mancuso, P.; Calleri, A.; Gregato, G.; Labanca, V.; Noonan, D.M.; Dallaglio, K.; Albini, A.; et al. The biguanides metformin and phenformin inhibit angiogenesis, local and metastatic growth of breast cancer by targeting both neoplastic and microenvironment cells. Int. J. Cancer Res. 2015, 136, E534–E544. [Google Scholar]
- Vara-Ciruelos, D.; Dandapani, M.; Russell, F.M.; Grzes, K.M.; Atrih, A.; Foretz, M.; Viollet, B.; Lamont, D.J.; Cantrell, D.A.; Hardie, D.G. Phenformin, but not metformin, delays development of T cell acute lymphoblastic leukemia/lymphoma via cell-autonomous AMPK activation. Cell Rep. 2019, 27, 690–698. [Google Scholar] [CrossRef]
- Kim, S.H.; Li, M.; Trousil, S.; Zhang, Y.; di Magliano, M.P.; Swanson, K.D.; Zheng, B. Phenformin inhibits myeloid-derived suppressor cells and enhances the anti-tumor activity of PD-1 blockade in melanoma. J. Investig. Dermatol. 2017, 137, 1740–1748. [Google Scholar]
- Divyapriya, C.; Kannan, A.; Raghavan, V. Expression of CD4, CD8 Biomarkers in Invasive Carcinoma of Breast with Clinicopathological Correlation. J. Pharm. Res. Int. 2021, 33, 65–73. [Google Scholar] [CrossRef]
- Baras, A.S.; Drake, C.; Liu, J.J.; Gandhi, N.; Kates, M.; Hoque, M.O.; Meeker, A.; Hahn, N.; Taube, J.M.; Schoenberg, M.P.; et al. The ratio of CD8 to Treg tumor-infiltrating lymphocytes is associated with response to cisplatin-based neoadjuvant chemotherapy in patients with muscle invasive urothelial carcinoma of the bladder. Oncoimmunology 2016, 5, e1134412. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors in cancer therapy: A focus on T-regulatory cells. Immunol. Cell. Biol. 2018, 96, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Fumet, J.D.; Richard, C.; Ledys, F.; Klopfenstein, Q.; Joubert, P.; Routy, B.; Truntzer, C.; Gagné, A.; Hamel, M.A.; Figueiredo Guimaraes, C.; et al. Prognostic and predictive role of CD8 and PD-L1 determination in lung tumor tissue of patients under anti-PD-1 therapy. Br. J. Cancer 2018, 119, 950–960. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Y.; Liu, S.; Wang, H.; Zhu, J.; Ou, L.; Xu, X. Targeting regulatory T cells for immunotherapy in melanoma. Mol. Biomed. 2021, 2, 11. [Google Scholar] [CrossRef]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; Van Der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.; Abu-Akeel, M.; et al. Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell 2018, 33, 843–852. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Geukes Foppen, M.H.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef]
- Omar, H.A.; Tolba, M.F. Tackling molecular targets beyond PD-1/PD-L1: Novel approaches to boost patients’ response to cancer immunotherapy. Crit. Rev. Oncol. Hematol. 2019, 135, 21–29. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Pan, R.Y.; Chung, W.H.; Chu, M.T.; Chen, S.J.; Chen, H.C.; Zheng, L.; Hung, S.I. Recent Development and Clinical Application of Cancer Vaccine: Targeting Neoantigens. J. Immunol. Res. 2018, 2018, 4325874. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.; Coukos, G.; Bassani-Sternberg, M. Identification of tumor antigens with immunopeptidomics. Nat. Biotechnol. 2022, 40, 175–188. [Google Scholar] [CrossRef]
- Thomas, C.; Tampé, R. MHC I assembly and peptide editing—Chaperones, clients, and molecular plasticity in immunity. Curr. Opin. Immunol. 2021, 70, 48–56. [Google Scholar] [CrossRef]
- Liu, C.C.; Steen, C.B.; Newman, A.M. Computational approaches for characterizing the tumor immune microenvironment. Immunology 2019, 158, 70–84. [Google Scholar] [CrossRef]
- Garcia-Garijo, A.; Fajardo, C.A.; Gros, A. Determinants for Neoantigen Identification. Front. Immunol. 2019, 10, 1392. [Google Scholar] [CrossRef] [PubMed]
- Dersh, D.; Hollý, J.; Yewdell, J.W. A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat. Rev. Immunol. 2021, 21, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef]
- Łuksza, M.; Riaz, N.; Makarov, V.; Balachandran, V.P.; Hellmann, M.D.; Solovyov, A.; Rizvi, N.A.; Merghoub, T.; Levine, A.J.; Chan, T.A.; et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 2017, 551, 517–520. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Neoantigen quality, not quantity. Sci. Transl. Med. 2019, 11, eaax7918. [Google Scholar] [CrossRef]
- Kim, T.K.; Herbst, R.S.; Chen, L. Defining and understanding adaptive resistance in cancer immunotherapy. Trends Immunol. 2018, 39, 624–631. [Google Scholar] [CrossRef]
- Disis, M.L. Mechanism of action of immunotherapy. Semin. Oncol. 2014, 41 (Suppl. 5), S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Webber, J.S.; et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Savoia, P.; Astrua, C.; Fava, P. Ipilimumab (Anti-Ctla-4 Mab) in the treatment of metastatic melanoma: Effectiveness and toxicity management. Hum. Vaccin. Immunother. 2016, 12, 1092–1101. [Google Scholar] [CrossRef]
- Tartari, F.; Santoni, M.; Burattini, L.; Mazzanti, P.; Onofri, A.; Berardi, R. Economic sustainability of anti-PD-1 agents nivolumab and pembrolizumab in cancer patients: Recent insights and future challenges. Cancer Treat. Rev. 2016, 48, 20–24. [Google Scholar] [CrossRef]
- Fumet, J.D.; Isambert, N.; Hervieu, A.; Zanetta, S.; Guion, J.F.; Hennequin, A.; Rederstorff, E.; Bertaut, A.; Ghiringhelli, F. Phase Ib/II trial evaluating the safety, tolerability and immunological activity of durvalumab (MEDI4736) (anti-PD-L1) plus tremelimumab (anti-CTLA-4) combined with FOLFOX in patients with metastatic colorectal cancer. ESMO Open 2018, 3, e000375. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl. J Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.S.; Chew, V.; Sieow, J.L.; Goh, S.; Yeong, J.P.; Soon, A.L.; Ricciardi-Castagnoli, P. PD-1 expression on dendritic cells suppresses CD8+ T cell function and antitumor immunity. Oncoimmunology 2015, 5, e1085146. [Google Scholar] [CrossRef] [PubMed]
- Gianchecchi, E.; Fierabracci, A. Inhibitory receptors and pathways of lymphocytes: The role of PD-1 in Treg development and their involvement in autoimmunity onset and cancer progression. Front. Immunol. 2018, 9, 2374. [Google Scholar] [CrossRef]
- Lee, H.T.; Lee, S.H.; Heo, Y.S. Molecular Interactions of Antibody Drugs Targeting PD-1, PD-L1, and CTLA-4 in Immuno-Oncology. Molecules 2019, 24, 1190. [Google Scholar] [CrossRef]
- Guo, L.; Wei, R.; Lin, Y.; Kwok, H.F. Clinical and recent patents applications of PD-1/PD-L1 targeting immunotherapy in cancer treatment—Current progress, strategy, and future perspective. Front. Immunol. 2020, 11, 1508. [Google Scholar] [CrossRef]
- Khalife, N.; Kordahi, M.; Felefly, T.; Saleh, K. Cemiplimab: A new option for the treatment of non-small-cell lung cancer. Future Oncol. 2021, 17, 2559–2562. [Google Scholar] [CrossRef]
- Wu, M.; Huang, Q.; Xie, Y.; Wu, X.; Ma, H.; Zhang, Y.; Xia, Y. Improvement of the anticancer efficacy of PD-1/PD-L1 blockade via combination therapy and PD-L1 regulation. J. Hematol. Oncol. 2022, 15, 24. [Google Scholar]
- Li, X.S.; Li, J.W.; Li, H.; Jiang, T. Prognostic value of programmed cell death ligand 1 (PD-L1) for hepatocellular carcinoma: A meta-analysis. Biosci. Rep. 2020, 40, BSR20200459. [Google Scholar]
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Inman, B.A.; Longo, T.A.; Ramalingam, S.; Harrison, M.R. Atezolizumab: A PD-L1-Blocking Antibody for Bladder Cancer. Clin. Cancer Res. 2017, 23, 1886–1890. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Bumbaca, D.; Pastuskovas, C.V.; Boswell, C.A.; West, D.; Cowan, K.J.; Chiu, H.; McBride, J.; Johnson, C.; Xin, Y.; et al. Preclinical pharmacokinetics, pharmacodynamics, tissue distribution, and tumor penetration of anti-PD-L1 monoclonal antibody, an immune checkpoint inhibitor. MAbs 2016, 8, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Durvalumab: First global approval. Drugs 2017, 77, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- FDA Medication Guide Reference ID: 5040342. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761069s035lbl.pdf (accessed on 11 June 2023).
- Wang, B.C.; Li, P.C.; Fan, J.Q.; Lin, G.H.; Liu, Q. Durvalumab and tremelimumab combination therapy versus durvalumab or tremelimumab monotherapy for patients with solid tumors: A systematic review and meta-analysis. Medicine 2020, 99, e21273. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Lau, G.; Kudo, M.; Chan, S.L.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.K.; Van Dao, T.; De Toni, E.N.; et al. Tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. NEJM Evid. 2022, 1, EVIDoa2100070. [Google Scholar] [CrossRef]
- Lee, K.-W.; Kim, H.K.; Rhy, M.-H.; Zang, D.Y.; Kim, J.-W.; Kim, B.J.; Kim, H.-D.; Kim, J.W.; Kang, Y.-K. Phase II study of the combination of durvalumab, tremelimumab, and paclitaxel as second-line chemotherapy in biomarker-selected patients with metastatic gastric cancer. J. Clin. Oncol. 2023, 41, 401. [Google Scholar] [CrossRef]
- Johnson, M.L.; Cho, B.C.; Luft, A.; Alatorre-Alexander, J.; Geater, S.L.; Laktionov, K.; Kim, S.-W.; Ursol, G.; Hussein, M.; Lim, F.L.; et al. Durvalumab With or Without Tremelimumab in Combination With Chemotherapy as First-Line Therapy for Metastatic Non–Small-Cell Lung Cancer: The Phase III POSEIDON Study. J. Clin. Oncol. 2023, 41, 1213–1227. [Google Scholar] [CrossRef]
- Mok, T.; Schmid, P.; Aren, O.A.; Arrieta, O.; Gottfried, M.; Jazieh, A.R.; Ramlau, R.; Timcheva, K.; Martin, C.; McIntosh, S. 192TiP: NEPTUNE: A global, phase 3 study of durvalumab (MEDI4736) plus tremelimumab combination therapy versus standard of care (SoC) platinum-based chemotherapy in the first-line treatment of patients (pts) with advanced or metastatic NSCLC. J. Thorac. Oncol. 2016, 11, S140–S141. [Google Scholar] [CrossRef]
- Stewart, R.; Morrow, M.; Hammond, S.A.; Mulgrew, K.; Marcus, D.; Poon, E.; Watkins, A.; Mullins, S.; Chodorge, M.; Andrews, J.; et al. Identification and characterization of MEDI4736, an antagonistic anti–PD-L1 monoclonal antibody. Cancer Immunol. Res. 2015, 3, 1052–1062. [Google Scholar] [CrossRef]
- Grenga, I.; Donahue, R.N.; Lepone, L.M.; Richards, J.; Schlom, J. A fully human IgG1 anti-PD-L1 MAb in an in vitro assay enhances antigen-specific T-cell responses. Clin. Transl. Immunol. 2016, 5, e83. [Google Scholar] [CrossRef]
- Pascolutti, R.; Sun, X.; Kao, J.; Maute, R.L.; Ring, A.M.; Bowman, G.R.; Kruse, A.C. Structure and dynamics of PD-L1 and an ultra-high-affinity PD-1 receptor mutant. Structure 2016, 24, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Pinchuk, I.V.; Powell, D.W. Immunosuppression by intestinal stromal cells. Adv. Exp. Med. Biol. 2018, 1060, 115–129. [Google Scholar] [CrossRef]
- Solinas, C.; Aiello, M.; Rozali, E.; Lambertini, M.; Willard-Gallo, K.; Migliori, E. Programmed cell death-ligand 2: A neglected but important target in the immune response to cancer? Transl. Oncol. 2020, 13, 100811. [Google Scholar] [CrossRef] [PubMed]
- Hong, I.S. Stimulatory versus suppressive effects of GM-CSF on tumor progression in multiple cancer types. Exp. Mol. Med. 2016, 48, e242. [Google Scholar] [CrossRef] [PubMed]
- Bankaitis, K.V.; Fingleton, B. Targeting IL4/IL4R for the treatment of epithelial cancer metastasis. Clin. Exp. Metastasis 2015, 32, 847–856. [Google Scholar] [CrossRef]
- Xiao, Y.; Yu, S.; Zhu, B.; Bedoret, D.; Bu, X.; Francisco, L.M.; Hua, P.; Duke-Cohan, J.S.; Umetsu, D.T.; Sharpe, A.H.; et al. RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J. Exp. Med. 2014, 211, 943–959. [Google Scholar] [CrossRef]
- Derks, S.; Nason, K.S.; Liao, X.; Stachler, M.D.; Liu, K.X.; Liu, J.B.; Sicinska, E.; Goldberg, M.S.; Freeman, G.J.; Rodig, S.J.; et al. Epithelial PD-L2 Expression Marks Barrett’s Esophagus and Esophageal Adenocarcinoma. Cancer Immunol. Res. 2015, 3, 1123–1129. [Google Scholar] [CrossRef]
- Masugi, Y.; Nishihara, R.; Hamada, T.; Song, M.; Da Silva, A.; Kosumi, K.; Gu, M.; Shi, Y.; Li, W.; Liu, L.; et al. Tumor PDCD1LG2 (PD-L2) expression and the lymphocytic reaction to colorectal cancer. Cancer Immunol. Res. 2017, 5, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Tanegashima, T.; Togashi, Y.; Azuma, K.; Kawahara, A.; Ideguchi, K.; Sugiyama, D.; Kinoshita, F.; Akiba, J.; Kashiwagi, E.; Takeuchi, A.; et al. Immune suppression by PD-L2 against spontaneous and treatment-related antitumor immunity. Clin. Cancer Res. 2019, 25, 4808–4819. [Google Scholar] [CrossRef]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef] [PubMed]
- van der Merwe, P.A.; Bodian, D.L.; Daenke, S.; Linsley, P.; Davis, S.J. CD80 (B7-1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J. Exp. Med. 1997, 185, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell 2017, 170, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Fu, T.; Suh, W.K.; Tsavachidou, D.; Wen, S.; Gao, J.; Ng Tang, D.; He, Q.; Sun, J.; Sharma, P. CD4 T cells require ICOS-mediated PI3K signaling to increase T-Bet expression in the setting of anti-CTLA-4 therapy. Cancer Immunol. Res. 2014, 2, 167–176. [Google Scholar] [CrossRef]
- Iwai, Y.; Hamanishi, J.; Chamoto, K.; Honjo, T. Cancer immunotherapies targeting the PD-1 signaling pathway. J. Biomed. Sci. 2017, 24, 26. [Google Scholar] [CrossRef]
- Yu, G.T.; Mao, L.; Wu, L.; Deng, W.W.; Bu, L.L.; Liu, J.F.; Chen, L.; Yang, L.L.; Wu, H.; Zhang, W.F.; et al. Inhibition of SRC family kinases facilitates anti-CTLA4 immunotherapy in head and neck squamous cell carcinoma. Cell. Mol. Life Sci. 2018, 75, 4223–4234. [Google Scholar] [CrossRef]
- Coutzac, C.; Jouniaux, J.; Vozy, A.; Ballas, M.; Brett, S.; Yadavilli, S.; Angevin, E.; Hoos, A.; Chaput, N. Agonistic T cell non depleting ICOS antibody strongly enhances anti-tumor activity with CTLA4 blocking monoclonal antibody without exacerbating colitis. Cancer Res. 2019, 79 (Suppl. 13), 2268. [Google Scholar] [CrossRef]
- Pillai, R.N.; Behera, M.; Owonikoko, T.K.; Kamphorst, A.O.; Pakkala, S.; Belani, C.P.; Khuri, F.R.; Ahmed, R.; Ramalingam, S.S. Comparison of the toxicity profile of PD-1 versus PD-L1 inhibitors in non–small cell lung cancer: A systematic analysis of the literature. Cancer 2018, 124, 271–277. [Google Scholar] [CrossRef]
- Camacho, L.H.; Antonia, S.; Sosman, J.; Kirkwood, J.M.; Gajewski, T.F.; Redman, B.; Pavlov, D.; Bulanhagui, C.; Bozon, V.A.; Gomez-Navarro, J.; et al. Phase I/II trial of tremelimumab in patients with metastatic melanoma. J. Clin. Oncol. 2009, 27, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Arooj, S.; Wang, H. Soluble B7-CD28 Family Inhibitory Immune Checkpoint Proteins and Anti-Cancer Immunotherapy. Front. Immunol. 2021, 12, 651634. [Google Scholar] [CrossRef]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar] [CrossRef]
- Wuerdemann, N.; Pütz, K.; Eckel, H.; Jain, R.; Wittekindt, C.; Huebbers, C.U.; Sharma, S.J.; Langer, C.; Gattenlöhner, S.; Büttner, R.; et al. LAG-3, TIM-3 and VISTA Expression on Tumor-Infiltrating Lymphocytes in Oropharyngeal Squamous Cell Carcinoma—Potential Biomarkers for Targeted Therapy Concepts. Int. J. Mol. Sci. 2021, 22, 379. [Google Scholar] [CrossRef]
- Lecocq, Q.; Keyaerts, M.; Devoogdt, N.; Breckpot, K. The Next-Generation Immune Checkpoint LAG-3 and Its Therapeutic Potential in Oncology: Third Time’s a Charm. Int. J. Mol Sci. 2020, 22, 75. [Google Scholar] [CrossRef]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A. LAG 3 (CD 223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef] [PubMed]
- Huard, B.; Tournier, M.; Hercend, T.; Triebel, F.; Faure, F. Lymphocyte-activation gene 3/major histocompatibility complex class II interaction modulates the antigenic response of CD4+ T lymphocytes. Eur. J. Immunol. 1994, 24, 3216–3221. [Google Scholar] [CrossRef] [PubMed]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef]
- Banerjee, H.; Kane, L.P. Immune regulation by Tim-3. F1000Research 2018, 7, 316. [Google Scholar] [CrossRef]
- Jin, H.T.; Anderson, A.C.; Tan, W.G.; West, E.E.; Ha, S.J.; Araki, K.; Freeman, G.J.; Kuchroo, V.K.; Ahmed, R. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 2010, 107, 14733–14738. [Google Scholar] [CrossRef]
- Yang, R.; Hung, M.C. The role of T-cell immunoglobulin mucin-3 and its ligand galectin-9 in antitumor immunity and cancer immunotherapy. Sci. China Life Sci. 2017, 60, 1058–1064. [Google Scholar] [CrossRef]
- Komohara, Y.; Morita, T.; Annan, D.A.; Horlad, H.; Ohnishi, K.; Yamada, S.; Nakayama, T.; Kitada, S.; Suzu, S.; Kinoshita, I.; et al. The coordinated actions of TIM-3 on cancer and myeloid cells in the regulation of tumorigenicity and clinical prognosis in clear cell renal cell carcinomas. Cancer Immunol. Res. 2015, 3, 999–1007. [Google Scholar] [CrossRef]
- Acharya, N.; Sabatos-Peyton, C.; Anderson, A.C. Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer 2020, 8, e000911. [Google Scholar] [CrossRef]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2020, 20, 173–185. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering car-t cells. Biomark. Res. 2017, 5, 1–6. [Google Scholar] [CrossRef]
- Wagner, D.L.; Fritsche, E.; Pulsipher, M.A.; Ahmed, N.; Hamieh, M.; Hegde, M.; Ruella, M.; Savoldo, B.; Shah, N.N.; Turtle, C.J.; et al. Immunogenicity of CAR T cells in cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 379–393. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Krause, A.; Guo, H.F.; Latouche, J.B.; Tan, C.; Cheung, N.K.; Sadelain, M. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J. Exp. Med. 1998, 188, 619–626. [Google Scholar] [CrossRef]
- Guedan, S.; Chen, X.; Madar, A.; Carpenito, C.; McGettigan, S.E.; Frigault, M.J.; Lee, J.; Posey, A.D., Jr.; Scholler, J.; Scholler, N.; et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood 2014, 124, 1070–1080. [Google Scholar] [CrossRef]
- Rosado-Sánchez, I.; Herrero-Fernández, I.; Genebat, M.; Del Romero, J.; Riera, M.; Podzamczer, D.; Olalla, J.; Vidal, F.; Muñoz-Fernández, M.; Leal, M.; et al. HIV-infected subjects with Poor CD4 T-cell recovery despite effective therapy express high levels of OX40 and α4β7 on CD4 T-cells prior therapy initiation. Front. Immunol. 2018, 9, 1673. [Google Scholar] [CrossRef]
- Kerkar, S.P.; Muranski, P.; Kaiser, A.; Boni, A.; Sanchez-Perez, L.; Yu, Z.; Palmer, D.C.; Reger, R.N.; Borman, Z.A.; Zhang, L.; et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 2010, 70, 6725–6734. [Google Scholar] [CrossRef]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef]
- Li, Y.; Ming, Y.; Fu, R.; Li, C.; Wu, Y.; Jiang, T.; Li, Z.; Ni, R.; Li, L.; Su, H.; et al. The pathogenesis, diagnosis, prevention, and treatment of CAR-T cell therapy-related adverse reactions. Front. Pharmacol. 2022, 13, 950923. [Google Scholar] [CrossRef]
- Chu, J.; Gao, F.; Yan, M.; Zhao, S.; Yan, Z.; Shi, B.; Liu, Y. Natural killer cells: A promising immunotherapy for cancer. J. Transl. Med. 2022, 20, 240. [Google Scholar] [CrossRef]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 85–100. [Google Scholar] [CrossRef]
- Liu, S.; Galat, V.; Galat4, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 2021, 14, 7. [Google Scholar] [CrossRef]
- Rees, R.C. MHC restricted and non-restricted killer lymphocytes. Blood Rev. 1990, 4, 204–210. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Chen, L. A paradigm shift in cancer immunotherapy: From enhancement to normalization. Cell 2018, 175, 313–326. [Google Scholar] [CrossRef]
- Parkhurst, M.R.; Riley, J.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin. Cancer Res. 2011, 17, 6287–6297. [Google Scholar] [CrossRef]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric antigen receptor-engineered NK-92 cells: An off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front. Immunol. 2017, 8, 533. [Google Scholar] [CrossRef]
- Oberschmidt, O.; Kloess, S.; Koehl, U. Redirected primary human chimeric antigen receptor natural killer cells as an “off-the-shelf immunotherapy” for improvement in cancer treatment. Front. Immunol. 2017, 8, 654. [Google Scholar] [CrossRef]
- Khawar, M.B.; Sun, H. CAR-NK Cells: From Natural Basis to Design for Kill. Front. Immunol. 2021, 12, 707542. [Google Scholar] [CrossRef]
- Jong, A.Y.; Wu, C.H.; Li, J.; Sun, J.; Fabbri, M.; Wayne, A.S.; Seeger, R.C. Large-scale isolation and cytotoxicity of extracellular vesicles derived from activated human natural killer cells. J. Extracell. Vesicles 2017, 6, 1294368. [Google Scholar] [CrossRef]
- Mandó, P.; Rivero, S.G.; Rizzo, M.M.; Pinkasz, M.; Levy, E.M. Targeting ADCC: A different approach to HER2 breast cancer in the immunotherapy era. Breast 2021, 60, 15–25. [Google Scholar] [CrossRef]
- Yu, J.; Sun, H.; Cao, W.; Song, Y.; Jiang, Z. Research progress on dendritic cell vaccines in cancer immunotherapy. Exp. Hematol. Oncol. 2022, 11, 3. [Google Scholar] [CrossRef]
- Constantino, J.; Gomes, C.; Falcão, A.; Cruz, M.T.; Neves, B.M. Antitumor dendritic cell-based vaccines: Lessons from 20 years of clinical trials and future perspectives. Transl. Res. 2016, 168, 74–95. [Google Scholar] [CrossRef]
- Huber, A.; Dammeijer, F.; Aerts, J.G.J.V.; Vroman, H. Current State of Dendritic Cell-Based Immunotherapy: Opportunities for in vitro Antigen Loading of Different DC Subsets? Front. Immunol. 2018, 9, 2804. [Google Scholar] [CrossRef]
- Wimmers, F.; Schreibelt, G.; Sköld, A.E.; Figdor, C.G.; De Vries, I.J. Paradigm shift in dendritic cell-based immunotherapy: From in vitro generated monocyte-derived DCs to naturally circulating DC subsets. Front. Immunol. 2014, 5, 165. [Google Scholar] [CrossRef]
- Okamoto, M.; Kobayashi, M.; Yonemitsu, Y.; Koido, S.; Homma, S. Dendritic cell-based vaccine for pancreatic cancer in Japan. World J. Gastrointest. Pharmacol. Ther. 2016, 7, 133–138. [Google Scholar] [CrossRef]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef]
- Steinman, R.M. Decisions about dendritic cells: Past, present, and future. Annu. Rev. Immunol. 2012, 30, 1–22. [Google Scholar] [CrossRef]
- Palucka, K.; Banchereau, J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013, 39, 38–48. [Google Scholar] [CrossRef]
- Melief, C.J.; Kast, W.M. Cytotoxic T lymphocyte therapy of cancer and tumor escape mechanisms. Semin. Cancer Biol. 1991, 2, 347–354. [Google Scholar]
- Martínez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef]
- Zhang, N.; Bevan, M.J. CD8+ T cells: Foot soldiers of the immune system. Immunity 2011, 35, 161–168. [Google Scholar] [CrossRef]
- Sutherland, S.I.; Ju, X.; Horvath, L.G.; Clark, G.J. Moving on From Sipuleucel-T: New Dendritic Cell Vaccine Strategies for Prostate Cancer. Front. Immunol. 2021, 12, 641307. [Google Scholar] [CrossRef]
- Anassi, E.; Ndefo, U.A. Sipuleucel-T (provenge) injection: The first immunotherapy agent (vaccine) for hormone-refractory prostate cancer. Pharmacol. Ther. 2011, 36, 197–202. [Google Scholar]
- Ou, X.; Ma, Q.; Yin, W.; Ma, X.; He, Z. CRISPR/Cas9 Gene-Editing in Cancer Immunotherapy: Promoting the Present Revolution in Cancer Therapy and Exploring More. Front. Cell Dev. Biol. 2021, 9, 674467. [Google Scholar] [CrossRef] [PubMed]
- Azangou-Khyavy, M.; Ghasemi, M.; Khanali, J.; Boroomand-Saboor, M.; Jamalkhah, M.; Soleimani, M.; Kiani, J. CRISPR/Cas: From Tumor Gene Editing to T Cell-Based Immunotherapy of Cancer. Front. Immunol. 2020, 11, 2062. [Google Scholar] [CrossRef] [PubMed]
- Xia, A.L.; He, Q.F.; Wang, J.C.; Zhu, J.; Sha, Y.Q.; Sun, B.; Lu, X.J. Applications and advances of CRISPR-Cas9 in cancer immunotherapy. J. Med. Genet. 2019, 56, 4–9. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, X.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 2017, 8, 17002–17011. [Google Scholar] [CrossRef] [PubMed]
- Künkele, A.; Johnson, A.J.; Rolczynski, L.S.; Chang, C.A.; Hoglund, V.; Kelly-Spratt, K.S.; Jensen, M.C. Functional Tuning of CARs Reveals Signaling Threshold above Which CD8+ CTL Antitumor Potency Is Attenuated due to Cell Fas-FasL-Dependent AICD. Cancer Immunol. Res. 2015, 3, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef] [PubMed]
- Masoomikarimi, M.; Garmabi, B.; Alizadeh, J.; Kazemi, E.; Jafari, A.A.; Mirmoeeni, S.; Dargahi, M.; Taheri, N.; Jafari, R. Advances in immunotherapy for COVID-19: A comprehensive review. Int. Immunopharmacol. 2021, 93, 107409. [Google Scholar] [CrossRef] [PubMed]
- Saini, K.S.; Lanza, C.; Romano, M.; de Azambuja, E.; Cortes, J.; de Las Heras, B.; de Castro, J.; Lamba Saini, M.; Loibl, S.; Curigliano, G.; et al. Repurposing anticancer drugs for COVID-19-induced inflammation, immune dysfunction, and coagulopathy. Br. J. Cancer 2020, 123, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Temena, M.A.; Acar, A. Increased TRIM31 gene expression is positively correlated with SARS-CoV-2 associated genes TMPRSS2 and TMPRSS4 in gastrointestinal cancers. Sci. Rep. 2022, 12, 11763. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.W.; Joshi, I.; Leal, L.; Ooi, E.E. Immune immunomodulation in coronavirus disease 2019 (COVID-19), strategic considerations for personalized therapeutic intervention. Clin. Infect. Dis. 2022, 74, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.S.; de Sá, K.S.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Goncalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2021, 218, e20201707. [Google Scholar]
- Ratajczak, M.Z.; Kucia, M. SARS-CoV-2 infection and overactivation of Nlrp3 inflammasome as a trigger of cytokine “storm” and risk factor for damage of hematopoietic stem cells. Leukemia 2020, 34, 1726–1729. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, M.; Li, Y.; Yuen, H.H.; He, M.L. The effects of SARS-CoV-2 infection on modulating innate immunity and strategies of combating inflammatory response for COVID-19 therapy. J. Biomed. Sci. 2022, 29, 27. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [PubMed]
- Lievre, A.; Turpin, A.; Ray-Coquard, I.; Le Malicot, K.; Thariat, J.; Ahle, G.; Neuzillet, C.; Paoletti, X.; Bouche, O.; Aldabbagh, K.; et al. GCO-002 CACOVID-19 collaborators/investigators. Risk factors for coronavirus disease 2019 (COVID-19) severity and mortality among solid cancer patients and impact of the disease on anticancer treatment: A French nationwide cohort study (GCO-002 CACOVID-19). Eur. J. Cancer 2020, 141, 62–81. [Google Scholar] [CrossRef]
- Tan, R.; Yun, C.; Seetasith, A.; Sheinson, D.; Walls, R.; Ngwa, I.; Reddy, J.C.; Zhang, Q.; Secrest, M.H.; Lambert, P.; et al. Impact of immune checkpoint inhibitors on COVID-19 severity in patients with cancer. Oncologist 2022, 27, 236–243. [Google Scholar]
- Varnai, C.; Palles, C.; Arnold, R.; Curley, H.M.; Purshouse, K.; Cheng, V.W.; Booth, S.; Campton, N.A.; Collins, G.P.; Hughes, D.J.; et al. Mortality among adults with cancer undergoing chemotherapy or immunotherapy and infected with COVID19. JAMA Netw. Open 2022, 5, e220130. [Google Scholar] [CrossRef] [PubMed]
- Robilotti, E.V.; Babady, N.E.; Mead, P.A.; Rolling, T.; Perez-Johnston, R.; Bernardes, M.; Bogler, Y.; Caldararo, M.; Figueroa, C.J.; Glickman, M.S.; et al. Determinants of COVID-19 disease severity in patients with cancer. Nat Med. 2020, 26, 1218–1223. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.D.; Gonzalez, M.A.; Rüthrich, M.M.; Sharon, E.; von Lilienfeld-Toal, M. COVID-19 and Cancer: Special Considerations for Patients Receiving Immunotherapy and Immunosuppressive Cancer Therapies. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Hensley, M.K.; Bain, W.G.; Jacobs, J.; Nambulli, S.; Parikh, U.; Cillo, A.; Staines, B.; Heaps, A.; Sobolewski, M.D.; Renicck, L.J.; et al. Intractable coronavirus disease 2019 (COVID-19) and prolonged severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) replication in a chimeric antigen receptor-modified T-cell therapy recipient: A case study. Clin. Infect. Dis. 2021, 73, e815–e821. [Google Scholar] [CrossRef]
- Aydillo, T.; Gonzalez-Reiche, A.S.; Aslam, S.; van de Guchte, A.; Khan, Z.; Obla, A.; Dutta, J.; van Bakel, H.; Aberg, J.; Garcia-Sastre, A.; et al. Shedding of viable SARS-CoV-2 after immunosuppressive therapy for cancer. N. Engl. J. Med. 2020, 383, 2586–2588. [Google Scholar] [CrossRef]
- Avanzato, V.A.; Matson, M.J.; Seifert, S.N.; Pryce, R.; Williamson, B.N.; Anzick, S.L.; Barbian, K.; Judson, S.D.; Fischer, E.R.; Martens, C.; et al. Case study: Prolonged infectious SARS-CoV-2 shedding from an asymptomatic immunocompromised individual with cancer. Cell 2020, 183, 1901–1912.e9. [Google Scholar] [CrossRef]
- Baang, J.H.; Smith, C.; Mirabelli, C.; Valesano, A.L.; Manthei, D.M.; Bachman, M.A.; Wobus, C.E.; Adams, M.; Washer, L.; Martin, E.T.; et al. Prolonged severe acute respiratory syndrome coronavirus 2 replication in an immunocompromised patient. J. Infect. Dis. 2021, 223, 23–27. [Google Scholar] [CrossRef]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.A.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Ushiro Lumb, I.; et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Nussenblatt, V.; Roder, A.E.; Das, S.; de Wit, E.; Youn, J.H.; Banakis, S.; Mushegian, A.; Mederos, C.; Wang, W.; Chung, M.; et al. Yearlong COVID-19 Infection reveals within-host evolution of SARS-CoV-2 in a patient with B-cell depletion. J. Infect. Dis. 2022, 225, 1118–1123. [Google Scholar] [PubMed]
- Corey, L.; Beyrer, C.; Cohen, M.S.; Michael, N.L.; Bedford, T.; Rolland, M. SARS-CoV-2 variants in patients with immunosuppression. N. Engl. J. Med. 2021, 385, 562–566. [Google Scholar]
- Ruiz, J.I.; Lopez-Olivo, M.A.; Geng, Y.; Suarez-Almazor, M.E. COVID-19 vaccination in patients with cancer receiving immune checkpoint inhibitors: A systematic review and meta-analysis. J. Immunother. Cancer 2023, 11, e006246. [Google Scholar] [CrossRef] [PubMed]
- Said, S.S.; Ibrahim, W.N. Cancer Resistance to Immunotherapy: Comprehensive Insights with Future Perspectives. Pharmaceutics 2023, 15, 1143. [Google Scholar] [CrossRef] [PubMed]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of resistance to immune checkpoint blockade: Why does checkpoint inhibitor immunotherapy not work for all patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar]
- Bai, R.; Chen, N.; Li, L.; Du, N.; Bai, L.; Lv, Z.; Tian, H.; Cui, J. Mechanisms of Cancer Resistance to Immunotherapy. Front. Oncol. 2020, 10, 1290. [Google Scholar] [CrossRef]
- Ponomarev, A.; Gilazieva, Z.; Solovyeva, V.; Allegrucci, C.; Rizvanov, A. Intrinsic and extrinsic factors impacting cancer stemness and tumor progression. Cancers 2022, 14, 970. [Google Scholar]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Rieth, J.; Subramanian, S. Mechanisms of Intrinsic Tumor Resistance to Immunotherapy. Int. J. Mol. Sci. 2018, 19, 1340. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [PubMed]
- Chen, X.; Zhang, W.; Yang, W.; Zhou, M.; Liu, F. Acquired resistance for immune checkpoint inhibitors in cancer immunotherapy: Challenges and prospects. Aging 2022, 14, 1048. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Han, Y.; Zhang, Y.; Zhao, Q.; Wang, H.; Wei, J.; Meng, L.; Xin, Y.; Jiang, X. Overcoming acquired resistance to cancer immune checkpoint therapy: Potential strategies based on molecular mechanisms. Cell Biosci. 2023, 13, 120. [Google Scholar]
- Kvistborg, P.; van Buuren, M.M.; Schumacher, T.N. Human cancer regression antigens. Curr. Opin. Immunol. 2013, 25, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar]
- Paulson, K.G.; Voillet, V.; McAfee, M.S.; Hunter, D.S.; Wagener, F.D.; Perdicchio, M.; Valente, W.J.; Koelle, S.J.; Church, C.D.; Vandeven, N.; et al. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat. Commun. 2018, 9, 3868. [Google Scholar]
- Lee, J.H.; Shklovskaya, E.; Lim, S.Y.; Carlino, M.S.; Menzies, A.M.; Stewart, A.; Pedersen, B.; Irvine, M.; Alavi, S.; Yang, J.Y.; et al. Transcriptional downregulation of MHC class I and melanoma de-differentiation in resistance to PD-1 inhibition. Nat. Commun. 2020, 11, 1897. [Google Scholar] [CrossRef]
- Thommen, D.; Uhlenbrock, F.; Herzig, P.; Prince, S.S.; Moersig, W.; Lardinois, D.; Zippelius, A. 66P Highly exhausted PD-1hi T cell subsets in human NSCLC are co-defined by the predominant expression of distinct inhibitory receptors and correlate with clinical outcome. J. Thorac. Oncol. 2016, 11, S83. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949. [Google Scholar] [CrossRef] [PubMed]
- Essa, M.M.; Qoronfleh, M.W. (Eds.) Personalized Food Intervention and Therapy for Autism Spectrum Disorder Management; Springer: Cham, Switzerland, 2020. [Google Scholar] [CrossRef]
- Piper, M.; Kluger, H.; Ruppin, E.; Hu-Lieskovan, S. Immune Resistance Mechanisms and the Road to Personalized Immunotherapy. Am. Soc. Clin. Oncol. Educ. Book 2023, 43, e390290. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e9. [Google Scholar]
- The, L.; Capra, E.; Amin, P.; Alford, J.; Thompson, D.; Davies, N.; Griffiths, G. Realizing the Promise of Immunotherapy: A Global Plan for Action; World Innovation Summit for Health: Doha, Qatar, 2020; ISBN 978-1-913991-05-0. [Google Scholar]
- Qoronfleh, M.W. Pathway to excellence in cancer care: Learning from Qatar’s experience. Precis. Med. Sci. 2020, 9, 51–61. [Google Scholar] [CrossRef]
- Zhai, K.; Masoodi, N.A.; Zhang, L.; Yousef, M.S.; Qoronfleh, M.W. Healthcare Fusion: An Innovative Framework for Health Information Management. Electron. J. Knowl. Manag. 2022, 20, 179–192. [Google Scholar] [CrossRef]
- Qoronfleh, M.W.; Chouchane, L.; Mifsud, B.; Al Emadi, M.; Ismail, S. The future of medicine, healthcare innovation through precision medicine: Policy case study of Qatar. Life Sci. Soc. Policy 2020, 16, 12. [Google Scholar] [CrossRef] [PubMed]
- Al-Dewik, N.I.; Younes, S.N.; Essa, M.M.; Pathak, S.; Qoronfleh, M.W. Making Biomarkers Relevant to Healthcare Innovation and Precision Medicine. Processes 2022, 10, 1107. [Google Scholar] [CrossRef]
- Miller, M.J.; Foy, K.C.; Kaumaya, P.T. Cancer immunotherapy: Present status, future perspective, and a new paradigm of peptide immunotherapeutics. Discov. Med. 2013, 15, 166–176. [Google Scholar]
- Tsiatas, M.; Mountzios, G.; Curigliano, G. Future perspectives in cancer immunotherapy. Ann. Transl. Med. 2016, 4, 273. [Google Scholar] [CrossRef]
- Hildebrandt, M.; Peggs, K.; Uharek, L.; Bollard, C.M.; Heslop, H.E. Immunotherapy: Opportunities, risks and future perspectives. Cytotherapy 2014, 16 (Suppl. 4), S120–S129. [Google Scholar] [CrossRef] [PubMed]
- Zhai, K.; Yousef, M.S.; Mohammed, S.; Al-Dewik, N.I.; Qoronfleh, M.W. Optimizing Clinical Workflow Using Precision Medicine and Advanced Data Analytics. Processes 2023, 11, 939. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adhikary, S.; Pathak, S.; Palani, V.; Acar, A.; Banerjee, A.; Al-Dewik, N.I.; Essa, M.M.; Mohammed, S.G.A.A.; Qoronfleh, M.W. Current Technologies and Future Perspectives in Immunotherapy towards a Clinical Oncology Approach. Biomedicines 2024, 12, 217. https://doi.org/10.3390/biomedicines12010217
Adhikary S, Pathak S, Palani V, Acar A, Banerjee A, Al-Dewik NI, Essa MM, Mohammed SGAA, Qoronfleh MW. Current Technologies and Future Perspectives in Immunotherapy towards a Clinical Oncology Approach. Biomedicines. 2024; 12(1):217. https://doi.org/10.3390/biomedicines12010217
Chicago/Turabian StyleAdhikary, Subhamay, Surajit Pathak, Vignesh Palani, Ahmet Acar, Antara Banerjee, Nader I. Al-Dewik, Musthafa Mohamed Essa, Sawsan G. A. A. Mohammed, and M. Walid Qoronfleh. 2024. "Current Technologies and Future Perspectives in Immunotherapy towards a Clinical Oncology Approach" Biomedicines 12, no. 1: 217. https://doi.org/10.3390/biomedicines12010217
APA StyleAdhikary, S., Pathak, S., Palani, V., Acar, A., Banerjee, A., Al-Dewik, N. I., Essa, M. M., Mohammed, S. G. A. A., & Qoronfleh, M. W. (2024). Current Technologies and Future Perspectives in Immunotherapy towards a Clinical Oncology Approach. Biomedicines, 12(1), 217. https://doi.org/10.3390/biomedicines12010217