
Single-Cell DNA Sequencing and Immunophenotypic Profiling to Track Clonal Evolution in an Acute Myeloid Leukemia Patient
 ,
,  ,
,  and
and
Abstract
:1. Introduction
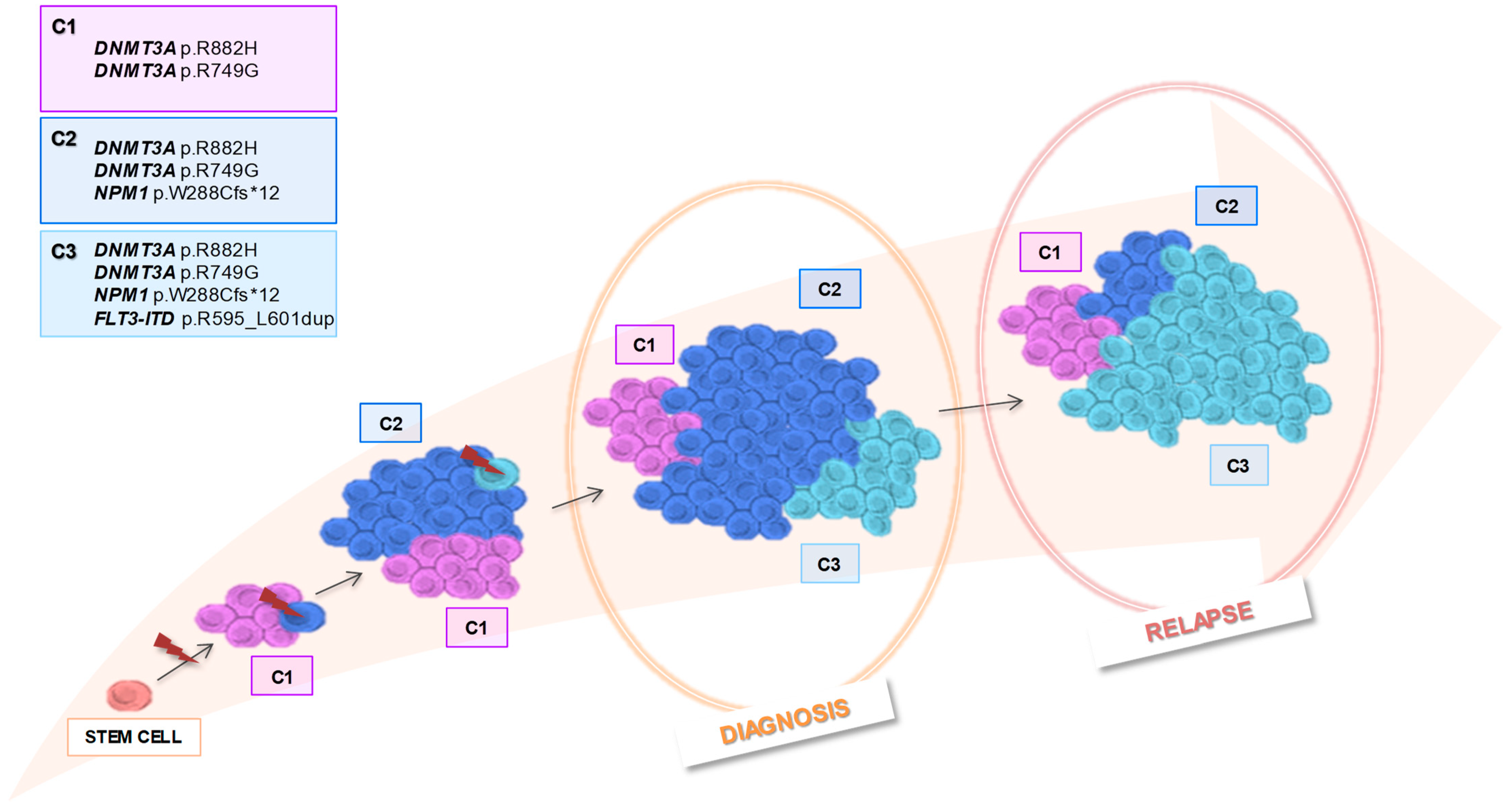
2. Case Presentation
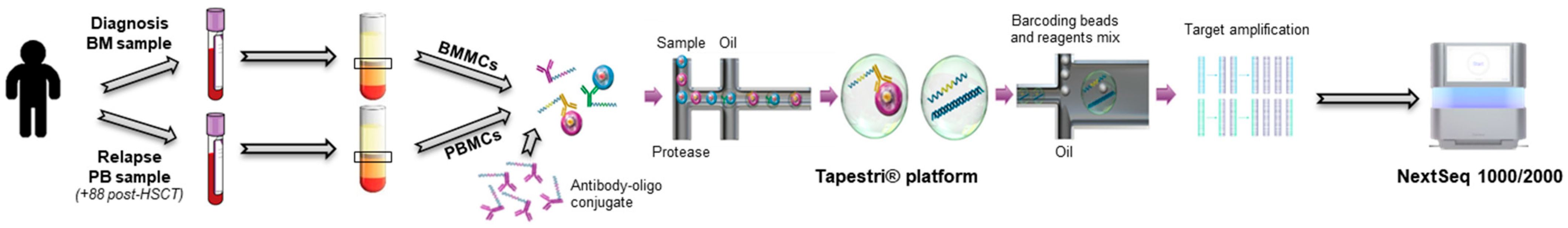
2.1. Single-Cell Analysis
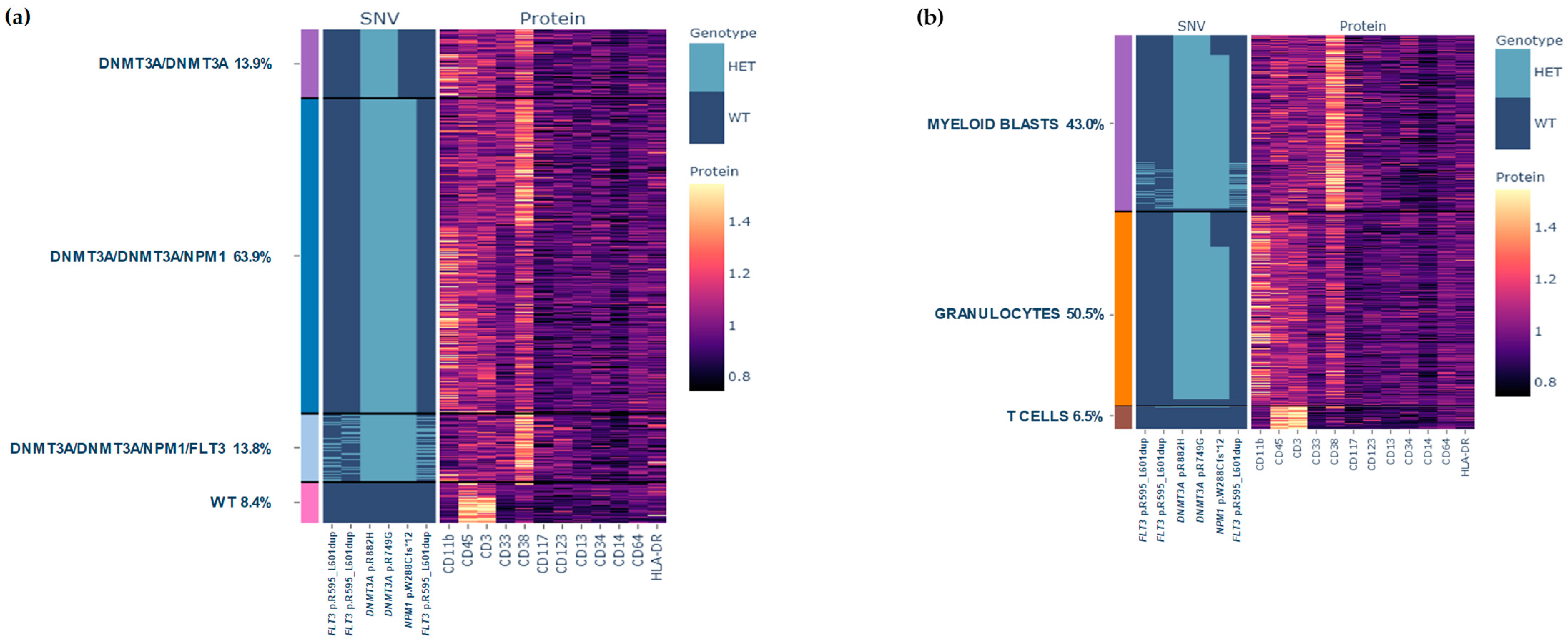
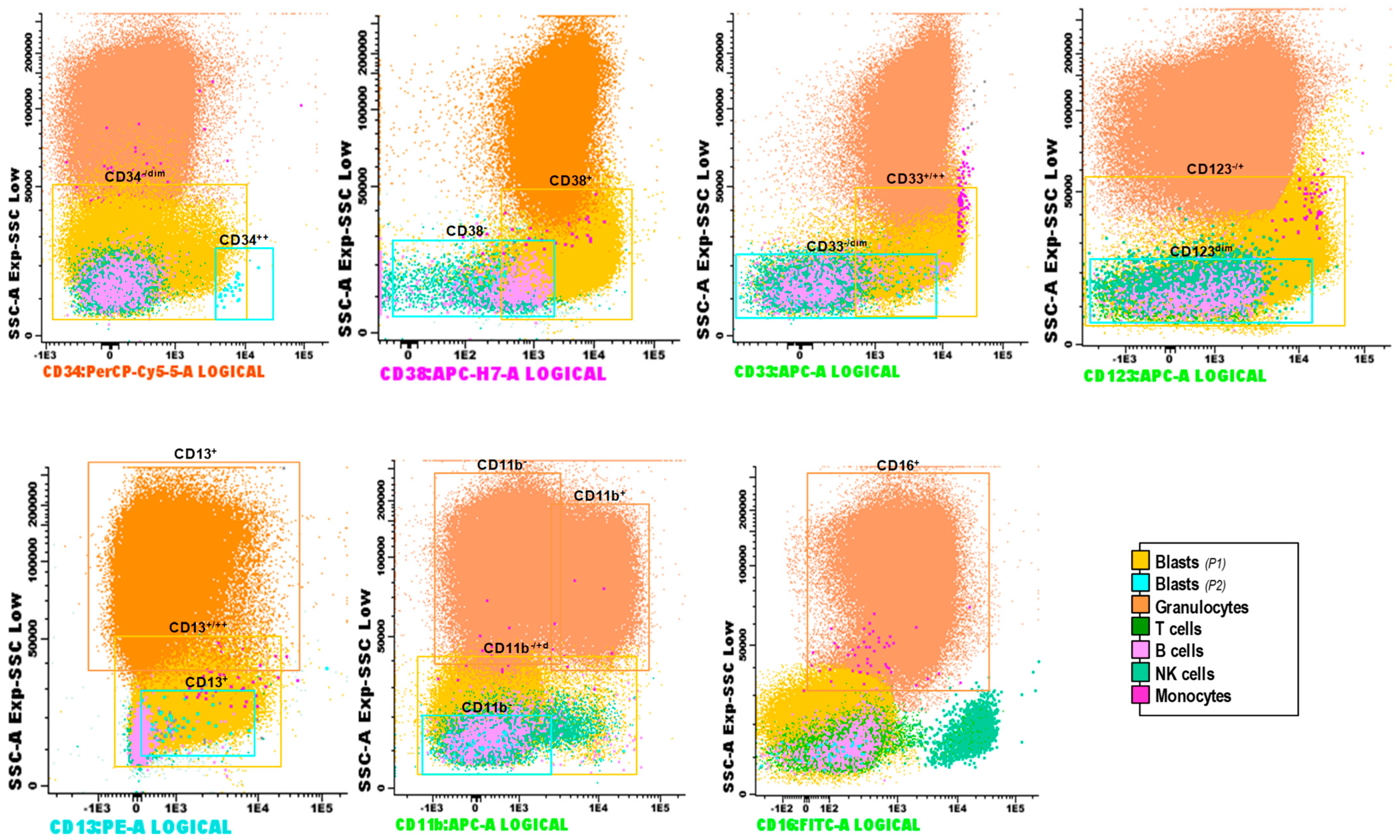
2.1.1. Results at Diagnosis
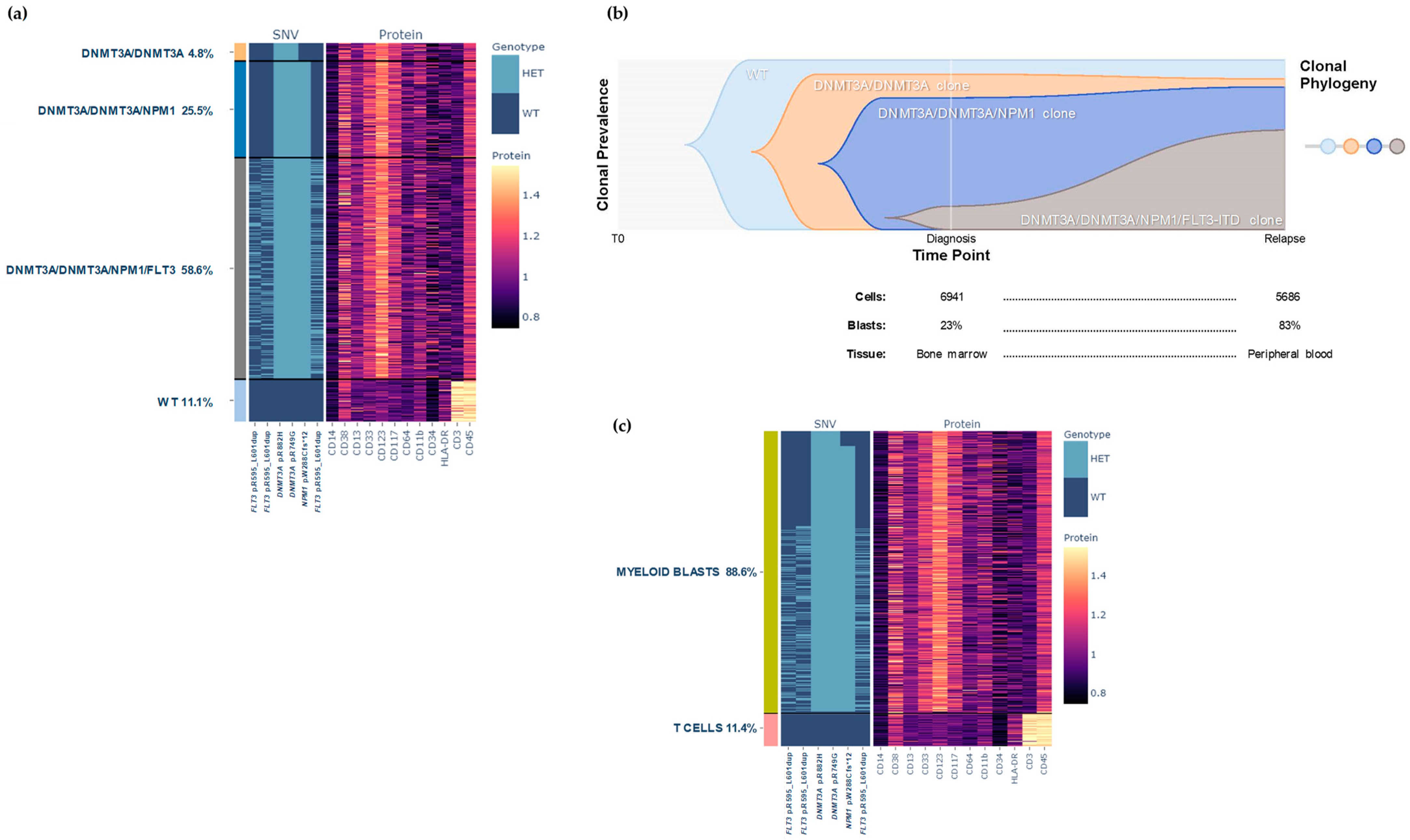
2.1.2. Results at Relapse
3. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [PubMed]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Bullinger, L.; Döhner, K.; Döhner, H. Genomics of Acute Myeloid Leukemia Diagnosis and Pathways. J. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Döhner, K.; Thiede, C.; Jahn, N.; Panina, E.; Gambietz, A.; Larson, R.A.; Prior, T.W.; Marcucci, G.; Jones, D.; Krauter, J.; et al. Impact of NPM1/FLT3-ITD genotypes defined by the 2017 European LeukemiaNet in patients with acute myeloid leukemia. Blood 2020, 135, 371–380. [Google Scholar] [CrossRef]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2019, 9, 1050–1063. [Google Scholar] [CrossRef]
- Romer-Seibert, J.S.; Meyer, S.E. Genetic heterogeneity and clonal evolution in acute myeloid leukemia. Curr. Opin. Hematol. 2021, 28, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Wang, F.; Jahn, K.; Hu, T.; Tanaka, T.; Sasaki, Y.; Kuipers, J.; Loghavi, S.; Wang, S.A.; Yan, Y.; et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat. Commun. 2020, 11, 5327. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Navin, N.E. Advances and applications of single-cell sequencing technologies. Mol. Cell 2015, 58, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.; Sciambi, A.; Treusch, S.; Durruthy-Durruthy, R.; Gokhale, K.; Jacob, J.; Chen, T.X.; Geis, J.A.; Oldham, W.; Matthews, J.; et al. High-throughput single-cell DNA sequencing of acute myeloid leukemia tumors with droplet microfluidics. Genome Res. 2018, 28, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Demaree, B.; Delley, C.L.; Vasudevan, H.N.; Peretz, C.A.C.; Ruff, D.; Smith, C.C.; Abate, A.R. Joint profiling of DNA and proteins in single cells to dissect genotype-phenotype associations in leukemia. Nat. Commun. 2021, 12, 1583. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.G.; Gralnick, H.R.; Sultan, C. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann. Intern. Med. 1985, 103, 620–625. [Google Scholar] [CrossRef]
- Kussick, S.J.; Stirewalt, D.L.; Yi, H.S.; Sheets, K.M.; Pogosova-Agadjanyan, E.; Braswell, S.; Norwood, T.H.; Radich, J.P.; Wood, B.L. A distinctive nuclear morphology in acute myeloid leukemia is strongly associated with loss of HLA-DR expression and FLT3 internal tandem duplication. Leukemia 2004, 18, 1591–1598. [Google Scholar] [CrossRef]
- Park, B.G.; Chi, H.-S.; Jang, S.; Park, C.-J.; Kim, D.-Y.; Lee, J.-H.; Lee, J.-H.; Lee, K.-H. Association of cup-like nuclei in blasts with FLT3 and NPM1 mutations in acute myeloid leukemia. Ann. Hematol. 2013, 92, 451–457. [Google Scholar] [CrossRef]
- van Dongen, J.J.M.; Lhermitte, L.; Böttcher, S.; Almeida, J.; van der Velden, V.H.J.; Flores-Montero, J.; Rawstron, A.; Asnafi, V.; Lécrevisse, Q.; Lucio, P.; et al. EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia 2012, 26, 1908–1975. [Google Scholar] [CrossRef]
- Kalina, T.; Flores-Montero, J.; van der Velden, V.H.J.; Martin-Ayuso, M.; Böttcher, S.; Ritgen, M.; Almeida, J.; Lhermitte, L.; Asnafi, V.; Mendonça, A.; et al. EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia 2012, 26, 1986–2010. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Aguilera-Diaz, A.; Vazquez, I.; Ariceta, B.; Mañú, A.; Blasco-Iturri, Z.; Palomino-Echeverría, S.; Larrayoz, M.J.; García-Sanz, R.; Prieto-Conde, M.I.; Chillón, M.d.C.; et al. Assessment of the clinical utility of four NGS panels in myeloid malignancies. Suggestions for NGS panel choice or design. PLoS ONE 2020, 15, e0227986. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Smith, M.A.; Nielsen, C.B.; Chan, F.C.; McPherson, A.; Roth, A.; Farahani, H.; Machev, D.; Steif, A.; Shah, S.P. E-scape: Interactive visualization of single-cell phylogenetics and cancer evolution. Nat. Methods 2017, 14, 549–550. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.J.; Shen, D.; Ding, L.; Shao, J.; Koboldt, D.C.; Chen, K.; Larson, D.E.; McLellan, M.D.; Dooling, D.; Abbott, R.; et al. Clonal architecture of secondary acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1090–1098. [Google Scholar] [CrossRef]
- Saeed, B.R.; Manta, L.; Raffel, S.; Pyl, P.T.; Buss, E.C.; Wang, W.; Eckstein, V.; Jauch, A.; Trumpp, A.; Huber, W.; et al. Analysis of nonleukemic cellular subcompartments reconstructs clonal evolution of acute myeloid leukemia and identifies therapy-resistant preleukemic clones. Int. J. Cancer 2021, 148, 2825–2838. [Google Scholar] [CrossRef]
- Xu, L.; Durruthy-Durruthy, R.; Eastburn, D.J.; Pellegrino, M.; Shah, O.; Meyer, E.; Zehnder, J. Clonal Evolution and Changes in Two AML Patients Detected with A Novel Single-Cell DNA Sequencing Platform. Sci. Rep. 2019, 9, 11119. [Google Scholar] [CrossRef]
- Corces-Zimmerman, M.R.; Hong, W.-J.; Weissman, I.L.; Medeiros, B.C.; Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad. Sci. USA 2014, 111, 2548–2553. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Date | Sample Origen | Blasts by Morphology (%) | Blasts by MFC (%) | FLT3 Ratio | NPM1 Copies | NGS Analysis (VAF, %) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| FLT3 p.R595_L601dup | NPM1 p.W288Cfs*12 | DNMT3A p.R882H | DNMT3 p.R749G | |||||||
| Diagnosis | 17 August 2020 | BM | 23.00 | 12.50 | 0.07 | 157,196.2 | 4.70 | 48.00 | 44.50 | 47.20 |
| Induction therapy (idarubicin + cytarabine + midostaurin) | 19 August 2020 | |||||||||
| Day +21 | 9 September 2020 | BM | 0 | 0.01 | NA | 55,275.9 | NA | NA | NA | NA |
| Day +29 | 17 September 2020 | BM | 0 | 0.21 | NA | 27,653.7 | NA | NA | NA | NA |
| Consolidation (intermediate-dose cytarabine + midostaurin) | 26 September 2020 | |||||||||
| Cycle 1 | 29 October 2020 | BM | 0 | 0 | NA | 1528.1 | NA | NA | NA | NA |
| Cycle 2 | 26 November2020 | BM | 0 | 0.13 | Unmutated | 4586.1 | NA | NA | NA | NA |
| Relapse | 21 Decemebr 2020 | BM | 6.00 | 2.70 | 0.03 | 108,460.8 | NA | NA | NA | NA |
| Rescue therapy (demivistat + HiDAC + mitoxantrone) | 23 Decemebr 2020 | |||||||||
| Day +21, progression | 13 January 2021 | BM | 35.00 | 21.10 | 0.10 | 328,495.3 | 12.40 | 13.20 | 25.10 | 25.90 |
| Allo-HSCT using sequential conditioning | ||||||||||
| Pre-HSCT | 21 January 2021 | BM | Aplasia | Aplasia | NA | 63,591.9 | NA | NA | NA | NA |
| Day +24 post-HSCT | 23 February 2021 | BM | 0 | 0 | Unmutated | 19.3 | NA | NA | NA | NA |
| Day +56 post-HSCT | 29 March 2021 | BM | 0 | 0 | Unmutated | 9555.6 | NA | NA | NA | NA |
| Day +88 post-HSCT, relapse | 27 April 2021 | PB | 83.00 | - | 0.84 | 335,980.0 | 46.80 | 41.70 | 45.10 | 46.30 |
| Rescue therapy (gilteritinib) | 12 May 2021 | |||||||||
| Day +30 | 11 June 2021 | PB | 3.00 | - | - | - | - | - | - | - |
| Day +61, progression | 13 July 2021 | PB | 43.60 | - | 0.89 | 275,722.5 | NA | NA | NA | NA |
| Day +74 | 26 July 2021 | PB | 10.00 | - | - | - | - | - | - | - |
| Day +77 | 29 July 2021 | PB | 13.90 | - | - | - | - | - | - | - |
| Day +80 | 1 August 2021 | PB | 16.70 | - | - | - | - | - | - | - |
| Death | 20 August 2021 | PB | NA | - | 0.87 | 480,703.3 | NA | NA | NA | NA |
| Mutations | Diagnosis | Relapse | ||
|---|---|---|---|---|
| Bulk NGS (%) | scDNAseq (%) | Bulk NGS (%) | scDNAseq (%) | |
| DNMT3A p.R882H | 44.5 | 48.3 | 45.1 | 46.8 |
| DNMT3A p.R749G | 47.2 | 46.9 | 46.3 | 46.3 |
| NPM1 p.W288Cfs*12 | 48.0 | 42.9 | 41.7 | 43.7 |
| FLT3 p.R595_L601dup | 4.6 | 7.4 | 46.8 | 50.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Álvarez, M.; Yeguas, A.; Jiménez, C.; Medina-Herrera, A.; González-Calle, V.; Hernández-Ruano, M.; Maldonado, R.; Aires, I.; Casquero, C.; Sánchez-Villares, I.; et al. Single-Cell DNA Sequencing and Immunophenotypic Profiling to Track Clonal Evolution in an Acute Myeloid Leukemia Patient. Biomedicines 2024, 12, 66. https://doi.org/10.3390/biomedicines12010066
García-Álvarez M, Yeguas A, Jiménez C, Medina-Herrera A, González-Calle V, Hernández-Ruano M, Maldonado R, Aires I, Casquero C, Sánchez-Villares I, et al. Single-Cell DNA Sequencing and Immunophenotypic Profiling to Track Clonal Evolution in an Acute Myeloid Leukemia Patient. Biomedicines. 2024; 12(1):66. https://doi.org/10.3390/biomedicines12010066
Chicago/Turabian StyleGarcía-Álvarez, María, Ana Yeguas, Cristina Jiménez, Alejandro Medina-Herrera, Verónica González-Calle, Montserrat Hernández-Ruano, Rebeca Maldonado, Irene Aires, Cristina Casquero, Inmaculada Sánchez-Villares, and et al. 2024. "Single-Cell DNA Sequencing and Immunophenotypic Profiling to Track Clonal Evolution in an Acute Myeloid Leukemia Patient" Biomedicines 12, no. 1: 66. https://doi.org/10.3390/biomedicines12010066