
Correlation of Myeloid-Derived Suppressor Cell Expansion with Upregulated Transposable Elements in Severe COVID-19 Unveiled in Single-Cell RNA Sequencing Reanalysis
 , and
, and
Abstract
:1. Introduction
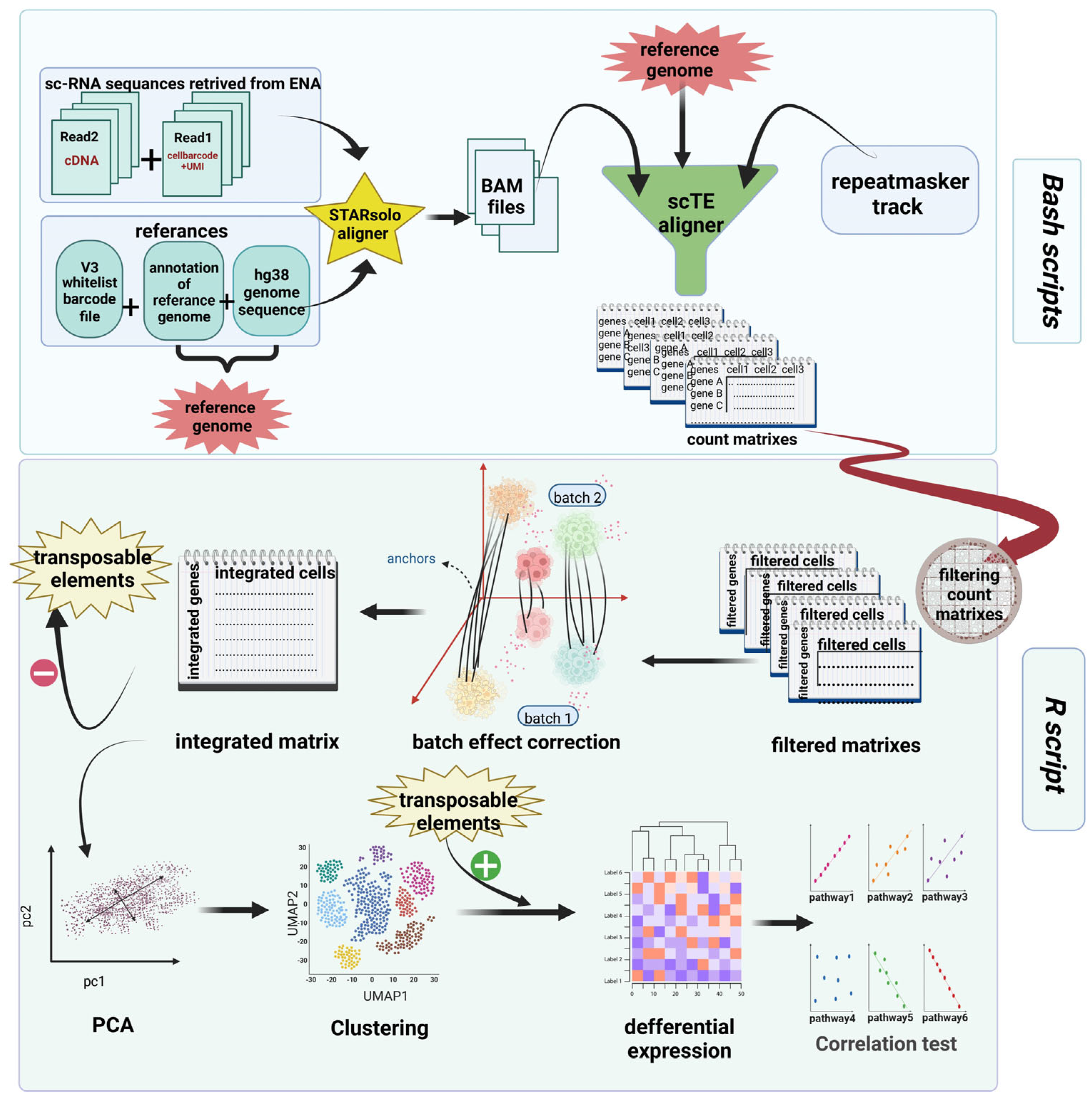
2. Materials and Methods
2.1. Aligning Reads
2.2. Quantifying TE Expression
2.3. Preprocessing, Analysis, and Exploration of scRNA-seq Data
2.4. Principal Component Analysis (PCA) and Clustering
2.5. Differential Expression (DE)
2.6. Scoring Pathways and Correlation Test
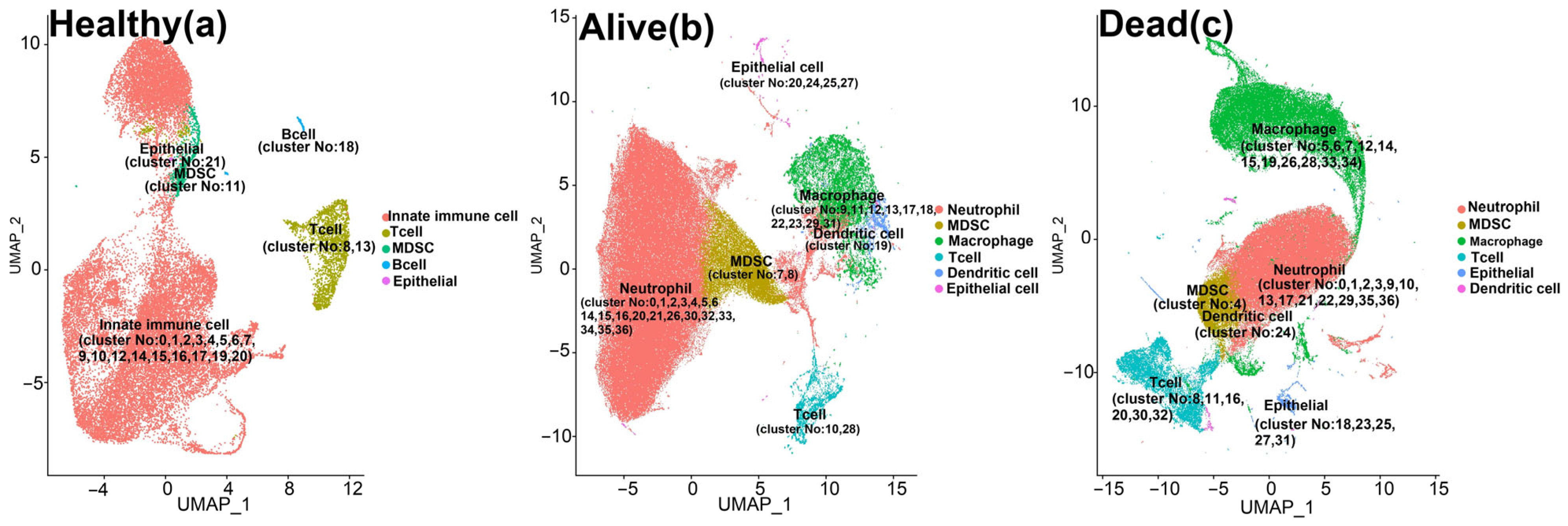
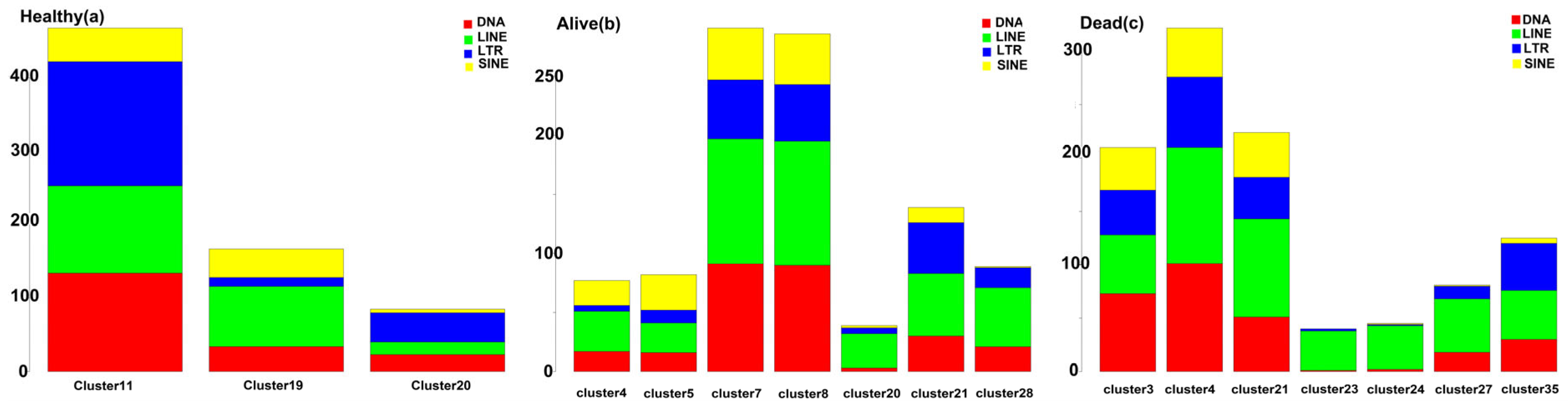
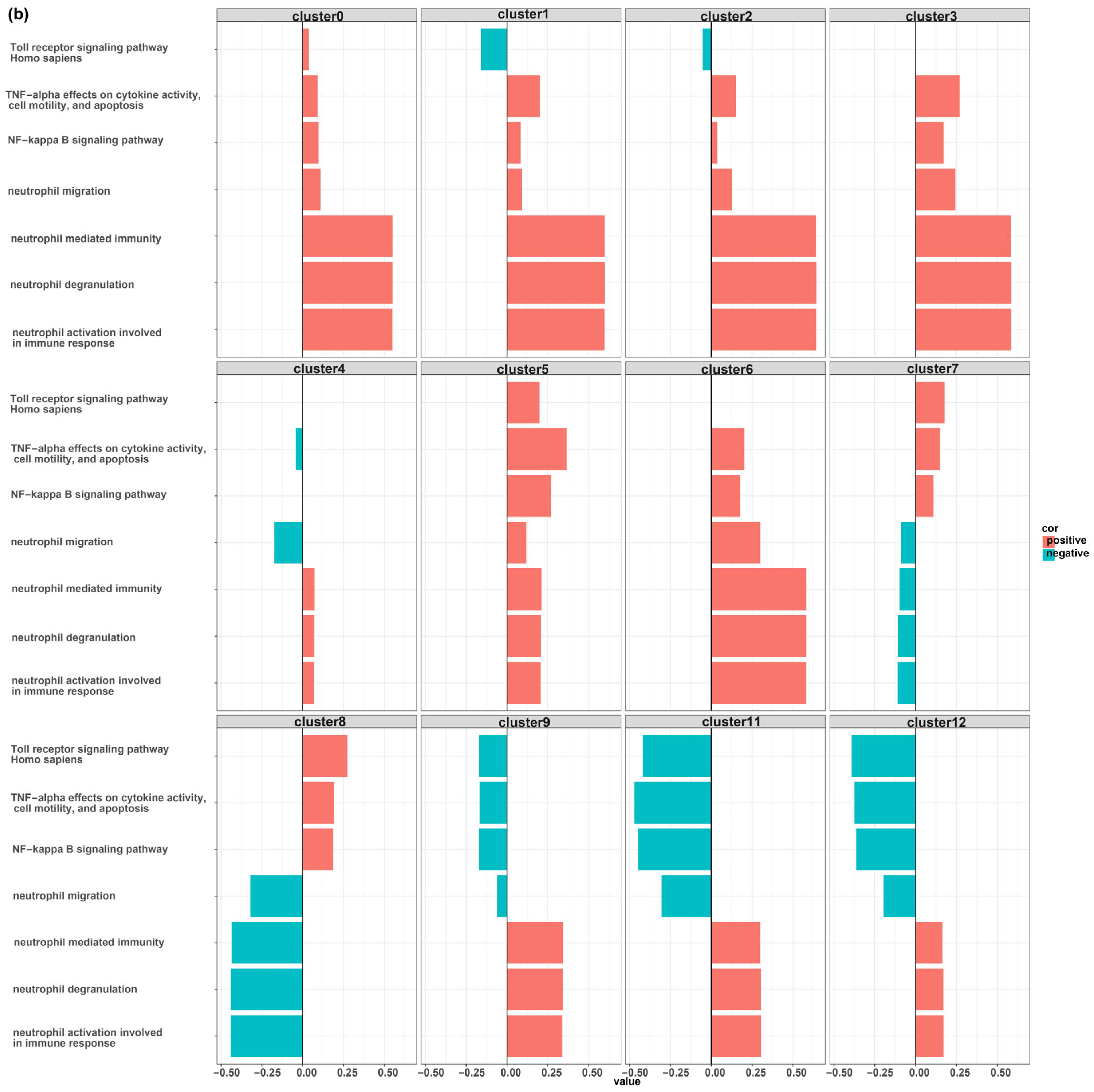
3. Results
MDSCs Reveal Distinct Immunological Functions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Parasher, A. COVID-19: Current understanding of its pathophysiology, clinical presentation and treatment. Postgrad. Med. J. 2021, 97, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, A.; Moezzi, S.M.I.; Mosaddeghi, P.; Nadimi Parashkouhi, S.; Fazel Hoseini, S.M.; Badakhshan, F.; Negahdaripour, M. Interferon-inducer antivirals: Potential candidates to combat COVID-19. Int. Immunopharmacol. 2021, 91, 107245. [Google Scholar] [CrossRef] [PubMed]
- Parashkouhi, S.N.; Mosaddeghi, P.; Bagheri, A.; Moezzi, S.M.I.; Hoseini, S.M.F.; Farahmandnejad, M.; Maleki, A.; Negahdaripour, M. The dual sides of interferon induction in COVID-19 treatment. Trends Pharm. Sci. 2021, 7, 9–14. [Google Scholar]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Jönsson, M.E.; Garza, R.; Johansson, P.A.; Jakobsson, J. Transposable Elements: A Common Feature of Neurodevelopmental and Neurodegenerative Disorders. Trends Genet. 2020, 36, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Andrenacci, D.; Cavaliere, V.; Lattanzi, G. The role of transposable elements activity in aging and their possible involvement in laminopathic diseases. Ageing Res. Rev. 2020, 57, 100995. [Google Scholar] [CrossRef] [PubMed]
- Larsen, P.A.; Lutz, M.W.; Hunnicutt, K.E.; Mihovilovic, M.; Saunders, A.M.; Yoder, A.D.; Roses, A.D. The Alu neurodegeneration hypothesis: A primate-specific mechanism for neuronal transcription noise, mitochondrial dysfunction, and manifestation of neurodegenerative disease. Alzheimer’s Dement. 2017, 13, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Burns, K.H. Our conflict with transposable elements and its implications for human disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 51–70. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, R.; Yu, J. New understanding of the relevant role of LINE-1 retrotransposition in human disease and immune modulation. Front. Cell Dev. Biol. 2020, 8, 657. [Google Scholar] [CrossRef]
- Burns, K.H. Transposable elements in cancer. Nat. Rev. Cancer 2017, 17, 415–424. [Google Scholar] [CrossRef]
- Mosaddeghi, P.; Farahmandnejad, M.; Zarshenas, M.M. The role of transposable elements in aging and cancer. Biogerontology 2023, 24, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Macchietto, M.G.; Langlois, R.A.; Shen, S.S. Virus-induced transposable element expression up-regulation in human and mouse host cells. Life Sci. Alliance 2020, 3, e201900536. [Google Scholar] [CrossRef]
- Hale, B.G. Antiviral immunity triggered by infection-induced host transposable elements. Curr. Opin. Virol. 2022, 52, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Pacis, A.; Aracena, K.A.; Gona, S.; Kwan, T.; Groza, C.; Lin, Y.L.; Sindeaux, R.; Yotova, V.; Pramatarova, A.; et al. Transposable elements are associated with the variable response to influenza infection. Cell Genom. 2023, 3, 100292. [Google Scholar] [CrossRef]
- Ferrarini, M.G.; Lal, A.; Rebollo, R.; Gruber, A.J.; Guarracino, A.; Gonzalez, I.M.; Floyd, T.; de Oliveira, D.S.; Shanklin, J.; Beausoleil, E. Genome-wide bioinformatic analyses predict key host and viral factors in SARS-CoV-2 pathogenesis. Commun. Biol. 2021, 4, 590. [Google Scholar] [CrossRef] [PubMed]
- Kitsou, K.; Kotanidou, A.; Paraskevis, D.; Karamitros, T.; Katzourakis, A.; Tedder, R.; Hurst, T.; Sapounas, S.; Kotsinas, A.; Gorgoulis, V. Upregulation of Human Endogenous Retroviruses in Bronchoalveolar Lavage Fluid of COVID-19 Patients. Microbiol. Spectr. 2021, 9, e0126021. [Google Scholar] [CrossRef] [PubMed]
- Mustafin, R.; Khusnutdinova, E. COVID-19, Retroelements, and Aging. Adv. Gerontol. 2021, 11, 83–92. [Google Scholar] [CrossRef]
- El-Shehawi, A.M.; Alotaibi, S.S.; Elseehy, M.M. Genomic study of COVID-19 corona virus excludes its origin from recombination or characterized biological sources and suggests a role for HERVS in its wide range symptoms. Cytol. Genet. 2020, 54, 588–604. [Google Scholar] [CrossRef]
- Zhang, L.; Richards, A.; Barrasa, M.I.; Hughes, S.H.; Young, R.A.; Jaenisch, R. Reverse-transcribed SARS-CoV-2 RNA can integrate into the genome of cultured human cells and can be expressed in patient-derived tissues. Proc. Natl. Acad. Sci. USA 2021, 118, e2105968118. [Google Scholar] [CrossRef]
- Sorek, M.; Meshorer, E.; Schlesinger, S. Impaired activation of transposable elements in SARS-CoV-2 infection. EMBO Rep. 2022, 23, e55101. [Google Scholar] [CrossRef]
- Bost, P.; De Sanctis, F.; Canè, S.; Ugel, S.; Donadello, K.; Castellucci, M.; Eyal, D.; Fiore, A.; Anselmi, C.; Barouni, R.M.; et al. Deciphering the state of immune silence in fatal COVID-19 patients. Nat. Commun. 2021, 12, 1428. [Google Scholar] [CrossRef]
- Stephenson, E.; Reynolds, G.; Botting, R.A.; Calero-Nieto, F.J.; Morgan, M.D.; Tuong, Z.K.; Bach, K.; Sungnak, W.; Worlock, K.B.; Yoshida, M.; et al. Single-cell multi-omics analysis of the immune response in COVID-19. Nat. Med. 2021, 27, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Qi, F.; Li, H.; Yang, Q.; Wang, H.; Wang, X.; Liu, X.; Zhao, J.; Liao, X.; Liu, Y.; et al. The differential immune responses to COVID-19 in peripheral and lung revealed by single-cell RNA sequencing. Cell Discov. 2020, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Mould, K.J.; Moore, C.M.; McManus, S.A.; McCubbrey, A.L.; McClendon, J.D.; Griesmer, C.L.; Henson, P.M.; Janssen, W.J. Airspace Macrophages and Monocytes Exist in Transcriptionally Distinct Subsets in Healthy Adults. Am. J. Respir. Crit. Care Med. 2021, 203, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Babarinde, I.A.; Sun, L.; Xu, S.; Chen, R.; Shi, J.; Wei, Y.; Li, Y.; Ma, G.; Zhuang, Q.; et al. Identifying transposable element expression dynamics and heterogeneity during development at the single-cell level with a processing pipeline scTE. Nat. Commun. 2021, 12, 1456. [Google Scholar] [CrossRef]
- Fernandes, J.D.; Zamudio-Hurtado, A.; Clawson, H.; Kent, W.J.; Haussler, D.; Salama, S.R.; Haeussler, M. The UCSC repeat browser allows discovery and visualization of evolutionary conflict across repeat families. Mob. DNA 2020, 11, 13. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., III; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., III; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Finak, G.; McDavid, A.; Yajima, M.; Deng, J.; Gersuk, V.; Shalek, A.K.; Slichter, C.K.; Miller, H.W.; McElrath, M.J.; Prlic, M.; et al. MAST: A flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015, 16, 278. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Martens, M.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.; Miller, R.A.; Digles, D.; Lopes, E.N.; Ehrhart, F.; et al. WikiPathways: Connecting communities. Nucleic Acids Res. 2021, 49, D613–D621. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. A Publ. Protein Soc. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Huang, R.; Grishagin, I.; Wang, Y.; Zhao, T.; Greene, J.; Obenauer, J.C.; Ngan, D.; Nguyen, D.-T.; Guha, R.; Jadhav, A.; et al. The NCATS BioPlanet—An Integrated Platform for Exploring the Universe of Cellular Signaling Pathways for Toxicology, Systems Biology, and Chemical Genomics. Front. Pharmacol. 2019, 10, 445. [Google Scholar] [CrossRef]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2021, 50, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Ben-Meir, K.; Twaik, N.; Baniyash, M. Plasticity and biological diversity of myeloid derived suppressor cells. Curr. Opin. Immunol. 2018, 51, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhong, J.; Deng, X.; Guo, X.; Lu, Y.; Lin, J.; Huang, X.; Wang, C. Targeting Myeloid-Derived Suppressor Cells to Enhance the Antitumor Efficacy of Immune Checkpoint Blockade Therapy. Front. Immunol. 2021, 12, 754196. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, C.; Liu, T.; Dai, X.; Bazhin, A.V. Myeloid-Derived Suppressor Cells in Tumors: From Mechanisms to Antigen Specificity and Microenvironmental Regulation. Front. Immunol. 2020, 11, 1371. [Google Scholar] [CrossRef] [PubMed]
- Grassi, G.; Notari, S.; Gili, S.; Bordoni, V.; Casetti, R.; Cimini, E.; Tartaglia, E.; Mariotti, D.; Agrati, C.; Sacchi, A. Myeloid-Derived Suppressor Cells in COVID-19: The Paradox of Good. Front. Immunol. 2022, 13, 842949. [Google Scholar] [CrossRef] [PubMed]
- Perfilyeva, Y.V.; Ostapchuk, Y.O.; Tleulieva, R.; Kali, A.; Abdolla, N.; Krasnoshtanov, V.K.; Perfilyeva, A.V.; Belyaev, N.N. Myeloid-derived suppressor cells in COVID-19: A review. Clin. Immunol. 2022, 238, 109024. [Google Scholar] [CrossRef]
- Vetsika, E.-K.; Koukos, A.; Kotsakis, A. Myeloid-derived suppressor cells: Major figures that shape the immunosuppressive and angiogenic network in cancer. Cells 2019, 8, 1647. [Google Scholar] [CrossRef]
- Trikha, P.; Carson, W.E., III. Signaling pathways involved in MDSC regulation. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2014, 1846, 55–65. [Google Scholar] [CrossRef]
- Wu, Y.; Pu, C.; Fu, Y.; Dong, G.; Huang, M.; Sheng, C. NAMPT-targeting PROTAC promotes antitumor immunity via suppressing myeloid-derived suppressor cell expansion. Acta Pharm. Sin. B 2022, 12, 2859–2868. [Google Scholar] [CrossRef] [PubMed]
- Travelli, C.; Consonni, F.M.; Sangaletti, S.; Storto, M.; Morlacchi, S.; Grolla, A.A.; Galli, U.; Tron, G.C.; Portararo, P.; Rimassa, L. Nicotinamide Phosphoribosyltransferase Acts as a Metabolic Gate for Mobilization of Myeloid-Derived Suppressor CellsNAMPT Orchestrates Immunometabolism of MDSCs. Cancer Res. 2019, 79, 1938–1951. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Yang, Y.; Li, X.; Yao, X.; Zhu, Y.; Zhang, H.; Wang, H.; Ma, Q.; Zhang, J.; Shi, H.; et al. Granulocytic myeloid-derived suppressor cells contribute to IFN-I signaling activation of B cells and disease progression through the lncRNA NEAT1-BAFF axis in systemic lupus erythematosus. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165554. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Han, Y.; Zhang, Y.; Lv, T.; Peng, X.; Huang, J. LncRNAs has been identified as regulators of Myeloid-derived suppressor cells in lung cancer. Front. Immunol. 2023, 14, 1067520. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Chakraborty, K.; Ray, P. Immunosuppressive MDSCs induced by TLR signaling during infection and role in resolution of inflammation. Front. Cell. Infect. Microbiol. 2013, 3, 52. [Google Scholar] [CrossRef]
- Lee, H.; Zhang, Z.; Krause, H.M. Long Noncoding RNAs and Repetitive Elements: Junk or Intimate Evolutionary Partners? Trends Genet. TIG 2019, 35, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Wang, C.; Vagts, C.; Raguveer, V.; Finn, P.W.; Perkins, D.L. Long non-coding RNAs (lncRNAs) NEAT1 and MALAT1 are differentially expressed in severe COVID-19 patients: An integrated single-cell analysis. PLoS ONE 2022, 17, e0261242. [Google Scholar] [CrossRef]
- Meydan, C.; Madrer, N.; Soreq, H. The Neat Dance of COVID-19: NEAT1, DANCR, and Co-Modulated Cholinergic RNAs Link to Inflammation. Front. Immunol. 2020, 11, 590870. [Google Scholar] [CrossRef]
- Moazzam-Jazi, M.; Lanjanian, H.; Maleknia, S.; Hedayati, M.; Daneshpour, M.S. Interplay between SARS-CoV-2 and human long non-coding RNAs. J. Cell Mol. Med. 2021, 25, 5823–5827. [Google Scholar] [CrossRef]
- He, W.-P.; Shu, C.-L.; Li, B.-A.; Jun, Z.; Cheng, Y. Human LINE1 endonuclease domain as a putative target of SARS-associated autoantibodies involved in the pathogenesis of severe acute respiratory syndrome. Chin. Med. J. 2008, 121, 608–614. [Google Scholar] [CrossRef]
- Ivancevic, A.; Chuong, E.B. Transposable elements teach T cells new tricks. Proc. Natl. Acad. Sci. USA 2020, 117, 9145–9147. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, D.R.; Swigut, T.; Wysocka, J. Systematic perturbation of retroviral LTRs reveals widespread long-range effects on human gene regulation. eLife 2018, 7, e35989. [Google Scholar] [CrossRef] [PubMed]
- Römer, C.; Singh, M.; Hurst, L.D.; Izsvák, Z. How to tame an endogenous retrovirus: HERVH and the evolution of human pluripotency. Curr. Opin. Virol. 2017, 25, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 2016, 351, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Romanish, M.T.; Lock, W.M.; de Lagemaat, L.N.v.; Dunn, C.A.; Mager, D.L. Repeated recruitment of LTR retrotransposons as promoters by the anti-apoptotic locus NAIP during mammalian evolution. PLoS Genet. 2007, 3, e10. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Vicente-García, C.; Seruggia, D.; Moltó, E.; Fernandez-Miñán, A.; Neto, A.; Lee, E.; Gómez-Skarmeta, J.L.; Montoliu, L.; Lunyak, V.V. MIR retrotransposon sequences provide insulators to the human genome. Proc. Natl. Acad. Sci. USA 2015, 112, E4428–E4437. [Google Scholar] [CrossRef]
- Chénais, B. Transposable Elements and Human Diseases: Mechanisms and Implication in the Response to Environmental Pollutants. Int. J. Mol. Sci. 2022, 23, 2551. [Google Scholar] [CrossRef]
- Bhat, A.; Ghatage, T.; Bhan, S.; Lahane, G.P.; Dhar, A.; Kumar, R.; Pandita, R.K.; Bhat, K.M.; Ramos, K.S.; Pandita, T.K. Role of Transposable Elements in Genome Stability: Implications for Health and Disease. Int. J. Mol. Sci. 2022, 23, 7802. [Google Scholar] [CrossRef]
- Zhou, H.; Jiang, M.; Yuan, H.; Ni, W.; Tai, G. Dual roles of myeloid-derived suppressor cells induced by Toll-like receptor signaling in cancer. Oncol. Lett. 2021, 21, 149. [Google Scholar] [CrossRef]
- Gazquez-Gutierrez, A.; Witteveldt, J.; Heras, S.R.; Macias, S. Sensing of transposable elements by the antiviral innate immune system. RNA 2021, 27, 735–752. [Google Scholar] [CrossRef]
- O’Donnell, T.; Vabret, N. Repeat elements amplify TLR signaling. Nat. Rev. Immunol. 2021, 21, 760. [Google Scholar] [CrossRef]
- Clapes, T.; Polyzou, A.; Prater, P.; Sagar; Morales-Hernández, A.; Ferrarini, M.G.; Kehrer, N.; Lefkopoulos, S.; Bergo, V.; Hummel, B.; et al. Chemotherapy-induced transposable elements activate MDA5 to enhance haematopoietic regeneration. Nat. Cell Biol. 2021, 23, 704–717. [Google Scholar] [CrossRef]
- Lanciano, S.; Cristofari, G. Measuring and interpreting transposable element expression. Nat. Rev. Genet. 2020, 21, 721–736. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO Accession | Clinical Outcome | Clinical Status | Tissue |
|---|---|---|---|
| GSM4593888 | - | Healthy | BAL |
| GSM4593891 | - | Healthy | BAL |
| GSM4593890 | - | Healthy | BAL |
| GSM4593892 | - | Healthy | BAL |
| GSM4762143 | Alive | Severe COVID | BAL |
| GSM4762155 | Alive | Severe COVID | BAL |
| GSM4762159 | Alive | Severe COVID | BAL |
| GSM4762144 | Alive | Severe COVID | BAL |
| GSM4762140 | Dead | Severe COVID | BAL |
| GSM4762152 | Dead | Severe COVID | BAL |
| GSM4762150 | Dead | Severe COVID | BAL |
| GSM4762147 | Dead | Severe COVID | BAL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farahmandnejad, M.; Mosaddeghi, P.; Dorvash, M.; Sakhteman, A.; Negahdaripour, M.; Faridi, P. Correlation of Myeloid-Derived Suppressor Cell Expansion with Upregulated Transposable Elements in Severe COVID-19 Unveiled in Single-Cell RNA Sequencing Reanalysis. Biomedicines 2024, 12, 315. https://doi.org/10.3390/biomedicines12020315
Farahmandnejad M, Mosaddeghi P, Dorvash M, Sakhteman A, Negahdaripour M, Faridi P. Correlation of Myeloid-Derived Suppressor Cell Expansion with Upregulated Transposable Elements in Severe COVID-19 Unveiled in Single-Cell RNA Sequencing Reanalysis. Biomedicines. 2024; 12(2):315. https://doi.org/10.3390/biomedicines12020315
Chicago/Turabian StyleFarahmandnejad, Mitra, Pouria Mosaddeghi, Mohammadreza Dorvash, Amirhossein Sakhteman, Manica Negahdaripour, and Pouya Faridi. 2024. "Correlation of Myeloid-Derived Suppressor Cell Expansion with Upregulated Transposable Elements in Severe COVID-19 Unveiled in Single-Cell RNA Sequencing Reanalysis" Biomedicines 12, no. 2: 315. https://doi.org/10.3390/biomedicines12020315